

Summary
Notch signaling is broadly used to regulate cell fate decisions. We have identified a novel gene, rumi, with a temperature-sensitive Notch phenotype. At 28-30°C, rumi clones exhibit a full-blown loss of Notch signaling in all tissues tested. However, at 18°C only a mild Notch phenotype is evident. In vivo analyses reveal that the target of Rumi is the extracellular domain of Notch. Notch accumulates intracellularly and at the cell membrane of rumi cells, but fails to be properly cleaved, despite normal binding to Delta. Rumi is an endoplasmic reticulum-retained protein with a highly conserved CAP10 domain. Our studies show that Rumi is a protein O-glucosyltransferase, capable of adding glucose to serine residues in Notch EGF repeats with the consensus C1-X-S-X-P-C2 sequence. These data indicate that by O-glucosylating Notch in the ER, Rumi regulates its folding and/or trafficking and allows signaling at the cell membrane.
Keywords: temperature-sensitive, Notch signaling, glucose, EGF repeats, glycosyltransferase, Endoplasmic reticulum, Drosophila
Introduction
Notch signaling is one of the most widely used signaling pathways in animals (Artavanis-Tsakonas et al., 1999). It is required for maintenance of the undifferentiated state, lateral inhibition, asymmetric cell divisions, vertebrate somitogenesis, cortical neurite outgrowth, and differentiation. Aberrant Notch signaling has been implicated in human diseases including cerebrovascular dementia (CADASIL) (Joutel et al., 1996), cancer (Bolos et al., 2007) as well as developmental disorders of liver, heart, skeleton, eye, and kidney (Li et al., 1997; Oda et al., 1997). It has also been shown to play important roles in stem cell biology (Carlson and Conboy, 2007).
The core components of the Notch pathway are the transmembrane ligands (Delta and Serrate in flies) and receptor (Notch), and CSL transcription factors (Suppressor of Hairless in flies) (Lai, 2004; Schweisguth, 2004). Upon ligand binding, Notch is cleaved by an ADAM metalloprotease (Kuzbanian in flies), followed by an intramembranous cleavage mediated by the gamma-secretase complex (Brou et al., 2000; De Strooper et al., 1999; Mumm et al., 2000; Pan and Rubin, 1997; Struhl and Greenwald, 1999). The latter cleavage leads to translocation of the Notch intracellular domain (NICD) to the nucleus, where it binds CSL proteins to activate downstream effectors (Jarriault et al., 1995; Lecourtois and Schweisguth, 1995; Struhl and Adachi, 1998). In addition, there are many important proteins involved in the regulation of the pathway which function to regulate endocytosis, ubiquitination, intracellular trafficking, degradation and glycosylation of various components (Haines and Irvine, 2003; Hori et al., 2004; Le Borgne et al., 2005).
The extracellular domain of Notch (NECD) is approximately 200 kDa and contains 36 tandem Epidermal Growth Factor-like (EGF) repeats. The EGF repeats undergo O-fucosylation and O-glucosylation (Moloney et al., 2000b). The O-fucosyltransferase-1 (Pofut1 in mammals, Ofut1 in flies) adds fucose (Shao and Haltiwanger, 2003; Wang et al., 2001) and is required for folding of Notch in the ER (Okajima et al., 2005), for Notch-ligand interaction (Okajima et al., 2003), and for intracellular trafficking of Notch (Sasaki et al., 2007; Sasamura et al., 2007). Interestingly, some of these roles do not seem to require enzymatic function (Okajima et al., 2005; Sasamura et al., 2007). Moreover, loss of Ofut1 (Pofut1 in mice) results in Notch loss-of-function phenotypes in flies and mice (Okajima and Irvine, 2002; Sasamura et al., 2003; Shi and Stanley, 2003). The fucose residue added to Notch by Pofut1 can be further modified by Fringe proteins, another glycosyltransferase family that add N-acetylglucosamine to O-fucose residues linked to specific EGF repeats (Moloney et al., 2000a; Rampal et al., 2005; Shao et al., 2003). This modification alters the binding of Notch to Delta and Serrate and regulates Notch signaling in specific contexts (Bruckner et al., 2000; Okajima et al., 2003; Panin et al., 1997).
Notch is also O-glucosylated at serine residues between the first and second cysteine residues of EGF repeats that contain a C1-X-S-X-P-C2 consensus (Moloney et al., 2000b; Shao et al., 2002). Protein O-glucosylation is a rare modification that occurs on EGF repeats of a few proteins including coagulation factors VII and IX, protein Z, Delta-like protein, and Thrombospondin (Shao et al., 2002). Even though an enzymatic activity able to O-glucosylate EGF repeats is present in cell extracts from a variety of species (Shao et al., 2002), no specific protein has been identified that O-glucosylates Notch or any other protein. Although Drosophila Notch carries 19 putative O-glucosylation sites, many of which are evolutionarily conserved (Haines and Irvine, 2003; Moloney et al., 2000b; Shao et al., 2002), the in vivo role of O-glucosylation is unknown.
In a mosaic genetic screen designed to identify mutants that affect bristle development in flies (Jafar-Nejad et al., 2005), we have isolated a novel gene named rumi that causes a temperature-sensitive (ts) loss of bristles. Loss of rumi affects Notch signaling in all tissues tested. rumi encodes a soluble, ER protein with a CAP10 domain, which is involved in capsule formation and virulence in Cryptococcus neoformans (Chang and Kwon-Chung, 1999). Rumi has highly conserved homologues in species from yeast to human, but its role is unknown in multicellular organisms (Chang and Kwon-Chung, 1999; Teng et al., 2006). Our data indicate that Rumi regulates Notch signaling by modifying Notch in the ER, and that Rumi is a protein O-glucosyltransferase (Poglut). We propose that lack of O-glucosylation of Notch in rumi mutants results in a ts defect in Notch folding and signaling.
Results
rumi mutations cause a temperature-dependent loss of Notch signaling
We performed a chemical mutagenesis screen to identify novel genes that affect adult bristle development (Jafar-Nejad et al., 2005) (Figure 1A). One of the complementation groups, named rumi (after a 13th century poet), showed severe bristle loss in mitotic clones when raised at 25°C (Figure 1B). However, when grown at 18°C, mutant clones did not show bristle loss (Figure 1C) but exhibited an increase in bristle density, suggesting a mild lateral inhibition defect (Figure 1D). To determine the cause of bristle loss, we stained rumi pupae raised at 25°C or 18°C for Cut, a protein which marks the nuclei of all cells of sensory clusters and for ELAV, which marks neurons. As shown in Figures 1E and 1E’, all cells in a rumi sensory cluster raised at 25°C express ELAV, indicating a Notch-like cell fate specification defect. However, rumi pupae raised at 18°C contain a single neuron in each sensory cluster (Figures 1F and 1F’), similar to wild-type pupae.
Figure 1. rumi mutations cause a ts Notch phenotype.
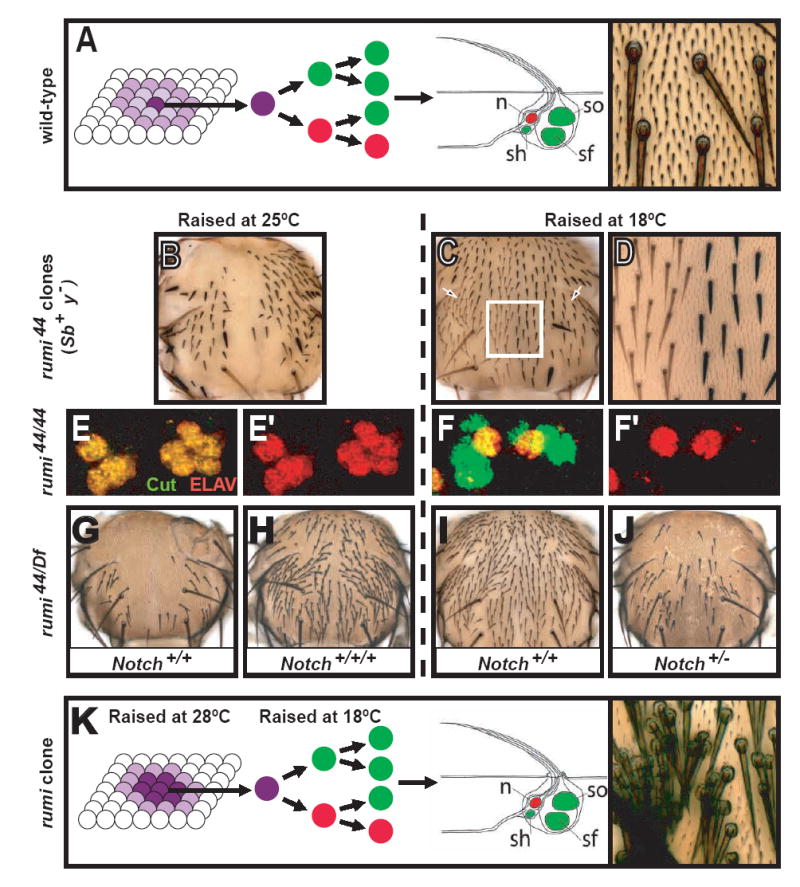
(A) Formation of an external sensory (es) organ from a single sensory organ precursor (SOP) cell is shown. Once the SOP is specified, it undergoes a series of asymmetric cell divisions to form the sensory organ structure. Only a single neuron forms in each sensory organ (red). The right picture shows wild-type microchaetae located on the thorax. n: neuron, so: socket cell, sh: sheath cell, sf: shaft cell. Bristle schematic adapted from (Lai and Orgogozo, 2004).
(B) Bristle development is impaired in rumi clones at 25°C. Heterozygote bristles are marked with Sb and y+ markers. Note that not all bristles are lost at this temperature.
(C and D) Bristles form in a denser pattern in rumi clones raised at 18°C. Non-Sb and yellow bristles mark rumi clones (compare the regions marked by the two arrows in (C)).
(D) is a close-up of the white square.
(E-F’) Cell fate specification is impaired in the adult microchaetae lineages of rumi mutants raised at 25°C, as evidenced by presence of multiple ELAV+ cells (red) per cluster (E and E’). Note that when raised at 18°C, only one of the Cut+ cells (green) per cluster expresses ELAV (F and F’).
(G-J) Genetic interaction between Notch and rumi.
(G) Hemizygous rumi44 flies that eclose at 25°C lose most of their microchaetae. (H) Adding an extra copy of Notch rescues many of the lost bristles (Compare G and H).
(I) At 18°C, rumi mutant flies do not exhibit a significant bristle loss.
(J) Flies heterozygous for Notch and hemizygous for rumi44, however, lose many bristles at 18°C (Compare I and J).
(K) Tufts of bristles form in rumi mutant clones in flies that are kept at 28°C during SOP specification and at 18°C during asymmetric cell divisions, indicating an important role for rumi during lateral inhibition.
To provide a more direct link between rumi and Notch signaling, we performed genetic interaction experiments. Some rumi mutant animals reach adulthood at 25°C. These flies show a severe loss of microchaetae (Figure 1G). Adding one copy of Notch+ restores most microchaetae at 25°C, indicating that the phenotype is sensitive to Notch dosage (Figure 1H). When raised at 18°C, rumi mutant animals do not show a bristle loss (Figure 1I), but removing a copy of Notch in these females results in a loss of microchaetae (Figure 1J). These data indicate that increasing the temperature results in a worsening of the Notch phenotype in rumi animals. Indeed, a complete loss of microchaetae in rumi animals raised at 29°C during early pupal stage cannot be rescued with an additional copy of Notch (Figure S1).
To demonstrate that rumi affects lateral inhibition, we performed temperature shift experiments. Pupae harboring rumi clones were raised at room temperature, shifted to 28°C during lateral inhibition, and shifted back to 18°C during the asymmetric divisions (Figure 1K). Under this regimen, flies show a large excess of sensory bristles in mutant clones (Figure 1K). Hence, rumi regulates Notch signaling during lateral inhibition and asymmetric divisions of sensory precursors.
To determine if rumi affects Notch signaling in other contexts, we examined the embryonic nervous systems. As shown in Figures 2A-D, embryos lacking maternal and zygotic Rumi raised at 28°C have a neurogenic phenotype, similar to Notch embryos. Clonal analysis in the wing showed that ‘inductive signaling’ (Lai, 2004) is also affected (Figure 2F, asterisks). Immunohistochemical staining shows a loss of Cut and Wingless expression in rumi clones (Figures 2G-J’). Moreover, genetic studies reveal a strong dosage-sensitive interaction between rumi and Delta in wing, eye and leg development (Figures 2K-M and Figure S2). These data indicate that Rumi is a general regulator of Notch signaling.
Figure 2. Loss of rumi causes loss of Notch signaling in various contexts.
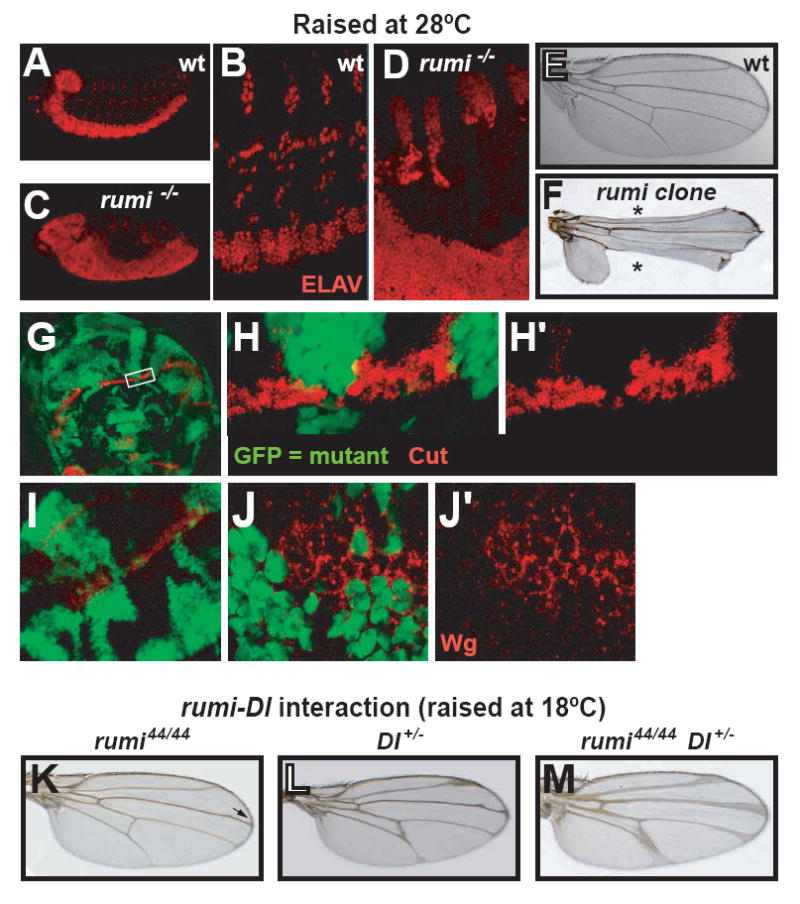
(A-D) rumi embryos laid by homozygous rumi flies show a neurogenic phenotype at 28°C (compare A and B with C and D). Neurons are marked with ELAV (red).
(E-F) Wing margin development is impaired in rumi clones, causing Notching of the wings (asterisks in F). Panel (F) shows the wing of an adult who was subjected to the temperature-shift experiments described in Figure 1K.
(G-J’) Expression of the Notch downstream targets Cut (red in G-H’) and Wg (red in I-J’) is lost in rumi clones at the dorso-ventral boundary of the third instar wing imaginal disc at 28°C in a cell-autonomous manner. GFP (green) marks rumi mutant cells.
(K-M) Genetic interaction between Delta and rumi. All flies were raised at 18°C.
(K) Wings of the homozygous rumi flies exhibit a mild Delta phenotype (arrow).
(L) A wing of a fly which is heterozygous mutant for Delta.
(M) There is a synergistic increase in wing vein expansion in flies that are homozygous mutant for rumi and heterozygous for Delta, indicating that rumi and Delta genetically interact.
rumi encodes a CAP10-like protein
To identify rumi, we mapped the locus (Zhai et al., 2003) (Figure 3A) and identified lesions in CG31152, which encodes a conserved protein (Figure 3C) with a signal peptide, a CAP10 domain, and a C-terminal KDEL ER-retention motif (Figure 3B). Allele 44 contains an in-frame deletion and allele79 harbors a missense mutation, G189E (Figure 3B). All homo- and transheterozygous combinations of these alleles in combination with Df(3R)Exel6192 produce viable progeny and exhibit a ts Notch phenotype.
Figure 3. rumi corresponds to CG31152.
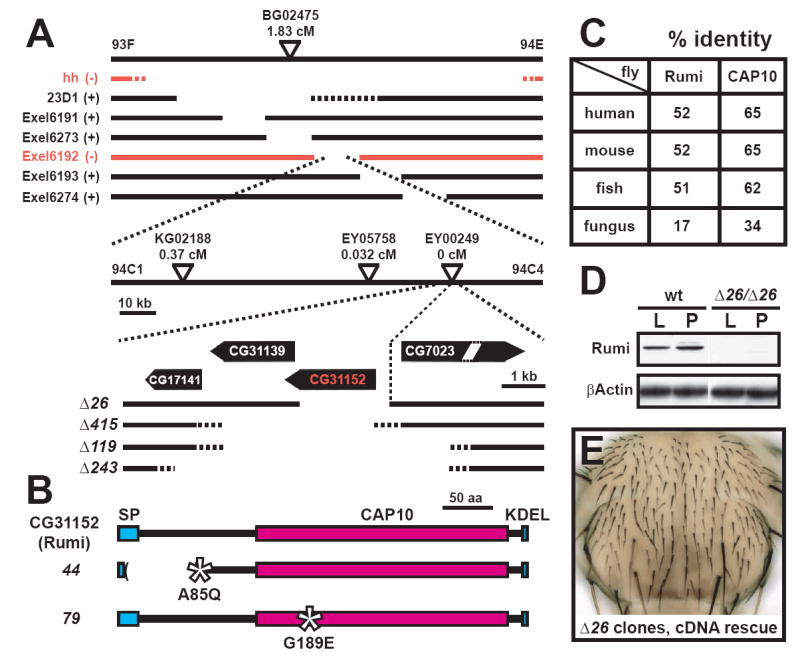
(A) The mapping strategy for rumi. The genomic region containing the rumi locus is shown. The broken lines depict deficiencies used to map rumi. hh and Exel6192 deletions (red lines) failed to complement rumi alleles. The genetic distance between the P-elements used for fine mapping and rumi is shown in centiMorgans (cM). Imprecise excision of EY00249 generated the additional alleles that remove rumi (CG31152) alone or in combination with CG31139, the only other fly gene capable of encoding a CAP10 domain.
(B) rumi encodes a protein with a CAP10 domain, a KDEL ER-retention signal located at the C-terminus and a signal peptide (SP) located at the N terminus. Molecular lesions in alleles 44 and 79 are shown. Asterisks denote missense mutations.
(C) Rumi protein, especially the CAP10 domain is highly conserved across different species.
(D) Western blotting on the protein extracts from wild-type (wt) and rumiΔ26 larvae (L) and pupae (P) with a polyclonal α-Rumi antibody indicates that rumiΔ26 is a protein-null allele.
(E) A UAS-rumi transgene can rescue the bristles lost in rumi clones. The bristles rescued by the transgene are yellow.
The temperature-sensitivity of the rumi alleles may be due to an abnormal Rumi protein that fails to function at high temperatures. Alternatively, rumi’s neighbor, CG31139 (Figure 3A)—the only other fly gene encoding a CAP10 domain protein—may compensate in part for the lack of rumi, resulting in a ts phenotype. We therefore excised P-element EY00249 inserted 238 bps upstream of CG31152 (Bellen et al., 2004). All deletions generated by imprecise excisions lack most of the rumi ORF (Figure 3A), and an antibody raised against Rumi failed to detect the protein in Δ26/Δ26 animals, indicating that Δ26 is a null allele (Figure 3D). Complementation analysis of the excisions and EMS induced alleles showed that all alleles in combination with each other or with Df(3R)Exel6192 exhibit the ts phenotype. Hence, loss of rumi per se is responsible for the temperature sensitivity. These data also indicate that the partner of rumi (CG31139) is not redundant, as flies that carry a deletion of both genes are viable at 18°C and exhibit a ts phenotype. Finally, all allelic combinations can be rescued with a UAS-CG31152 or a genomic transgene only containing CG31152 (Figure 3E and data not shown). Hence, loss of CG31152 is the cause of the loss-of-function phenotypes of rumi mutants and Rumi regulates a ts aspect of Notch signaling.
rumi is required in the signal-receiving cell
To assess whether rumi is required in the signal-sending and/or receiving cell, we used the MARCM system (Lee and Luo, 2001) to overexpress Delta, Serrate, and Notch in rumi clones (28°C). If Rumi is essential for Delta or Serrate to induce Notch signaling in the neighboring cells, then expression of Delta and Serrate should not be able to induce Cut expression in cells along the border of the MARCM clones, as reported for epsin mutations (Wang and Struhl, 2004). As shown in Figures S3A-B’, overexpression of Delta or Serrate in rumi clones results in expression of Cut, suggesting that the signal-sending cell does not require Rumi. Moreover, wing imaginal discs harboring rumi clones raised at 28°C and stained with anti-Delta or anti-Serrate show no alteration in the expression of these proteins (Figures S3C-D’). These data argue against a role for Rumi in the signal-sending cell and against a requirement for rumi for the function of Delta or Serrate.
To assess the function of Rumi in the signal-receiving cell, we performed similar experiments with full-length Notch (NFL). When overexpressed in clones homozygous for a wild-type chromosome, NFL induces Cut expression in proximity of the wing margin (Figures 4A and 4A’), as reported (Sasamura et al., 2003). However, NFL failed to induce Cut expression in rumi clones (Figures 4B-B’). These observations indicate a requirement for Rumi in the signal-receiving cells.
Figure 4. rumi is required in the signal-receiving cell upstream of the S3 cleavage.
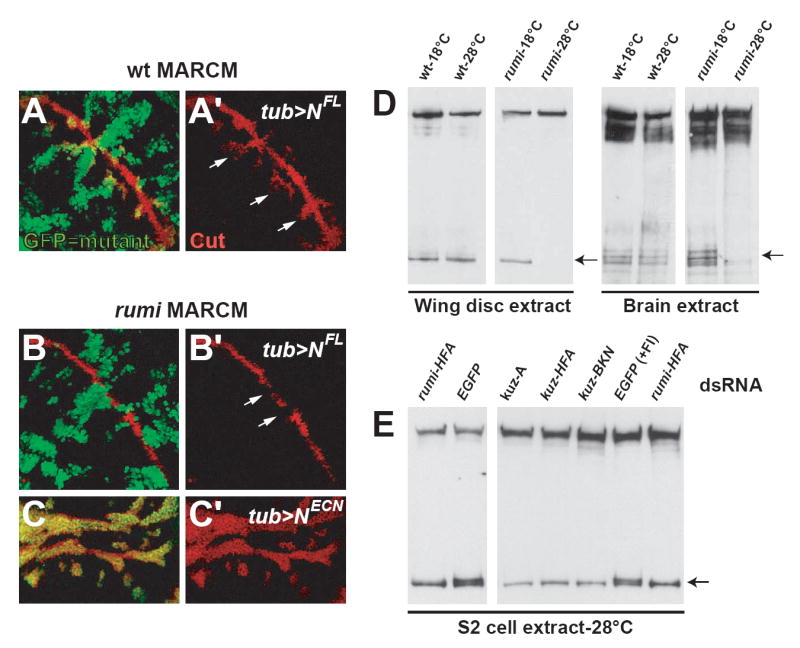
(A and A’) Ectopic expression of the full length Notch (NFL) induces aberrant Cut (red) expression in MARCM clones of a wild-type chromosome that are close to the dorso-ventral boundary (arrows in A’). GFP (green) marks the clones that ectopically express NFL in (A) and (B).
(B and B’) Ectopic expression of NFL in MARCM rumi clones does not induce Notch signaling. Note that in rumi mutant cells Cut (red) expression is lost in the prospective wing margin despite NFL overexpression (arrows in B’).
(C and C’) Ectopic expression of NECN induces Cut (red) expression in rumi cells. GFP marks MARCM rumi clones that ectopically express NECN, suggesting that the Rumi function is required upstream of the S3 cleavage of Notch.
(D) Western blots showing that Notch processing is altered in the absence of Rumi function at the restrictive temperature. Anti-NICD antibody was used in all blots. The top bands in each blot correspond to full length Notch, which is around 300 kDa. In wing disc extracts one predominant band that corresponds to Notch cleavage product is visible in both wild-type (wt) and in rumΔ26/Δ26 (rumi) larvae kept at 18°C. This band is not detected in extracts of rumi larvae kept at 28°C for 10 hours prior to dissection (left arrow). In brain extracts four ~120 kDa fragments are visible in wt larvae and in rumi larvae that were kept at 18°C. The upper two bands however are lost in rumi larvae kept at 28°C (right arrow).
(E) RNAi-mediated knockdown of Rumi and Kuz results in similar Notch processing defects in S2 cells. Western blotting with anti-NICD on protein extracts from S2 cells raised at 28°C is used to determine the pattern of Notch cleavage. Control cells (EGFP dsRNA) show two cleavage products (arrow). Addition of a Furin inhibitor (FI) does not alter this pattern. However, treatment of S2 cells with dsRNA against Rumi or Kuz results in the loss of the upper cleavage product, strongly suggesting that the Kuz-mediated S2 cleavage of Notch is affected by the loss of Rumi at the restrictive temperature. Note that three different kuz dsRNAs produce the same results.
Binding of ligands to the NECD induces S2 cleavage of Notch by ADAM/TACE/Kuzbanian proteases (Brou et al., 2000; Lieber et al., 2002). This generates an active membrane-bound form of Notch, which undergoes S3 cleavage mediated by Presenilin and its binding partners (De Strooper, 2003; Struhl and Greenwald, 1999). To refine the step in the Notch transduction cascade in which Rumi is required, we overexpressed a membrane-bound, active version of Notch called NECN (Struhl et al., 1993) in rumi clones raised at 28°C, and observed a robust induction of downstream targets (Figure 4C and 4C’). Since the activity of NECN depends on the Presenilin function, these data place the function of rumi upstream of the S3 cleavage of Notch in the signal-receiving cell and suggest that the NECD is the target of Rumi.
To address if Notch processing is impaired in rumi mutants, we performed Western blots by using a anti-NICD antibody (Hu et al., 2002; Pan and Rubin, 1997). Reduction of Kuzbanian (Kuz) or Presenilin function alters the pattern of the Notch cleavage products detected by western blots of protein extracts prepared in a hypotonic, detergent-free lysis buffer. We tested protein extracts from wing discs and brains of late third instar wild-type (wt) and rumi mutant larvae reared at 18°C. One set was shifted to 28°C (third instars), whereas the other set was maintained at 18°C for 10 hrs. The Notch cleavage product was detectable in wing disc extracts of both wt and rumi larvae kept at 18°C (see arrow in Figure 4D), but not in wing disc extracts of rumi mutants at 28°C (Figure 4D). Note that the full-length Notch protein serves as an internal control for protein loading. This is very similar to what has been observed in wing discs that express a dominant negative form of kuz (Pan and Rubin, 1997). Defects in Notch processing were also observed in brain extracts. Four ~120 kDa fragments are detected in Western blots using the extracts from rumi mutants kept at 18°C and from wt larvae, as reported (Hu et al., 2002). The top two bands of the quadruplet are strongly reduced in rumi larvae at 28°C (Figure 4D). These upper bands were shown to be lost when brains express a dominant negative Kuz construct (Hu et al., 2002). These results provide strong evidence that the rumi function is important for Notch processing.
To further assess the role of Rumi in Notch processing, we performed RNAi experiments in Drosophila S2 cells raised at 28°C. As shown in Figure 4E, when treated with double-stranded RNA (dsRNA) against EGFP, Western blots with anti-NICD antibody show two cleavage products. However, RNAi-mediated knockdown of Rumi or Kuz results in loss of the upper cleavage product (Figure 4E, arrow). Also, adding a Furin inhibitor does not alter the cleavage product pattern, consistent with the observation that the Furin-mediated S1 cleavage is not required for Notch signaling in flies (Kidd and Lieber, 2002). These data strongly suggest that Rumi is required for the function of Kuz at the restrictive temperature.
Notch accumulates intracellularly and at the cell surface in rumi mutant clones
Our data indicate that Rumi is retained in the ER by its C-terminal KDEL sequence and that ER-retention is required for the function of Rumi in vivo (Supplemental data and Figure S4) and that Rumi is required for proper folding of the NECD. Loss of Rumi may lead to accumulation of Notch in the ER, or an inability of Notch to be recognized by proteins like Fringe, Delta, Serrate, or Kuz. Indeed, staining of third instar discs with anti-NECD shows an accumulation of Notch in rumi clones raised at 25°C but not at 18°C (Figures 5A-B’) which is not due to an increase in Notch transcription (Figure S5).
Figure 5. High levels of Notch accumulate inside and at the membrane of rumi mutant cells at the restrictive temperature.
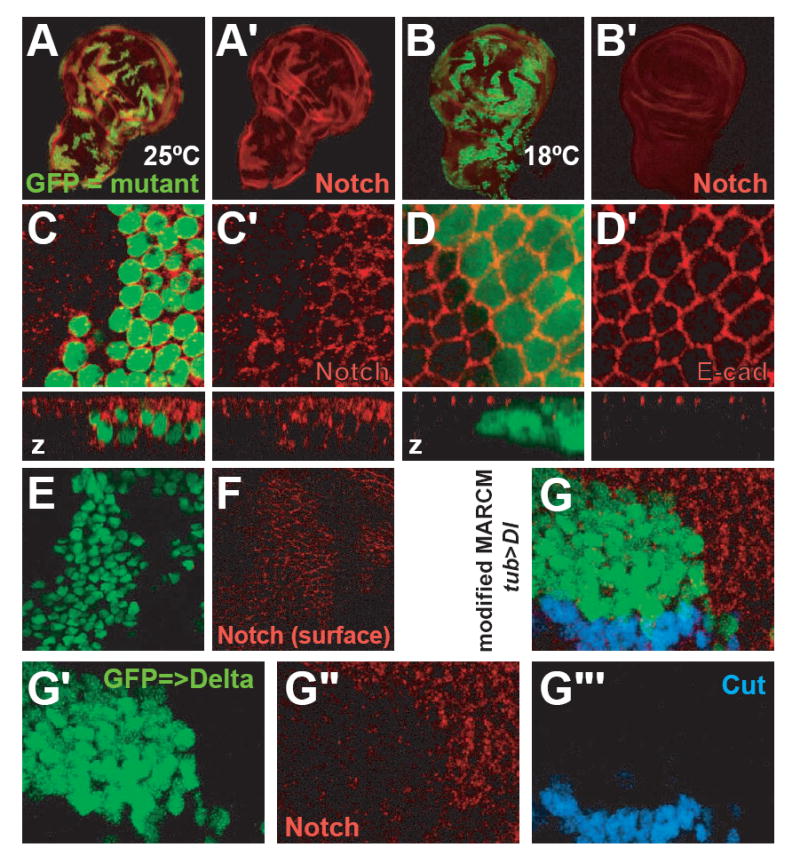
(A-C’) rumi clones marked by GFP (green in A, B and C) show an accumulation of Notch (red) when raised at 25°C (A, A’, C, C’) but not when raised at 18°C (B, B’). The accumulation is evident with antibodies against both NECD (A, A’) and NICD (C, C’). A-B’ show third instar wing imaginal discs; C and C’ show a close-up from a pupa 12 hours after puparium formation (APF). Note the cell-autonomous increase in Notch levels (C, C’). Also, optical z sectioning shows that in mutant cells Notch is not mainly in the apical regions anymore (z in C, C’).
(D and D’) E-Cadherin (E-cad, red) localization and levels do not change in rumi cells at 25°C.
(E and F) Notch protein levels at the cell membrane increase in rumi cells. Shown are two different sections of the wing disc of a third instar larva with MARCM rumi clones (green) raised at the restrictive temperature, and fixed and stained with the α-NECD (red) antibody in the absence of detergent. (F) is close to the apical surface, and (E) is 700 nm basal to (F). Note the accumulation of Notch at apical regions (F) overlaying the clones marked by the nuclear GFP in (E). Absence of specific α-NECD staining in (E) indicates that the antibody has not entered the cell because of the lack of detergent.
(G-G’’’) Ectopic over-expression of Delta in wild-type cells does not induce Notch signaling in adjacent cells that are mutant for rumi, despite the accumulation of Notch in these cells. Shown is an example of a modified MARCM clone located in the dorsal part of the wing pouch away from the wing margin (not shown), in which GFP (green) marks rumi+/+ cells that overexpress Delta, and Notch accumulation (red) marks rumi-/- cells. Note that Delta from green cells is able to induce Cut expression (blue) in rumi+/- cells but not in rumi-/- cells.
To examine the subcellular localization of Notch in rumi clones, we stained with an α-NICD antibody. Notch accumulates in a cell-autonomous manner in rumi mutant cells in basal and apical areas, unlike in wild-type cells, where Notch is mainly localized apically (Figures 5C and 5C’). To ensure that rumi mutation does not disrupt apical-basal polarity, we examined the distribution of adherens junction marker E-Cadherin (Tepass et al., 1996). E-Cadherin is expressed at normal levels and is localized to adherens junctions in rumi clones (Figures 5D and 5D’), suggesting that accumulation and mislocalization of Notch is not due to polarity defects.
The above data suggest that lack of Rumi prevents proper trafficking and may affect surface expression of Notch at the restrictive temperature. To test this possibility, we used a no-detergent protocol to label the surface Notch with α-NECD (Wang and Struhl, 2004). With this protocol intracellular Notch is not detected (Figure 5E vs Figure 5C), but Notch accumulates at the surface of rumi mutant cells (Figure 5F). Moreover, the unfolded protein response is not induced in rumi clones, as evidenced by normal levels of HSC3(BiP) (Ryoo et al., 2007) (Figures S6A-A”). Finally, we do not observe an increase in the size of the ER in rumi clones (Figures S6B-B”). Together, these data indicate that accumulation of Notch in rumi clones is not due to ER entrapment, and that Notch is present at high levels at the surface of the rumi mutant cells.
Lack of Rumi does not decrease binding of Notch to Delta
Lack of Rumi may render Notch sensitive to temperature changes, and it may therefore be unable to bind its ligands at high temperatures. To address this issue, we first used a modified MARCM strategy (Wang and Struhl, 2004) to test whether increasing Delta levels in the signal-sending cell can overcome the inefficient reception of signal by rumi mutant cells. In this experiment, clones of wild-type cells overexpressing Delta flank homozygous mutant clones of rumi. As shown in Figure 5G-G’”, overexpression of Delta results in induction of Cut in wild-type neighboring cells. However, despite the accumulation of Notch, rumi mutant cells fail to express Cut. Hence, overexpression of Delta in the signal-sending cell cannot suppress the rumi mutant phenotype in the signal-receiving cell.
To test if receptor-ligand interaction is impaired we used assays based on a secreted Notch-alkaline phosphatase (N-AP) fusion protein (Bruckner et al., 2000; Okajima et al., 2003; Sasamura et al., 2003; Xu et al., 2005). Since loss of rumi causes a ts phenotype, we performed receptor-ligand interaction assays at room temperature and at 28°C. As shown in Figure S7, the binding of N-AP to Delta is not decreased by addition of rumi dsRNA to the N-AP producing cells.
It has been recently shown that mutations in lethal giant discs (lgd) affect proper trafficking of Notch, causing ectopic activation of Notch in a ligand-independent manner (Childress et al., 2006; Gallagher and Knoblich, 2006; Jaekel and Klein, 2006). We therefore decided to carry out epistatic experiments between lgd and rumi. As shown in Figure S8, loss of rumi suppresses the ectopic activation of Notch in lgd mutant cells. Hence, loss of rumi affects the ligand dependent and independent Notch signaling.
Loss of Rumi affects O-glucosylation of the EGF repeats of Notch
The CAP proteins (CAP10, 59, 60 and 64) are referred to as putative polysaccharide modifiers as they affect extracellular polysaccharide capsule formation (Okabayashi et al., 2007). Since Rumi contains a CAP10 domain (Figure 3) it may be a glycosyltransferase that modifies Notch. Two unusual forms of O-linked carbohydrate modifications occur on Notch EGF repeats: O-fucosylation and O-glucosylation (Haines and Irvine, 2003; Moloney et al., 2000b). The enzymes involved in the synthesis of O-fucose glycans are known (Bruckner et al., 2000; Moloney et al., 2000a; Wang et al., 2001) but the enzymes involved in addition of O-glucose glycans are unknown. To examine whether Rumi plays a role in synthesis of O-glucose glycans, Rumi was knocked down in S2 cells using RNAi. A portion of the NECD encoding EGF repeat 7 up to the transmembrane domain (EGF7-TM) was expressed in control and Rumi knockdown cells and purified from the medium. The presence of O-fucose and O-glucose glycans on EGF7-TM was then assessed using mass spectral analysis of tryptic peptides generated from EGF7-TM protein (Figure S9). No changes in O-fucosylation were detected. In contrast, reduction of O-glucose on several peptides from the Rumi RNAi samples was seen. Comparison of the relative amounts of a peptide from EGF repeat 14 in the two samples shows that while the glycopeptide can be detected in both samples, significantly less is seen in the Rumi knockdown sample (Figure 6B). Also, significant amounts of the unglycosylated peptide are seen only in the sample where Rumi was knocked down (Figure 6A). Rumi RNAi also caused reduction in the level of glycosylated form of EGF repeats 16, 17, 19, and 35 (Figure S10). These data strongly suggest that reduction in Rumi is causing a reduction in O-glucose levels on Notch.
Figure 6. Rumi is a protein O-glucosyltransferase.
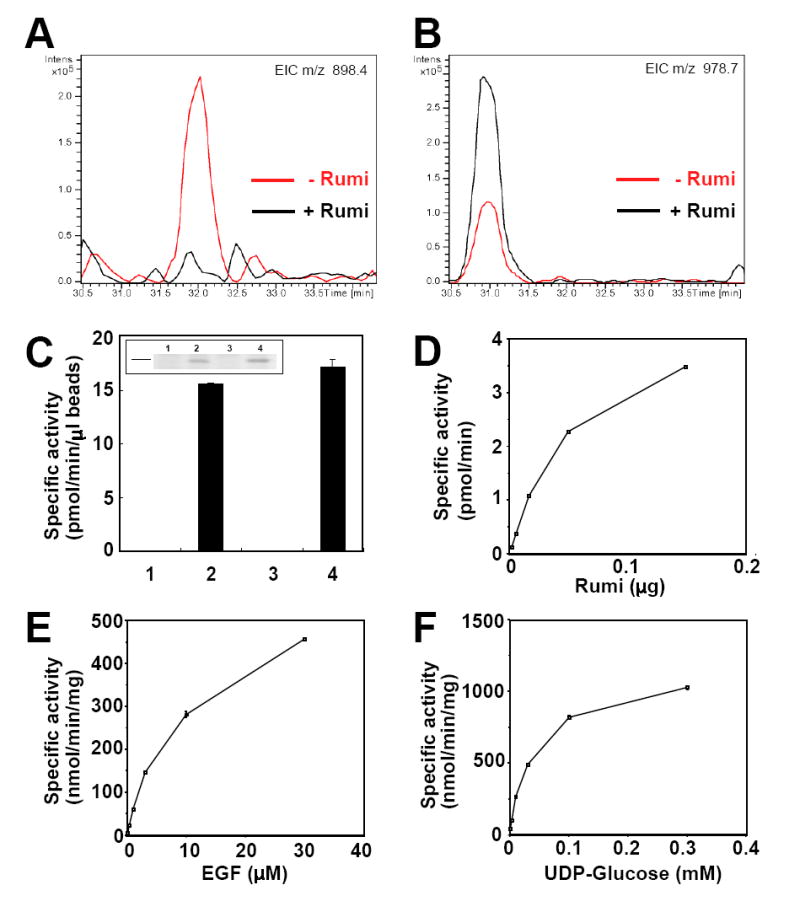
(A) The unglycosylated form of 561CQINIDDCQSQPCR574 is present in the Rumi knockdown sample, but not in the control sample. The MS data from both samples were searched for the doubly charged form of the peptide, m/z 898.4 (see Figure S9A for MS and MS/MS spectra at 32.0 min). The ion can be clearly seen eluting at 32.0 minutes in the Rumi knockdown sample (-Rumi), but it cannot be detected above the noise in the control sample (+Rumi).
(B) The O-glucosylated form of 561CQINIDDCQSQPCR574 is more abundant in the control sample than in the Rumi knockdown sample. The MS data from both samples was searched for the doubly charged form of the glycopeptide, m/z 978.7 (see Figure S9B for MS and MS/MS spectra at 30.8 min). The ion can be seen in both samples at 30.8 minutes, but more is present in the control sample (+Rumi) than in the Rumi knockdown sample (-Rumi).
(C) Both cell extracts and culture media of Rumi-overexpressing S2 cells showed an O-glucosyltransferase activity in vitro. Inset: Coomassie staining after 10% SDS-PAGE of the equivalent amounts of the beads. Line indicates 50 kDa size marker. 1: cell extract from control cells, 2: cell extract from Rumi-overexpressing cells, 3: media from control cells, 4: media from Rumi-overexpressing cells.
(D) O-glucosyltransferase activity was dependent on the amount of the purified Rumi protein in vitro.
(E) O-glucosyltransferase activity of the purified Rumi protein was dependent on the concentration of factor VII EGF as acceptor substrate.
(F) O-glucosyltransferase activity of the purified Rumi protein was dependent on the concentration of UDP-glucose as donor substrate.
All O-glucosyltransferase assays were performed in duplicate. Error bars represent the range of duplicates.
Rumi encodes a protein O-glucosyltransferase
To directly examine whether Rumi has protein O-glucosyltransferase activity toward EGF repeats, a FLAG-tagged version of Rumi was overexpressed in S2 cells, affinity-purified from cell extracts and media, and utilized in an in vitro O-glucosyltransferase assay (Shao et al., 2002). A factor VII EGF repeat containing an O-glucose consensus site was used as acceptor substrate, and UDP-[3H]glucose as donor. Rumi samples showed O-glucosyltransferase activity compared to controls (Figure 6C). Chromatographic analyses confirmed that the product in the assays consisted of a glucose residue covalently attached to the factor VII EGF repeat in an O-linkage (Figure S11). These results show that Rumi is capable of adding a single glucose in O-linkage to an EGF repeat containing an O-glucose consensus sequence. Further assays showed that the O-glucosyltransferase activity was dependent on the amount of Rumi, the concentration of factor VII EGF repeat, and the concentration of UDP-glucose (Figure 6D-F). Taken together, these results demonstrate that Rumi is a protein O-glucosyltransferase.
The O-glucosyltransferase activity mediated by Rumi is required for proper Notch signaling
Our data show that Rumi is an O-glucosyltransferase that adds glucose residues to EGF repeats of Notch and that Rumi function is essential for Notch signaling in a ts manner. O-glucosyltransferase activity may be required in some contexts for Notch signaling, but Rumi may also function as a chaperone independently of its enzymatic function, as reported for Ofut1 (Okajima et al., 2005; Sasamura et al., 2007). To examine the importance of Notch O-glucosylation for signaling, we took advantage of one of our severe EMS induced mutants, rumi79, that has a G189E mutation (Figure 3B). We examined whether G189E possesses O-glucosyltransferase activity or not. Rumi-G189E is expressed at normal levels in rumi79/79 flies (Figure 7A), and the protein is expressed and secreted as efficiently as the wild-type Rumi protein in S2 cells (Figure 7B). Moreover, the intracellular localization of Rumi-G189E in S2 cells is indistinguishable from wild-type Rumi (data not shown). These observations indicate that the G189E mutation does not impair Rumi expression or stability. However, Rumi-G189E showed no enzymatic activity (Figure 7C). These data do not support the presence of a key non-enzymatic role for Rumi, unlike what has been reported for Ofut1 (Okajima et al., 2005; Sasamura et al., 2007). The data also indicate that O-glucosylation mediated by Rumi is essential for Notch signaling. These conclusions are supported by in vitro and in vivo structure-function analyses of rumi (see Supplemental Data).
Figure 7. O-glucosylation mediated by Rumi is required for Notch signaling.
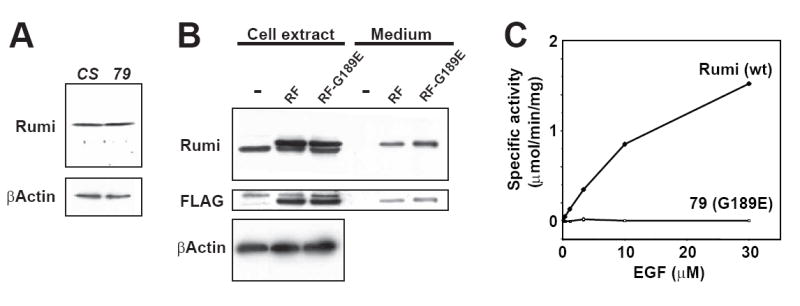
(A) Western blots showing the relative levels of Rumi and β-Actin proteins in wild-type (CS) and homozygous rumi79 (79) larval protein extracts. Late third instar larvae were used to prepare protein extracts for each genotype. Note that the G189E mutation in rumi79 larvae does not affect the level of the Rumi protein.
(B) The G189E mutation does not affect the expression level of Rumi in S2 cells. Western blots showing the expression and secretion of Rumi-FLAG (RF) and Rumi-FLAG with the G189E mutation (RF-G189E) in S2 cells. RF and RF-G189E were expressed in S2 cells using pUAST and pAc-Gal4 vectors. Note that the RF and RF-G189E proteins expressed in S2 cells run slightly slower than the endogenous Rumi protein due to the presence of the FLAG tag (see anti-Rumi blot). Endogenous Rumi is not expressed at levels that lead to its secretion in the medium. Some of the RF and RF-G189E proteins are secreted into the medium when expressed in S2 cells.
(C) The G189E mutation causes a complete loss of O-glucosyltransferase activity. Both wild-type and G189E Rumi proteins were overexpressed in S2 cells and purified from culture media by using the FLAG epitope as described in Experimental Procedures. The purified proteins were assayed for protein O-glucosyltransferase activity using increasing concentrations of the acceptor substrate, factor VII EGF repeat (wild-type Rumi, diamonds; G189E mutant, open squares). All assays were performed using 100 μM of UDP-glucose in duplicate. Error bars represent the range of duplicates.
Discussion
Loss of Rumi causes a loss of Notch signaling in a ts manner
In all contexts that we have examined, rumi is essential for Notch signaling in a ts manner, i.e. lateral inhibition, asymmetric division and inductive signaling. Homozygous rumi animals are viable and fertile when kept at 18°C and exhibit a mild lateral inhibition defect and a modest Delta wing vein phenotype (Figures 1 and 2). rumi animals raised at 25°C show a very significant decrease in viability and fertility. At this temperature, there is a failure in the cell fate specification process (Figure 1). At 28-30°C we observe a full-blown Notch phenotype in all tissues examined, and homozygous mutants can only reach the third instar stage because of the wild-type maternal component. The difference between the requirements for Rumi at 25°C and 28-30°C is also reflected in our genetic interaction studies, as an extra copy of Notch is able to improve Notch signaling in rumi mutants raised at 25°C (partial requirement), but not at 28-30°C (full requirement). Altogether these observations indicate that loss of rumi phenocopies loss of Notch in a temperature-dependent fashion.
Rumi targets the extracellular domain of Notch
Multiple lines of evidence suggest that Rumi functions in the signal-receiving cell. Our MARCM experiments indicate that overexpression of Notch ligands in rumi mutant cells is able to induce signaling, suggesting that Rumi function is not required in the signal-sending cell (Figure S3). However, cells that are mutant for rumi are not able to receive the signal, even when ligands are overexpressed in adjacent cells (Figure 5). Of note, the only component of the Notch signaling pathway in flies with multiple O-glucosylation sites is the Notch protein itself, besides Delta which contains a single predicted site.
As we observed an upregulation of Notch protein in rumi mutant clones we hypothesized that Notch might be trapped in the ER and fail to reach the membrane at the restrictive temperature. However, we observe an accumulation of Notch at the surface of rumi mutant cells. In addition, we find a lack of an unfolded protein response (Patil and Walter, 2001; Ryoo et al., 2007), and a lack of expansion of the ER in rumi clones raised at the restrictive temperature (Figure S6). These data raised the possibility that Notch present at the cell surface may not interact with its ligands at the restrictive temperature. However, our data suggest that the Notch-Delta interaction is not decreased at 28°C (Figure S7), but rather that the cleavage of Notch at the membrane is impaired (Figure 4). Hence our data indicate that the S2 cleavage of Notch is impaired in rumi mutant signal-receiving cells.
Rumi is a protein O-glucosyltransferase
Most proteins with a CAP10 domain contain a signal peptide and an ER retention signal. The CAP10 gene was first discovered in the fungus Cryptococcus neoformans (Chang and Kwon-Chung, 1999). The CAP proteins (CAP10, 59, 60 and 64) are referred to as putative polysaccharide modifiers as they affect extracellular polysaccharide capsule formation (Okabayashi et al., 2007). Our data indicate that knockdown of Rumi in S2 cells results in loss of O-glucosylation at Serines in C1-X-S-X-P-C2 sites on numerous EGF repeats. No effects were seen on levels of O-fucosylation. In vitro assays with purified Rumi demonstrate that it can catalyze the transfer of glucose from UDP-glucose to an EGF repeat with the consensus sequence. Hence, Rumi encodes the first identified protein O-glucosyltransferase. Rumi shares several common features with enzymes responsible for addition of O-fucose to EGF repeats and thrombospondin type 1 repeats (TSRs), Pofut1 and Pofut2, respectively. These proteins are soluble, ER localized and only modify properly folded structures (EGF repeats for Pofut1, TSRs for Pofut2) (Luo et al., 2006a; Luo et al., 2006b; Wang and Spellman, 1998). Preliminary studies using crude lysates suggest that the mammalian form of the protein O-glucosyltransferase (presumably a Rumi homologue) can also distinguish folded from unfolded structures (Shao et al., 2002). The ER localization and ability to distinguish folded from unfolded structures suggests that all of these enzymes may function in folding and/or quality control.
Unlike Ofut1, which is reported to have important non-enzymatic functions (Okajima et al., 2005; Sasamura et al., 2007), our results indicate that the function of Rumi resides in the O-glucosyltransferase activity (Figure 7 and Supplemental Data). We therefore propose that preventing the addition of O-glucose to Notch causes a ts phenotype. We propose that the O-glucose glycans may function to hold the NECD in a stable conformation needed for proper function, especially at higher temperatures. For example, O-glucosylation of Notch might be a prerequisite for conformational changes in the NECD that are proposed to promote the S2 cleavage (Malecki et al., 2006; Parks et al., 2000). Alternatively, addition of O-glucose might be required for another posttranslational modification. The importance of O-glucosylation of Notch is also supported by studies showing that elimination of individual O-glucosylation sites in mouse Notch1 impairs activation in cell-based Notch signaling assays (Nita-Lazar et al., in revision).
Lack of O-glucosylation at the restrictive temperature does not block the ER-to-membrane transport and ligand interactions but disrupts Notch cleavage. These data, together with accumulation of Notch intracellularly and at the cell membrane in rumi cells suggest that lack of O-glucose modification causes a folding problem which impairs Notch function. Trafficking problems upstream of S3 cleavage have been documented to cause accumulation of Notch and ectopic activation of Notch signaling (Le Borgne, 2006). For example, loss of Lethal giant discs (Lgd), a protein required for proper trafficking of Notch, causes ectopic activation of Notch in a ligand-independent manner (Childress et al., 2006; Gallagher and Knoblich, 2006; Jaekel and Klein, 2006). Our data show that the loss of rumi suppresses the ectopic activation of Notch in lgd mutant cells (Figure S8), suggesting that the lack of O-glucosylation prevents the ligand-independent activation of Notch in the absence of Lgd.
In summary, our data uncover a novel mechanism for enzymatic regulation of Notch signaling in Drosophila by a protein O-glucosyltransferase, and provide an in vivo model to study the role of O-glucosylation in developmental signaling. Given the evolutionary conservation of Notch signaling and the presence of conserved O-glucosylation motifs in other Notch proteins, addition of glucose may be required for proper folding and cleavage in many species.
Experimental Procedures (See also Supplemental Data)
Analysis of O-glucosylation of NECD in Rumi knockdown S2 cells
The region that encodes the signal peptide of the Drosophila Acetylcholine esterase protein (CG17907) was amplified by PCR and cloned into the pMT/V5-HisB vector (Invitrogen) in-frame with the V5 and His tags. A CAAC optimal translation start sequence was incorporated before the start codon ATG via the 5’ PCR primer. This vector is then called pMT/V5B-ACE. The region that covers from the 7th EGF repeat to the transmembrane domain of Notch (EGF7-TM) was amplified by PCR using primers with additional EcoRI sites. The EGF7-TM fragment was then inserted into the pMT/V5B-ACE vector using the EcoRI site and in-frame with the signal peptide and the C-terminal V5 and His tags. S2 cells that are adapted to serum free media (SFM, Invitrogen) were transfected with the pMT/V5B-EGF7-TM construct by using the FuGENE-HD transfection reagent (Roche). One day after transfection, the S2 cells were divided into two groups that contain equal amount of cells. One group was treated with dsRNA against EGFP and the other was treated with dsRNA against rumi. Two days after dsRNA treatment, expression of the pMT/V5B-EGF7-TM construct was induced using 0.7 mM CuSO4. Three days after induction, media from both groups were collected after spinning down the cells at 300g for 10 minutes. The media were dialyzed three times using 1X binding buffer for His Bind Resin (Novagen). After dialysis, the His-tagged EGF7-TM protein secreted in the media was purified using His Bind Resin (Novagen) according to the manufacturer’s protocol. After purification, the eluted protein was TCA precipitated and washed with cold acetone before mass spectral analysis.
Analysis of O-glucosylation of tryptic peptides from EGF7-TM protein was performed by LC-MS/MS essentially as described (Nita-Lazar and Haltiwanger, 2006; Ricketts et al., 2007; Wang et al., 2007). Briefly, approximately 500 ng of EGF7-TM protein purified from the medium of control and Rumi knockdown cells were reduced, alkylated, separated by SDS-PAGE, and subjected to in-gel tryptic digestion. The resulting peptides were separated by reverse-phase HPLC and sprayed directly into an Agilent XCT ion trap mass spectrometer. Low energy CID fragmentation was performed on the two most abundant ions in each MS scan. Unglycosylated peptides were identified by searching databases with the MS/MS data using the X! Tandem (Global Proteome Machine) search engine (http://h777.thegpm.org/tandem/thegpm_tandem.html) or by manually searching the MS data for ions matching predicted masses of tryptic peptides containing O-glucose consensus sequences. O-fucosylated peptides were identified by performing neutral loss scans of the data for ions losing 146 Da upon CID fragmentation. Similarly, O-glucosylated peptides were identified by performing neutral loss scans for ions losing 162 Da upon fragmentation. Relative amounts of individual molecular ions (representing either glycosylated or unglycosylated forms of specific peptides) in control or Rumi knockdown samples were compared by performing extracted ion searches of the MS data for the ion of interest.
Supplementary Material
Acknowledgments
We thank Mark Fortini for very valuable advice with the Notch cleavage assay, Vafa Bayat for noticing the ts phenotype, Yu-Chun He for transgenic injections, and members of the Haltiwanger and Bellen labs for discussions. We thank Michael Tiemeyer for suggestions, Georg Halder, Jennifer Childress, Bassem Hassan, Gary Struhl, Marc Muskavitch, Ken Irvine, Steve Cohen, Richard Mann, Spyros Artavanis-Tsakonas, Hyung Don Ryoo, Kenji Matsuno, Hermann Steller, Richard Mann, Tobby Lieber, Simon Kidd, the Bloomington Stock Center and the Developmental Studies Hybridoma Bank for reagents, Kenneth Dunner, Jr. for assistance with SEM, and the Confocal Microscopy Core of BCM MRDDRC. We acknowledge support from NIH grant 5R01GM061126-07 to R.S.H., and NIH Medical Genetics Research Fellowship Program grant T32-GMO7526 to H.J.-N. H.T. was supported in part by a fellowship of Astellas Foundation for Research on Metabolic Disorders. H.J.B. is an investigator of the HHMI.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–776. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- Bellen HJ, Levis RW, Liao G, He Y, Carlson JW, Tsang G, Evans-Holm M, Hiesinger PR, Schulze KL, Rubin GM, et al. The BDGP gene disruption project: single transposon insertions associated with 40% of Drosophila genes. Genetics. 2004;167:761–781. doi: 10.1534/genetics.104.026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolos V, Grego-Bessa J, de la Pompa JL. Notch signaling in development and cancer. Endocrine reviews. 2007;28:339–363. doi: 10.1210/er.2006-0046. [DOI] [PubMed] [Google Scholar]
- Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israel A. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Molecular cell. 2000;5:207–216. doi: 10.1016/s1097-2765(00)80417-7. [DOI] [PubMed] [Google Scholar]
- Bruckner K, Perez L, Clausen H, Cohen S. Glycosyltransferase activity of Fringe modulates Notch-Delta interactions. Nature. 2000;406:411–415. doi: 10.1038/35019075. [DOI] [PubMed] [Google Scholar]
- Carlson ME, Conboy IM. Regulating the Notch pathway in embryonic, adult and old stem cells. Current opinion in pharmacology. 2007;7:303–309. doi: 10.1016/j.coph.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Chang YC, Kwon-Chung KJ. Isolation, characterization, and localization of a capsule-associated gene, CAP10, of Cryptococcus neoformans. Journal of bacteriology. 1999;181:5636–5643. doi: 10.1128/jb.181.18.5636-5643.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childress JL, Acar M, Tao C, Halder G. Lethal giant discs, a novel C2-domain protein, restricts notch activation during endocytosis. Curr Biol. 2006;16:2228–2233. doi: 10.1016/j.cub.2006.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–522. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- Gallagher CM, Knoblich JA. The conserved c2 domain protein lethal (2) giant discs regulates protein trafficking in Drosophila. Dev Cell. 2006;11:641–653. doi: 10.1016/j.devcel.2006.09.014. [DOI] [PubMed] [Google Scholar]
- Haines N, Irvine KD. Glycosylation regulates Notch signalling. Nat Rev Mol Cell Biol. 2003;4:786–797. doi: 10.1038/nrm1228. [DOI] [PubMed] [Google Scholar]
- Hori K, Fostier M, Ito M, Fuwa TJ, Go MJ, Okano H, Baron M, Matsuno K. Drosophila deltex mediates suppressor of Hairless-independent and late-endosomal activation of Notch signaling. Development. 2004;131:5527–5537. doi: 10.1242/dev.01448. [DOI] [PubMed] [Google Scholar]
- Hu Y, Ye Y, Fortini ME. Nicastrin is required for gamma-secretase cleavage of the Drosophila Notch receptor. Dev Cell. 2002;2:69–78. doi: 10.1016/s1534-5807(01)00105-8. [DOI] [PubMed] [Google Scholar]
- Jaekel R, Klein T. The Drosophila Notch inhibitor and tumor suppressor gene lethal (2) giant discs encodes a conserved regulator of endosomal trafficking. Dev Cell. 2006;11:655–669. doi: 10.1016/j.devcel.2006.09.019. [DOI] [PubMed] [Google Scholar]
- Jafar-Nejad H, Andrews HK, Acar M, Bayat V, Wirtz-Peitz F, Mehta SQ, Knoblich JA, Bellen HJ. Sec15, a component of the exocyst, promotes notch signaling during the asymmetric division of Drosophila sensory organ precursors. Dev Cell. 2005;9:351–363. doi: 10.1016/j.devcel.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cecillion M, Marechal E, et al. Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature. 1996;383:707–710. doi: 10.1038/383707a0. [DOI] [PubMed] [Google Scholar]
- Kidd S, Lieber T. Furin cleavage is not a requirement for Drosophila Notch function. Mech Dev. 2002;115:41–51. doi: 10.1016/s0925-4773(02)00120-x. [DOI] [PubMed] [Google Scholar]
- Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131:965–973. doi: 10.1242/dev.01074. [DOI] [PubMed] [Google Scholar]
- Lai EC, Orgogozo V. A hidden program in Drosophila peripheral neurogenesis revealed: fundamental principles underlying sensory organ diversity. Dev Biol. 2004;269:1–17. doi: 10.1016/j.ydbio.2004.01.032. [DOI] [PubMed] [Google Scholar]
- Le Borgne R. Regulation of Notch signalling by endocytosis and endosomal sorting. Curr Opin Cell Biol. 2006;18:213–222. doi: 10.1016/j.ceb.2006.02.011. [DOI] [PubMed] [Google Scholar]
- Le Borgne R, Bardin A, Schweisguth F. The roles of receptor and ligand endocytosis in regulating Notch signaling. Development. 2005;132:1751–1762. doi: 10.1242/dev.01789. [DOI] [PubMed] [Google Scholar]
- Lecourtois M, Schweisguth F. The neurogenic suppressor of hairless DNA-binding protein mediates the transcriptional activation of the enhancer of split complex genes triggered by Notch signaling. Genes Dev. 1995;9:2598–2608. doi: 10.1101/gad.9.21.2598. [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 2001;24:251–254. doi: 10.1016/s0166-2236(00)01791-4. [DOI] [PubMed] [Google Scholar]
- Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, Qi M, Trask BJ, Kuo WL, Cochran J, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nature genetics. 1997;16:243–251. doi: 10.1038/ng0797-243. [DOI] [PubMed] [Google Scholar]
- Lieber T, Kidd S, Young MW. kuzbanian-mediated cleavage of Drosophila Notch. Genes Dev. 2002;16:209–221. doi: 10.1101/gad.942302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Koles K, Vorndam W, Haltiwanger RS, Panin VM. Protein O-fucosyltransferase 2 adds O-fucose to thrombospondin type 1 repeats. J Biol Chem. 2006a;281:9393–9399. doi: 10.1074/jbc.M511975200. [DOI] [PubMed] [Google Scholar]
- Luo Y, Nita-Lazar A, Haltiwanger RS. Two distinct pathways for O-fucosylation of epidermal growth factor-like or thrombospondin type 1 repeats. J Biol Chem. 2006b;281:9385–9392. doi: 10.1074/jbc.M511974200. [DOI] [PubMed] [Google Scholar]
- Malecki MJ, Sanchez-Irizarry C, Mitchell JL, Histen G, Xu ML, Aster JC, Blacklow SC. Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Molecular and cellular biology. 2006;26:4642–4651. doi: 10.1128/MCB.01655-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, Wang Y, Stanley P, Irvine KD, Haltiwanger RS, et al. Fringe is a glycosyltransferase that modifies Notch. Nature. 2000a;406:369–375. doi: 10.1038/35019000. [DOI] [PubMed] [Google Scholar]
- Moloney DJ, Shair LH, Lu FM, Xia J, Locke R, Matta KL, Haltiwanger RS. Mammalian Notch1 is modified with two unusual forms of O-linked glycosylation found on epidermal growth factor-like modules. J Biol Chem. 2000b;275:9604–9611. doi: 10.1074/jbc.275.13.9604. [DOI] [PubMed] [Google Scholar]
- Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, Ray WJ, Kopan R. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Molecular cell. 2000;5:197–206. doi: 10.1016/s1097-2765(00)80416-5. [DOI] [PubMed] [Google Scholar]
- Nita-Lazar A, Haltiwanger RS. Methods for analysis of O-linked modifications on epidermal growth factor-like and thrombospondin type 1 repeats. Methods in enzymology. 2006;417:93–111. doi: 10.1016/S0076-6879(06)17008-1. [DOI] [PubMed] [Google Scholar]
- Nita-Lazar A, Rana NA, Orhue R, Myers MP, Luther KB, Takeuchi H, Haltiwanger R. O-Glucose is essential for both stability and function of mouse Notch1. J Biol Chem in revision. [Google Scholar]
- Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nature genetics. 1997;16:235–242. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- Okabayashi K, Hasegawa A, Watanabe T. Microreview: Capsule-associated genes of Cryptococcus neoformans. Mycopathologia. 2007;163:1–8. doi: 10.1007/s11046-006-0083-0. [DOI] [PubMed] [Google Scholar]
- Okajima T, Irvine KD. Regulation of notch signaling by o-linked fucose. Cell. 2002;111:893–904. doi: 10.1016/s0092-8674(02)01114-5. [DOI] [PubMed] [Google Scholar]
- Okajima T, Xu A, Irvine KD. Modulation of notch-ligand binding by protein O-fucosyltransferase 1 and fringe. J Biol Chem. 2003;278:42340–42345. doi: 10.1074/jbc.M308687200. [DOI] [PubMed] [Google Scholar]
- Okajima T, Xu A, Lei L, Irvine KD. Chaperone activity of protein O-fucosyltransferase 1 promotes notch receptor folding. Science. 2005;307:1599–1603. doi: 10.1126/science.1108995. [DOI] [PubMed] [Google Scholar]
- Pan D, Rubin GM. Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell. 1997;90:271–280. doi: 10.1016/s0092-8674(00)80335-9. [DOI] [PubMed] [Google Scholar]
- Panin VM, Papayannopoulos V, Wilson R, Irvine KD. Fringe modulates Notch-ligand interactions. Nature. 1997;387:908–912. doi: 10.1038/43191. [DOI] [PubMed] [Google Scholar]
- Parks AL, Klueg KM, Stout JR, Muskavitch MA. Ligand endocytosis drives receptor dissociation and activation in the Notch pathway. Development. 2000;127:1373–1385. doi: 10.1242/dev.127.7.1373. [DOI] [PubMed] [Google Scholar]
- Patil C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol. 2001;13:349–355. doi: 10.1016/s0955-0674(00)00219-2. [DOI] [PubMed] [Google Scholar]
- Rampal R, Li AS, Moloney DJ, Georgiou SA, Luther KB, Nita-Lazar A, Haltiwanger RS. Lunatic fringe, manic fringe, and radical fringe recognize similar specificity determinants in O-fucosylated epidermal growth factor-like repeats. J Biol Chem. 2005;280:42454–42463. doi: 10.1074/jbc.M509552200. [DOI] [PubMed] [Google Scholar]
- Ricketts LM, Dlugosz M, Luther KB, Haltiwanger RS, Majerus EM. O-Fucosylation is Required for ADAMTS13 Secretion. J Biol Chem. 2007 doi: 10.1074/jbc.M700317200. in press. [DOI] [PubMed] [Google Scholar]
- Ryoo HD, Domingos PM, Kang MJ, Steller H. Unfolded protein response in a Drosophila model for retinal degeneration. Embo J. 2007;26:242–252. doi: 10.1038/sj.emboj.7601477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki N, Sasamura T, Ishikawa HO, Kanai M, Ueda R, Saigo K, Matsuno K. Polarized exocytosis and transcytosis of Notch during its apical localization in Drosophila epithelial cells. Genes Cells. 2007;12:89–103. doi: 10.1111/j.1365-2443.2007.01037.x. [DOI] [PubMed] [Google Scholar]
- Sasamura T, Ishikawa HO, Sasaki N, Higashi S, Kanai M, Nakao S, Ayukawa T, Aigaki T, Noda K, Miyoshi E, et al. The O-fucosyltransferase O-fut1 is an extracellular component that is essential for the constitutive endocytic trafficking of Notch in Drosophila. Development. 2007 doi: 10.1242/dev.02811. [DOI] [PubMed] [Google Scholar]
- Sasamura T, Sasaki N, Miyashita F, Nakao S, Ishikawa HO, Ito M, Kitagawa M, Harigaya K, Spana E, Bilder D, et al. neurotic, a novel maternal neurogenic gene, encodes an O-fucosyltransferase that is essential for Notch-Delta interactions. Development. 2003;130:4785–4795. doi: 10.1242/dev.00679. [DOI] [PubMed] [Google Scholar]
- Schweisguth F. Notch signaling activity. Curr Biol. 2004;14:R129–138. [PubMed] [Google Scholar]
- Shao L, Haltiwanger RS. O-fucose modifications of epidermal growth factor-like repeats and thrombospondin type 1 repeats: unusual modifications in unusual places. Cell Mol Life Sci. 2003;60:241–250. doi: 10.1007/s000180300019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao L, Luo Y, Moloney DJ, Haltiwanger R. O-glycosylation of EGF repeats: identification and initial characterization of a UDP-glucose: protein O-glucosyltransferase. Glycobiology. 2002;12:763–770. doi: 10.1093/glycob/cwf085. [DOI] [PubMed] [Google Scholar]
- Shao L, Moloney DJ, Haltiwanger R. Fringe modifies O-fucose on mouse Notch1 at epidermal growth factor-like repeats within the ligand-binding site and the Abruptex region. J Biol Chem. 2003;278:7775–7782. doi: 10.1074/jbc.M212221200. [DOI] [PubMed] [Google Scholar]
- Shi S, Stanley P. Protein O-fucosyltransferase 1 is an essential component of Notch signaling pathways. Proc Natl Acad Sci U S A. 2003;100:5234–5239. doi: 10.1073/pnas.0831126100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struhl G, Adachi A. Nuclear access and action of notch in vivo. Cell. 1998;93:649–660. doi: 10.1016/s0092-8674(00)81193-9. [DOI] [PubMed] [Google Scholar]
- Struhl G, Fitzgerald K, Greenwald I. Intrinsic activity of the Lin-12 and Notch intracellular domains in vivo. Cell. 1993;74:331–345. doi: 10.1016/0092-8674(93)90424-o. [DOI] [PubMed] [Google Scholar]
- Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature. 1999;398:522–525. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- Teng Y, Liu Q, Ma J, Liu F, Han Z, Wang Y, Wang W. Cloning, expression and characterization of a novel human CAP10-like gene hCLP46 from CD34(+) stem/progenitor cells. Gene. 2006;371:7–15. doi: 10.1016/j.gene.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Tepass U, Gruszynski-DeFeo E, Haag TA, Omatyar L, Torok T, Hartenstein V. shotgun encodes Drosophila E-cadherin and is preferentially required during cell rearrangement in the neurectoderm and other morphogenetically active epithelia. Genes Dev. 1996;10:672–685. doi: 10.1101/gad.10.6.672. [DOI] [PubMed] [Google Scholar]
- Wang LW, Dlugosz M, Somerville RP, Raed M, Haltiwanger RS, Apte SS. O-Fucosylation of thrombospondin type 1 repeats in ADAMTS like-1/punctin-1 regulates secretion: Implications for the ADAMTS superfamily. J Biol Chem. 2007 doi: 10.1074/jbc.M701065200. in press. [DOI] [PubMed] [Google Scholar]
- Wang W, Struhl G. Drosophila Epsin mediates a select endocytic pathway that DSL ligands must enter to activate Notch. Development. 2004;131:5367–5380. doi: 10.1242/dev.01413. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shao L, Shi S, Harris RJ, Spellman MW, Stanley P, Haltiwanger RS. Modification of epidermal growth factor-like repeats with O-fucose. Molecular cloning and expression of a novel GDP-fucose protein O-fucosyltransferase. J Biol Chem. 2001;276:40338–40345. doi: 10.1074/jbc.M107849200. [DOI] [PubMed] [Google Scholar]
- Wang Y, Spellman MW. Purification and characterization of a GDP-fucose:polypeptide fucosyltransferase from Chinese hamster ovary cells. J Biol Chem. 1998;273:8112–8118. doi: 10.1074/jbc.273.14.8112. [DOI] [PubMed] [Google Scholar]
- Xu A, Lei L, Irvine KD. Regions of Drosophila Notch that contribute to ligand binding and the modulatory influence of Fringe. J Biol Chem. 2005;280:30158–30165. doi: 10.1074/jbc.M505569200. [DOI] [PubMed] [Google Scholar]
- Zhai RG, Hiesinger PR, Koh TW, Verstreken P, Schulze KL, Cao Y, Jafar-Nejad H, Norga KK, Pan H, Bayat V, et al. Mapping Drosophila mutations with molecularly defined P element insertions. Proc Natl Acad Sci U S A. 2003;100:10860–10865. doi: 10.1073/pnas.1832753100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.