

Abstract
Polymorphisms in αIIbβ3 are important genetic factors that alter platelet biology and have been associated with susceptibility to thromboembolic disorders. To define the molecular mechanisms that lead to variance in thrombotic diathesis dictated by the β3 polymorphism, we examined regulation of intracellular signaling by αIIbβ3, and studied the effects of a common β subunit PlA2 polymorphism. We found that PP2A regulates αIIbβ3 control of the ERK signaling in a polymorphism specific fashion. In CHO cells, exogenous expression of αIIbβ3 reduced ATP-stimulated ERK phosphorylation and more so for PlA2 than PlA1. Interestingly, reduced level of ERK phosphorylation correlated with an increase in PP2A activity, with higher activity associated with PlA2 than PlA1. We tested the effect of PP2A on αIIbβ3-dependent adhesion, and found that PP2A overexpression increased cell adhesion, while phosphatase inhibitors decreased cell adhesion. We propose that PlA2 alters cell signaling at least in part by increasing β3-associated PP2A activity.
Keywords: PlA2 polymorphism, integrin αIIbβ3, PP2A
INTRODUCTION
Platelet integrin αIIbβ3 plays a pivotal role in platelet mediated haemostasis and thrombosis. Sequence polymorphisms affecting αIIbβ3 are important genetic factors associated with susceptibility for immune disorders such as neonatal alloimmune thrombocytopenia, post-transfusion purpura, and thromboembolic disorders in adults. A common polymorphism of the β-subunit of αIIbβ3 termed PlA2, resulting from a single amino acid substitution (PlA2 Pro33/Leu33 PlA1), is associated with coronary events, arterial thrombosis and sudden cardiac death [1–3],. While many clinical studies have confirmed the association between PlA2 and susceptibility for arterial thrombosis, not all studies are consistent [3, 4]. Discrepancies among reports for the association of single nucleotide polymorphisms and phenotypes or traits are not unusual and in the case of PlA2 have been linked to various modifiers that promote phenotypic changes including age, lipids, smoking, and drugs like aspirin and other platelet inhibitors [1, 5].
To seek further understanding the PlA2 impact on thrombotic diathesis, we established a model of exogenous expression of αIIbβ3 displaying either the PlA1 or PlA2 variant in Chinese Hamster Ovary (CHO) cells. In this study, we sought to contrast the effect of the PlA2 versus the PlA1 of β3 on the regulation of the intracellular signaling pathway to define the molecular mechanisms by which PlA1 or PlA2 might contribute to arterial thrombosis.
MATERIALS AND METHODS
Materials
Monoclonal mouse anti-CD61 (clone Y2/51) antibody was from Dako (Denmark). Antibodies to phospho-p44/42 MAPK (Thr202/Tyr204), ERK 1/2, phospho-MEK 1/2 (Ser217/221) were from Cell Signaling (Beverly, MA). Antibody to pT183 MAPK was from Promega. Fibrinogen, fibronectin, poly-L-lysine, and protease inhibitors were from Sigma-Aldrich. The Fugene 6 transfection reagent was from Roche Applied Science (Basel, Switzerland). Human PP2A cDNA was subcloned into pcDNA4/HisMax (B) under the control of CMV promoter. All cloning reagents and cell culture media were from Invitrogen.
Cell adhesion assays
Cells were grown to 70–80% confluency, harvested by trypsinization and resuspended in Tyrode's buffer. Cell adhesion assays were carried out between 24 h-48 h post transfection in the phosphatase overexpression studies. Six-well tissue culture plates were prepared for adhesion assays by coating with fibrinogen (10 µg/ml) for 1–2 h at 37°C, blocked with 1% BSA for 1 h, washed , and then 0.8 ml Tyrode’s buffer was added to each well. Plates were placed on a shaker to introduce continuous motion at 58 rotations per min (rpm), and cells were allowed to attach for 25–40 min. Unattached cells were removed and adherent cells quantified based on nucleic acid concentration. In some experiments, cells were incubated with CA (2 nM) or OA (1 µM) for 30 min prior to the adhesion assay.
MEK1 kinase assay
Cells were starved for 16 hours in serum-free media. Resuspended cells were seeded on fibrinogen-coated plates. Adherent cells were treated either with or without 40uM ATP, washed once with PBS, and lysed in RIPA buffer supplemented with protease inhibitors and MEK1 protein was immunoprecipitated. MEK1 kinase activity was assayed following manufactory instruction provided by MEK1 immunoprecipitation kinase assay kit (Cat# 17–159) from Upstate.
Phosphatase assay
Phosphatase activity was determined using a non-radioactive serine/threonine phosphatase assay kit (Upstate Biotechnology). Cells plated on fibrinogen were lysed in phosphatase reaction buffer. Immune complexes were prepared using antibody to PP2A or β3, washed with TBS, and serine/threonine assay buffer. The immune complexes were resuspended in 60 µl of Ser/Thr assay buffer containing 60 ng of phosphopeptide (KRpTIRR), and the reaction incubated for 10 min at 30°C. After brief centrifugation, 5 µl- 25 µl supernatant was incubated with 100 µl of malachite green phosphate detection solution, and the reaction quantified at OD650nm using a plate reader.
RESULTS
Our experimental system consisting of polyclonal stable transgenic CHO cells that express equal levels of αIIb and β3 subunits of platelet integrin αIIbβ3, displaying either the PlA1 or PlA2 polymorphism of β3, has been characterized previously in a study that examined polymorphism specific regulation of αIIbβ3-dependent adhesion to fibrinogen [6]. Stable transformants of CHO cells with vector only were used as controls (LK cells). Flow cytometric analysis with an antibody (clone Y2/51) that specifically recognizes β3 of both PlA1 and PlA2 equally revealed equivalent levels of β3 expression on the cell surface of PlA1 and PlA2 cells and absence for LK cells (Figure 1A). By Western blotting, antibodies to β3 or αIIb subunit recognized the expected protein moiety at a molecular weight of 90 kD and 120kD, respectively in both PlA1 and PlA2 expressing cells (Figure 1B).
Figure 1. Polymorphism specific difference in adhesion and inhibition of ERK phosphorylation.
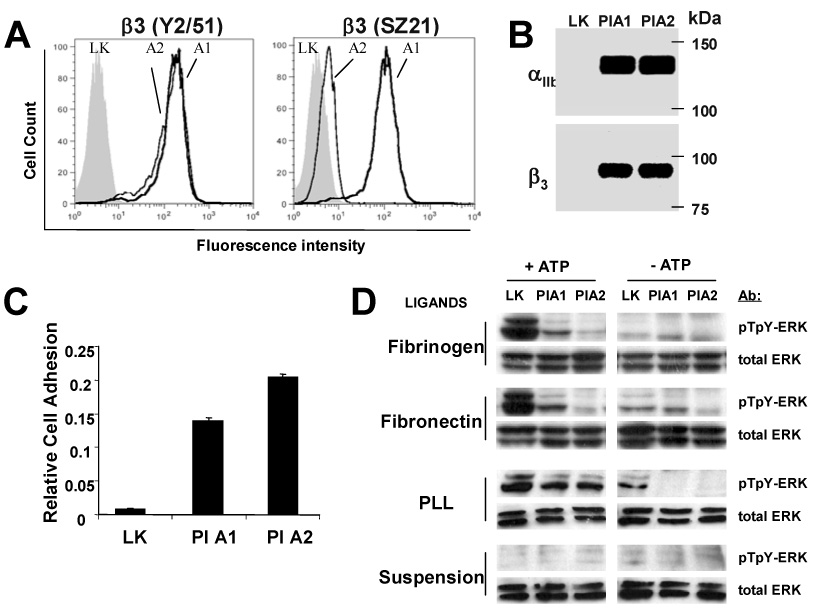
(A) CHO cells stably transfected with either αIIbβ3 integrin carrying β3-PlA1 (solid line), β-PlA2(dotted line), or vector only (LK, gray area), were analyzed by flow cytometry. Cells were incubated with an antibody to β3 (clone Y2/51) or an antibody to β3 (clone SZ21) that binds PlA1 with a higher affinity than PlA2. (B) WB showed expression of the αIIb and β3 in PlA1 and PlA2 cells, but abscent in LK cells. (C) Cells were subject to cell adhesion assay with fibrinogen-coated plates under continuous motion created by rotation of the plates at 58 rpm. Numbers of adherent cells were quantified by nucleic acid absorbance. The results are representative of ten independent experiments. Presented results are the mean ± SD of triplicates. The difference is significant (P <0.001) between LK vs PlA1, LK vs PlA2, PlA1 vs PlA2 (n = 10). (D) PlA2 inhibits ERK activation more strongly than PlA1. Starved CHO cells were suspended, then plated to fibrinogen, fibronectin, poly-L-lysine (PLL), or were maintained in suspension. Cells were then stimulated with 40 µM ATP for 5 minutes and analyzed by WB with antibody specific to the dual-phosphorylated active form of ERK (pTpY-ERK) or with antibody to ERK protein (total ERK). The results are representative of four independent experiments.
We developed an in vitro functional assay to assess the physiological impact of β3 polymorphisms on αIIbβ3-mediated cell adhesion. PlA1, PlA2 or LK cells were allowed to attach to plates coated with the αIIbβ3 ligand fibrinogen under continuous motion created by rotation of plates at 58 rpm and with a radius of 17.5 mm. In a typical experiment, few LK cells adhered to the fibrinogen-coated plates, compared to 30–80% of PlA1 and PlA2 cells (Figure 1C). Approximately 50% more PlA2 cells adhered over time than did PlA1 cells. These results confirm that the PlA polymorphism affects the adhesiveness of αIIbβ3 for fibrinogen and in the context of an assay where all other variables (cellular and extracellular) remain constant, suggest that PlA2 is a “gain of function” allele variant.
The Pro33 substitution of Leu33 (consequence of the PlA2 polymorphism) may alter the interface between an N-terminal PSI domain and EGF2 domain of this integrin and promote integrin activation [7]. However, previous studies showed that binding constant, for soluble fibrinogen, comparing PlA1 and PlA2 polymorphism, are not significantly different [6]. Hence, we hypothesized that polymorphism-specific changes in cell adhesion may result from differential regulation of signaling pathways downstream from αIIbβ3 engagement with ligands. We examined the effect of PlA1 versus PlA2 on the ATP-dependent activation of the ERK signaling pathway, which is known to be regulated by the ligand engagement of integrins [8]. Serum-starved cells were allowed to attach to either fibrinogen or fibronectin, and then assayed for ATP stimulated ERK activation. ATP-dependent ERK phosphorylation was intact in LK cells, but significantly reduced in PlA1 cells and nearly absent in PlA2 cells (Figure 1D). In cells attached to the non-specific charged substrate poly-L-lysine, ATP-dependent ERK phosphorylation does not vary significantly. ERK wasn’t activated in non-adherent cells. These results support the idea that binding of αIIbβ3 to a physiological ligand is required for αIIbβ3-dependent down-regulation of ERK phosphorylation. Importantly, PlA2 decreases the levels of phosphorylated ERK to a greater extent than does PlA1.
Inhibition of ERK phosphorylation could result from reduced upstream kinase activity or increased phosphatase activity, or a combination of the two. We found that PlA cells showed increased levels of MEK1 activity (Figure 2A) and active phosphorylated MEK1 (p-MEK1, Figure 2B) relative to LK; in contrast, PlA1 or PlA2 cells showed reduced levels of ERK phosphorylation relative to LK cells (Figure 1D and 2B). These results suggest that reduced ERK phosphorylation in αIIbβ3 expressing cells is not due to a suppression of upstream MEK1 activation by αIIbβ3.
Figure 2. αIIbβ3-dependent ERK dephosphorylation is not due to reduction in MEK1 kinase activity.
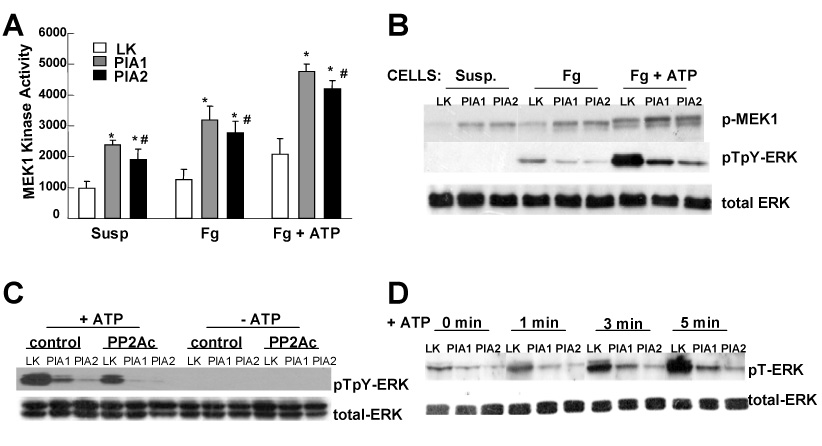
(A) Starved cells were plated on fibrinogen coated dishes (Fg, Fg + ATP) or left in suspension (Susp). After treatment with or without 40 µM ATP (Fg + ATP, Fg) for 5 min, cells were washed, MEK1 kinase activity quantified. Results are representative of three independent experiments. Presented data are the mean ± SD of triplicates. (*) P-value < 0.03 for PlA1 vs LK and PlA2 vs LK. (#) insignificant P-value for PlA1 vs PlA2. (B) WB analysis for the same cell extracts as described in (A) for the active forms of ERK (pTpY-ERK) and MEK1 (p-MEK1). (C) Cells were transfected with vectors containing a cDNA encoding for PP2A catalytic subunit (PP2Ac), or vector only (control). Twelve hours after transfection, cells were starved for 16 hours, and then plated on fibrinogen coated plates, and stimulated with 40 µM ATP (+ATP). WB was performed using antibody against active ERK (pTpY-ERK) or total ERK protein (total-ERK) as control. (D) Serum-starved cells were plated on fibrinogen, and stimulated with 40 µM ATP for the indicated time. Phosphorylation level on the Thr 183 residue of endogenous ERK was measured using an antibody specific to ERK phosphorylated at Thr 183 (pT-ERK). In both C and D, the results are representative of three independent experiments.
The discrepancy between upregulated MEK1 activity and reduced ERK phosphorylation level in PlA cells lead to our investigation whether PP2A is critical to ERK phosphorylation status. PP2A inactivates ERK through selective dephosphorylation of the threonine residue on a TEY sequence in the ERK catalytic domain [9]. The effect of PP2A overexpression on ERK phosphorylation was assayed. Following stimulation with ATP, ERK dephosphorylation was more pronounced in cells overexpressing PP2A relative to that in cells transfected with vector alone (Figure 2C). ERK inactivation/dephosphorylation during αIIbβ3-mediated platelet aggregation is through the dephosphorylation of the Thr-183 residue [10]. We found that phosphor-Thr183 in ERK (pT-ERK) increases with time of ATP stimulation in LK cells, whereas in PlA1 or PlA2 cells, ERK phosphor-Thr183 accumulates to a far less extent than in LK cells (Figure 2D). These results suggest that ERK phosphorylation-reduction induced by αIIbβ3/fibrinogen engagement might occur via the upregulation of PP2A activity.
Endogenous PP2A expression levels were not detectably different among LK, PlA1 and PlA2 cells when assayed with WB (not shown) and IP-WB (Figure 3A). However, when assayed for PP2A activity, PlA cells on fibrinogen present a higher level PP2A activity relative to LK; furthermore, PP2A activity was significantly higher in PlA2 cells relative to that in PlA1 cells, increased by 9.5% and 26% in PlA1 and PlA2 respectively relative to LK cells (Figure 3B). However, when αIIbβ3 was not engaged to its ligands (cells in suspension), PP2A activity was not significant different among LK, PlA1 and PlA2 cells (not shown) which indicate that upregulation of PP2A activity was specific response to αIIbβ3-engagement. Consistently, PP2A activity was also upregulated by αIIbβ3 engagement in platelets treated with αIIbβ3 ligands (fibrinogen, integrilin). The addition of 50ng/ml fibrinogen and integrilin, a cyclic heptapeptide with strong affinity to αIIbβ3, significantly increased PP2A activity in platelets (Figure 3C). The integrilin effect on PP2A activation suggests that an integrin conformational change is enough to activate PP2A without requirement of integrin clustering mediated by divalent ligands, which integrilin does not permit.
Figure 3. Endogenous PP2A activity is increased upon αIIbβ3 engagement, with higher increase in PlA2 than PlA1.
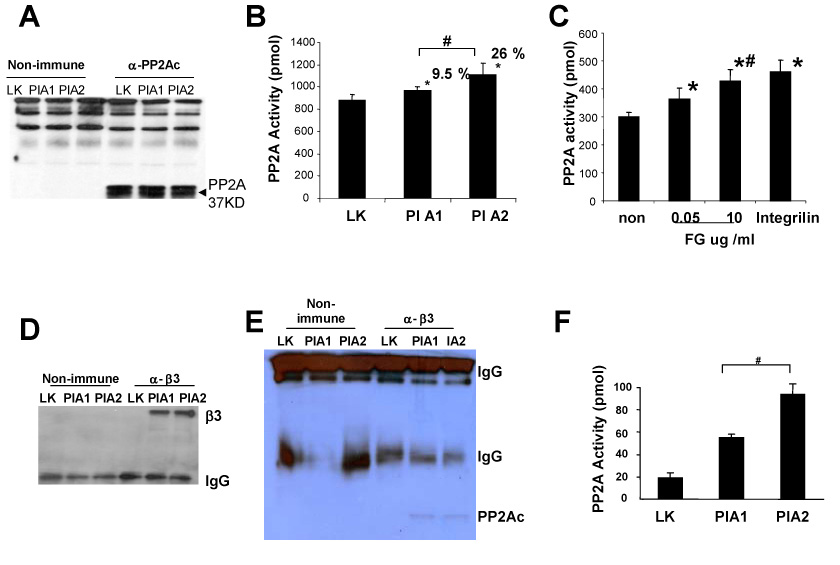
Cells were plated on fibrinogen coated plates. Adhered cells were then lysed and cell extract used for IP-PP2A phosphatase assay. (A) IP-WB with antibody against PP2Ac. (B) Endogenous PP2A activity was measured by assaying PP2A activity in immune complexes formed with antibody to PP2Ac. The graph indicates the percentage increase of PP2A activity in PlA1, PlA2 relative to that in LK cells. The results are presented as average of released phosphate value from five independent experiments, with SD as indicated. (*) P < 0.02 for PlA1 vs LK and PlA2 vs LK; (#) P < 0.02 for PlA1 vs PlA2. (C) Washed platelets were exposed to either fibrinogen (FG), integrilin (20ug/ml), or without adding ligand (non) on the presence of 1mM Ca2+ for 6 min. After aggregation, platelets were solubilized and IP-PP2A phosphatase activity assay was performed. The results are representative of three independent experiments. The presented data are means ± SD of triplicates in one experiment. (*) P < 0.05 for FG or integrilin treated vs platelets without ligand; (#) represents insignificant P value for FG 0.05 ug/ml vs FG 10 ug/ml. (D-E) CHO cell lysates were immunoprecipitated with antibody to β3 (α -β3) or its isotype control (non-immune), WB were probed with either β3 antibody (in D) or PP2Ac antibody (in E). (F) PP2A activity was measured in β3 immunoprecipitates. The results are presented as mean of released phosphate values from three independent experiments with SD as indicated. The difference between PlA1 and PlA2 is significant where (#) represents P < 0.04.
We then tested for association of PP2A with αIIbβ3-organized complexes by immunoprecipitating the β3-subunit and then quantified associated PP2A activity. Same amount of β3 was immunoprecipitated using β3 antibody from PlA1 and PlA2 cells (Figure 3D). Equal amount of PP2Ac were detected in β3- immune complex in PlA1 and PlA2 cell (Figure 3E). Strikingly, while total PP2A activity was only marginally stronger in PlA2 cells (26% increase relative to LK) as compared to PlA1 cells (9.5% increase relative to LK), PP2A activity was twice as high in PlA2-immune complexes compared to PlA1 (Figure 3F). These results are consistent with the concept that for the observed αIIbβ3-dependent inhibition of ERK phosphorylation, where stronger inhibition is induced by PlA2 compared to PlA1, such differences could be accounted for, at least in part, by differential levels of associated PP2A activity (PlA2 greater than PlA1).
To assess whether PP2A regulates αIIbβ3-mediated cell adhesion, cells were transiently transfected with PP2A or treated cells with PP2A inhibitor, then subjected to adhesion assay using fibrinogen as immobilized ligand. Overexpression of PP2A in PlA1 cells increased adhesion by approximately 2.6-fold, whereas overexpression of PP2A in PlA2 cells increased adhesion by approximately 1.9-fold over that of cells transfected with vector alone (Figure 4A). Due to its rather poor cell membrane permeability, Okadaic acid treatment of cells and tissues was reported to require at 1uM concentration to inhibit PP2A specifically, and keep PP1 activity intact [11, 12]. Okadaic acid reduced adhesion of PlA1 cells relative to control by 40% and PlA2 cells by 14%. Likewise, calyculin A reduced adhesion of PlA1 by 35% and PlA2 by 17% relative to controls. Our data supports the concept that PP2A interacts with αIIbβ3, and that, in turn, affects the impact of inhibitors on phosphatase activity in a polymorphism-dependent fashion. Taken together, our data are consistent with a unique interaction between PP2A and αIIbβ3 that results in enhancing αIIbβ3-mediated cell adhesion, and that the associated phenotypes of PlA2 polymorphism may be attributable to a relative increase in associated PP2A activity to the PlA2 variant of β3.
Figure 4. PP2A regulates αIIbβ3-dependent adhesion under motion in vitro.
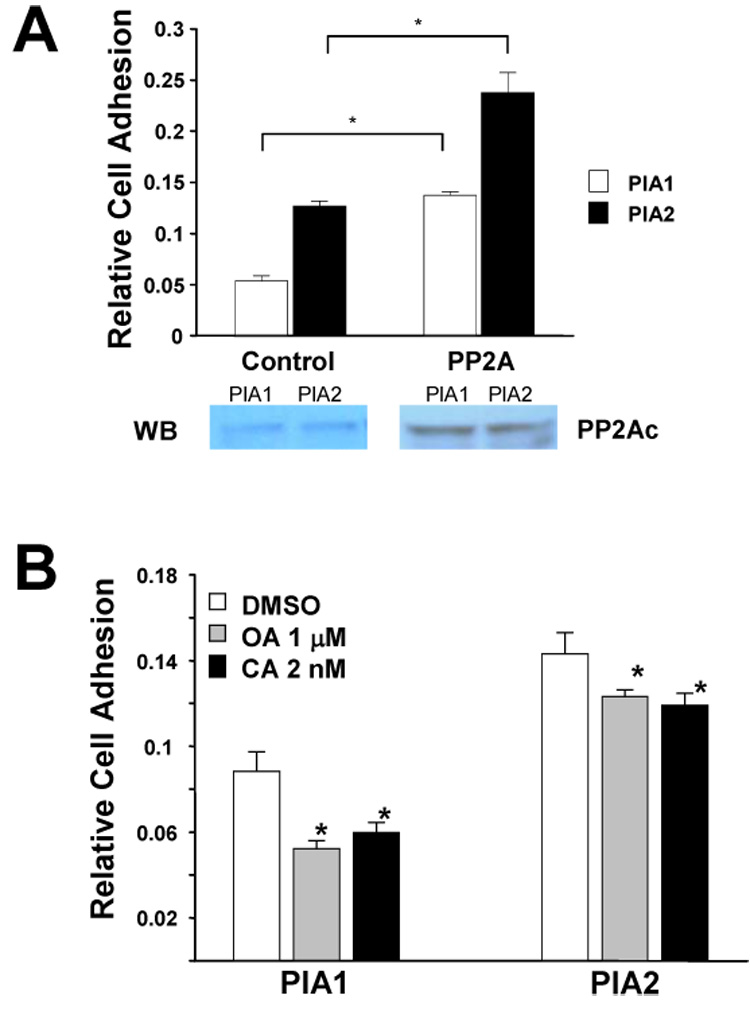
(A) PlA1 and PlA2 CHO cells were transiently transfected with PP2Ac (PP2A) or control vector (Control) and assayed for adhesion on fibrinogen-coated plates 36 hours post-transfection. The results are representative of three independent experiments. The presented data are the mean ± SD of triplicates in one experiment where (*) represents P < 0.01 (n = 3). Western blot showed the cell expression level of PP2A. (B) PlA1 and PlA2 CHO cells were treated with either okadaic acid (OA), or calyculin A (CA), or with DMSO control (DMSO) for 30 min, and then analyzed by adhesion assay on fibrinogen-coated plates. The results are representative of three independent experiments. The presented data are means ± SD of triplicates in one experiment. The difference between inhibitor treated vs DMSO-treated cells is significant where (*) represents P < 0.01, n = 3.
DISCUSSION
PlA2 polymorphism is a genetic risk factor associated with immunoreactivity to β3 and with susceptibility for thromboembolic cardiovascular disorders. PlA2 polymorphism is also associated with increased cancer risk including breast cancer, ovarian cancer and melanoma, which likely reflects a functional modification of the αvβ3 integrin in endothelial cells or both αIIbβ3 and αvβ3 integrin expressed on tumor cells [13–15]. The PlA2 polymorphism of αIIbβ3 on platelets leads to increased aggregability compared to PlA2 negative platelets [16]. In patients with atherosclerosis, the PlA2 allele may confer heightened risk of thrombosis and sudden thrombotic death [5, 17]. Thus, we sought to refine our understanding of the molecular mechanism by which PlA2 contributes to thromboembolic disorders so that improved therapeutic strategies could be developed. Furthermore, studies of the PlA2 effect are likely to lead to an accrued understanding of the biology of this essential integrin.
The use of a reconstituted cell system, instead of platelets, allowed us to minimize variance inherent to platelet assays, and thus allows to quantify the polymorphism-dependent intracellular signaling changes with high precision. Central to our findings, while αIIbβ3 binding to its ligands increased the activation of phosphatase PP2A, the PlA2 polymorphism of β3 did so to a greater extent than PlA1 did. PP2A, in turn, plays a critical role in αIIbβ3-mediated cell adhesion Integrin mediated cell adhesion is known to induce changes in PP2A activity [18] and localization within integrin-organized focal adhesion complex through its interaction with paxillin [19]. Instructively, the activity of αIIbβ3 has been shown to regulate phosphatase PP1 [20]. The association of both PP1 and PP2A with αIIb and β3, respectively, highlights the importance of phosphatases in the regulation of integrin signaling. Actually, collectively our data and data from other laboratories suggest that control of phosphatase activity could be a major mechanism for “outside-in signaling” led by αIIbβ3.
Though the engagement of integrins usually promote the activation of ERK/MAPK, our data indicate that αIIbβ3 engagement, especially the PlA2 allele of this integrin, reduced ERK phosphorylation, with specific dephosphorylation of threonine 183 of ERK. This is consistent with previous report that association of αIIbβ3 with fibrinogen inhibits ERK activation in platelet [21]. It was reported that the increase in PP2A activity induced by EGF results in inhibition of ERK2 activity in A431 cells [9]. In our study, enhanced PP2A activity induced by either integrin αIIbβ3 expression or PP2A overexpression resulted in ERK dephosphorylation. While the specific physiologic impact of the strongest dephosphorylation of ERK upon engagement of PlA2-β3 remains uncertain, our data provide the first link between PP2A/ERK signaling and the PlA2 polymorphism of integrin β3. The functional and close spatial relationship of PP2A to αIIbβ3 indicates the importance of PP2A in the regulation of integrin in a PlA polymorphism-responsive manner.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Weiss EJ, Bray PF, Tayback M, Schulman SP, Kickler TS, Becker LC, Weiss JL, Gerstenblith G, Goldschmidt-Clermont PJ. A polymorphism of a platelet glycoprotein receptor as an inherited risk factor for coronary thrombosis. N Engl J Med. 1996;334:1090–1094. doi: 10.1056/NEJM199604253341703. [DOI] [PubMed] [Google Scholar]
- 2.Cooke GE, Bray PF, Hamlington JD, Pham DM, Goldschmidt-Clermont PJ. PlA2 polymorphism and efficacy of aspirin. Lancet. 1998;351:1253. doi: 10.1016/S0140-6736(05)79320-X. [DOI] [PubMed] [Google Scholar]
- 3.Bray PF. Bray, Integrin polymorphisms as risk factors for thrombosis. Thromb Haemost. 1999;82:337–344. [PubMed] [Google Scholar]
- 4.Ridker PM, Hennekens CH, Schmitz C, Stampfer MJ, Lindpaintner K. PIA1/A2 polymorphism of platelet glycoprotein IIIa and risks of myocardial infarction, stroke, and venous thrombosis. Lancet. 1997;349:385–388. doi: 10.1016/S0140-6736(97)80010-4. [DOI] [PubMed] [Google Scholar]
- 5.Goldschmidt-Clermont PJ, Cooke GE, Eaton GM, Binkley PF. PlA2, a variant of GPIIIa implicated in coronary thromboembolic complications. J Am Coll Cardiol. 2000;36:90–93. doi: 10.1016/s0735-1097(00)00710-5. [DOI] [PubMed] [Google Scholar]
- 6.Vijayan KV, Goldschmidt-Clermont PJ, Roos C, Bray PF. The Pl(A2) polymorphism of integrin beta(3) enhances outside-in signaling and adhesive functions. J Clin Invest. 2000;105:793–802. doi: 10.1172/JCI6982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xiong JP, Stehle T, Goodman SL, Arnaout MA. A novel adaptation of the integrin PSI domain revealed from its crystal structure. J Biol Chem. 2004;279:40252–40254. doi: 10.1074/jbc.C400362200. [DOI] [PubMed] [Google Scholar]
- 8.Short SM, Boyer JL, Juliano RL. Integrins regulate the linkage between upstream and downstream events in G protein-coupled receptor signaling to mitogen-activated protein kinase. J Biol Chem. 2000;275:12970–12977. doi: 10.1074/jbc.275.17.12970. [DOI] [PubMed] [Google Scholar]
- 9.Chajry N, Martin PM, Cochet C, Berthois Y. Regulation of p42 mitogen-activated-protein kinase activity by protein phosphatase 2A under conditions of growth inhibition by epidermal growth factor in A431 cells. Eur J Biochem. 1996;235:97–102. doi: 10.1111/j.1432-1033.1996.00097.x. [DOI] [PubMed] [Google Scholar]
- 10.Pawlowski M, Ragab A, Rosa JP, Bryckaert M. Selective dephosphorylation of the threonine(183) residue of ERK2 upon (alpha)llb(beta)3 engagement in platelets. FEBS Lett. 2002;521:145–151. doi: 10.1016/s0014-5793(02)02862-4. [DOI] [PubMed] [Google Scholar]
- 11.Favre B, Turowski P, Hemmings BA. Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with calyculin-A, okadaic acid, and tautomycin. J Biol Chem. 1997;272:13856–13863. doi: 10.1074/jbc.272.21.13856. [DOI] [PubMed] [Google Scholar]
- 12.Resjo S, Oknianska A, Zolnierowicz S, Manganiello V, Degerman E. Phosphorylation and activation of phosphodiesterase type 3B (PDE3B) in adipocytes in response to serine/threonine phosphatase inhibitors: deactivation of PDE3B in vitro by protein phosphatase type 2A. Biochem J. 1999;341(Pt 3):839–845. [PMC free article] [PubMed] [Google Scholar]
- 13.Bojesen SE, Tybjaerg-Hansen BG. Nordestgaard, Integrin beta3 Leu33Pro homozygosity and risk of cancer. J Natl Cancer Inst. 2003;95:1150–1157. doi: 10.1093/jnci/djg005. [DOI] [PubMed] [Google Scholar]
- 14.Bojesen SE, Kjaer SK, Hogdall EV, Thomsen BL, Hogdall CK, Blaakaer J, Tybjaerg-Hansen A, Nordestgaard BG. Increased risk of ovarian cancer in integrin beta3 Leu33Pro homozygotes. Endocr Relat Cancer. 2005;12:945–952. doi: 10.1677/erc.1.01083. [DOI] [PubMed] [Google Scholar]
- 15.Wang-Gohrke S, Chang-Claude J. Integrin beta3 Leu33Pro polymorphism and breast cancer risk: a population-based case-control study in Germany. Breast Cancer Res Treat. 2004;88:231–237. doi: 10.1007/s10549-004-0782-5. [DOI] [PubMed] [Google Scholar]
- 16.Feng D, Lindpaintner K, Larson MG, Rao VS, O'Donnell CJ, Lipinska I, Schmitz C, Sutherland PA, Silbershatz H, D'Agostino RB, Muller JE, Myers RH, Levy D, Tofler GH. Increased platelet aggregability associated with platelet GPIIIa PlA2 polymorphism: the Framingham Offspring Study. Arterioscler Thromb Vasc Biol. 1999;19:1142–1147. doi: 10.1161/01.atv.19.4.1142. [DOI] [PubMed] [Google Scholar]
- 17.Mikkelsson J, Perola M, Laippala P, Penttila A, Karhunen PJ. Glycoprotein IIIa Pl(A1/A2) polymorphism and sudden cardiac death. J Am Coll Cardiol. 2000;36:1317–1323. doi: 10.1016/s0735-1097(00)00871-8. [DOI] [PubMed] [Google Scholar]
- 18.Ivaska J, Nissinen L, Immonen N, Eriksson JE, Kahari VM, Heino J. Integrin alpha 2 beta 1 promotes activation of protein phosphatase 2A and dephosphorylation of Akt and glycogen synthase kinase 3 beta. Mol Cell Biol. 2002;22:1352–1359. doi: 10.1128/mcb.22.5.1352-1359.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito A, Kataoka TR, Watanabe M, Nishiyama K, Mazaki Y, Sabe H, Kitamura Y, Nojima H. A truncated isoform of the PP2A B56 subunit promotes cell motility through paxillin phosphorylation. Embo J. 2000;19:562–571. doi: 10.1093/emboj/19.4.562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vijayan KV, Liu Y, Li TT, Bray PF. Protein phosphatase 1 associates with the integrin alphaIIb subunit and regulates signaling. J Biol Chem. 2004;279:33039–33042. doi: 10.1074/jbc.C400239200. [DOI] [PubMed] [Google Scholar]
- 21.Nadal F, Levy-Toledano S, Grelac F, Caen JP, Rosa JP, Bryckaert M. Negative regulation of mitogen-activated protein kinase activation by integrin alphaIIbbeta3 in platelets. J Biol Chem. 1997;272:22381–22384. doi: 10.1074/jbc.272.36.22381. [DOI] [PubMed] [Google Scholar]