

Abstract
The analysis of reactions involving amino acids esterified to tRNAs traditionally uses radiolabeled amino acids. We describe here an alternative assay involving [3’-32P] labeled tRNA followed by nuclease digestion and TLC analysis that permits aminoacylation to be monitored in an efficient, quantitative manner while circumventing many of the problems faced when using radiolabeled amino acids. We also describe a similar assay using [3’-32P] labeled aa-tRNAs to determine the rate of peptide bond formation on the ribosome. This type of assay can also potentially be adapted to study other reactions involving an amino acid or peptide esterified to tRNA.
Keywords: ribosomes, synthetases
1. Introduction
The most common assay used to monitor the aminoacylation of tRNAs by aminoacyl tRNA synthetases (aaRSs) employs 3H or 14C labeled amino acids and unlabeled tRNAs [1]. The extent of the reaction is determined by separating the aa-tRNA from the excess free amino acid, usually by acid precipitation onto filters, and quantifying the fraction of aa-tRNA by liquid scintillation counting. This assay has been extensively used to determine the steady state kinetic parameters of many aaRSs as well as to determine the relative activities of mutant aaRSs or mutant tRNA substrates [2]. However, this assay has several shortcomings. First, radiolabeled amino acids are typically expensive and not always chemically stable. Second, the relatively weak KM of aaRSs for their cognate amino acids (10−6 to 10−4 M [3-6]) generally requires the use of subsaturating concentrations of amino acid or inconveniently high tRNA concentrations to avoid using excessive radioactivity. Third, determining the fraction of tRNA molecules that have been aminoacylated at a given time point is calculated from the amount of radioactive aa-tRNA. This measurement requires accurately knowing the specific activity of the amino acid, the scintillation counting efficiency on the filters, the fraction of active tRNA molecules present in the reaction, and the aliquot volume. Uncertainties in any of these values will propagate through the calculation resulting in a higher error in the final aminoacylation value. Finally, the relatively tight KM of aaRSs for their cognate tRNAs makes it difficult to adapt the assay to measure pre-steady state kinetics since not enough radioactivity will be present in the product [2]. Pre-steady state kinetics are essential for studying the mechanism of aaRSs since product release is often rate limiting for these enzymes [7].
An improved assay for following tRNA aminoacylation uses tRNA nucleotidyltransferase to introduce a 32P label on the 3’ terminal nucleotide of tRNA [8]. These [3’-32P] labeled tRNAs are then used in aaRS reactions containing unlabeled amino acids to form aa-tRNAs containing a 32P label on the 3’ terminus. By digesting the tRNA with S1 or P1 nuclease after the aminoacylation reaction, the [32P] aa-AMP and unreacted [32P] AMP can be separated by thin layer chromatography and quantitated on a phosphoimager. The fraction of aa-tRNA is then calculated directly. This assay is both more accurate and more sensitive than the one using radioactive amino acids. In addition, very low tRNA concentrations can be used, permitting pre-steady state kinetic measurements [9]. The amino acid choice is also unlimited and therefore permits using non-natural amino acids [10].
In this paper we review the protocol for radiolabeling tRNA using tRNA nucleotidyltransferase to exchange the endogenous terminal adenosine for [α-32P] ATP. We also present an example of using radiolabeled tRNAs to monitor the extent of aminoacylation and review examples of how this method has been used to monitor aaRS activity. Since radiolabeled tRNAs can be used to follow modifications to anything that is esterified to the tRNA, we also describe how the assay has recently been adapted to measure the rate of aa-tRNA conversion to dipeptidyl-tRNA catalyzed by the ribosome (Ledoux and Uhlenbeck, submitted). Finally, other enzymes which could be studied by similar assays using [3’-32P] labeled aa-tRNAs are discussed.
2. [3’-32P] Labeling tRNA using tRNA nucleotidyltransferase
2.1 tRNA nucleotidyltransferase
Radiolabeling the 3’ terminus of tRNA is performed utilizing the natural function of tRNA nucleotidyltransferase to exchange the endogenous A76 of tRNA with [α-32P] ATP. tRNA nucleotidyltransferase is responsible for the addition of the universally conserved 3’ terminal CCA sequence to tRNAs which do not encode this sequence within their genes [11]. Since this enzyme also catalyzes the reverse pyrophosphorolysis reaction, it can both degrade and reform the CCA sequence and thus is thought to maintain the 3’ end of all tRNAs in organisms such as E. coli which do encode the CCA sequence [12]. tRNA nucleotidyltransferase only recognizes the backbone of the acceptor and T stems of tRNA so that it can function with any tRNA including many mutants and minihelices [13-15]. tRNA nucleotidyltransferase has also been shown to catalyze the addition of several modified nucleotides onto tRNA [16, 17]. Crystal structures of the Archeoglobus fulgidus tRNA nucleotidyltransferase at various stages of the polymerization cycle show that it is able to carry out template-independent, sequence specific nucleotide addition using the conformational changes of the NTP binding pocket which sequentially stabilizes only CTP or ATP depending on the 3’ sequence of the tRNA [18]. tRNA nucleotidyltransferases from several different organisms have been purified and studied by many different labs [19-23] including the His-tagged E. coli enzyme [24].
2.2 [α-32P] ATP exchange labeling of tRNA
The exchange reaction catalyzed by the tRNA nucleotidyltransferase reaches equilibrium fairly slowly, so the protocol is divided into two steps to first favor the removal of the unlabeled endogenous adenosine and then second to favor the addition of the [α-32P] ATP (Fig. 1A). The first step involves adding pyrophosphate to a reaction containing a tRNA and tRNA nucleotidyltransferase to stimulate the removal of A76 to form ATP. The equilibrium is then shifted in the opposite direction by adding pyrophosphatase to degrade the free pyrophosphate and stimulate the nucleotidyltransferase catalyzed addition of [α-32P] ATP to the tRNA. The advantage of this two step procedure is that it greatly accelerates the equilibration of the [α-32P] ATP with the tRNA. The disadvantage is the possibility for side reactions that lead to alternative products is also enhanced. Thus it is critical to consider substrate concentrations and enzyme activities as well as reaction times to ensure proper product formation. The following protocol is based on a preparation of N terminally His6-tagged E. coli tRNA nucleotidyltransferase purified in our laboratory which is stored at a stock concentration of 10 μM at -20°C in 50% glycerol with 50 mM Tris (pH 7.8), 300 mM KCl, 1 mM MgCl2, 10 mM imidizole, and 1 mM β-Me. Since it is likely that the enzyme activity will vary depending on the individual preparation, it is essential to initially test the following protocol with several different enzyme concentrations to optimize the label incorporation while minimizing aberrant products.
Figure 1.
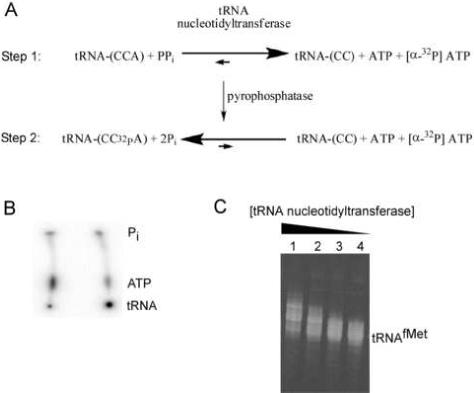
[3’-32P] labeling tRNA by nucleotide exchange. (A) Two step reaction scheme for nucleotide exchange by tRNA nucleotidyltransferase. (B) PEI cellulose TLC plate showing [α-32P] ATP before (left) and after (right) incorporation into tRNA with a yield of 76%. (C) 10% denaturing polyacrylamide gel showing 20 μM tRNAfMet after treatment different concentrations of tRNA nucleotidyltransferase: Lane 1: 6 μM; Lane 2: 2 μM; Lane 3: 0.67 μM; Lane 4: 0.22 μM.
Exchange labeling begins with pyrophosphorolysis of the 3’ terminal adenosine of tRNA by the tRNA nucleotidyltransferase (Fig. 1A, top). A typical 50 μL reaction consists of 1 μM tRNA, 50 μM sodium pyrophosphate (Sigma), 0.2 μM tRNA nucleotidyltransferase, 0.3 μM [α-32P] ATP (specific activity ∼3,000 Ci/mMole), 10 mM MgCl2, and 50 mM glycine (pH 9.0). Lower specific activity [α-32P] ATP or [3H] ATP can be used if desired, but the end product will be less radioactive. The reaction is incubated at 37°C for 5 minutes in order to complete the removal of the endogenous A76 residue. It is crucial that the reaction time remains short to minimize the subsequent removal of C74 and C75 of the tRNA. [α-32P] ATP is added at this step for convenience and is not necessary until the second phase of the reaction. It is important to keep the concentration of [α-32P] ATP lower than that of tRNA to prevent aberrant or excess ATP incorporation.
In the second step of the reaction, 1 μM CTP and 10 u/mL baker's yeast inorganic pyrophosphatase is added and the reaction is incubated at 37°C for an additional 2 minutes (Fig. 1A, bottom). The pyrophosphatase is supplied as a lyophilized powder by Sigma and should be is resuspended in 10 mM Tris (pH 7.0) to 10 u/mL. The pyrophosphatase is in sufficient excess to fully degrade the pyrophosphate added in the first step in less than a second and therefore does not have to be optimized. The CTP is added to ensure that those tRNAs missing C74 or C75 are properly repaired. If no CTP is added, labeling yields are often lower, but significantly higher CTP concentrations lead to aberrantly incorporated additional cytidines or CCA sequences [25].
Once the reaction conditions have been optimized to give a high yield of the proper product formation, the reaction must be phenol/chloroform extracted to remove the proteins from the reaction. The aqueous layer of the extraction is then run over two consecutive Micro Bio-Spin P-6 columns (Bio-Rad) to remove phenol and unreacted [α-32P] ATP which will contribute to a high background signal in the product analysis. The reaction is then ethanol precipitated and resuspended in a buffer appropriate for the experimental use of the radiolabeled tRNA. Gel purification is generally unnecessary.
2.3 Troubleshooting
Samples should be analyzed to ensure efficient labeling as well as proper product formation. According to the protocol above, 1 μM tRNA is equilibrated with 0.3 μM [α-32P] ATP and so if equilibration is complete the predicted yield of 32P incorporation into tRNA is 1/1.3, or 77%. This yield can be easily determined by spotting a small aliquot (< 1 μL) of the reaction onto a polyethylimine (PEI) cellulose thin layer chromatography (TLC) plate. By running the plate in saturated ammonium sulfate, [32P] ATP (Rf = 0.3) can be separated from [32P] tRNA which remains at the origin (Fig. 1B). If the [α-32P] ATP incorporation is low, the concentration of tRNA nucleotidyltransferase should be increased or the ratio of tRNA to [α-32P] ATP can be adjusted to favor a higher yield of [32P] tRNA. Since the TLC analysis will also separate any [32P] AMP and free inorganic phosphate [26], it is possible to check if the stock [32P] ATP is degraded or if phosphatase contamination is present in the tRNA nucleotidyltransferase or the pyrophosphatase preparations.
In order to check for aberrant nucleotide incorporation it is necessary to analyze the reaction products on a 10 to 20% denaturing polyacrylamide gel followed by phosphorimaging or staining. Alternatively, the reaction can first be digested with T1 nuclease which cleaves specifically after guanisine residues so that the 3’ terminally radiolabeled oligonucleotide can be visualized on a high percentage denaturing gel to check that only a single product is formed. If the radiolabeled tRNA shows length heterogeneity (Fig. 1C), lowering the CTP or tRNA nucleotidyltransferase concentrations or reducing incubation times is recommended.
tRNAs radiolabeled by this protocol have been used to examine many different aspects of protein synthesis. [3’-32P] tRNAs have been used to examine the interactions between tRNA and modification enzymes [27], the kinetics of tRNA binding to the ribosome [28-32], and the “structure mapping” of tRNAs bound to the ribosome [33].
3. Aminoacylation of [3’-32P] tRNAs
3.1 Aminoacylation
Since our laboratory is primarily interested in how aminoacyl-tRNAs function on the ribosome, the aminoacylation protocol described below is designed to maximize the fraction of aa-tRNA formed. However, the aminoacylation of [3’-32P] labeled tRNAs with unlabeled amino acids has been used to measure the reaction rate over a range of enzyme, amino acid, and tRNA concentrations [9, 34, 35]. The aminoacylation reaction is followed by digesting the tRNA with S1 or P1 nuclease and separating [32P] AMP from [32P] aa-AMP by TLC. Therefore the fraction of aminoacylated tRNA can be directly quantitated.
First, to ensure proper folding of the ethanol precipitated [3’-32P] tRNA, 2 uM [32P] tRNA in 30 mM HEPES (pH 7.0) and 30 mM KCl is heated to 70°C for 5 minutes after which 15 mM MgCl2 is added and the reaction is then allowed to cool to room temperature over a period of 30 minutes. Separately, an amino acid mix is assembled on ice by combining 250 μM amino acid, 30 mM HEPES (pH 7.0), 30 mM KCl, 15 mM MgCl2, 20 u/mL inorganic pyrophosphatase from baker's yeast, 10 mM DTT, 8 mM ATP, and 2 μM of the appropriate aaRS. Equal volumes of the tRNA and amino acid mixes, typically 25 μL each, are combined and incubated at 37°C for 10 minutes. 1/10 volume of 3 M NaOAc (pH 5.2) is then added both in preparation for ethanol precipitation and also to lower the pH in order to stabilize the labile ester linkage between amino acid and tRNA. The mixture is then phenol/chloroform extracted to remove protein and precipitated before resuspending in 5 mM NaOAc (pH 5.2). A low pH buffer is necessary to maintain the labile aminoacyl linkage. For the peptide bond formation reaction described later in this paper, tRNAs are resuspended to a final concentration of 500 nM.
3.2 Analysis of aminoacylated [3’-32P] tRNAs
The percentage of aminoacylated [3’-32P] tRNA is determined by digesting the tRNA with S1 or P1 nuclease which fully degrades the tRNA to yield 5’ monophosphates and [32P] aa-AMP. Both enzymes can function at low pH which is essential to avoid deacylation during the digestion reaction. For the analysis described below these nucleases are interchangeable and will give the same result. The digested products are then run on a PEI cellulose TLC plate to separate the [32P] AMP from the [32P] aa-AMP which permits quantitation of the fraction of aminoacylated tRNA using only a small amount of the original reaction (Fig. 2). Nuclease S1 is available as a 50% glycerol 100 u/μL stock from Fermentas with an acidic 5× reaction buffer. Nuclease P1 is available as a lyophilized powder from Sigma which can be resuspended in water to 100 u/μL and stored in single use aliquots at -20°C.
Figure 2.

Aminoacylation analysis of [3’-32P] tRNAAla by AlaRS. S1 nuclease digestion of Ala-tRNAAla results in free [32P] AMP and [32P] Ala-AMP that can be separated on a PEI cellulose TLC plate in glacial acetic acid/1 M NH4Cl/water (5:10:85). Reaction yields as a function of AlaRS concentration: Lane 1: 100 pM; Lane 2: 1 nM; Lane 3: 10 nM; Lane 4: 100 nM, Lane 5: 1 μM.
First, 4 μL of either nuclease S1, diluted in its 1× buffer, or P1 is mixed with 1 μL of 500 nM aa-tRNA and allowed to react at room temperature for 10 minutes. The sample should be completely digested since the enzyme is in vast. 1 μL of the reaction is then spotted on a PEI cellulose TLC plate that has been pre-run in water and dried. It is important to avoid drying the sample applied to the plate at temperatures above 25°C to prevent deacylation which would artifactually reduce the aminoacylation yield. Once the reaction spots have dried on the plate, it is run in glacial acetic acid/1 M NH4Cl/water (5:10:85). The Rf of AMP is approximately 0.5 and the Rf for most aminoacyl-AMPs is approximately 0.9, meaning that running the solvent approximately 10 cm will give excellent separation between the two bands in under half an hour. Figure 2 shows the aminoacylation yield of tRNA2Ala as a function of the concentration of AlaRS.
This direct determination of the fraction of aminoacylated tRNAs makes it easy to optimize several different reactions on a single TLC plate. For example, similar protocols have been used to test the esterification of multiple non-natural amino acids to tRNAs by synthetases [36-38] as well as to test rates and extents of misacylation of different tRNAs [35, 37, 39, 40]. This assay is especially beneficial for the study of non-natural amino acids since radioactive derivatives are generally not available [10]. Our laboratory has used this method with 13 different aaRSs with either cognate or non-cognate tRNAs [8, 30, 32] (Ledoux and Uhlenbeck, submitted). Other groups have reported that 15 of the 20 canonical amino acids, as well as pyrolysine, have been esterified to tRNAs using this approach (Table 1) [8, 30, 34-37, 39-44].
Table 1.
Synthetases used with 3′ 32P labeled tRNA
| aaRS | tRNA | aa | References |
|---|---|---|---|
| mtLeu | mt tRNALeu | Leu | [42] |
| Gln | tRNAGln | Gln | [34] |
| Ala | tRNAAla | Gly, Ala | [35] |
| mtPhe, mtTrp | tRNATrp, tRNAPhe | Trp, Phe, Tyr, Leu | [40] |
| Lys | tRNALys | Lys, Lys analogs, Arg, Glu, Gln | [37] |
| mtSer | mt tRNAAla, mt tRNAAsn | Ser | [39] |
| Pyl | tRNAPyl, tRNASer | Pyl analog | [41, 44] |
| Phe | tRNAPhe | Phe analogs | [36] |
| Trp | tRNATrp | Trp | [43] |
| Ala | tRNAAla | Ala | [8] |
| Gly, Arg, Val, His, Glu, Tyr, Phe, Lys | Cognate tRNAs | Cognate aa's | [30] |
| Ile, Met | tRNAIle, tRNAfMet | Ile, Met | [Ledoux and Uhlenbeck, submitted] |
4. Measuring rate of peptide bond formation using [3’-32P] aa-tRNAs
Aside from studying aminoacyl-tRNA synthetase reactions using [3’-32P] labeled tRNAs, this assay is useful for studying other reactions involving aa-tRNAs. Described below is an assay based on [3’-32P] labeled aa-tRNAs which measures kpep, the observed rate of peptide bond formation on the ribosome. This rate reflects the rate of the much slower aa-tRNA accommodation into the A site of the ribosome [45]. [3’-32P] labeled aa-tRNAs allow us to measure this step since [32P] aa-AMP and [32P] dipeptidyl-AMP can be separated on TLC plates.
4.1 Measuring the observed rate of peptide bond formation
The standard assay for measuring kpep uses [35S] fMet-tRNAfMet in the P site and an unlabeled, or 14C labeled, aa-tRNA in the A site [43, 45]. After peptidyl transfer, the dipeptide is cleaved from the A site tRNA by alkaline hydrolysis and the products are separated either by HPLC and analyzed by scintillation counting [45] or by paper electrophoresis followed by phosphorimaging [43]. The standard assay therefore indirectly measures the activity of the A site aa-tRNA on the ribosome by monitoring the P site amino acid. Both methods of product analysis use alkaline hydrolysis which can result in byproducts other than [35S] fMet and [35S] fMet-aa due to the oxidation of fMet. We have developed a kpep assay that uses [3’-32P] labeled aa-tRNAs in the A site and unlabeled fMet-tRNAfMet in the P site (Ledoux and Uhlenbeck, submitted). The reaction is otherwise very similar to the standard assay, although the high specific activity of the [3’-32P] aa-tRNA permits its use at lower concentrations. In addition, product analysis is somewhat faster and more easily quantified.
The first step in measuring kpep is to convert the stable EF-Tu/GDP to the active EF-Tu/GTP complex by adding 0.55 μM EF-Tu, 55 μM GTP, 3 mM phosphoenolpyruvate (Sigma), and 12 u/mL pyruvate kinase (Sigma) in ribosome buffer (50 mM HEPES (pH 7.0), 70 mM NH4Cl, 30 mM KCl, 10 mM MgCl2, 1 mM DTT) and incubating at 37°C for 20 minutes. The addition of EF-Tu's nucleotide exchange factor, EF-Ts, can enhance the rate of GTP exchange, but it is not necessary. The ternary complex is then formed simply by adding the EF-Tu/GTP mix to aa-tRNA for final concentrations of 50 nM [32P] aa-tRNA and 0.5 μM EF-Tu in ribosome buffer and allowing the complex to form on ice for 20 minutes. It is unnecessary to purify the ternary complex away from any deacyl tRNA since the data analysis of the kpep reaction includes the separation of deacyl tRNA from dipeptidyl-tRNA. While the ternary complex is being formed, ribosomes are heat activated by incubating at 42°C for 10 minutes, 37°C for 30 minutes, and then kept at room temperature. Ribosomes programmed with mRNA and fMet-tRNAfMet are obtained by incubating 0.5 μM of the heat activated ribosomes, 1.5 μM mRNA, and 1 μM fMet-tRNAfMet in ribosome buffer at room temperature for 20 minutes to allow for non-enzymatic binding of mRNA and fMet-tRNAfMet. Since kpep is approximately 2 s−1 for initiating ribosomes (Ledoux and Uhlenbeck submitted), a rapid quench device (KinTek) must be used to take timepoints. Equal volumes of ternary complex and programmed ribosomes, approximately 16 μL each, are incubated for set times ranging between 5 milliseconds and 30 seconds before quenching with 100mM EDTA, 5mM NaOAc (pH 4.5). Due to the nature of the rapid quench apparatus the quench volume ranges from 50 μL to 200 μL based on the length of the timepoint. The quench serves to both stabilize the aminoacyl- and dipeptidyl-tRNA bonds as well as to facilitate complex dissociation.
4.2 Analysis of products
The timepoints are then analyzed by the same method of determining aminoacylation yields described in 3.2. 1 μL of each quenched timepoint is added to 4 μL of either 100 u/μL S1 or P1 nuclease, incubated at room temperature for 10 minutes, and spotted on PEI cellulose TLC plates that have been pre-run in water and dried. TLC plates analyzing reactions containing small amino acids (Gly, Ala, Val, Ile) are run in glacial acetic acid/1 M NH4Cl/water (5:10:85) and separate into three bands, [32P] AMP, [32P] aa-AMP, and [32P] dipeptidyl-AMP as shown in Figure 3 for the reaction with Alanyl-tRNAAla. Reactions with glutaminyl-tRNA separate three bands in isopropanol/HCl/water (70:15:15). Reactions involving Phe and Tyr amino acids only resolve two bands in this solvent; [32P] AMP and [32P] aminoacyl-AMP comigrate while [32P] dipeptidyl-AMP runs separately which still allows for quantitation of dipeptide formation. The rate of dipeptide formation using this new assay is similar to the rate determined by the standard method [46, 47]. Table 2 shows the different TLC solvents used for this analysis along with the approximate Rf's for each band.
Figure 3.
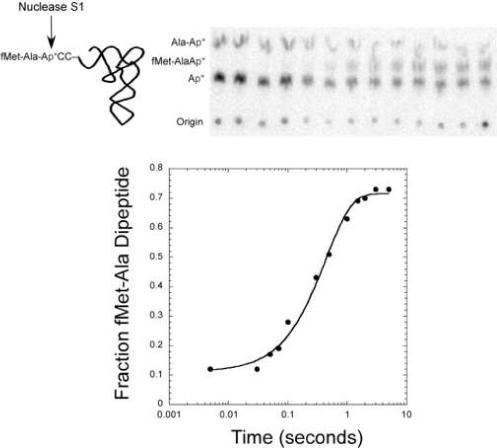
Analysis of kpep with fMet-tRNAfMet in the P site and [3’-32P] Ala-tRNAAla in the A site. S1 nuclease digestion of reaction mix results in free [32P] AMP, [32P] Ala-AMP, and [32P] fMet-Ala-AMP. Kinetic data is shown as digestion products developed on a PEI cellulose TLC plate in glacial acetic acid/1 M NH4Cl/water (5:10:85) and graphically to give kpep of 2.3 s−1.
Table 2.
aa-tRNAs Used to Measure kpep (Ledoux and Uhlenbeck, submitted)
| Amino acid esterified to A Site tRNA | TLC Solvent | Approximate Rf | ||
|---|---|---|---|---|
| AMP | aa-AMP | dipeptidyl-AMP | ||
| Ala, Gly, Ile, Val | A* | 0.5 | 0.9 | 0.7 |
| Glu | B** | 0.4 | 0.2 | 0.8 |
| Phe, Tyr | B | 0.4 | 0.4 | 0.8 |
A* Glacial acetic acid/1 M NH4Cl/water (5:10:85)
B** Ispropanol/HCl/water (70:15:15)
This assay is not only easy and requires low amounts of reactants, but the TLC analysis is faster and more convenient than the HPLC or electrophoretic analysis. This permits comparison of multiple aa-tRNAs in a single experiment.
5. Other potential applications for [3’-32P] tRNAs
In addition to studying tRNA function with synthetases and the ribosome, similar assays could be developed for several other enzymes that use aa-tRNAs as substrates. We have found that methionyl-AMP and formylmethionyl-AMP migrate differently on the PEI cellulose TLC plates. In the glacial acetic acid/1 M NH4Cl/water solvent AMP comigrates with formylmethionyl-AMP while in the isopropanol/HCl/water solvent methionyl-AMP comigrates with AMP. Thus either of these solvents could be used to monitor the activity of methionyl-tRNA formyltransferase. It is likely that a similar assay could be used to monitor the transamidation reactions that converts Gln-tRNAGln from Glu-tRNAGln or Asn-tRNAAsn from Asp-tRNAAsn [48]. In addition, Ser-tRNASec conversion to Sec-tRNASec by SelA [49], the interaction between O-phosphoserine-tRNACys and Cys synthase [50], or Glutamyl-tRNA reductase's modification of Glu-tRNAGlu [48] could all be assayed similarly. Indeed any enzyme that modifies what is esterified to the 3’ terminus of tRNA could be studied using a variation of this assay.
Acknowledgements
The project described above was supported by R01-GM37552-21 from the National Institutes of Health, and 1C06RR018850-01from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of NCRR or NIH. The authors would also like to thank: Dr. Ying Zhao for allowing us to include unpublished data, and Dr. Alexey Wolfson for the original development of the labeling assay.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hoben P, Soll D. Glutaminyl-tRNA synthetase of Escherichia coli. Methods Enzymol. 1985;113:55–9. doi: 10.1016/s0076-6879(85)13011-9. [DOI] [PubMed] [Google Scholar]
- 2.Giege R. The early history of tRNA recognition by aminoacyl-tRNA synthetases. J Biosci. 2006;31(4):477–88. doi: 10.1007/BF02705187. [DOI] [PubMed] [Google Scholar]
- 3.Willison JC, Hartlein M, Leberman R. Isolation and characterization of an Escherichia coli seryl-tRNA synthetase mutant with a large increase in Km for serine. J Bacteriol. 1995;177(11):3347–50. doi: 10.1128/jb.177.11.3347-3350.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ibba M, et al. Interactions between tRNA identity nucleotides and their recognition sites in glutaminyl-tRNA synthetase determine the cognate amino acid affinity of the enzyme. Proc Natl Acad Sci U S A. 1996;93(14):6953–8. doi: 10.1073/pnas.93.14.6953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hill K, Schimmel P. Evidence that the 3' end of a tRNA binds to a site in the adenylate synthesis domain of an aminoacyl-tRNA synthetase. Biochemistry. 1989;28(6):2577–86. doi: 10.1021/bi00432a035. [DOI] [PubMed] [Google Scholar]
- 6.Eriani G, et al. Role of dimerization in yeast aspartyl-tRNA synthetase and importance of the class II invariant proline. Proc Natl Acad Sci U S A. 1993;90(22):10816–20. doi: 10.1073/pnas.90.22.10816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang CM, et al. Distinct kinetic mechanisms of the two classes of Aminoacyl-tRNA synthetases. J Mol Biol. 2006;361(2):300–11. doi: 10.1016/j.jmb.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 8.Wolfson AD, Uhlenbeck OC. Modulation of tRNAAla identity by inorganic pyrophosphatase. Proc Natl Acad Sci U S A. 2002;99(9):5965–70. doi: 10.1073/pnas.092152799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uter NT, Perona JJ. Long-range intramolecular signaling in a tRNA synthetase complex revealed by pre-steady-state kinetics. Proc Natl Acad Sci U S A. 2004;101(40):14396–401. doi: 10.1073/pnas.0404017101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartman MC, Josephson K, Szostak JW. Enzymatic aminoacylation of tRNA with unnatural amino acids. Proc Natl Acad Sci U S A. 2006;103(12):4356–61. doi: 10.1073/pnas.0509219103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schurer H, et al. This is the end: processing, editing and repair at the tRNA 3'-terminus. Biol Chem. 2001;382(8):1147–56. doi: 10.1515/BC.2001.144. [DOI] [PubMed] [Google Scholar]
- 12.Zhu L, Deutscher MP. tRNA nucleotidyltransferase is not essential for Escherichia coli viability. Embo J. 1987;6(8):2473–7. doi: 10.1002/j.1460-2075.1987.tb02528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Z, Sun Y, Thurlow DL. RNA minihelices as model substrates for ATP/CTP:tRNA nucleotidyltransferase. Biochem J. 1997;327(Pt 3):847–51. doi: 10.1042/bj3270847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xiong Y, Steitz TA. A story with a good ending: tRNA 3'-end maturation by CCA-adding enzymes. Curr Opin Struct Biol. 2006;16(1):12–7. doi: 10.1016/j.sbi.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 15.Weiner AM. tRNA maturation: RNA polymerization without a nucleic acid template. Curr Biol. 2004;14(20):R883–5. doi: 10.1016/j.cub.2004.09.069. [DOI] [PubMed] [Google Scholar]
- 16.Cho HD, et al. Use of nucleotide analogs by class I and class II CCA-adding enzymes (tRNA nucleotidyltransferase): deciphering the basis for nucleotide selection. Rna. 2003;9(8):970–81. doi: 10.1261/rna.2110903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sprinzl M, et al. Enzymatic incorporation of ATP and CTP analogues into the 3' end of tRNA. Eur J Biochem. 1977;81(3):579–89. doi: 10.1111/j.1432-1033.1977.tb11985.x. [DOI] [PubMed] [Google Scholar]
- 18.Xiong Y, Steitz TA. Mechanism of transfer RNA maturation by CCA-adding enzyme without using an oligonucleotide template. Nature. 2004;430(7000):640–5. doi: 10.1038/nature02711. [DOI] [PubMed] [Google Scholar]
- 19.Yue D, Maizels N, Weiner AM. CCA-adding enzymes and poly(A) polymerases are all members of the same nucleotidyltransferase superfamily: characterization of the CCA-adding enzyme from the archaeal hyperthermophile Sulfolobus shibatae. Rna. 1996;2(9):895–908. [PMC free article] [PubMed] [Google Scholar]
- 20.Shi PY, Maizels N, Weiner AM. Recovery of soluble, active recombinant protein from inclusion bodies. Biotechniques. 1997;23(6):1036–8. doi: 10.2144/97236bm15. [DOI] [PubMed] [Google Scholar]
- 21.Shi PY, Maizels N, Weiner AM. CCA addition by tRNA nucleotidyltransferase: polymerization without translocation? Embo J. 1998;17(11):3197–206. doi: 10.1093/emboj/17.11.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cudny H, et al. Cloning, sequencing, and species relatedness of the Escherichia coli cca gene encoding the enzyme tRNA nucleotidyltransferase. J Biol Chem. 1986;261(14):6444–9. [PubMed] [Google Scholar]
- 23.Aebi M, et al. Isolation of a temperature-sensitive mutant with an altered tRNA nucleotidyltransferase and cloning of the gene encoding tRNA nucleotidyltransferase in the yeast Saccharomyces cerevisiae. J Biol Chem. 1990;265(27):16216–20. [PubMed] [Google Scholar]
- 24.Shi PY, Weiner AM, Maizels N. A top-half tDNA minihelix is a good substrate for the eubacterial CCA-adding enzyme. Rna. 1998;4(3):276–84. [PMC free article] [PubMed] [Google Scholar]
- 25.Hou YM. Unusual synthesis by the Escherichia coli CCA-adding enzyme. Rna. 2000;6(7):1031–43. doi: 10.1017/s1355838200000686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bochner BR, Ames BN. Complete analysis of cellular nucleotides by two-dimensional thin layer chromatography. J Biol Chem. 1982;257(16):9759–69. [PubMed] [Google Scholar]
- 27.Sabina J, Soll D. The RNA-binding PUA domain of archaeal tRNA-guanine transglycosylase is not required for archaeosine formation. J Biol Chem. 2006;281(11):6993–7001. doi: 10.1074/jbc.M512841200. [DOI] [PubMed] [Google Scholar]
- 28.Fahlman RP, Olejniczak M, Uhlenbeck OC. Quantitative analysis of deoxynucleotide substitutions in the codon-anticodon helix. J Mol Biol. 2006;355(5):887–92. doi: 10.1016/j.jmb.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 29.Olejniczak M, et al. Idiosyncratic tuning of tRNAs to achieve uniform ribosome binding. Nat Struct Mol Biol. 2005;12(9):788–93. doi: 10.1038/nsmb978. [DOI] [PubMed] [Google Scholar]
- 30.Fahlman RP, Dale T, Uhlenbeck OC. Uniform binding of aminoacylated transfer RNAs to the ribosomal A and P sites. Mol Cell. 2004;16(5):799–805. doi: 10.1016/j.molcel.2004.10.030. [DOI] [PubMed] [Google Scholar]
- 31.Fahlman RP, Uhlenbeck OC. Contribution of the esterified amino acid to the binding of aminoacylated tRNAs to the ribosomal P- and A-sites. Biochemistry. 2004;43(23):7575–83. doi: 10.1021/bi0495836. [DOI] [PubMed] [Google Scholar]
- 32.Dale T, Uhlenbeck OC. Binding of misacylated tRNAs to the ribosomal A site. Rna. 2005;11(11):1610–5. doi: 10.1261/rna.2130505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huggins W, Wollenzien P. A 16S rRNA-tRNA product containing a nucleotide phototrimer and specific for tRNA in the P/E hybrid state in the Escherichia coli ribosome. Nucleic Acids Res. 2004;32(22):6548–56. doi: 10.1093/nar/gkh1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bullock TL, et al. Amino acid discrimination by a class I aminoacyl-tRNA synthetase specified by negative determinants. J Mol Biol. 2003;328(2):395–408. doi: 10.1016/s0022-2836(03)00305-x. [DOI] [PubMed] [Google Scholar]
- 35.Beebe K, Merriman E, Schimmel P. Structure-specific tRNA determinants for editing a mischarged amino acid. J Biol Chem. 2003;278(46):45056–61. doi: 10.1074/jbc.M307080200. [DOI] [PubMed] [Google Scholar]
- 36.Bentin T, et al. Photoreactive bicyclic amino acids as substrates for mutant Escherichia coli phenylalanyl-tRNA synthetases. J Biol Chem. 2004;279(19):19839–45. doi: 10.1074/jbc.M401278200. [DOI] [PubMed] [Google Scholar]
- 37.Levengood J, et al. Divergence in noncognate amino acid recognition between class I and class II lysyl-tRNA synthetases. J Biol Chem. 2004;279(17):17707–14. doi: 10.1074/jbc.M313665200. [DOI] [PubMed] [Google Scholar]
- 38.Josephson K, Hartman MC, Szostak JW. Ribosomal synthesis of unnatural peptides. J Am Chem Soc. 2005;127(33):11727–35. doi: 10.1021/ja0515809. [DOI] [PubMed] [Google Scholar]
- 39.Shimada N, et al. Quality control of translation through the kinetic discrimination of tRNAs in the network of aminoacyl-tRNA synthetases. Nucleic Acids Res Suppl. 2002;(2):79–80. doi: 10.1093/nass/2.1.79. [DOI] [PubMed] [Google Scholar]
- 40.Roy H, et al. Loss of editing activity during the evolution of mitochondrial phenylalanyl-tRNA synthetase. J Biol Chem. 2005;280(46):38186–92. doi: 10.1074/jbc.M508281200. [DOI] [PubMed] [Google Scholar]
- 41.Ambrogelly A, et al. Pyrrolysine is not hardwired for cotranslational insertion at UAG codons. Proc Natl Acad Sci U S A. 2007;104(9):3141–6. doi: 10.1073/pnas.0611634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sohm B, et al. Recognition of human mitochondrial tRNALeu(UUR) by its cognate leucyl-tRNA synthetase. J Mol Biol. 2004;339(1):17–29. doi: 10.1016/j.jmb.2004.03.066. [DOI] [PubMed] [Google Scholar]
- 43.Cochella L, Green R. An active role for tRNA in decoding beyond codon:anticodon pairing. Science. 2005;308(5725):1178–80. doi: 10.1126/science.1111408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herring S, et al. Recognition of pyrrolysine tRNA by the Desulfitobacterium hafniense pyrrolysyl-tRNA synthetase. Nucleic Acids Res. 2007;35(4):1270–8. doi: 10.1093/nar/gkl1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pape T, Wintermeyer W, Rodnina MV. Complete kinetic mechanism of elongation factor Tu-dependent binding of aminoacyl-tRNA to the A site of the E. coli ribosome. Embo J. 1998;17(24):7490–7. doi: 10.1093/emboj/17.24.7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gromadski KB, Daviter T, Rodnina MV. A uniform response to mismatches in codon-anticodon complexes ensures ribosomal fidelity. Mol Cell. 2006;21(3):369–77. doi: 10.1016/j.molcel.2005.12.018. [DOI] [PubMed] [Google Scholar]
- 47.Cochella L, Brunelle JL, Green R. Mutational analysis reveals two independent molecular requirements during transfer RNA selection on the ribosome. Nat Struct Mol Biol. 2007;14(1):30–6. doi: 10.1038/nsmb1183. [DOI] [PubMed] [Google Scholar]
- 48.Feng L, et al. Aminoacyl-tRNA synthesis by pre-translational amino acid modification. RNA Biol. 2004;1(1):16–20. [PubMed] [Google Scholar]
- 49.Allmang C, Krol A. Selenoprotein synthesis: UGA does not end the story. Biochimie. 2006;88(11):1561–71. doi: 10.1016/j.biochi.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 50.Sauerwald A, et al. RNA-dependent cysteine biosynthesis in archaea. Science. 2005;307(5717):1969–72. doi: 10.1126/science.1108329. [DOI] [PubMed] [Google Scholar]