

Abstract
The leukocyte integrins have critical roles in host defense and inflammatory tissue injury. We found that integrin αDβ2, a novel but largely uncharacterized member of this family, is restricted to subsets of macrophages and a small population of circulating leukocytes in wild-type mice in the absence of inflammatory challenge and is expressed in regulated fashion during cytokine-induced macrophage differentiation in vitro. αDβ2 is highly displayed on splenic red pulp macrophages and mediates their adhesion to local targets, identifying key functional activity. In response to challenge with Plasmodium berghei, a malarial pathogen that models systemic infection and inflammatory injury, new populations of αD+ macrophages evolved in the spleen and liver. Unexpectedly, targeted deletion of αD conferred a survival advantage in P. berghei infection over a 30-day observation period. Mechanistic studies demonstrated that the increased survival of αD−/− animals at these time points is not attributed to differences in magnitude of anemia or parasitemia or to alterations in splenic microanatomy, each of which is a key variable in the natural history of P. berghei infection, and indicated that an altered pattern of inflammatory cytokines may contribute to the difference in mortality. In contrast to the outcome in malarial challenge, death of αD−/− animals was accelerated in a model of Salmonella sepsis, demonstrating differential rather than stereotyped roles for αDβ2 in systemic infection. These studies identify previously unrecognized and unique activities of αDβ2, and macrophages that express it, in host defense and injury.
Integrins are plasma membrane heterodimers that are broadly distributed on metazoan cells and mediate critical functions, including adhesion, homing, signaling, and gene expression. The leukocyte integrin subfamily (also called β2 or CD11/CD18 integrins) is expressed on myeloid and lymphoid leukocytes and their precursors and consists of four members (1): αLβ2 (CD11aCD18; LFA-1), αMβ2 (CD11b/CD18; MAC-1; CR3), αxβ2 (CD11c/CD18), and αDβ2 (CD11d/CD18). Leukocyte integrins are required for host defense against invasion by many pathogens and for wound surveillance and repair, demonstrated by heritable deficiency syndromes that cause recurrent and often life-threatening infections in humans and domesticated animals (2). Experimental models in which leukocyte integrins are genetically deleted or are blocked demonstrate critical and complex activities relevant to this function (1, 3–6). In contrast, unregulated accumulation and signaling of leukocytes mediated by β2 integrins also contribute to inflammatory tissue injury (1, 2, 6, 7).
Integrin αDβ2 is the most recently discovered β2 integrin (1, 8). Molecular characterization of the αD subunit suggested that αDβ2 has novel activation pathways and modes of regulation (8). αDβ2 recognizes ICAM-3, VCAM-1, and several other ligands in assays with primary and surrogate cells (8–11), although adhesive interactions in vivo have not been identified. Sequence similarity of the I domains of αD and αM (8) predicted this diversity in ligand recognition: the I domain is critical in binding interactions of integrins, and αMβ2 also has multiple ligand partners in experiments using transfected cell lines (1, 10, 11). Limited observations suggest that αDβ2 is preferentially expressed by macrophages in normal human tissues and in clinical syndromes of dysregulated inflammation, including atherosclerosis, rheumatoid arthritis, and acute lung inflammation (Refs. 8, 12, and 13 and our unpublished data), and that it is involved in immune regulation and inflammatory tissue injury in rodent models (14–16). Nevertheless, the distribution, regulation, and activities of αDβ2 in physiologic and pathologic inflammation are largely undefined, and phenotypes in mice made genetically deficient in αD have not been reported (1, 3, 16). In this study, we examined the expression and functions of αDβ2 in naive conditions and in response to inflammatory stimulation in models of pathologic infectious syndromes. We found that it is selectively expressed by subsets of splenic red pulp, thymic, and marrow macrophages in wild-type (WT)8 animals in the basal state. This pattern of expression suggested roles in systemic infectious challenge. We found that new populations of αD+ macrophages emerge in response to infection with Plasmodium berghei, a blood-borne malarial protozoan recognized by splenic red pulp macrophages, demonstrating that αDβ2 is dynamically modulated in systemic inflammation. Targeted deletion of αD did not cause a spontaneous phenotype but, unexpectedly, conferred a survival advantage in lethal P. berghei infection. In contrast, deletion of αDβ2 reduced survival in animals infected with Salmonella typhimurium, an intracellular bacterial pathogen that also causes a lethal systemic infection in mice and is recognized by splenic marginal zone macrophages and other macrophage subsets. Our findings demonstrate unique functional roles for αDβ2 and the macrophage populations that express it in innate responses to microbial invasion and systemic infectious syndromes.
Materials and Methods
Targeting vector construction, functional disruption of αD by homologous recombination, and generation of αD−/− mice
Genomic clones were isolated from a murine 129Sv genomic library using probes specific for the αD I domain, which is critical for ligand recognition (1, 10, 11), and targeted deletion of αD was accomplished using previously described strategies (17–19) (Fig. 1).
FIGURE 1.

Functional disruption of αD by homologous recombination. A, The murine αD genomic locus containing exons 1–10 is represented (WT). Genomic clones were isolated from a 129Sv mouse genomic λ DNA library using oligonucleotide probes specific to the αD I domain. The largest (clone 3.1) was 14 kb in length and contained exons 1–6 as determined by Southern blot analysis and nucleic acid sequencing. A second 10-kb genomic clone (4.1) that overlapped with the 3′ end of 3.1 and contained exons 6–10 was also isolated. A 7.5-kb genomic fragment containing exons 1–5 and a 5′ region of exon 6 were cloned from 3.1 and inserted upstream of three in-frame stop codons in a polylinker modified pBlueScript vector. A 3-kb Xmn1-BglII fragment containing the 3′ end of exons 6 and 7 was cloned downstream of this fragment to generate a novel αD allele leading to termination of translation in the I domain following residue 153(Ile). The novel αD allele was cloned between two herpes simplex TK genes and a floxed neomycin selection cassette was inserted 3′ to the stop codons in exon 6 to generate the targeting vector. The targeting vector was linearized with XhoI and electroporated into TC1 embryonic stem cells. Recombinant embryonic stem cells were selected in the presence of G418 and FIAU as previously described (18) and were identified by Southern blot analysis using BglII and a 5′ flanking BamHi/BgIII probe located outside the targeted region. Positive cell lines were confirmed using XmnI digestion and hybridization to a probe corresponding to the neomycin gene. A single recombinant cell line (1e-1) was used for blastocyst injection to create recombinant mice. The floxed neomycin selection cassette was removed by mating to a transgenic mouse line that expresses Cre recombinase (19). Mice containing αDloxP and WT alleles were genotyped by PCR using isolated tail DNA. The forward (5′-GGACCCCAGGACACAGTTGAG-3′) and reverse (5′-CACAGGCCACAGTGTACAGTATT-3′) primers amplify a 268-bp band including the inserted loxP site from the recombinant allele and 215 bp from the WT allele. The recombinant mouse line was subsequently backcrossed and maintained on a C57BL/6 background. B, Southern blot analysis of genomic DNA isolated from WT embryonic stem cells and the αD-recombinant embryonic stem cell line (1e-1). Genomic DNA digested with BglII was probed with a 600-bp BamH1-BglII fragment from the αD genomic clone. This 5′ probe hybridized to both the 11-kb fragment derived from the WT allele and the 8-kb fragment corresponding to the recombinant allele. The genomic structure of the 3′ end of the αDneo allele was confirmed in the 1e-1 clone by hybridization of the targeting vector-specific neomycin probe to the 6.5-kb XmnI genomic fragment.
Immunohistochemistry
The following were used as primary mAbs in immunocytochemical reactions: αD, 205c, and 279F (hamster IgG; ICOS) (14); F4/80, A3-1 (rat IgG2; Serotec); MARCO, ED31 (rat IgG1; Serotec); and MOMA-1 (rat IgG2; Serotec) (20). Tissues from various organs, usually from 8- to 12-wk-old animals in studies of distribution of αDβ2 in WT mice, were frozen in Tissue-Tek (Sakura Finetek) and 8-μm cryostat sections were deposited on Superfrost Plus slides (Fisher Scientific). The sections were then fixed in cold acetone for 10 min. Immunoperoxidase staining was based on a previously described method (21) (Fig. 2). For immunofluorescent staining, sections were blocked with 5% normal goat serum, 1% BSA, and avidin solution (Vector Laboratories) for 30 min and subsequently incubated with primary Abs overnight at 4°C. Biotin-conjugated goat anti-hamster Ab was applied at a 1/1000 dilution and incubated for 30 min followed by incubation with Alexa 488-conjugated streptavidin and Alexa 568-conjugated anti-rat IgG (Molecular Probes) for 45 min. The sections were then layered with anti-fade medium (Biomeda) and sealed with a coverslip. Fluorescent staining was analyzed using a FluoView 300 microscope (Olympus) in the University of Utah Health Science Center’s Core Cell Imaging Facility.
FIGURE 2.
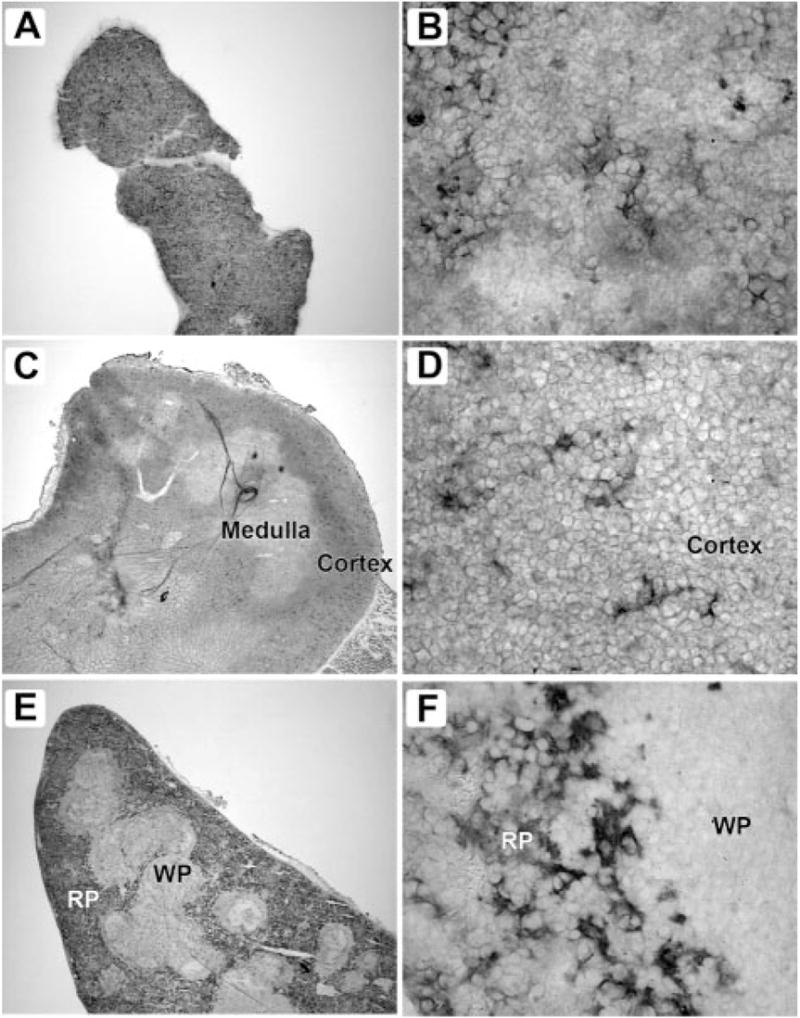
αDβ2 is expressed by distinct populations of macrophages in bone marrow, thymus, and spleen. Frozen sections of freshly isolated tissues were stained with mAb 205c using an immunoperoxidase detection method. Tissues from 8- to 12-wk-old C57BL/6 mice were frozen in Tissue-Tek (Sakura Finetek) and 8-μm cryostat sections were deposited on Superfrost Plus slides (Fisher Scientific). The sections were then fixed in cold acetone for 10 min. Endogenous tissue peroxidase was eliminated by incubation with 0.01% hydrogen peroxide for 15 min. The sections were blocked by incubating with 5% normal goat serum, 1% BSA and avidin solution (Vector Laboratories) for 30 min, and primary Abs (hamster mAbs 205c or 279f raised against mouse αD) were applied to the slides and incubated overnight at 4°C. A secondary Ab (biotin-conjugated goat anti-hamster; Jackson ImmunoResearch Laboratories) was applied at a 1/1000 dilution and incubated for 30 min. Negative controls were performed by omitting the primary Ab or by applying an irrelevant isotype-matched primary Ab as a control. ABC reagent was prepared according to the manufacturer’s instructions (Vector Laboratories) and applied to the sections for 30 min. Staining was developed using a NovaRed kit (Vector Laboratories), followed by counterstaining with Gill’s hematoxylin, dehydration, and mounting using Cytoseal 60 medium (VWR). In each pair of figures, the panel on the left is magnified ×40 and on the right ×800. These distributions of αD are representative of multiple studies. A and B, Bone marrow; C and D, thymus; E and F, spleen; WP, white pulp; RP, red pulp.
Isolation of mouse splenocytes and F4/80-positive splenic macrophages
Single-cell suspensions from freshly isolated spleen were obtained as described previously (22), with minor modifications. F4/80-positive splenic macrophages were further isolated using an AutoMACS cell sorter and positive selection using PE-conjugated anti-F4/80 Ab (Serotec) and anti-PE magnetic beads according to the manufacturer’s instructions (Miltenyi Biotec).
Flow cytometric analysis
Isolated F4/80-positive splenocytes or cultured M1 cells were incubated on ice for 30 min in the presence of Alexa 488-conjugated mAb 205c or a control Ab. Cells were washed with FACS medium (HBSS without Ca2+ and Mg2+/0.1% NaN3/0.5% human serum albumin) and fixed in 1% paraformaldehyde. They were then analyzed by flow cytometry as previously described (23), with minor modifications.
Culture of murine M1 cells
The murine M1 myeloid leukemic cell line was obtained from American Type Culture Collection. Macrophage differentiation was induced as described elsewhere (24). PCR assays for αD mRNA were done essentially as described previously (17). The primers used (5′-CAATCCCTGCTACCATGCCAGA-3′ and 5′-CTCCTGTAGGGCAGTGGGTTC-3′) amplified a 490-bp product. Immunocytochemical analysis was accomplished as previously outlined (25).
Modified Stamper-Woodruff adhesion assay
An assay for in vitro binding of splenic macrophages to freshly isolated spleen sections was developed based on previously described methods (26, 27). All steps were conducted in a cold room or on ice. Incubations were done using freshly cut, unfixed, 10-μm sections of WT or αD knockout spleen prepared using a cryostat (Leica CM 3050). F4/80-positive splenocytes were prepared and labeled with PE, suspended in RPMI 10 at a final concentration of 1 × 107 cells/ml, and layered over each section. The slides were immediately placed on a rotating platform at 60 rpm. After incubation for 30 min, the sections were washed by dipping the slides repeatedly in cold PBS and fixed for 10 min in 2% paraformaldehyde at 4°C. The sections were then washed again and mounted on glass slides with anti-fade medium. Images of the adherent PE-positive cells detected by fluorescent microscopy were captured and analyzed using a FluoView 300 microscope and the number of PE-positive cells bound to each section was counted using NIH ImageJ.
Characterization of αDβ2-deficient and WT mice in models of systemic infectious challenge
The experiments in these studies were approved by the Animal Welfare Committee of the Oswaldo Cruz Institute and by the University of Utah Animal Care and Use Committee. C57BL/6 WT and αD−/− mice from the Oswaldo Cruz Foundation breeding unit weighing 20–25g were used for most studies. A/J, SJL/J, and Swiss mice from B&K were also used in some experiments. The animals were kept at constant temperature (25°C) with free access to chow and water in a room with a 12-h light/dark cycle.
C57BL/6 mice were infected by an i.p. injection of 200 μl of PBS containing 105 RBC parasitized with the Pasteur strain of P. berghei ANKA (28). In parallel experiments, we established that this inoculum is lethal in a high percentage of animals after 20–30 days of infection, consistent with studies using other P. berghei strains (29). Samples of spleen and liver were obtained at different times postinfection of WT mice after sacrificing the animals in a CO2 chamber. The tissue was frozen and later analyzed by immunohistochemistry. For survival studies, groups of 10 WT and 10 αD−/− mice were studied together. Surviving animals were sacrificed at 30 days postinfection. The numbers of parasitized RBC and the hematocrits were determined as previously described (28) in separate groups of animals.
For studies of Salmonella infection, Salmonella enterica serovar typhimurium ISC 5302/2004, a generous gift from the Department of Microbiology of the Instituto Oswaldo Cruz, was cultured in Luria-Bertani broth (Guria Broth Miller; Sigma-Aldrich) for 16–18 h at 37°C to obtain stationary growth phase cultures. The bacteria were then centrifuged (200 rpm) for 10 min at 4°C and the pellets were resuspended in PBS to an OD of 0.1 at 660 nm, corresponding to 108 CFU/ml. WT and αD−/− mice were infected by i.p. injection of 200 μl of bacterial suspension (105 CFU/mouse). Control WT mice were sham injected (saline alone) in parallel. Survival was monitored for 10 or 15 days, at which time animals remaining alive were sacrificed.
Log rank tests were used for comparison of Kaplan-Meier survival plots in studies of P. berghei and Salmonella infection. A p ≤ 0.05 was considered statistically significant for differences in the mortality data.
Multiplex cytokine and IL-12 determinations
Initial screening analysis of multiple cytokines in the serum of three αD−/− and three WT mice was done using a Luminex system (Bio-Plex; Bio-Rad). Subsequent measurements of IL-12(p40) levels in samples from larger groups of αD−/− and WT animals were done by commercial ELISA according to the manufacturer’s instructions (R&D Systems). Differences in mean IL-12 levels were examined by one-way ANOVA.
Results
Generation and characterization of αD knockout mice
αD-deficient (αD−/−) mice were generated by targeted deletion of the I domain and excision of the “floxed” selection cassette to avoid spurious in vivo consequences (Ref. 17 and Fig. 1). Progeny containing the αDloxP allele were backcrossed and maintained on the C57BL/6 background. Heterozygous and homozygous animals were born at expected Mendelian ratios and did not display gross physical abnormalities. Homozygous animals were fertile and did not have an increased incidence of spontaneous infection. Leukocyte counts and morphology were similar between WT and homozygous mutant animals. The mean weight of spleens from 10-to 11-wk-old αD−/− mice was greater than that of WT animals (105 vs 70 mg, n = 5 of each genotype), but the spleens were of similar size at 17–18 wk of age (90 vs 85 mg, n = 4).
Integrin αDβ2 is constitutively expressed by limited subsets of leukocytes in the absence of infectious challenge
A small population (<1%) of peripheral blood leukocytes from WT mice of C57BL/6, A/J, SJL/J, and Swiss backgrounds stained positively for αDβ2. Splenic macrophages were used as controls in the staining reactions (see below and Fig. 2, E and F). This feature was unexpected since other β2 integrins are highly expressed on circulating mouse leukocytes and are involved in their emigration from the blood in response to inflammatory stimuli (5, 6) and because αDβ2 is more broadly displayed on human leukocytes (Ref. 8 and our unpublished observations). In previous studies, αD was reported to be absent from circulating murine leukocytes (16) and to be present on only a small fraction of circulating canine leukocytes (30).
When we examined tissues, αD staining was restricted to cells of the splenic red pulp, thymus, and bone marrow in adult WT C57BL/6 mice (Figs. 2 and 3 and Table I). By flow cytometry, αD+ splenocytes were positive for β2 (CD18), as expected (1, 3). αDβ2 is also expressed in rat splenic red pulp (data not shown) and in the red pulp of canine and human spleen (8, 30), indicating that its localization in this domain is conserved across species. Scattered groups of cells in the bone marrow and thymus of WT mice express αD at lower staining intensity compared with that in spleen but with similar cellular morphology (Table I and Fig. 2). These cells likely represent specific macrophage populations (see below). There was no staining of tissues from αD−/− mice, documenting specificity of the immunohistochemical reactions and null phenotype of the αD−/− animals (Table I, Fig. 3F, and data not shown).
FIGURE 3.
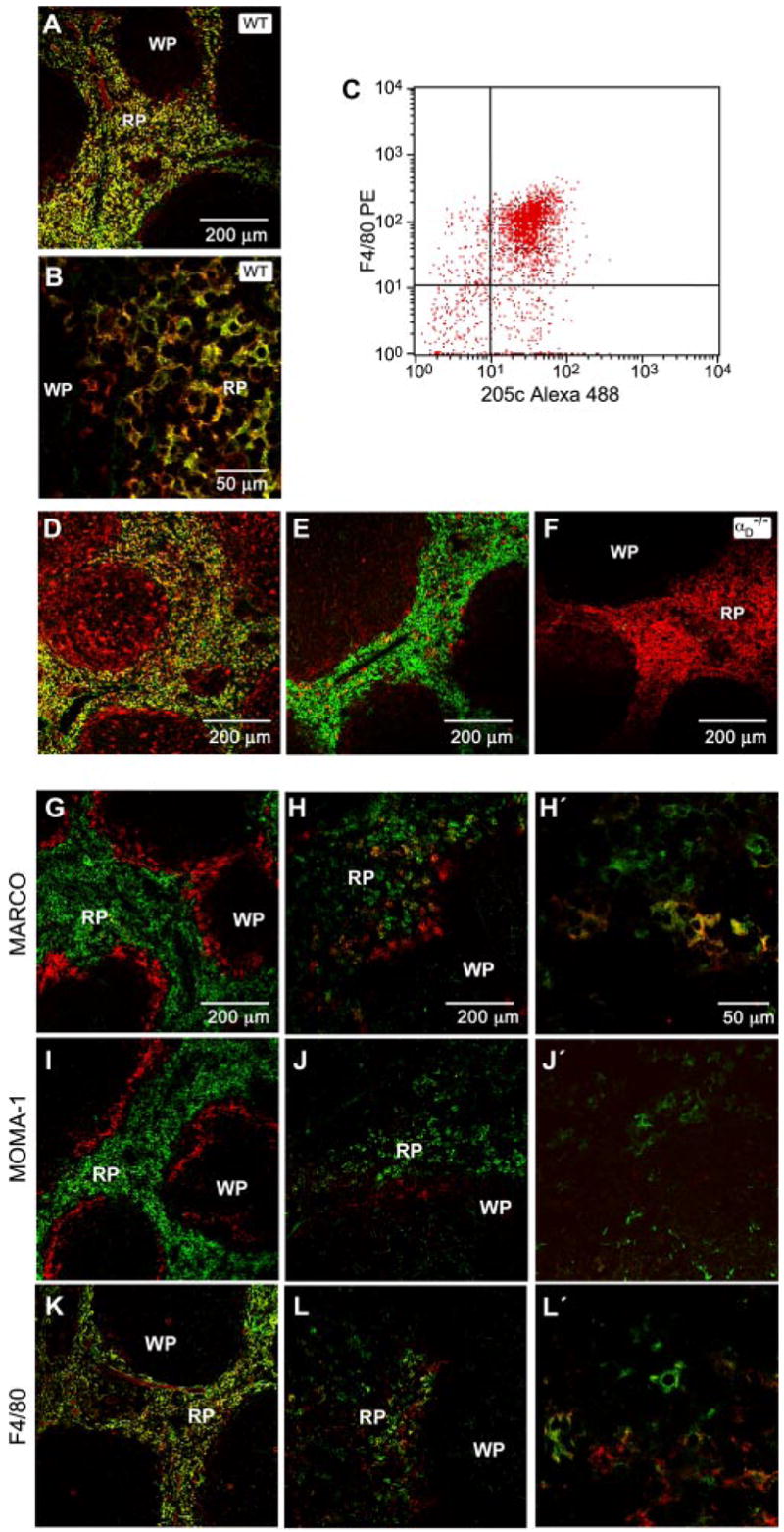
αDβ2 is expressed by a unique population of splenic red pulp macrophages that is altered in P. berghei infection. Frozen sections of freshly isolated spleen from WT mice were costained using anti-αD mAb (mAb 205c, green fluorescent labeling) and mAb against F4/80 (A and B), macrosialin (D), or αm (E; red labeling). F, Spleen sections from αD−/− mice were costained for αD and F4/80 as in A and B. The staining patterns shown are representative of tissue from multiple WT and αD−/− mice. C, Splenocytes were selected based on binding of PE-labeled anti-F4/80 and examined for expression of αD by flow cytometry using mAb 205c conjugated to Alexa 488. A similar pattern was seen in two additional experiments. G–L, Frozen sections of spleen from control (G, I, and K) or P. berghei-infected (7 days postinfection; H, J, and L) WT mice were costained for αD and MARCO, MOMA-1, or F4/80. In each set of panels, αD was detected by green fluorescence and the second marker was detected by red fluorescence. The right panels (H′, J′, and L′) show higher magnification detail. The staining patterns were similar in tissue from three infected animals. RP, Red pulp; WP, white pulp.
Table I.
αDβ2 is restricted to macrophages in a limited number of tissues in WT C57BL/6 mice in the absence of infection or inflammation and is absent in αD−/− animalsa
| Tissue | Samples from WT Mice | Samples from αD−/− Mice |
|---|---|---|
| Lung | 0 | 0 |
| Thoracic lymph node | 0 | 0 |
| Liver | 0 | 0 |
| Kidney | 0 | 0 |
| Peyer’s patch (gut) | 0 | 0 |
| Alveolar macrophages | 0 | ND |
| Brain | 0 | 0 |
| Skin | 0 | 0 |
| Spleen | ++++ | 0 |
| Thymus | ++ | 0 |
| Bone marrow | ++ | 0 |
| Blood leukocytes | 0−+ | 0 |
| Peritoneal macrophages | 0−+ | ND |
The levels of αDβ2 expression analyzed by immunocytochemistry were estimated based on staining intensity as strong (++++), intermediate (++), weak (0–+), or absent (0). In tissues with positive staining, αDβ2 was expressed by specific cells with morphologic characteristics of macrophages (see text and Figs. 2 and 3). These results summarize the findings in analysis of samples from multiple animals. Alveolar macrophages were obtained by bronchoalveolar lavage (n = 2). Resident peritoneal macrophages collected in the absence of agonist stimulation were largely negative for αDβ2, a feature that was confirmed by flow cytometry (0.23–4.7% of cells positive for αD, n = 4). Expression of αD was not increased in peritoneal macrophages at 48 h after thioglycolate injection into the peritoneal cavity as assayed by flow cytometry (n = 3).
Specific populations of splenic macrophages differentially express integrin αDβ2
When splenocytes were first sorted based on their display of F4/80, a determinant that identifies murine monocytes and subpopulations of macrophages in normal and inflamed tissues (20, 31, 32), >80% were also positive for αD (Fig. 3C). Consistent with the flow cytometric results, the majority of red pulp splenocytes costained for αD and F4/80 (Fig. 3, A, B, and K), although a small number of cells were brightly positive for one or the other marker. We detected F4/80 in the absence of αDβ2 in WT liver Kupffer cells, alveolar macrophages, and peripheral blood monocytes (data not shown) and in red pulp splenocytes from αDβ2−/− mice (Fig. 3F), demonstrating differential regulation of the two surface factors.
The majority of αD+ red pulp splenocytes also stained for macrosialin (Fig. 3D), the murine ortholog of human CD68, confirming their identity as macrophages (20). CD68 and αD are also coexpressed on a subset of macrophages in human atherosclerotic tissue (8). Costaining for MARCO, a scavenger receptor that identifies the splenic marginal zone macrophage, and MOMA-1, which identifies the marginal metallophilic macrophage (20, 33), demonstrated distinct populations of cells and localization patterns (Fig. 3, G and I). Thus, αDβ2 is expressed by a specific population of macrophages in the red pulp that is distinct from the marginal zone macrophage subsets. Each macrophage subtype has particular roles in blood surveillance, antimicrobial defense, and organization of splenic microarchitecture, and is influenced by unique differentiation and retention signals (34). Analysis of expression of other α subunits (αM, αL, αX) indicated that αDβ2 is the dominant β2 integrin heterodimer on red pulp macrophages (Fig. 3E and data not shown).
αDβ2 mediates adhesive interactions of splenic red pulp macrophages
The functions of αDβ2 in vivo are uncharacterized, although it is assumed to mediate adhesive interactions (1, 3, 6, 8, 10, 11, 16). We established a modified Stamper-Woodruff assay system (26, 27) using sections of spleen from WT and αD-deficient mice and suspensions of isolated splenocytes. Suspended F4/80-positive macrophages from spleens of αD−/− animals were much less adherent than those from WT mice (Fig. 4). In parallel, incubation of suspended F4/80-positive splenocytes from WT mice with Abs against αD inhibited adhesion to WT spleen sections by 65–90%; in addition, an Ab against VCAM-1, a ligand for human αDβ2 (9, 10), reduced adhesion by a mean of 22% (10–43%; n = 2, data not shown). These results demonstrate that αDβ2 mediates adhesive interactions of red pulp macrophages and that it may influence their distribution. Furthermore, although VCAM-1 appears to be a ligand for αDβ2 in mouse tissues, these experiments also indicate that there are additional binding partners on target spleen cells and/or matrix. Although αDβ2 mediates binding of a critical subset of splenic macrophages (Figs. 2–4), it is not required for establishment of the basal topography of the red and white pulp (34) of the adult murine spleen or for retention of macrophage subsets in the splenic red pulp (Fig. 3F).
FIGURE 4.
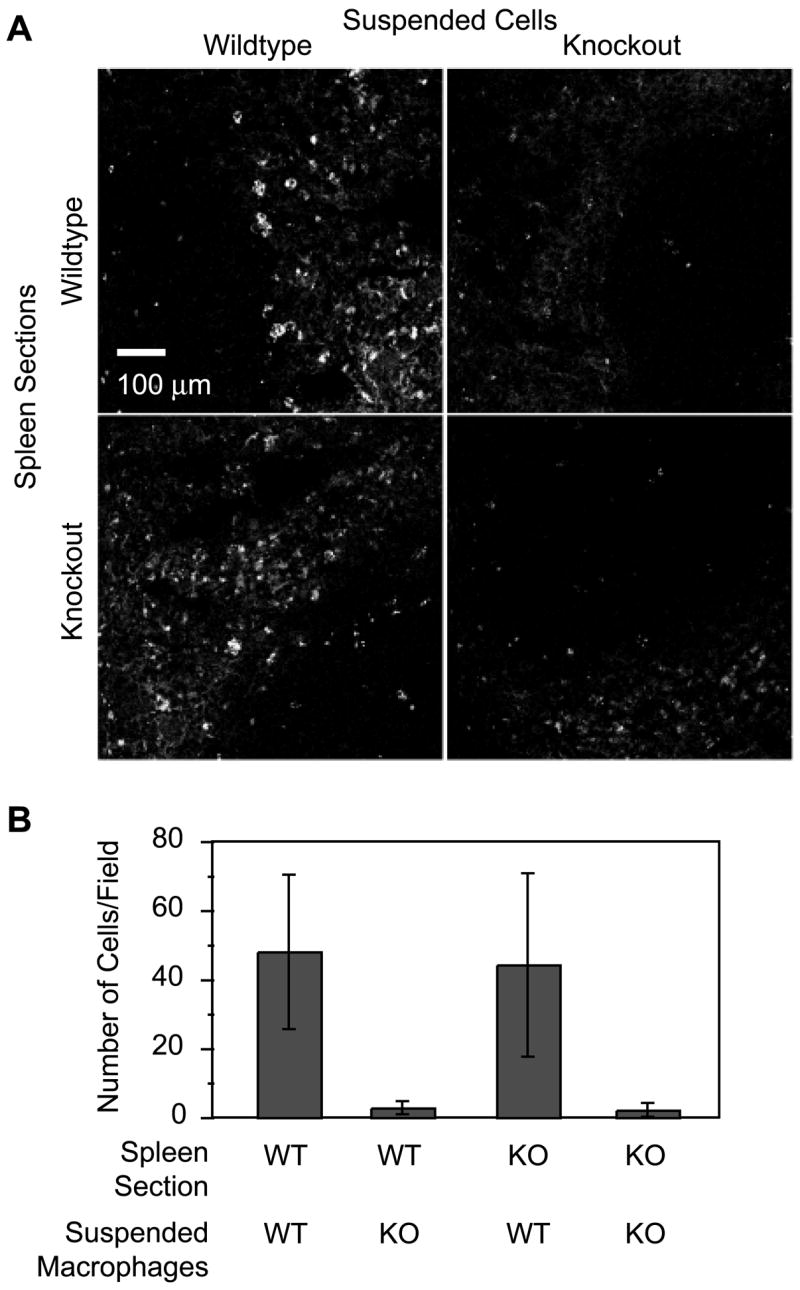
Integrin αDβ2 mediates adhesive interactions of splenic red pulp macrophages. Suspensions of PE-labeled F4/80-positive macrophages (suspended cells) from WT or αD−/− (knockout (KO)) mice were incubated with spleen sections from WT or αD−/− animals, and the adherent splenocytes were imaged (A) and counted (B) as described in Materials and Methods. A and B, Representative of three separate experiments.
αDβ2 expression is dynamically altered on inflamed macrophages in response to P. berghei challenge and is regulated by cytokine signaling
Because red pulp macrophages have major effector activities in blood-borne infection and antigenemia (33, 34), we chose models of systemic infectious challenge to further characterize regulation and functions of αDβ2. The spleen, an anatomic interface between innate and acquired immune effector cells (34), is critical in experimental malaria in all animals studied and red pulp macrophages have unique roles in recognition and clearance of infected RBC (35, 36). Macrophages in other organs are also activated as a consequence of recognition of PBRC and by inflammatory signals generated by the systemic infection (36–38). Therefore, we first examined αDβ2 distribution and function in experimental malaria.
The distribution of splenic macrophages was dramatically altered by challenge with intraerythrocytic P. berghei, a lethal rodent malarial strain that induces pathophysiologic features similar to those in severe human malaria (28, 37–39). Spleens of the infected animals were also enlarged (data not shown). Serial examinations over 5–20 days after infection demonstrated progressive reorganization of the splenic architecture and extensive loss of geographic localization of αD+ and MARCO+ macrophages (Fig. 3, G, H, H′, K, L, and L′, and data not shown). MARCO+ cells were widely distributed in the red pulp of P. berghei-infected animals, in contrast to their strict compartmentalization in the marginal zone of the uninfected spleen (Fig. 3, G, H, and H′). Linear arrays of doubly stained αD+ and MARCO+ macrophages were present in red pulp (Fig. 3, H and H′), whereas expression of αD and MARCO was restricted under basal conditions (Fig. 3G). Functional characteristics of the new population of αD+ MARCO+ macrophages are not yet completely defined. Nevertheless, these cells contained heme pigment deposits, consistent with erythrophagocytosis (data not shown). The topography of MOMA-1+ macrophages was also altered (Fig. 3, J and J′). Redistribution of splenic leukocytes also occurs in infection with other malarial pathogens (40).
Parallel but distinct changes occurred in the liver, which is a target organ in P. berghei infection (29, 41). No αD+ cells were detected in the livers of uninfected animals (Table I, Fig. 5A, and data not shown), indicating that murine Kupffer cells, a macrophage subset (20), do not basally express αD. αD is also absent from human and canine Kupffer cells (8, 30). By day 10 of infection with P. berghei, a minor population of small αD+ cells was detected, primarily in the portal vein areas (Fig. 5B). These cells were macrosialin+ but negative for F4/80, αMβ2, and αXβ2. By day 20, large αD+ cells with morphology similar to that of splenic red pulp macrophages were numerous, were found in both the portal and central vein areas (Fig. 5C), and were again negative for F4/80, αMβ2, and αXβ2 while being positive for macrosialin. They were associated with deposits of heme pigment (Fig. 5, B and C, right panels, and data not shown), indicating that these cells participate in erythrophagocytosis as do splenic macrophages. There was also progressive hepatocyte necrosis and hepatic inflammation in the 10- to 20-day interval (data not shown).
FIGURE 5.
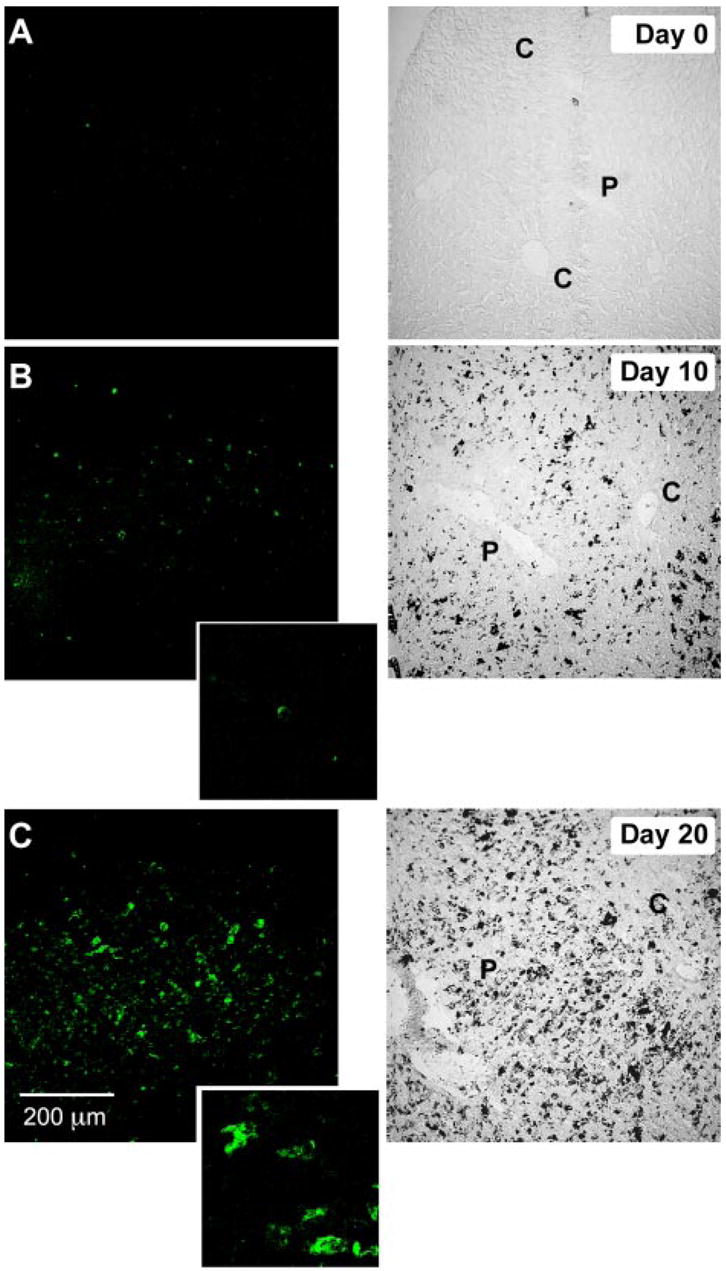
New populations of αD+ macrophages are induced in the livers of mice infected with P. berghei. Control mice or animals infected with P. berghei were sacrificed at day 0 (A; control), day 10 (B), or day 20 (C) followed by staining for αDβ2 with immunofluorescent detection (mAb 205c). Transmitted light images are shown in the right panels for comparison. Original magnification, ×200; ×1200 in insets in B and C. The black deposits in the right panels of B and C are heme pigments. The patterns shown are representative of those in liver tissue from three mice. C, Central vein; P, portal vein.
In parallel, we examined αD expression by murine M1 myeloid cells, an established model of terminal differentiation of myeloid precursors to macrophages. Treatment of M1 cells with LIF or IL 6, which are multifunctional cytokines and inducers of hemopoietic cell differentiation, causes them to differentiate to macrophages and undergo growth arrest (24). Undifferentiated M1 cells expressed no or low levels of αD mRNA and protein under basal conditions (Fig. 6, A, and B). LIF induced expression of αD mRNA and protein over 4 days of differentiation (Fig. 6, A–C, and data not shown). The pattern of staining of αD in clusters of differentiated M1 macrophages (Fig. 6B) was similar to that of macrophages in situ (Fig. 2). F4/80 was also up-regulated on M1 macrophages (data not shown). To determine whether IL-6, which is increased in the blood in experimental and clinical malaria (36), induces αD expression, we incubated M1 cells with IL-6 (1–100 ng/ml) or LIF (50 U/ml) for 4 days. IL-6 induced αD mRNA and protein expression in a concentration-dependent fashion with a maximal effect at 10 ng/ml. By immunocytochemistry, the staining pattern and number of cells positive for αD were equivalent when M1 were stimulated with 10 ng/ml IL-6 or 50 U/ml LIF. These experiments demonstrate that αD expression is regulated by cytokine-induced macrophage differentiation, providing a mechanism for its local induction on macrophage subsets in inflamed tissues such as the liver (Fig. 5).
FIGURE 6.
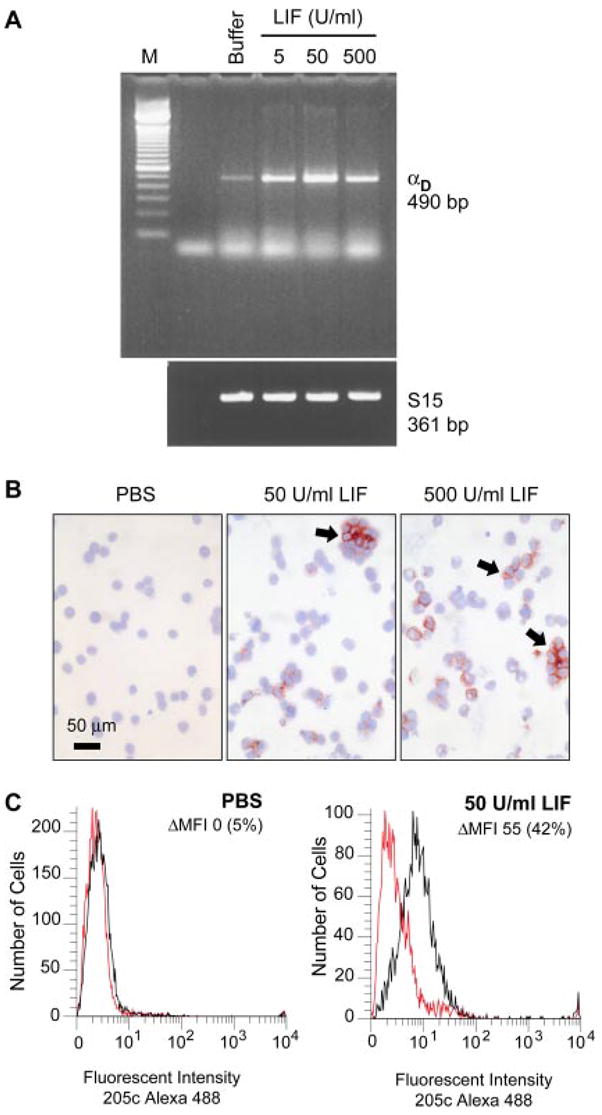
Cytokine-mediated differentiation of macrophage precursors induces expression of αD. M1 myeloid cells were cultured in the presence of the indicated concentrations of LIF for 4 days and then examined for expression of αD. A, mRNA was isolated and probed for the αD transcript by PCR. S15 served as a control for loading. B, Expression of αD protein was assayed by incubation with mAb 205c and detection with an alkaline phosphatase-labeled secondary Ab (arrows). C, M1 cells were cultured with PBS or LIF as additives to the medium for 4 days and analyzed for surface expression of αDβ2 by flow cytometry using Alexa 488-conjugated mAb 205c to detect αD (red tracings, control Ab; black tracings, mAb 205c). Change in mean fluorescent intensity is denoted by ΔMFI. These results are representative of three additional studies.
Targeted deletion of αD differentially influences survival in systemic infection with P. berghei and Salmonella
We found that targeted deletion of αD alters survival in P. berghei infection in an unexpected fashion. In three separate experiments, all of the WT animals infected with P. berghei were dead at 30 days (Fig. 7A). In two of the experiments, each of 10 WT mice succumbed by days 27 and 21, respectively, whereas a total of 5 αD−/− animals (3 in experiment 1 and 2 in experiment 2) survived to day 30. A third experiment demonstrated an even more malignant course, with 9 of 10 WT animals dead by day 14 compared with 7 of 10 αD−/− animals; 1 αD−/− mouse survived until day 30. The difference between the cumulative survival curves for each genotype in these experiments (Fig. 7A) was highly significant ( p = 0.0059). Thus, targeted deletion of αD alters the natural history and increases survival in the first 30 days of infection in this model, a period in which all WT animals succumbed. To confirm that the survival advantage during this time span represents a biologic feature selectively imposed by deletion of αDβ2, we performed two experiments in which other genetically modified mice were examined as controls. In the first, mice deficient in TLR2 (10 animals) were infected with P. berghei in parallel with WT mice (10 animals). In the second, mice deficient in inducible NO synthase (iNOS) (15 animals) and WT mice (24 animals) were studied. All iNOS and TLR2 animals were dead by days 18 and 19, respectively, whereas mortality of the WT mice was similar to that shown in Fig. 7A, with a progressive decrease in the number of surviving animals to day 28. Thus, deletion of neither iNOS nor TLR2, innate immune factors that influence the natural history of malaria (39), reproduced the mortality pattern imposed by genetic deletion of αD in these studies (Fig. 7A).
FIGURE 7.
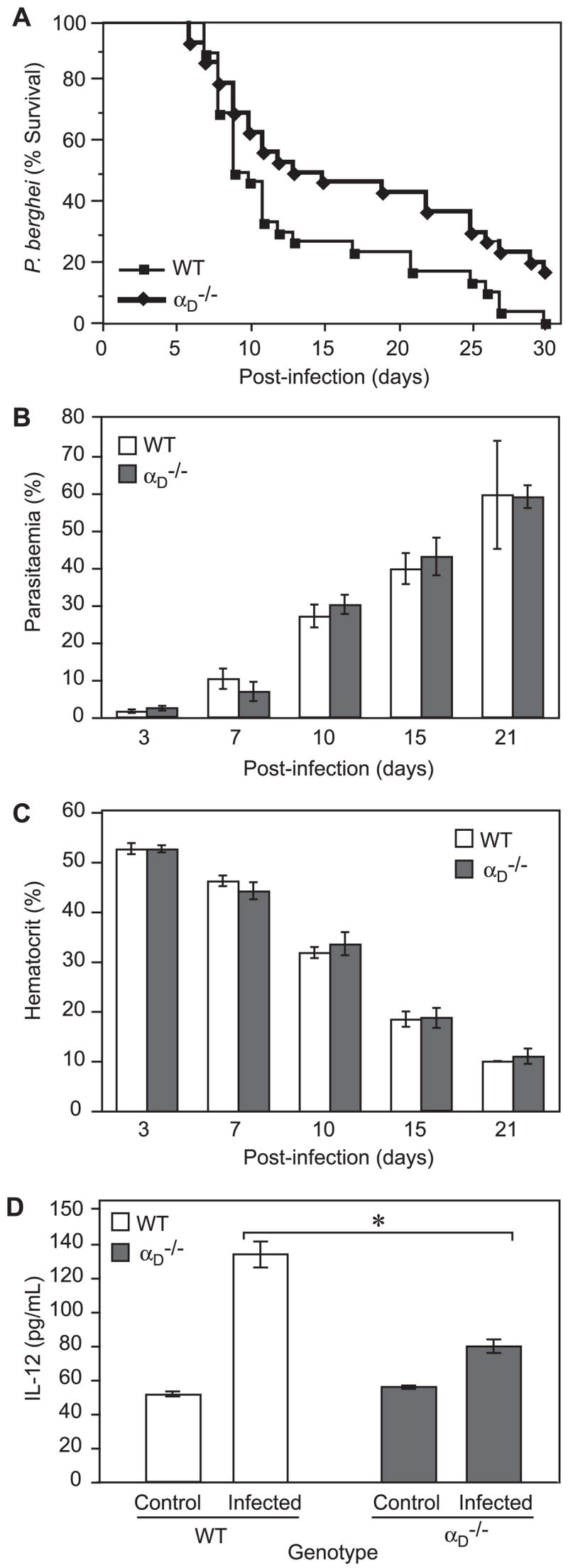
Deletion of αD provides a survival advantage in P. berghei infection. A, Mice were infected by i.p. challenge with P. berghei PRBC. Three separate experiments, each involving 10 WT and 10 αD−/− animals, were accomplished and the pooled results were analyzed. The difference between the cumulative survival curves was significant (p = 0.0059). B and C, The percentage of RBC infected by P. berghei was determined by microscopy and the hematocrit by RBC sedimentation. B, Results from one of three experiments and C, results from one of two experiments. Samples from variable numbers of surviving animals were studied at each time point. D, Serum samples were collected from WT control mice (n = 5), WT animals infected with P. berghei (n = 10), αD−/− control mice (n = 8), and P. berghei-infected αD−/− animals (n = 7) at 10 days after i.p. inoculation with PRBC. IL-12(p 40) concentrations were measured by ELISA. The differences between levels in samples from infected WT and αD−/− mice (*) was highly significant (p < 0.0001).
The survival curves of WT and αD−/− mice diverged at days 8–10 after P. berghei infection and significantly more αD−/− animals were alive over the succeeding 3 wk (Fig. 7A). To begin to examine the mechanisms involved, we focused on the period of divergence and first examined erythrocyte parasite burden and magnitude of anemia, which are key determinants of outcome in severe malaria (36–39). Between days 10 and 21, the mice became hyperparasitemic and profoundly anemic but there were no consistent differences in samples from αD−/− and WT mice (Fig. 7, B and C), excluding differences in these two central mechanisms (39) as causes for the difference in survival (Fig. 7A). In addition, these results indicate that αDβ2 is not required for clearance of PRBC. The splenic microarchitecture is essential for rapid resolution of primary infection with Plasmodium species and influences key aspects of the host response (35, 40). We found that the organization of splenic red and white pulp is dramatically altered in P. berghei infection (Fig. 3, H, J, and L). Nevertheless, this rearrangement of splenic architecture also occurred in αD−/− animals with loss of distinct boundaries between the red pulp and marginal zone. This indicated that differences in splenic macroarchitecture, which might occur due to alteration in adhesive properties of red pulp macrophages (Fig. 4) as a result of deletion of αDβ2, do not account for the differences in survival (Fig. 7A).
Systemic cytokines are proposed to be key regulators of the pathogenesis of complicated malaria (35, 40). Therefore, we next performed an initial survey of multiple cytokines in the blood of three animals of each genotype at 10 days of infection using a multiplex screening assay. This analysis suggested diverse alterations in systemic levels of cytokines in αD−/− and WT mice, including variations in IL-12, IFN-γ, IL-6, MCP-1, and KC. To explore this initial finding further, we examined levels of IL-12 in serum samples from a second group of animals studied on day 10 of infection. We chose IL-12 as an initial candidate for further analysis because it is implicated in the pathogenesis of severe human malaria and has been identified as a mediator of tissue injury in lethal P. berghei infection (29, 41). Consistent with the preliminary analysis, we found that IL-12 concentrations were lower in the blood of infected αD−/− mice compared with levels in samples from WT mice (Fig. 7D). Thus, alterations in the pattern and levels of inflammatory cytokines may contribute to the survival advantage in αD−/− animals in this phase of the systemic infection (Fig. 7A).
To further define the phenotype of αD-deficient mice and to determine whether deletion of αD has a stereotyped, or conversely, differential impact on systemic infections, we examined αD−/− and WT mice in a model of S. typhimurium sepsis. This model was chosen for comparison because, although it is a bacterial pathogen, it also involves blood-borne phases and clearance by splenic, hepatic, and marrow macrophages (35, 44), features that are similar to P. berghei infection. A preliminary experiment (eight mice of each genotype) with 105 bacteria per inoculum suggested greater mortality in αD−/− animals. A second experiment with 10 animals of each genotype but with a 10-fold lower inoculum supported the initial result ( p = 0.008). We then performed three additional experiments with 10 WT and 10 αD−/− mice, an inoculum of 105 Salmonella, and a 10-day study period in each. (The change in study period was required because of animal housing constraints.) These studies replicated the original finding and again documented greater mortality in the αDβ2-deficient animals (Fig. 8; p = 0.0004). Thus, survival patterns of αD−/− mice were clearly different in systemic infection with P. berghei (Fig. 7A) and S. typhimurium (Fig. 8). This difference in natural history demonstrates that the effect of genetic deletion of αD in murine models of systemic infection varies with the nature of the pathogen, indicating specialized functions of integrin αDβ2 and the immune effector cells that express it in host defense against invading microbes.
FIGURE 8.
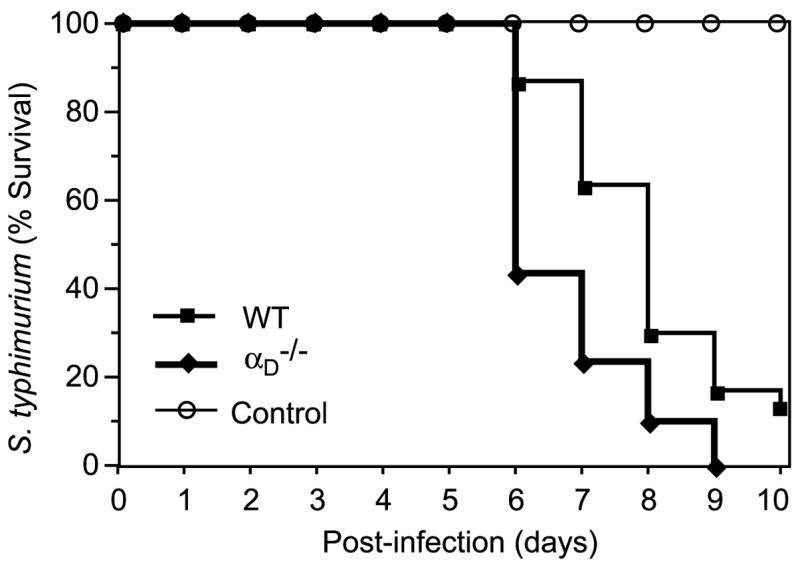
Mortality is accelerated in αDβ2-deficient mice infected with S. typhimurium. WT or αD−/− mice were infected with S. typhimurium by the i.p. route (105 bacteria/animal). Groups of 10 αD−/− and 10 WT animals were studied in each of three separate experiments. The pooled survival data for 30 animals of each genotype are shown. The difference between the cumulative survival plots was significant (p = 0.0004). Control sham-infected mice that received vehicle alone were studied in parallel; no sham-infected animals died.
Discussion
Leukocyte integrins have essential but incompletely defined activities in host defense and conserved functions in mice and humans (1–6). Our observations provide the first detailed characterization of αDβ2 in vivo, information that is currently lacking in the field (1, 3), along with new insights into its regulation and functions. The studies clearly demonstrate that it has unique features in inflammation and host defense. Interestingly, although other β2 integrins are highly expressed on circulating murine leukocytes and are critical in their trafficking from blood to tissues, αDβ2 is present on only a minor and as yet uncharacterized population of these cells in WT animals in naive conditions (see Results; Y. Miyazaki and G. A. Zimmerman, unpublished data). In contrast, αDβ2 is constitutively expressed on macrophages of splenic red pulp, bone marrow, and thymus of WT mice in the absence of infectious or inflammatory challenge (Table I and Figs. 2 and 3) indicating that, in this species, αDβ2 has major functions apart from emigration of leukocytes from the blood. We found that αDβ2 mediates adhesion of red pulp macrophages, identifying one of its extravascular activities and demonstrating a functional role for the αDβ2 heterodimer on macrophages for the first time. Previously, adhesive functions for αDβ2 have only been observed in human eosinophils and surrogate cell lines (8–11). We also found that in response to erythrocytic infection with a malarial pathogen, P. berghei, new and previously unrecognized populations of αD+ macrophages emerge in the spleen and liver, demonstrating dynamic regulation of αDβ2 in this cell type and illustrating the daedal nature of macrophage heterogeneity (42, 43). In contrast, bacterial and immune challenge to the murine lung, another macrophage-rich organ, did not induce expression of αD under several experimental conditions (Y. Miyazaki, H. Castro Faria Neto, and G. A. Zimmerman, unpublished data) and αD was not increased on peritoneal macrophages in response to thioglycolate instillation (Table I). This indicates that selective signals are required for its display on macrophage subpopulations, pointing to context-specific functions of αDβ2. Targeted deletion of αD unexpectedly yielded a survival advantage in lethal malarial infection. Conversely, deletion of αD had the opposite effect in a model of Salmonella sepsis, indicating complex roles for αDβ2 in defense against systemic pathogens. The latter findings are consistent with both beneficial and injurious activities of leukocyte integrins (2, 7) and macrophages (42, 44) in innate immune responses.
The roles of leukocyte integrins in responses to microbial invasion are incompletely characterized, although this is clearly a chief defensive function of these specialized effector proteins (1, 2, 6). Targeted deletion of β2, which causes deficiency of all leukocyte integrin heterodimers, increases susceptibility to many bacterial pathogens but affords resistance to Listeria monocytogenes and improves control of local infection by Leishmania major, a protozoan parasite of macrophages, in murine models (5, 6). These patterns of susceptibility and resistance demonstrate complex functions of leukocyte integrins in innate and acquired immune responses to infection (6), a feature further indicated by variable contributions of individual heterodimers in specific experimental models (45, 46). Participation of integrins in the pathobiology of complicated malaria is suggested (37), but largely unexplored. mAbs against αL improved survival in models of cerebral malaria, suggesting a role for αLβ2 (LFA-1), whereas Abs against αm or ICAM-1, a ligand for αLβ2, did not protect (47, 48). In contrast to Ab administration, targeted deletion of ICAM-1 increased survival in a cerebral malaria model (49). A β3 integrin (3) regulates the pattern of cytokines released by human macrophages and myeloid dendritic cells activated by PRBC (36); nevertheless, contributions of β2 integrins to this process are undefined. Thus, the regulation and activities of leukocyte integrins in malarial infection are uncharacterized. We found extensive display of αD on red pulp macrophages of the WT spleen (Figs. 2 and 3), suggesting that αDβ2 mediates responses to systemic pathogens (34), potentially including malarial parasites (35). We then explored this possibility using a model that mimics many features of severe human malaria, P. berghei infection (28, 29, 37–39).
Challenge of WT mice with intraerythrocytic P. berghei resulted in a dramatic alteration in the microarchitecture of the splenic red pulp and marginal zone with loss of demarcation of these regions and appearance of a new subset of αD+MARCO+ macrophages (Fig. 3 and data not shown). The distribution and activities of splenic macrophages and other leukocytes influence the inflammatory and immune responses to malarial invaders and may alter susceptibility to concomitant infection by bacterial and viral pathogens (35, 36, 39, 40, 50, 51). In lethal P. falciparum infection, the marginal zone is dramatically altered with almost complete histologic “dissolution” (50), similar to the microarchitectural disruption in P. berghei infection (Fig. 3). In parallel with changes in splenic microarchitecture and macrophage populations, a new subset of αD+ macrosialin+ macrophages emerged in the livers of WT mice infected with P. berghei (Fig. 5). It is likely that these new macrophage subpopulations in spleen and liver mediate responses to malarial infection that are in some cases injurious or maladaptive (36, 37, 42, 43). In addition, they clearly demonstrate dynamic expression of αDβ2 induced by pathogen challenge and inflammatory signals.
It is not clear whether new populations of αDβ2-expressing macrophages in the spleen and liver in P. berghei infection are due to migration, proliferation, and differentiation of blood leukocytes that express αD or, conversely, to induction of αDon resident macrophages or macrophage precursors. To begin to explore this issue, we used the murine M1 myeloid cell model, in which agonist-induced macrophage differentiation can be dissociated from cellular proliferation (24). LIF and IL-6, which are multifunctional cytokines released into the blood in murine endotoxemia and, in the case of IL-6, malaria (37, 38), trigger terminal differentiation of M1 cells to macrophages (24). We found that LIF and IL-6 induced new expression of αD on M1 cell-derived macrophages, providing a potential mechanism for dynamic regulation of αDβ2 on specific macrophage subsets in tissues such as the liver or spleen in response to inflammatory signals (Figs. 3 and 5). As noted previously, unique patterns of cytokines or other signaling factors are likely required for the highly restricted expression of αDβ2 on individual macrophage subsets in healthy animals (Table I and Figs. 2 and 3) and changes in expression in response to inflammatory challenge (Figs. 3 and 5). Additional characterization of the minor subpopulation of αD+ blood leukocytes will be required to further define regulation of αD on this subset and whether it is altered by signals for migration and/or proliferation.
Improved survival of αD−/− mice infected by P. berghei was unexpected and indicated that pathophysiologic events mediated by αDβ2 have deleterious consequences in lethal malarial infection. Although at first glance the survival advantage at 30 days appears relatively small, significant differences over an approximate 3-wk period represent a substantial benefit in a syndrome with 100% mortality (Fig. 7A). Mice infected with P. berghei die secondary to cerebral, hepatic, pulmonary, renal, and cardiac complications that are superimposed on hyperparasitemia and severe malarial anemia (28, 29, 37–39, 41, 52, 53). The pathobiologic mechanisms that mediate systemic inflammation and critical organ injury in experimental and clinical malaria are complex and multifactorial, and their precise identification has remained elusive (36–39). One or multiple organ-specific or multiorgan components could be influenced by deletion of αDβ2. To begin to explore these possibilities, we first examined two key determinants of malarial pathogenesis, severe malarial anemia, and intraerythrocyte parasite burden (39). Splenic red pulp macrophages, which are rich in αDβ2 (Figs. 2 and 3 and Table I), are critical for removal of PRBC and for parasite killing, and changes in splenic function and splenectomy alter anemia parasitemia and outcomes (35, 50, 51). Profound anemia in murine malaria is due in part to hemolysis and hyperphagocytosis of infected and uninfected RBC by splenic red pulp and/or hepatic macrophages and to ineffective erythropoiesis (39, 52, 53). The latter process is influenced by marrow macrophages (54, 55) and, therefore, potentially by αDβ2 (Table I and Fig. 2). Nevertheless, we found similar numbers of PRBC and degrees of anemia in αD−/− and WT mice (Fig. 7, B and C), indicating that differences in intraerythrocytic parasite burden, clearance of PBRC by macrophage subsets, and ineffective erythropoiesis are not the pivotal mechanisms that determined altered survival of αD−/− animals. In addition, we evaluated differences in splenic microarchitecture, which influences early and late host responses to malarial pathogens (36, 40, 50, 51) (also see above). We found that dramatic alterations in red pulp and marginal zone topography, which were histologically similar to those reported in P. falciparum infection in humans (50), occurred in both WT and αD−/− animals (Fig. 3 and data not shown). Therefore, differences in this variable do not account for the survival advantage of αD−/− animals. In addition to hyperparasitemia, severe anemia, and splenic reorganization, cytoadhesive vasculopathy, in which PRBC adhere to endothelium and occlude microvessels of critical organs, is also a proposed mechanism of injury and death in severe malarial syndromes (37–39). We have not observed expression of αDβ2 on murine erythrocytes or endothelial cells (Table I and data not shown), consistent with the highly restricted distribution of leukocyte integrins (1, 3), making it unlikely that altered interactions of PRBC with endothelium account for improved survival of αD−/− animals. Therefore our studies, in aggregate, excluded several major determinants of mortality in P. berghei infection and pointed to other mechanisms.
Cytokine signaling is central to the systemic manifestations of experimental and clinical malaria (37–39, 53), and outside-in signals mediated by integrins both trigger and modify synthesis of cytokines by macrophages and monocytes (1, 3, 56–59). In preliminary studies, we found altered patterns of plasma cytokines in αD−/− mice infected with P. berghei compared with those in WT animals at 10 days after challenge. In additional assays to validate these preliminary data, we found that IL-12 levels in the blood of infected αDβ2-deficient mice were substantially lower than those in samples from WT controls (Fig. 7D). IL-12 signaling induces hepatic injury in lethal erythrocytic P. berghei infection (35, 40), providing a rationale for examining this cytokine as a sentinel proinflammatory mediator under the conditions of our experiment. (Studies of additional cytokines are in progress.) Furthermore, synthesis of IL-12 by monocytes and macrophages is suppressed by engagement of β2 integrins (57–59), suggesting a mechanism for its regulation under these conditions, although this has not yet been examined for αDβ2. Nevertheless, we do not propose decreased plasma levels of IL-12 as the sole mechanism for improved survival in αD−/− animals, since excesses of individual cytokines seem unlikely to account for all multiorgan and systemic manifestations of severe malarial syndromes (38, 39). Furthermore, additional cellular complexity may be involved. αDβ2 influences responses of T cells, which are immune effectors in severe experimental malaria (37–39, 60), and is reported to determine functional ontogeny of T lymphocytic subsets and their responses to microbial Ags (16).
To further characterize the phenotype of αD−/− animals in systemic infectious models and to determine whether the activities of αDβ2 are stereotyped regardless of the nature of the pathogen, we examined αD-deficient mice in S. typhimurium infection. Macrophage activity, cytokine signaling, and immune cell interactions are central in responses to Salmonella invasion (44), as they are in malarial infection (36–39). Deletion of αDβ2 accelerated mortality in mice infected with S. typhimurium, in contrast to the survival pattern in infection with P. berghei (see Results and Fig. 8). The difference in outcomes in the P. berghei and Salmonella models may in part be due to recognition of Salmonella by splenic marginal zone macrophages rather than αDβ2-rich red pulp macrophages (35). Furthermore, endogenous IL-12 augments protective immune responses against Salmonella species (61), whereas it contributed to immune injury in P. berghei infection (29, 41). Thus, the altered levels of IL-12 in αD−/− mice may partially account for the differences in survival in the two infectious challenges. In addition, however, different patterns of survival in the malaria and Salmonella models may also be due to different functional roles played by αDβ2+ macrophage subsets in systemic infection by intracellular bacteria vs malarial parasites. It is not yet clear how effector characteristics of macrophages (42, 43) govern the balance between defense and injury in P. berghei and S. typhimurium infections (44, 62) or how αDβ2 influences activation patterns of specific macrophage populations. Further delineation of mechanisms by which αDβ2 and other leukocyte integrins regulate the localization and functional repertoires of macrophage subsets (42) will clarify this issue. This information may then be useful in understanding the effects of innate immune events in humans infected by malaria and Salmonella, which together kill >2.5 million subjects each year worldwide (36, 39, 44, 62). αDβ2 is expressed by leukocytes in human splenic red pulp (8) and is displayed in regulated fashion by primary human macrophages and dendritic cells (Y. Miyazaki, E. Harris, A. Shah, and G. A. Zimmerman, manuscript in preparation), suggesting that it has key roles in these and other human infectious syndromes.
Acknowledgments
We thank our laboratory staffs for technical assistance, Andressa Almeida for preparation of infectious agents for in vivo experiments, and Kate Anderson for skilled animal husbandry. Liping Chen made important contributions to the development of αD-deficient mice before her untimely death. Diana Lim prepared the figures and graphic art and Mary Madsen, Adrienne Triplett, and Linn Steele contributed invaluable effort in preparation of this manuscript. We thank Leslie Ostler for performing specific experiments, Kelley Murphy for advice regarding immunocytochemical analysis, and Marga Massey for samples of rat spleen. Monica Van der Vieren, Patricia Hoffman, and Michael Gallatin of ICOS Corporation generously provided critical reagents, including key anti-αD Abs and a murine αD probe template for genomic library screening. We also thank colleagues in our group and Dr. Merle Sande for valuable discussions.
Footnotes
This work was supported by National Institutes of Health Grant HL44525 (to G.A.Z.), a National Institutes of Health Special Center of Research in Acute Respiratory Distress Syndrome (P50 H150153), an Asthma Research Center Award funded by the American Lung Association, the Margolis Foundation, and an Established Investigator Award from the American Heart Association (to A.S.W.).
Abbreviations used in this paper: WT, wild type; PRBC, parasitized RBC; iNOS, inducible NO synthase.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Harris ES, McIntyre TM, Prescott SM, Zimmerman GA. The leukocyte integrins. J Biol Chem. 2000;275:23409–23412. doi: 10.1074/jbc.R000004200. [DOI] [PubMed] [Google Scholar]
- 2.Bunting M, Harris ES, McIntyre TM, Prescott SM, Zimmerman GA. Leukocyte adhesion deficiency syndromes: adhesion and tethering defects involving β2 integrins and selectin ligands. Curr Opin Hematol. 2002;9:30–35. doi: 10.1097/00062752-200201000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 4.Scharffetter-Kochanek K, Lu H, Norman K, van Nood N, Munoz F, Grabbe S, McArthur M, Lorenzo I, Kaplan S, Ley K, et al. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J Exp Med. 1998;188:119–131. doi: 10.1084/jem.188.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Etzioni A, Doerschuk CM, Harlan JM. Of man and mouse: leukocyte and endothelial adhesion molecule deficiencies. Blood. 1999;94:3281–3288. [PubMed] [Google Scholar]
- 6.Lee SH, Corry DB. Homing alone? CD18 in infectious and allergic disease. Trends Mol Med. 2004;10:258–262. doi: 10.1016/j.molmed.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 7.Yonekawa K, Harlan JM. Targeting leukocyte integrins in human diseases. J Leukocyte Biol. 2005;77:129–140. doi: 10.1189/jlb.0804460. [DOI] [PubMed] [Google Scholar]
- 8.Van der Vieren M, Le Trong H, Wood CL, Moore PF, St John T, Staunton DE, Gallatin WM. A novel leukointegrin, αdβ2, binds preferentially to ICAM-3. Immunity. 1995;3:683–690. doi: 10.1016/1074-7613(95)90058-6. [DOI] [PubMed] [Google Scholar]
- 9.Grayson MH, Van der Vieren M, Sterbinsky SA, Michael Gallatin W, Hoffman PA, Staunton DE, Bochner BS. αdβ2 integrin is expressed on human eosinophils and functions as an alternative ligand for vascular cell adhesion molecule 1 (VCAM-1) J Exp Med. 1998;188:2187–2191. doi: 10.1084/jem.188.11.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van der Vieren M, Crowe DT, Hoekstra D, Vazeux R, Hoffman PA, Grayson MH, Bochner BS, Gallatin WM, Staunton DE. The leukocyte integrin αDβ2 binds VCAM-1: evidence for a binding interface between I domain and VCAM-1. J Immunol. 1999;163:1984–1990. [PubMed] [Google Scholar]
- 11.Yakubenko VP, Yadav SP, Ugarova TP. Integrin αDβ2, an adhesion receptor up-regulated on macrophage foam cells, exhibits multiligand-binding properties. Blood. 2006;107:1643–1650. doi: 10.1182/blood-2005-06-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.El-Gabalawy H, Canvin J, Ma GM, Van der Vieren M, Hoffman P, Gallatin M, Wilkins J. Synovial distribution of αd/CD18, a novel leukointegrin: comparison with other integrins and their ligands. Arthritis Rheum. 1996;39:1913–1921. doi: 10.1002/art.1780391119. [DOI] [PubMed] [Google Scholar]
- 13.Noti JD. Expression of the myeloid-specific leukocyte integrin gene CD11d during macrophage foam cell differentiation and exposure to lipoproteins. Int J Mol Med. 2002;10:721–727. [PubMed] [Google Scholar]
- 14.Shanley TP, Warner RL, Crouch LD, Dietsch GN, Clark DL, O’Brien MM, Gallatin WM, Ward PA. Requirements for αd in IgG immune complex-induced rat lung injury. J Immunol. 1998;160:1014–1020. [PubMed] [Google Scholar]
- 15.Oatway MA, Chen Y, Bruce JC, Dekaban GA, Weaver LC. Anti-CD11d integrin antibody treatment restores normal serotonergic projections to the dorsal, intermediate, and ventral horns of the injured spinal cord. J Neurosci. 2005;25:637–647. doi: 10.1523/JNEUROSCI.3960-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu H, Rodgers JR, Perrard XY, Perrard JL, Prince JE, Abe Y, Davis BK, Dietsch G, Smith CW, Ballantyne CM. Deficiency of CD11b or CD11d results in reduced staphylococcal enterotoxin-induced T cell response and T cell phenotypic changes. J Immunol. 2004;173:297–306. doi: 10.4049/jimmunol.173.1.297. [DOI] [PubMed] [Google Scholar]
- 17.Bunting M, Bernstein KE, Greer JM, Capecchi MR, Thomas KR. Targeting genes for self-excision in the germ line. Genes Dev. 1999;13:1524–1528. doi: 10.1101/gad.13.12.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mansour SL, Thomas KR, Capecchi MR. Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature. 1988;336:348–352. doi: 10.1038/336348a0. [DOI] [PubMed] [Google Scholar]
- 19.Schwenk F, Baron U, Rajewsky K. A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995;23:5080–5081. doi: 10.1093/nar/23.24.5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McKnight AJ, Gordon S. Membrane molecules as differentiation antigens of murine macrophages. Adv Immunol. 1998;68:271–314. doi: 10.1016/s0065-2776(08)60562-3. [DOI] [PubMed] [Google Scholar]
- 21.Cao Y, Murphy KJ, McIntyre TM, Zimmerman GA, Prescott SM. Expression of fatty acid-CoA ligase 4 during development and in brain. FEBS Lett. 2000;467:263–267. doi: 10.1016/s0014-5793(00)01159-5. [DOI] [PubMed] [Google Scholar]
- 22.Ohteki T, Maki C, Koyasu S. Overexpression of Bcl-2 differentially restores development of thymus-derived CD4−8+ T cells and intestinal intraepithelial T cells in IFN-regulatory factor-1-deficient mice. J Immunol. 2001;166:6509–6513. doi: 10.4049/jimmunol.166.11.6509. [DOI] [PubMed] [Google Scholar]
- 23.Kessel JM, Hayflick J, Weyrich AS, Hoffman PA, Gallatin M, McIntyre TM, Prescott SM, Zimmerman GA. Coengagement of ICAM-3 and Fc receptors induces chemokine secretion and spreading by myeloid leukocytes. J Immunol. 1998;160:5579–5587. [PubMed] [Google Scholar]
- 24.Hoffman-Liebermann B, Liebermann DA. Interleukin-6- and leukemia inhibitory factor-induced terminal differentiation of myeloid leukemia cells is blocked at an intermediate stage by constitutive c-myc. Mol Cell Biol. 1991;11:2375–2381. doi: 10.1128/mcb.11.5.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pabla R, Weyrich AS, Dixon DA, Bray PF, McIntyre TM, Prescott SM, Zimmerman GA. Integrin-dependent control of translation: engagement of integrin αIIbβ3 regulates synthesis of proteins in activated human platelets. J Cell Biol. 1999;144:175–184. doi: 10.1083/jcb.144.1.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stamper HB, Jr, Woodruff JJ. Lymphocyte homing into lymph nodes: in vitro demonstration of the selective affinity of recirculating lymphocytes for high-endothelial venules. J Exp Med. 1976;144:828–833. doi: 10.1084/jem.144.3.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature. 1992;356:63–66. doi: 10.1038/356063a0. [DOI] [PubMed] [Google Scholar]
- 28.Cordeiro RS, Cunha FQ, Filho JA, Flores CA, Vasconcelos HN, Martins MA. Plasmodium berghei: physiopathological changes during infections in mice. Ann Trop Med Parasitol. 1983;77:455–465. doi: 10.1080/00034983.1983.11811739. [DOI] [PubMed] [Google Scholar]
- 29.Adachi K, Tsutsui H, Kashiwamura S, Seki E, Nakano H, Takeuchi O, Takeda K, Okumura K, Van Kaer L, Okamura H, et al. Plasmodium berghei infection in mice induces liver injury by an IL-12- and Toll-like receptor/myeloid differentiation factor 88-dependent mechanism. J Immunol. 2001;167:5928–5934. doi: 10.4049/jimmunol.167.10.5928. [DOI] [PubMed] [Google Scholar]
- 30.Danilenko DM, Rossitto PV, Van der Vieren M, Le Trong H, McDonough SP, Affolter VK, Moore PF. A novel canine leukointegrin, αdβ2, is expressed by specific macrophage subpopulations in tissue and a minor CD8+ lymphocyte subpopulation in peripheral blood. J Immunol. 1995;155:35–44. [PubMed] [Google Scholar]
- 31.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 32.Randolph GJ, Beaulieu S, Lebecque S, Steinman RM, Muller WA. Differentiation of monocytes into dendritic cells in a model of transendothelial trafficking. Science. 1998;282:480–483. doi: 10.1126/science.282.5388.480. [DOI] [PubMed] [Google Scholar]
- 33.Van der Laan LJ, Dopp EA, Haworth R, Pikkarainen T, Kangas M, Elomaa O, Dijkstra CD, Gordon S, Tryggvason K, Kraal G. Regulation and functional involvement of macrophage scavenger receptor MARCO in clearance of bacteria in vivo. J Immunol. 1999;162:939–947. [PubMed] [Google Scholar]
- 34.Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol. 2005;5:606–616. doi: 10.1038/nri1669. [DOI] [PubMed] [Google Scholar]
- 35.Yadava A, Kumar S, Dvorak JA, Milon G, Miller LH. Trafficking of Plasmodium chabaudi adami-infected erythrocytes within the mouse spleen. Proc Natl Acad Sci USA. 1996;93:4595–4599. doi: 10.1073/pnas.93.10.4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urban BC, Roberts DJ. Malaria, monocytes, macrophages, and myeloid dendritic cells: sticking of infected erythrocytes switches off host cells. Curr Opin Immunol. 2002;14:458–465. doi: 10.1016/s0952-7915(02)00368-0. [DOI] [PubMed] [Google Scholar]
- 37.de Souza JB, Riley EM. Cerebral malaria: the contribution of studies in animal models to our understanding of immunopathogenesis. Microbes Infect. 2002;4:291–300. doi: 10.1016/s1286-4579(02)01541-1. [DOI] [PubMed] [Google Scholar]
- 38.Hunt NH, Grau GE. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends Immunol. 2003;24:491–499. doi: 10.1016/s1471-4906(03)00229-1. [DOI] [PubMed] [Google Scholar]
- 39.Schofield L. Intravascular infiltrates and organ-specific inflammation in malaria pathogenesis. Immunol Cell Biol. 2007;85:130–137. doi: 10.1038/sj.icb.7100040. [DOI] [PubMed] [Google Scholar]
- 40.Beattie L, Engwerda CR, Wykes M, Good MF. CD8+ T lymphocyte-mediated loss of marginal metallophilic macrophages following infection with Plasmodium chabaudi chabaudi AS. J Immunol. 2006;177:2518–2526. doi: 10.4049/jimmunol.177.4.2518. [DOI] [PubMed] [Google Scholar]
- 41.Yoshimoto T, Takahama Y, Wang CR, Yoneto T, Waki S, Nariuchi H. A pathogenic role of IL-12 in blood-stage murine malaria lethal strain Plasmodium berghei NK65 infection. J Immunol. 1998;160:5500–5505. [PubMed] [Google Scholar]
- 42.Gordon S. Alternative activation of macrophages. Nat Immunol. 2002;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 43.Noel W, Raes G, Hassanzadeh Ghassabeh G, De Baetselier P, Beschin A. Alternatively activated macrophages during parasite infections. Trends Parasitol. 2004;20:126–133. doi: 10.1016/j.pt.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 44.Wijburg OL, Simmons CP, van Rooijen N, Strugnell RA. Dual role for macrophages in vivo in pathogenesis and control of murine Salmonella enterica var. Typhimurium infections. Eur J Immunol. 2000;30:944–953. doi: 10.1002/1521-4141(200003)30:3<944::AID-IMMU944>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 45.Guerau-de-Arellano M, Alroy J, Bullard D, Huber BT. Aggravated Lyme carditis in CD11a−/− and CD11c−/− mice. Infect Immun. 2005;73:7637–7643. doi: 10.1128/IAI.73.11.7637-7643.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghosh S, Chackerian AA, Parker CM, Ballantyne CM, Behar SM. The LFA-1 adhesion molecule is required for protective immunity during pulmonary Mycobacterium tuberculosis infection. J Immunol. 2006;176:4914–4922. doi: 10.4049/jimmunol.176.8.4914. [DOI] [PubMed] [Google Scholar]
- 47.Grau GE, Pointaire P, Piguet PF, Vesin C, Rosen H, Stamenkovic I, Takei F, Vassalli P. Late administration of monoclonal antibody to leukocyte function-antigen 1 abrogates incipient murine cerebral malaria. Eur J Immunol. 1991;21:2265–2267. doi: 10.1002/eji.1830210939. [DOI] [PubMed] [Google Scholar]
- 48.Falanga PB, Butcher EC. Late treatment with anti-LFA-1 (CD11a) antibody prevents cerebral malaria in a mouse model. Eur J Immunol. 1991;21:2259–2263. doi: 10.1002/eji.1830210938. [DOI] [PubMed] [Google Scholar]
- 49.Favre N, Da Laperousaz C, Ryffel B, Weiss NA, Imhof BA, Rudin W, Lucas R, Piguet PF. Role of ICAM-1 (CD54) in the development of murine cerebral malaria. Microbes Infect. 1999;1:961–968. doi: 10.1016/s1286-4579(99)80513-9. [DOI] [PubMed] [Google Scholar]
- 50.Urban BC, Hien TT, Day NP, Phu NH, Roberts R, Pongponratn E, Jones M, Mai NT, Bethell D, Turner GD, et al. Fatal Plasmodium falciparum malaria causes specific patterns of splenic architectural disorganization. Infect Immun. 2005;73:1986–1994. doi: 10.1128/IAI.73.4.1986-1994.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weiss L. Mechanisms of splenic control of murine malaria: cellular reactions of the spleen in lethal (strain 17XL) Plasmodium yoelii malaria in BALB/c mice, and the consequences of pre-infective splenectomy. Am J Trop Med Hyg. 1989;41:144–160. doi: 10.4269/ajtmh.1989.41.144. [DOI] [PubMed] [Google Scholar]
- 52.Evans KJ, Hansen DS, van Rooijen N, Buckingham LA, Schofield L. Severe malarial anemia of low parasite burden in rodent models results from accelerated clearance of uninfected erythrocytes. Blood. 2006;107:1192–1199. doi: 10.1182/blood-2005-08-3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McDevitt MA, Xie J, Shanmugasundaram G, Griffith J, Liu A, McDonald C, Thuma P, Gordeuk VR, Metz CN, Mitchell R, et al. A critical role for the host mediator macrophage migration inhibitory factor in the pathogenesis of malarial anemia. J Exp Med. 2006;203:1185–1196. doi: 10.1084/jem.20052398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yoshida H, Kawane K, Koike M, Mori Y, Uchiyama Y, Nagata S. Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature. 2005;437:754–758. doi: 10.1038/nature03964. [DOI] [PubMed] [Google Scholar]
- 55.Soni S, Bala S, Gwynn B, Sahr KE, Peters LL, Hanspal M. Absence of erythroblast macrophage protein (Emp) leads to failure of erythroblast nuclear extrusion. J Biol Chem. 2006;281:20181–20189. doi: 10.1074/jbc.M603226200. [DOI] [PubMed] [Google Scholar]
- 56.Rosales C, Juliano RL. Signal transduction by cell adhesion receptors in leukocytes. J Leukocyte Biol. 1995;57:189–198. doi: 10.1002/jlb.57.2.189. [DOI] [PubMed] [Google Scholar]
- 57.Marth T, Kelsall BL. Regulation of interleukin-12 by complement receptor 3 signaling. J Exp Med. 1997;185:1987–1995. doi: 10.1084/jem.185.11.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mosser DM, Karp CL. Receptor mediated subversion of macrophage cytokine production by intracellular pathogens. Curr Opin Immunol. 1999;11:406–411. doi: 10.1016/s0952-7915(99)80068-5. [DOI] [PubMed] [Google Scholar]
- 59.Sutterwala FS, Noel GJ, Clynes R, Mosser DM. Selective suppression of interleukin-12 induction after macrophage receptor ligation. J Exp Med. 1997;185:1977–1985. doi: 10.1084/jem.185.11.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jacobs T, Plate T, Gaworski I, Fleischer B. CTLA-4-dependent mechanisms prevent T cell induced-liver pathology during the erythrocyte stage of Plasmodium berghei malaria. Eur J Immunol. 2004;34:972–980. doi: 10.1002/eji.200324477. [DOI] [PubMed] [Google Scholar]
- 61.Kincy-Cain T, Clements JD, Bost KL. Endogenous and exogenous interleukin-12 augment the protective immune response in mice orally challenged with Salmonella dublin. Infect Immun. 1996;64:1437–1440. doi: 10.1128/iai.64.4.1437-1440.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stevenson MM, Riley EM. Innate immunity to malaria. Nat Rev Immunol. 2004;4:169–180. doi: 10.1038/nri1311. [DOI] [PubMed] [Google Scholar]