

Abstract
Vaccinia virus (VACV) C7L is a host-range gene that regulates cellular tropism of VACV. Distantly related C7L homologues are encoded by nearly all mammalian poxviruses, but whether they are host-range genes functioning similar to VACV C7L has not been determined. Here, we used VACV as a model system to analyze five different C7L homologues from diverse mammalian poxviruses for their abilities to regulate poxvirus cellular tropism. Three C7L homologues (myxoma virus M63R, M64R and cowpox virus 020), when expressed with an epitope tag and from a VACV mutant lacking the host-range genes K1L and C7L (vK1L−C7L−), failed to support productive viral replication in human and murine cells. In nonpermissive cells, these viruses did not synthesize viral late proteins, expressed a reduced level of the early protein E3L, and were defective at suppressing cellular PKR activation. In contrast, two other C7L homologues, myxoma virus (MYXV) M62R and Yaba-like disease virus (YLDV) 67R, when expressed with an epitope tag and from vK1L−C7L−, supported normal viral replication in human and murine cells and restored the ability of the virus to suppress PKR activation. Furthermore, M62R rescued the defect of vK1L−C7L− at replicating and disseminating in mice following intranasal inoculation. These results show that MYXV M62R and YLDV 67R function equivalently to C7L at supporting VACV replication in mammalian hosts and suggest that a C7L-like host-range gene is essential for the replication of many mammalian poxviruses in mammalian hosts.
Keywords: poxvirus, vaccinia virus, myxoma virus, C7L, host-range, PKR
Introduction
Poxviruses are a family of complex, double-stranded DNA viruses that replicate entirely in the cytoplasm (Moss, 2001). Members of the poxvirus family infect a wide variety of hosts, including insects, reptiles, birds, and mammals. The poxviruses that infect mammals are classified into seven genera, among which the orthopoxvirus genus has the greatest impact on human health. Orthopoxviruses include variola virus, monkeypox virus, cowpox virus and vaccinia virus (VACV). Variola virus causes smallpox, for which VACV serves as an effective vaccine. VACV has a broad host-range, infecting many mammalian species. Most other mammalian poxviruses have a narrower host-range, but many of them could infect cell lines derived from diverse mammalian species. For instance, myxoma virus (MYXV) of the leporipoxvirus genus has a limited host-range in rabbit, but it can replicate in many transformed human cell lines and has been tested as an oncolytic agent (Lun et al., 2005). In contrast, avian poxviruses replicate abortively in mammalian cells, although they are capable of gaining entry and expressing a limited set of viral genes in mammalian cells (Somogyi, Frazier, and Skinner, 1993). Indeed, unlike most other viruses, cellular tropism of poxviruses does not appear to be limited by the availability of specific receptors on host cell surface but by some intracellular factors (reviewed by McFadden, 2005).
The identities of the intracellular factors that determine poxvirus cellular tropism are unknown, but poxvirus genes that are required for viral replication in specific mammalian cell types have been identified from orthopoxviruses and myxoma virus (McFadden, 2005). Among these so-called “host-range” genes, K1L and C7L of VACV affect the widest range of cell lines (Perkus et al., 1990). The deletion of K1L and C7L from VACV results in abortive replication of the mutant in most mammalian cell lines, while it has no effect on viral replication in avian cells and a few specific mammalian cell lines (Drillien, Koehren, and Kirn, 1981; Gillard et al., 1986; Perkus et al., 1990). The replication block of the mutant was shown to occur at the translation of intermediate stage viral mRNA in human HeLa cells (Hsiao et al., 2004). NYVAC, a host-range restricted VACV strain lacking 18 genes including K1L and C7L, has been developed as a safe vaccine vector for infectious diseases and cancer (Tartaglia et al., 1992). The replication defect of K1L and C7L deficient VACV can be complemented in human cells by either K1L or C7L or the cowpox virus CP77 gene, while its defect in rabbit RK13 cells can be complemented by either K1L or CP77 but not by C7L (Perkus et al., 1990; Ramsey-Ewing and Moss, 1996). K1L was shown to prevent the degradation of IκBα and thus inhibit host NF-κB activation in rabbit RK-13 (Shisler and Jin, 2004). Both K1L and CP77 contain multiple ankyrin repeats, which typically serves as a structural scaffold for mediating protein-protein interactions (Sedgwick and Smerdon, 1999). K1L is able to bind ACAP2 (Bradley and Terajima, 2005), a GTPase-activating protein for ARF6, but this ability does not appear to be required for K1L’s host-range function (Meng and Xiang, 2006). CP77 is able to interact with a cellular protein HMG20A (Hsiao et al., 2006). However, the exact mechanism by which K1L, C7L or CP77 control VACV cellular tropism remains elusive.
Among these poxvirus host-range genes, the C7L gene appears to be the most conserved in mammalian poxviruses. The CP77 gene is only present in cowpox virus and monkeypox virus, while the K1L gene is present in a few orthopoxviruses including VACV, cowpox virus and monkeypox virus. They are either absent or fragmented in other orthopoxviruses. In contrast, an ORF that is almost identical to VACV C7L is present in the genome of every orthopoxvirus. Furthermore, homologues that are distantly related to VACV C7L are found in nearly all sequenced mammalian poxviruses except molluscum contagiosum virus and parapoxviruses. However, whether these C7L homologues are host-range genes functioning similarly to VACV C7L is unknown. In this study, we analyzed 5 distantly related C7L homologues from diverse mammalian poxviruses and found that MYXV of the leporipoxvirus genus and Yaba-like disease virus (YLDV) of the yatapoxvirus genus encode a gene that functions equivalently to VACV C7L, suggesting that a C7L-like host-range gene is essential for productive replication of many mammalian poxviruses in mammalian hosts.
Results
Many mammalian poxviruses encode putative C7L homologues
A direct counterpart for VACV C7L gene is present in the genome of every orthopoxvirus. In addition, some orthopoxviruses, including cowpox virus (CWPX) and camelpox virus, encode a putative protein that is 26% identical to the C7L protein. This C7L homologue is represented by open reading frame (ORF) 020 of CWPX strain Brighton Red. Besides orthopoxviruses, nearly all other sequenced mammalian poxviruses encode putative C7L homologues. In particular, myxoma virus (MYXV) encodes three C7L homologues in neighboring ORFs (M62R, M63R and M64R), which are 13% to 27% identical to the C7L protein. Other C7L homologues include ORF 67R of Yaba-like disease virus (YLDV), which is 28% identical to the C7L protein. The similarities between C7L and its homologues from several different poxvirus genera are listed in Table 1 and shown by their multiple amino acid sequence alignment in Figure 1. Compared to VACV C7L, all C7L homologues have additional C-terminal tails that share very little sequence similarity among each other except that they all contain many acidic residues. MYXV M63R has an additional 24-amino acids internal insertion that is absent from other C7L homologues. Unlike CP77 and K1L, C7L and its homologues contain no ankyrin repeat, as assessed by the REP program (http://www.embl-heidelberg.de/~andrade/papers/rep/search.html) (Andrade et al., 2000). No C7L homologue was found outside the poxvirus family.
Table I.
Percentage identity and similarity between C7L and its homologues from mammalian poxviruses.
| VACV C7La | MYXV M62R | YLDV 67R | SPPV 063 | SWPV 064 | DPV 076 | MYXV M63R | MYXV M64R | CWPX 020 | |
|---|---|---|---|---|---|---|---|---|---|
| VACV C7L | 27 (42)b | 28 (45) | 24 (39) | 28 (41) | 25 (37) | 13 (25) | 16 (33) | 26 (41) | |
| MYXV M62R | 37 (48) | 30 (43) | 35 (45) | 29 (43) | 20 (36) | 30 (43) | 16 (28) | ||
| YLDV 67R | 40 (51) | 42 (58) | 39 (55) | 24 (39) | 28 (43) | 23 (37) | |||
| SPPV 063 | 44 (56) | 56 (65) | 20 (41) | 30 (44) | 18 (32) | ||||
| SWPV 064 | 42 (59) | 21 (36) | 31 (49) | 17 (32) | |||||
| DPV 076 | 29 (57) | 28 (45) | 16 (29) | ||||||
| MYXV M63R | 20 (36) | 14 (27) | |||||||
| MYXV M64R | 15 (29) |
The names of the ORFs are as described in the legend to Figure 1.
The number outside and inside the parenthesis indicates respectively the percentage identity and similarity between the two compared amino acid sequences.
Figure 1.

A multiple-sequence alignment of C7L and its homologues from mammalian poxviruses. They are VACV (vaccinia virus, orthopoxvirus) C7L, MYXV (myxoma virus, leporipoxvirus) M62R, YLDV (Yaba-like diseases virus, yatapoxvirus) 67R, SPPV (sheeppox virus, capripoxvirus) 063, SWPV (swinepox virus, suipoxvirus) 064, DPV (deerpox virus, unclassified) 076, MYXV M63R, MYXV M64R and CWPX (cowpox virus, orthopoxvirus) 020. Amino acids that are identical or similar in five or more of the nine proteins are highlighted. The dark and light shading indicate amino acids that are identical or similar, respectively.
Construction of recombinant VACV with its K1L deleted and its C7L substituted with a C7L homologue
In the absence of the K1L gene, VACV depends on C7L to provide an essential function to replicate productively in human cells (Perkus et al., 1990). Therefore, to determine whether any C7L homologue could perform the same function as VACV C7L, we constructed recombinant VACV with its K1L deleted and its C7L substituted with a C7L homologue. The viruses were constructed by placing the coding sequence for the C7L homologue into the C7L locus of vK1L−C7L−, a previously constructed virus with precise deletion of K1L and C7L genes (Meng and Xiang, 2006) (Fig. 2A). Five representative C7L homologues, YLDV 67R, CWPX 020 and MYXV M62R, M63R and M64R, were chosen to substitute C7L. Through homologous recombination, each C7L homologue, along with a C-terminal tag and an adjacent selective marker consisting of the green fluorescent protein (GFP) controlled by a late stage promoter, was placed immediate downstream of the C7L promoter so that its expression was controlled by the authentic C7L promoter (Fig. 2A). For all the C7L homologues, their C-terminal tag has the V5 epitope to facilitate their detection. For YLDV 67R and CWPX020, their C-terminal tag includes the additional calmodulin-binding domain and biotin binding domain (referred together as V5-TAP tag), intended to facilitate their tandem affinity purification. As controls, recombinant viruses encoding C7L with either a V5 or a V5-TAP tag were constructed similarly. All the recombinant viruses were isolated on VERO cells where they form normal-size plaques (shown later). The viruses are referred hereafter by the name of the ORF present at the original C7L locus of the virus.
Figure 2.
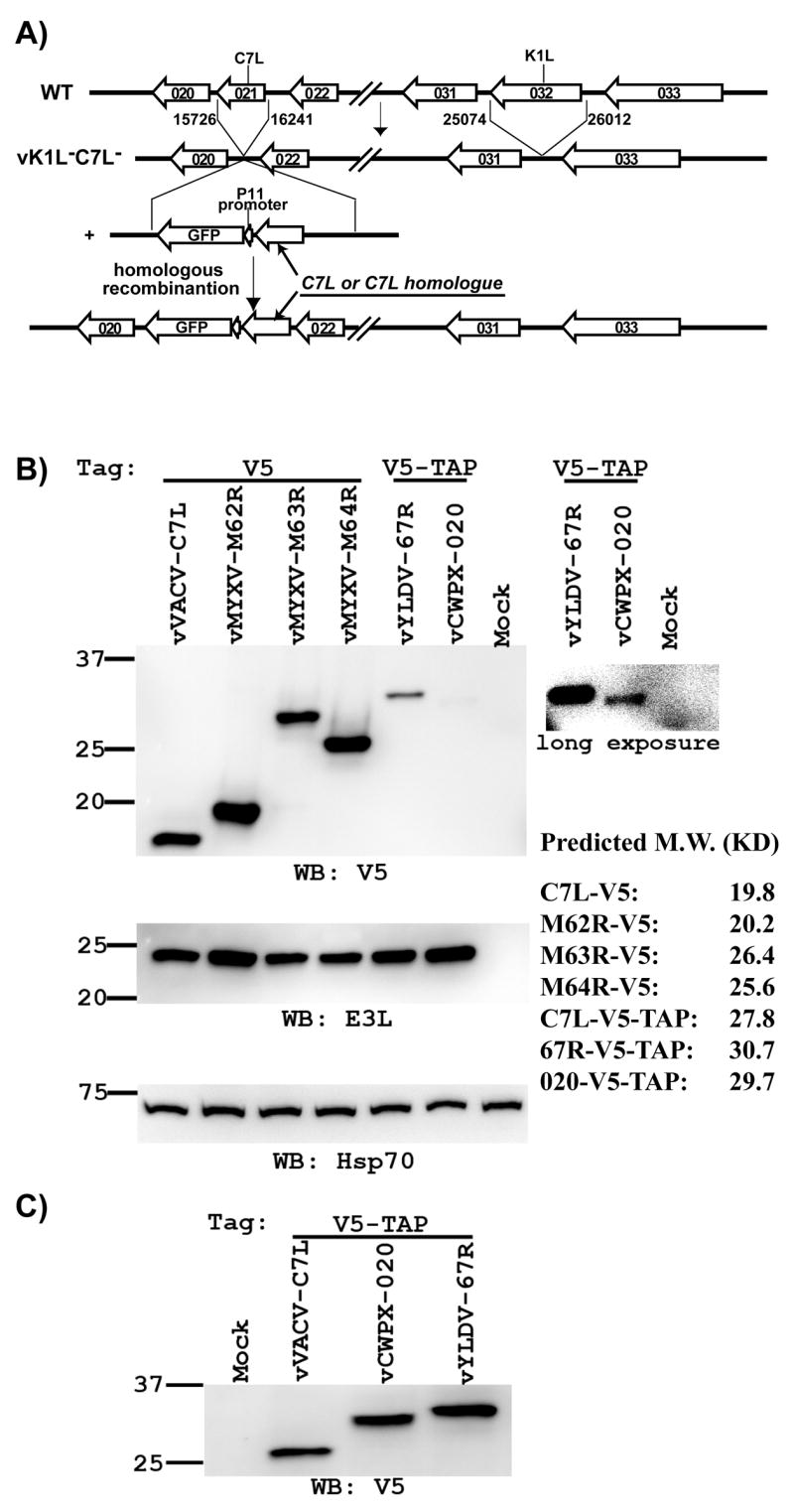
Construction of recombinant VACV with its K1L deleted and its C7L substituted with a C7L homologue. (A). Schematic illustration of the construction and genomic organization of the recombinant VACV. The construction of K1L and C7L deletion mutant vK1L−C7L− was described previously (15). Recombinant VACV encoding the C7L homologue were constructed by homologous recombination of vK1L−C7L− and a plasmid encoding the C7L homologue and the green fluorescence protein (GFP). P11 indicates the VACV promoter that normally regulates late expression of the gene encoding the 11K protein (WR F18R). (B & C). The recombinant VACV expresses the expected C7L homologue. VERO cells were infected at a MOI of 10 PFU (B) or 25 PFU (C) per cell with the indicated viruses, which are referred by the name of ORF present at the C7L locus. The C7L homologue is tagged at its C-terminus with either a V5 or V5-TAP tag. At 8 hpi, the level of C7L homologue in infected cells was determined by Western blot with a monoclonal antibody (mAb) against V5. A longer exposure of the blot was shown on the right to illustrate the weaker band. On the same blot, the levels of VACV early protein E3L and host cell protein Hsp70 were also determined with an anti-E3L polyclonal antiserum and an anti-Hsp70 mAb. Hsp70 serves as a control for gel loading, while E3L serves as an indicator of VACV early protein expression. The size of the molecular weight marker in kilodaltons is shown on the left. The predicted molecular weights of the tagged C7L-homologues are listed on the right.
To assess whether the recombinant viruses expressed the expected C7L homologue, VERO cells were infected with the viruses at a multiplicity of infection (MOI) of 10 PFU per cell, and then the cell lysates were analyzed by immunoblot. A protein band that is consistent with the predicated molecular mass of the respective C7L homologue was detected by anti-V5 antibody from cells that were infected by the recombinant viruses (Fig. 2B). The signal for YLDV 67R and CWPX 020 proteins, which were tagged with V5-TAP, was significantly weaker than that for the other C7L homologues, which were tagged with V5 epitope. This was not due to a reduced viral protein synthesis by the particular recombinant viruses, as the expression level of another VACV early protein E3L was similar for all the viruses. The signal for C7L tagged with V5-TAP was also weaker than that for C7L tagged with V5 only (data not shown), so the TAP tag sequence C-terminal to the V5 epitope probably made the protein less stable or less detectible by anti-V5 antibody in immunoblot. C7L, YLDV 67R and CWPX 020 that were tagged with V5-TAP were better detected when the cells were infected at a higher MOI (Fig. 2C).
YLDV 67R and MYXV M62R can substitute C7L for its function in human and murine cell lines while CWPX 020, MYXV M63R and M64R cannot
To determine whether the C7L homologues could function as a host-range gene for VACV, the abilities of the recombinant viruses to form plaques on various human and murine cell lines were analyzed. A-431, an epidermoid carcinoma cell line, and HeLa, a cervix adenocarcinoma cell line, were chosen as representative human cells. 3T3, an embryonic fibroblast cell line, was chosen as representative murine cells. On these cell types, the parental vK1L−C7L− did not form plaque (data not shown), but vVACV-C7L, encoding either the V5 or V5-TAP tagged C7L, formed normal-size plaques (Fig. 3A and data not shown), indicating that the tagged C7L protein is functional in murine as well as in human cells. vYLDV-67R and vMYXV-M62R also formed normal-size plaques on these cells, while vCWPX-020, vMYXV-M63R and vMYXV-M64R did not form any plaque (Fig. 3A), indicating that only YLDV 67R and MYXV M62R can function as a C7L-like host-range gene. This is further demonstrated by one-step growth analysis, which showed that vYLDV-67R and vMYXV-M62R replicated as efficiently as vVACV-C7L in human and murine cells (Fig. 3B and data not shown). None of the recombinant viruses was able to replicate productively in rabbit RK-13 cell (data not shown), indicating that YLDV 67R and MYXV M62R, similar to C7L, were not able to substitute K1L for the host-range function in RK-13 cells.
Figure 3.
YLDV 67R and MYXV M62R can substitute C7L for its function in human and murine cell lines while CPXV 020, MYXV M63R and M64R cannot. (A). Plaque morphology of the recombinant VACV on representative cell lines. VERO, 3T3, HeLa and A431 cells were infected at a MOI of 0.01 PFU/cell with the indicated viruses. At approximately 36 hpi, GFP-expressing cells were visualized with an inverted fluorescence microscope using a 10× objective and photographed. (B). Growth curves of the recombinant VACV in HeLa cells at MOI of 5 PFU/cell. Virus yields at 0, 24 and 48 hpi were determined by plaque assay on the permissive VERO cells. The growth curves of the recombinant VACV in 3T3 and A431 were similar to the ones in HeLa cells and thus not shown.
Host-range restricted viruses did not synthesize viral late proteins and expressed a reduced level of the early protein E3L in nonpermissive cells
In nonpermissive murine and human cells, vCWPX-020, vMYXV-M63R and vMYXV-M64R did not express GFP that was controlled by the late P11 promoter (Fig. 3A), suggesting that their late protein synthesis is defective. To further characterize the step at which the replication of these host-range restricted viruses was blocked, HeLa cells were infected by the recombinant viruses for various times and then subjected to immunoblots. At 8 hpi, the C7L protein or its homologue was detected for all viruses except vCWPX-020, whose expression of CWPX 020 in the permissive VERO cells was also difficult to detect (Fig. 4). Similarly, VACV early protein E3L was expressed by all the viruses. At 12 hpi, the VACV late protein encoded by ORF WR148 (Jones-Trower et al., 2005), a remnant of orthopoxvirus A-type inclusion (ATI) protein, was abundantly expressed by the replication competent viruses (vVACV-C7L, vMYXV-M62R and vYLDV-67R), but it was not expressed at all by the host-range restricted viruses (vCWPX-020, vMYXV-M63R and vMYXV-M64R), demonstrating a defect of the host-range restricted viruses at late protein synthesis in nonpermissive cells.
Figure 4.
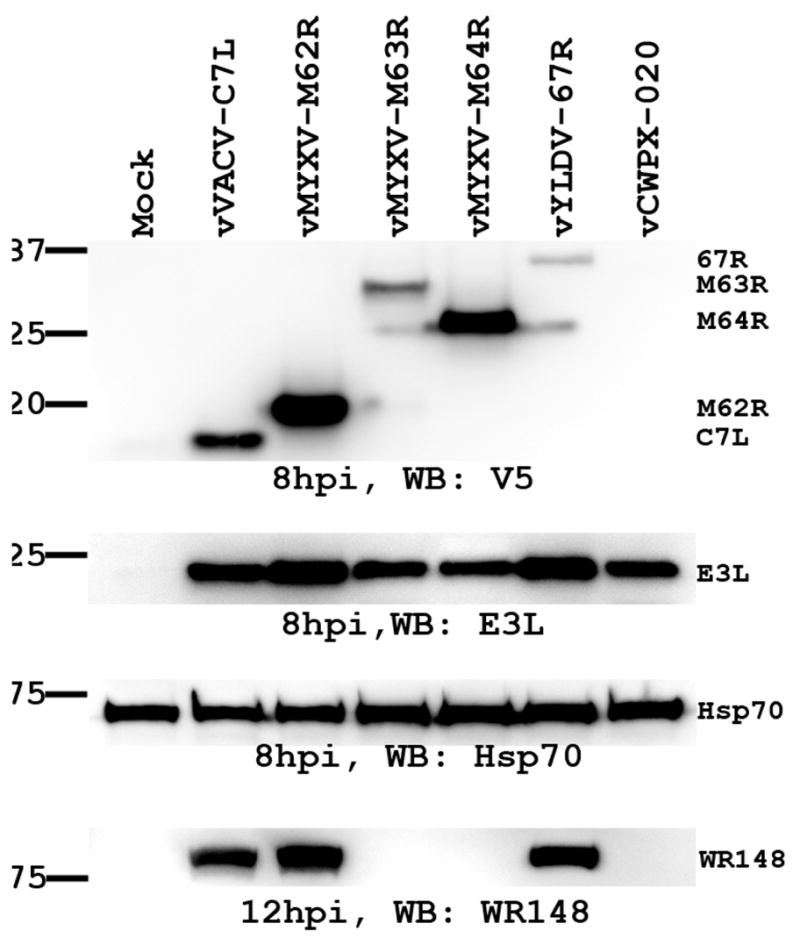
vMYXV-M63R, vMYXV-M64R and vCWPX-020 are defective at viral late protein synthesis in nonpermissive HeLa cells. HeLa cells were infected with the indicated viruses at MOI of 10 PFU/cell. At 8 hpi, the level of viral early proteins C7L (or its homologue) and E3L were determined by Western blot as described in Fig 2B. At 12 hpi, the level of a VACV late protein encoded by ORF WR148 (11) was determined by Western blot with a mAb against WR148. WR148 is a remnant of orthopoxvirus Atype inclusion (ATI) protein.
To define more precisely the difference in viral protein expression in permissive and nonpermissive cells by the host-range restricted viruses, a time course of viral protein expression was determined for vMYXV-M64R and vVACV-C7L. In the permissive VERO cells, the M64R protein was detected as early as 2 hpi, and its amount increased over time up to 12 hpi (Fig. 5A). This time course was similar to that of E3L. In contrast, VACV late protein WR148 was not detected until 8 hpi but its amount increased up to 24 hpi when the infection was stopped. A similar time course of viral protein expression was observed for vVACV-C7L. In nonpermissive HeLa cells, however, vMYXV-M64R did not express WR148 and expressed E3L at a reduced level (Fig. 5B). At 2 hpi, the level of E3L protein was similarly low in vMYXV-M64R- and vVACV-C7L-infected cells. While the level of E3L protein in vVACV-C7L-infected HeLa cells continued to increase up to 8 hpi, its level in vMYXV-M64R-infeced cells decreased after 4 hpi, indicating that vMYXV-M64R is defective at sustaining E3L protein synthesis. The level of M64R in vMYXV-M64R-infected cells continued to increase up to 8 hpi, suggesting that the expression of different viral early genes by vMYXV-M64R is affected by different degrees in nonpermissive cells.
Figure 5.
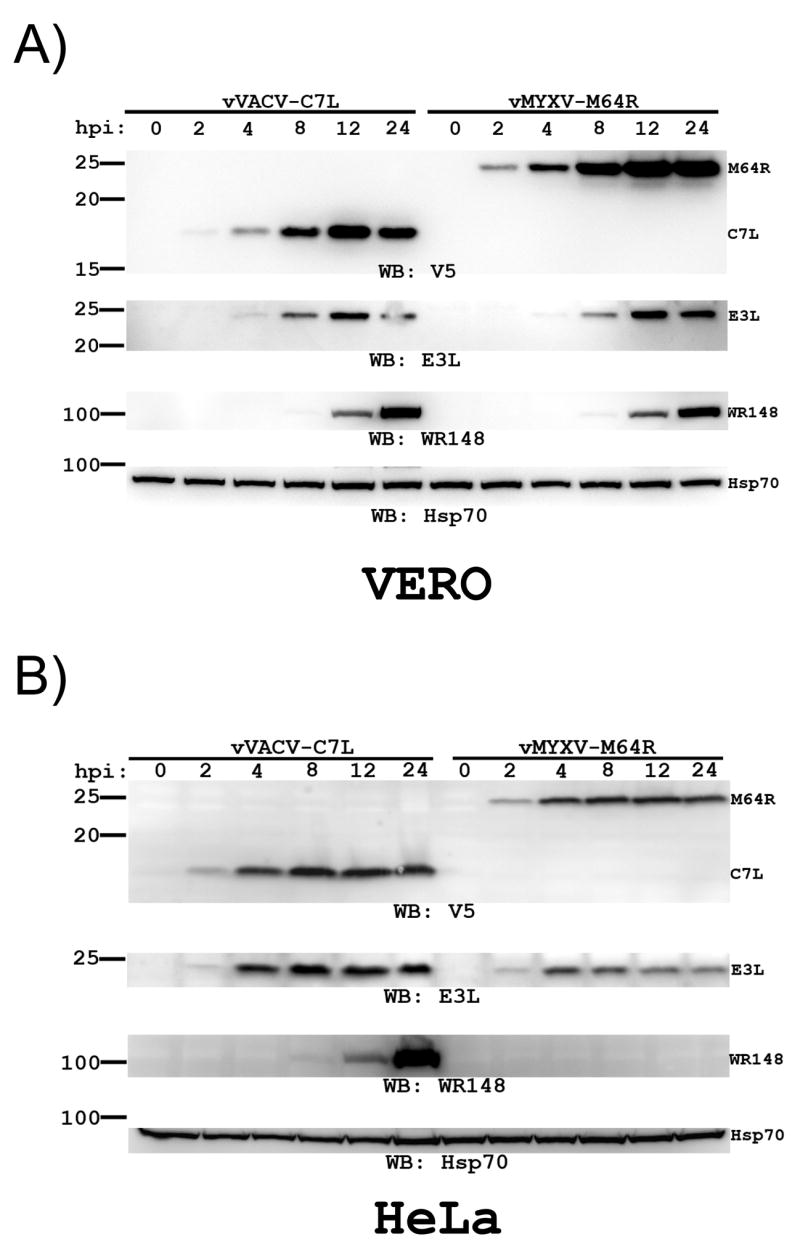
The temporal expression of viral proteins by vVACV-C7L and vMYXV-M64R in VERO and HeLa cells. VERO (A) and HeLa cells (B) were infected at a MOI of 10 PFU per cell with vVACV-C7L or vMYXV-M64R. At the indicated time after infection (hpi), the level of C7L or M64R proteins was determined by Western blot with an anti-V5 antibody. The levels of VACV proteins E3L, WR148 and host cell protein Hsp70 were also determined by Western blot as respective control for VACV early, late protein expression and gel loading.
The role of C7L in VACV-mediated suppression of cellular PKR and eIF2-α activities
It was previously reported that VACV mutants with neither K1L nor C7L genes induce PKR and eIF2-α phosphorylation in HeLa cells and that restoring C7L or CP77 to the mutants could prevent the phosphorylation (Hsiao et al., 2004; Najera et al., 2006). However, it was unclear how C7L or CP77 affects PKR and eIF2-α phosphorylation and whether this effect contributes to the ability of C7L and CP77 to support viral protein translation. The E3L protein is a well-known inhibitor of PKR and the consequent eIF2-α phosphorylation (Langland and Jacobs, 2004). As we found that C7L was required for sustaining E3L expression in HeLa cells, we then performed experiments to assess whether the effect of C7L on cellular PKR and eIF2-α phosphorylation could be attributed to its effect on E3L expression. First, we determined the time courses of PKR and eIF2-α phosphorylation in vMYXV-M64R- or vVACV-C7L-infected HeLa cells by immunoblotting with antibodies against total and phosphorylated PKR and eIF2-α. At 4 hpi, while phospho-PKR was not detected in vVACV-C7L-infected cells, it appeared in vMYXV-M64R-infected cells (Fig. 6A). No difference in the level of phospho-eIF2-α was observed at this time point. At 8 and 12 hpi, an increasing amount of phospho-PKR was detected in vMYXV-M64R-infected cells. Concomitantly, there was an increase in the amount of phospho-eIF2-α in the cells. In contrast, the amount of phospho-PKR and phospho-eIF2-α were relatively lower in vVACV-C7L infected cells at these time points. This result, taken together with the time course of E3L expression shown in Fig. 5B, indicates that the difference in the level of phospho-PKR and phospho-eIF2-α in vVACV-C7L- and vMYXV-M64R-infected HeLa cells correlates closely with the difference in the level of E3L protein in the cells.
Figure 6.
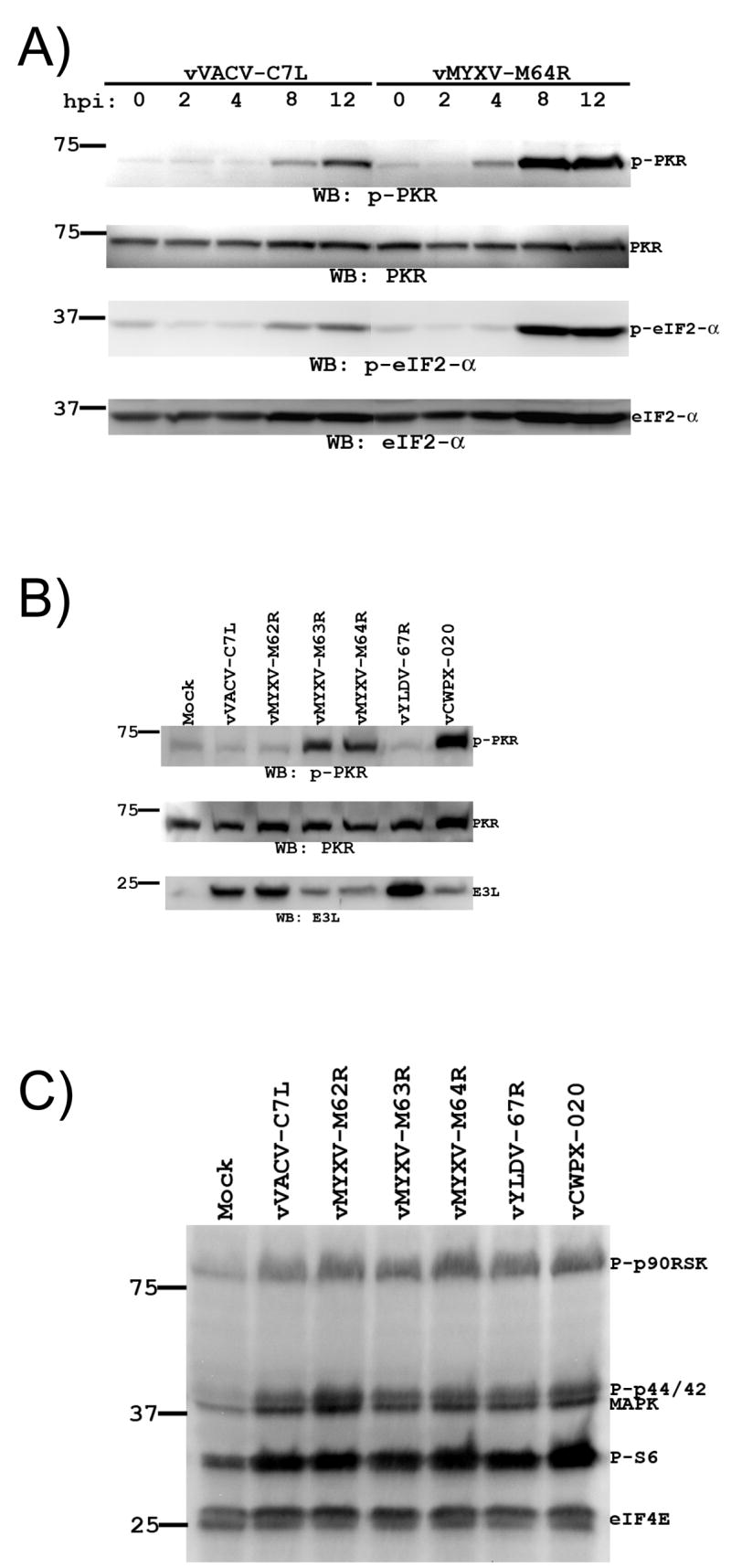
The defect of the host-range restricted viruses at suppressing cellular PKR and eIF2-α phosphorylation in HeLa cells correlates with a defect of the viruses at maintaining E3L expression. (A). The same cell lysates as described in Fig 5B were used to determine the levels of total and phosphorylated PKR and eIF2-α proteins in HeLa cells that were infected with vVACV-C7L or vMYXV-M64R for various time. The level of phosphorylated PKR and eIF2-α proteins were determined by Western blot with mAbs against phospho-PKR (Thr446) and phospho-eIF2α (Ser51). The blot was then stripped, and the levels of total PKR and eIF2-α were determined by Western blot with a mAb against total PKR and a polyclonal antibody against total eIF2-α. (B). HeLa cells were infected with the indicated viruses for 4 h and the level of total and phosphorylated PKR and VACV E3L proteins were determined by Western blot. (C). The same samples as shown in B were analyzed by Western blot with a multiplex of antibodies, including antibodies against phospho-p90RSK (Ser-308), phospho-Akt (Ser-473), phospho-p44/42 mitogen-activated protein kinase (Thr-202/Tyr-204), phospho-S6 ribosomal protein (Ser-235/236), and total eIF4E.
Next, to determine the effect of C7L homologues on PKR activation, we infected HeLa cells with viruses expressing different C7L homologues for 4 h and then assessed the level of phospho-PKR (Fig. 6B). In cells that are mock-infected or infected with replication competent virus (vVACV-C7L, vMYXV-M62R or vYLDV-67R), very little phospho-PKR was detected. In contrast, phospho-PKR was clearly detected in cells that were infected with host-range restricted viruses (vMYXV-M63R, vMXYV-M64R and vCPXV-020). The level of E3L proteins in these cells was also significantly lower than that in cells infected by replication competent viruses. This result shows that MYXV M62R and YLDV 67R can substitute C7L for its effect on the inhibition of PKR phosphorylation and is again consistent with the idea that C7L affects PKR phosphorylation indirectly through its effect on E3L expression.
In an effort to find additional cellular pathways that are affected by C7L-like host-range genes, we tested the effects of the recombinant viruses on several signaling molecules that are related to cellular growth and protein translation. HeLa cells were infected by the viruses for 4 h. Then, the cellular level of phospho-p90RSK, phospho-Akt, phospho-p44/42 mitogen-activated protein kinase, phospho-S6 ribosomal protein, and total eIF4E were determined together with a multiplex of antibodies in immunoblot (Fig. 6C). The level of total eIF4E was similar for mock- and virus-infected cells, indicating equal amount of proteins loaded on the gel. No or very low level of phospho-Akt was detected in the cells during repeated experiments. The levels of phospho-S6 ribosomal protein, phosphoylated p44/42 and its downstream effector p90RSK were elevated in virus-infected cells. Since the phosphorylation of these molecules was equally induced by all the recombinant VACVs, C7L is unlikely to play any role in this process and thus this phenomenon was not investigated further. Nevertheless, this experiment served as a control demonstrating that PKR is a specific pathway that is affected by the presence of C7L-like host-range gene in VACV.
vMYXV-M64R is highly attenuated in a mouse intranasal infection model while vMYXV-M62R is virulent
To assess whether the ability of the C7L homologue to support VACV replication in cell lines correlates with an ability to support VACV replication in animal hosts, the relative virulence of vVACV-C7L, vMYXV-M62R and vMYXV-M64R was determined with a mouse model. Mice were infected intranasally with 105 PFU of purified viruses and monitored daily for their body weights (Fig. 7A). Similar to mock-infected mice, vMYXV-M64R-infected mice gained some weight by day 5 post infection, indicating that vMYXV-M64R is highly attenuated and does not cause disease in mice. In contrast, both vVACV-C7L- and vMYXV-M62R-infected mice lost close to 30% of their original body weight by day 5 post infection (Fig. 7A), when they were euthanized and the viral loads in their organs were determined. While the mice infected by vMYXV-M64R had no detectable level of virus in their organs (data not shown), the mice infected by either vVACV-C7L or vMYXV-M62R had a similarly high level of virus in their lung and spleen (Fig. 7B), indicating that MYXV M62R can substitute VACV C7L at supporting VACV replication and dissemination in vivo while M64R cannot. In a separate experiment, the VACV mutant with a deletion in K1L and C7L, which was the parental virus for vVACV-C7L, vMYXV-M62R and vMYXV-M64R, was found to be highly attenuated in mice (data not shown), similar to vMYXV-M64R.
Figure 7.
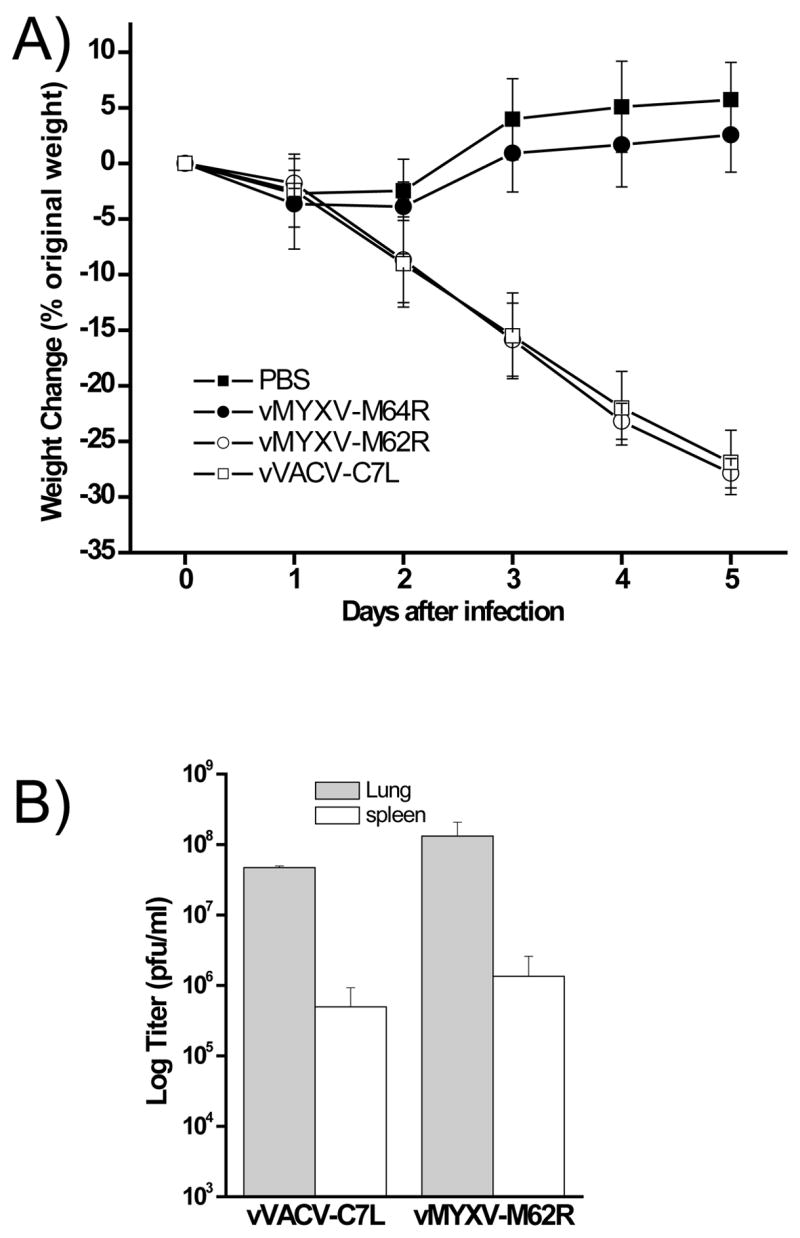
vMYXV-M64R is highly attenuated in a mouse intranasal infection model while vMYXV-M62R is virulent. (A). Groups of 5 Balb/c mice were infected intranasally with 105 PFU of indicated viruses or mock infected with the same volume of PBS. The mice were then monitored daily for their body weights. For each group, the average of the percentage of body weight change and standard deviation are shown. (B). At day 7 post infection, mice were euthanized and their lungs and spleen were harvested. Approximately 16 mg of spleen and 5 mg of lung from each mouse were homogenized and their viral loads were determined by plaque assay on VERO cells. Error bars represent standard deviation.
Discussion
Our current understanding of the mechanism by which poxvirus host tropism is controlled came mainly from the identification and analysis of poxvirus host-range genes. Previously, different sets of host-range genes were identified for orthopoxviruses and leporipoxviruses, suggesting that poxviruses of different genera may have evolved their own unique sets of host-range genes to adapt to their unique host environment. In this study, however, we found that the function encoded by VACV host-range gene C7L is not limited to orthopoxviruses and is not specific for host interaction in human cell lines as previous studies suggested (Perkus et al., 1990). We found that myxoma virus and yaba-like disease virus both encode a gene that functions equivalently to VACV C7L, suggesting that C7L is a poxvirus host-range gene whose function is essential for the replication of many mammalian poxviruses in mammalian hosts.
We characterized five distantly related C7L homologues and found that two of these, MYXV M62R and YLDV 67R, function equivalently to VACV C7L, while three others, MYXV M63R, M64R and CPXV 020, do not. Among the latter three, CPXV 020 was expressed at a reduced level by our recombinant VACV, so the possibility that a higher level of CPXV 020 could have provided a C7L-like function for poxvirus cannot be ruled out. However, it would have been redundant for CPXV 020 to encode a C7L-like function, since CPXV encodes a C7L orthologue that is 97–100% identical to VACV C7L. The identification of both functionally equivalent and non-equivalent C7L homologues offers us an opportunity to examine other poxvirus genomes for the presence of C7L-like host-range gene. Although all C7L homologues have overall similar sequence homology to C7L, we notice that the N-terminal 10 amino acid of C7L are more conserved in functionally equivalent C7L homologues (MYXV M62R and YLDV 67R) than in functionally non-equivalent C7L homologues (CPXV020, MYXV M63R and MYXV M64R). Whether these N-terminal residues play any functional role remains to be determined, but it suggests that sheeppox virus (SPPV) 063, swinepox virus (SWPV) 064 and deerpox virus (DPV) 076 may also function equivalently to C7L, as their N-terminus 10 amino acids are more similar to C7L than to the nonfunctional C7L homologues (Fig. 1). YLDV 67R and MYXV M62R share more than 40% amino acid similarity with each other and with SPPV 063, SWPV 064 and DPV 076 (Table 1). This level of sequence similarity is higher than that between these C7L homologues and VACV C7L. Moreover, the locations of SPPV 063, SWPV 064 and DPV 076 in the viral genomes are identical to that of YLDV 67R and MYXV M62R. All are present in the central region of the genome between the genes encoding thymidine kinase and the polyA polymerase regulatory protein. Thus, it appears that SPPV 063, SWPV 064 and DPV 076 are orthologues of YLDV 67R and MYXV M62R. Interestingly, the genomic location of the C7L gene differs from that of YLDV 67R or MYXV M62R. In orthopoxviruses, the C7L gene locates near the left end of the genome, and there is no ORF located between the genes encoding thymidine kinase and the polyA polymerase regulatory protein.
The identification of a confirmed or putative C7L-like host-range gene in orthopoxviruses, leporipoxviruses, yatapoxviruses, capripoxviruses, suipoxviruses and the unclassified deerpox virus suggests that this gene does not confer species-specific tropism to poxvirus, as these poxviruses have different host-ranges in mammalian species. Rather, the C7L-like host-range gene may represent an adaptation of mammalian poxviruses to mammalian hosts, as it is not encoded by avian poxviruses and not required for orthopoxviruses to replicate in avian cells. Among all sequenced mammalian poxviruses, the only viruses that do not encode a C7L homologue are molluscum contagiosum virus (MCV) and parapoxviruses. MCV has the narrowest host-range among mammalian poxviruses. It infects human exclusively and replicates only in keratinocytes in the basal epidermal layer of the skin (Buller et al., 1995). MCV does not replicate in any tissue culture cells. Parapoxviruses also have a limited host-range, causing localized skin lesion in ruminants. In tissue culture, they were usually propagated on primary muscle or testis cells and can be adapted to propagate on some mammalian cell lines including VERO cells (Fischer et al., 2003). It is possible that the lack of a C7L-like host-range gene may contribute to the very narrow host-range of these viruses in mammalian cells.
We showed here that, in the absence of the K1L gene, C7L or a C7L-like host-range gene is required for VACV to replicate and disseminate in mice. As some orthopoxviruses including variola virus do not have an intact K1L gene, our results suggest that the variola virus ORF corresponding to C7L is essential for variola virus to replicate in humans and thus cause smallpox. Similarly, as no functional homologue of K1L has been found outside the orthopoxviruses, the C7L-like host-range gene in leporipoxviruses, yatapoxviruses, capripoxviruses, suipoxviruses and deerpox virus is predicted to be essential for the replication of these viruses in their respective hosts. Among these viruses, myxoma virus is unique in that it encodes three C7L homologues. M62R is shown here to function equivalently to C7L, while M63R was recently found to be a host-range gene that is essential for myxoma virus replication in rabbits (Barrett et al., 2007). Our finding that M63R cannot rescue the host-range defect of the K1L and C7L deficient VACV is consistent with the previous report that VACV K1L cannot rescue the host-range defect of M63R deficient myxoma virus (Barrett et al., 2007). M62R and M63R may operate on two different cellular pathways that are both essential for the replication of myxoma virus. While the cellular pathway K1L/C7L/M62R operates on may be conserved in all mammalian cells, the pathway M63R operates on may be more specific to rabbit cells.
Our studies of K1L and C7L deficient VACV mutants revealed a defect of the mutants at maintaining the synthesis of VACV early protein E3L in nonpermissive cells. Previous studies of a K1L and C7L deficient VACV mutant suggested that the replication of the mutant in nonpermissive cells was blocked at the stage of intermediate mRNA translation (Hsiao et al., 2004). This was based on the finding that viral intermediate and late proteins were not synthesized in nonpermissive cells even though viral intermediate genes were transcribed. The synthesis of viral early proteins was presumed to be normal. In this study, we also found that viral late proteins were not synthesized by our host-range mutants in nonpermissive cells. In addition, we found that, while the level of VACV early protein E3L accumulated up to 8 hpi in HeLa cells infected by C7L positive VACV, its level in HeLa cells infected by K1L and C7L deficient VACV was low and did not increase after 4 hpi. We suggest that, in addition to a defect in viral intermediate and late protein synthesis, K1L and C7L deficient VACV also has a defect at maintaining viral early protein synthesis. While this defect is quite pronounced for the synthesis of E3L, it is subtle for C7L promoter directed expression of MYXV M64R. This may due to a difference in the mRNA levels of these two early genes, which may result in their different susceptibility to the translational block. Alternatively, it may due to a difference in the stability of their gene products, which may result in an apparent difference in the steady state protein levels. We speculate that the host-range mutant gradually elicits a general translational block in nonpermissive host cells and that the block reaches its full effect when viral intermediate mRNAs are translated. This may explain why the translational block may appear to affect predominantly viral intermediate protein synthesis.
Our finding that viral early protein levels are affected by the lack of a C7L-like host-range gene suggests that some of the reported defects associated with K1L and C7L deficient VACV may be the indirect consequence of reduced expression of some viral early genes, which include many host response modulators. Similar to previous reports (Hsiao et al., 2004; Najera et al., 2006), we observed that K1L and C7L deficient VACV mutants triggered PKR and eIF2-α phosphorylation in nonpermissive cells and that restoring C7L can reverse this phenotype. In addition, we found that MYXV M62R and YLDV 67R can substitute C7L for the same effect, while MYXV M63R, M64R and CWPX 020 cannot. This apparent effect of C7L-like host-range genes on PKR and eIF2-α phosphorylation appears to be specific as several other signaling pathways, including ERK1/2 MAPK pathway, are not affected. However, the inhibition of PKR and eIF2-α phosphorylation is unlikely to be the mechanism by which C7L-like host-range genes prevent a translational block on viral proteins, as we observed a decrease in E3L protein level at 4 hpi, which coincides with PKR phosphorylation and precedes eIF2-α phosphorylation. A more likely explanation is that PKR and eIF2-α phosphorylation is the consequences of diminished expression of E3L proteins by K1L and C7L deficient VACV. Similarly, the reported induction of cellular apoptosis by K1L and C7L deficient VACV (Hsiao et al., 2004; Najera et al., 2006) may be another consequence of diminished expression of viral early proteins, which include several inhibitors of apoptosis. This is consistent with the fact that inhibiting cellular apoptosis cannot rescue the host-range defect of the mutants (Hsiao et al., 2004; Ink, Gilbert, and Evan, 1995). The exact mechanism by which CP77/K1L/C7L regulates poxvirus tropism will have to await the identification of direct molecular targets of these host-range genes.
Materials and Methods
Cells and viruses
VERO (ATCC CCL-81), RK-13 (ATCC CCL-37) and CV-1 (ATCC CCL-70) cells were cultured in minimum essential medium with Earle’s balanced salts (Invitrogen) supplemented with 10% fetal bovine serum (FBS). HeLa 229 (ATCC CCL-2.1), A-431 (ATCC CRL-1555) and NIH/3T3 (ATCC CRL-1658) cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen) with 10% FBS. All recombinant VACVs were propagated on VERO cells. Yaba-like disease virus (ATCC VR-937) was obtained from ATCC and propagated on CV-1 cells.
Recombinant virus construction
Recombinant viruses used in this study were derived from two viruses with similar deletions in K1L and C7L genes. vK1L−C7L− was used initially in the study. It was derived from vTF7.3 (Fuerst et al., 1986) as reported previously (Meng and Xiang, 2006). WR.K1L−C7L− was used subsequently. It was constructed identically to vK1L−C7L− except that the wild type WR strain was used as the parental virus. WR.K1L−C7L− behaves identically to vK1L−C7L− in tissue culture, but it has the intact thymidine kinase (TK) gene, which is a virulence factor for VACV in mice (Buller et al., 1985). All studies involving mice were done with WR.K1L−C7L− derived viruses.
Viruses expressing V5-TAP-tagged C7L, YLDV 67R or CWPX 020 ORF were derived from vK1L−C7L− through homologous recombination with appropriate transfer plasmids. Each transfer plasmid contains (i) 300 bp of downstream flanking region of C7L, (ii) a GFP under the control of VACV late promoter P11, (iii) ORF for C7L or its homologue with a C-terminal V5 and TAP tag, and (iv) 300 bp of upstream flanking region of C7L including the C7L promoter. The transfer plasmids were assembled together through recombinant PCR followed by cloning into pCR2.1 (Invitrogen). The GFP cassette was derived from pYW31 (Meng and Xiang, 2006) and was inserted between the downstream flanking region of C7L and the stop codon for C7L (or its homologue). The TAP tag was derived from pCTAP (Stratagene) and placed immediately after the V5 tag and before the stop codon. YLDV 67R or CWPX 020 ORF was derived from genomic DNA of YLDV or CWPX and inserted immediately downstream of the C7L promoter. The recombinant virus construction was done according to standard protocols (Earl et al., 1998). Briefly, lipofectamine 2000 (Invitrogen) was used to transfect the transfer plasmids into VERO cells that had been infected with vK1L−C7L−. Recombinant viruses expressing GFP were picked under the fluorescence microscope and purified through four rounds of plaque isolation on VERO cells.
Viruses expressing V5-tagged C7L, MYXV M62R, M63R and M64R were constructed similarly as described above except that WR.K1L−C7L− was used as the parental virus and only the V5 tag was appended to C-terminus of C7L (or its homologue) in the transfer plasmids. MYXV M62R, M63R and M64R were derived from MYXV genomic DNA.
To confirm that the recombinant viruses had the desired ORF inserted at the C7L locus, viral DNA was exacted from infected cells (Qiagen Blood Kit) and the region of the viral genome from 300 bp downstream and 300 bp upstream of the original C7L ORF was PCR-amplified. The PCR product was further digested with the restriction enzyme that recognized the inserted ORF.
Growth curve analysis
Cells in 12-well plates were incubated with 0.01 or 5 PFU per cell of the viruses for 2 h at room temperature. Following adsorption, the cells were washed twice with phosphate-buffered saline (PBS) and moved to 37°C incubator to initiate viral entry and replication. The cells were harvest at 0, 24 and 48 hpi. The viral titers in the cell lysates were determined by plaque assays on VERO cells. The results were confirmed by repeating the same experiment one time. Infected cells expressing GFP were also visualized under an inverted fluorescence microscopy using a 10× objective and photographed with a digital camera.
Western blot analysis
The Western blot analysis was performed essentially as described previously (Meng and Xiang, 2006). Briefly, the same amount of cell lysates, as measured by Bradford protein assay, were solubilized in sodium dodecyl sulfate (SDS) sample buffer, resolved by SDS-polyacrylamide gel electrophoresis (PAGE), transferred to nitrocellulose membranes, blocked, incubated with appropriate antibodies and analyzed with chemiluminescence reagent (Pierce). The same membrane was sometimes analyzed with additional antibodies after striping the membrane with the Restore Western Blot Stripping Buffer (Pierce).
Antibodies
The mouse monoclonal antibody (mAb) against V5 was obtained from Sigma-Aldrich. The mAbs against phospho-PKR (Thr446), phospho-eIF2α (Ser51) and the PathScan Multiplex Western Mixture I were from Cell Signaling technology. The mAbs against PKR and Hsp70, and the polyclonal antibody against total eIF2α were from Santa Cruz biotechnology. Rabbit anti-E3L polyclonal antiserum was a kind gift of Dr. Bertram Jacobs. The mouse mAb against WR148 was from a hybridoma developed with a WR-infected mouse (Meng X., G. Zhong and Y. Xiang, unpublished).
Animals
Groups of five female BALB/c 5-week-old mice were anesthetized and infected intranasally with 105 PFU of viruses in 20 μl PBS. The viruses used were previously purified through a sucrose cushion sedimentation according to the standard protocol (Earl et al., 1998). Individual mice were weighed every day. At day 7 post infection, all mice were euthanized and their lung and spleen were harvested. The organs were homogenized in 1.5-ml Eppendorf tube with a battery operated pestle grinder (Fisher) and sonicated with a cup horn sonicator. Titration of virus was done with VERO cells.
Acknowledgments
We thank Dr. Bertram Jacobs for providing anti-E3L antiserum and Dr. Grant McFadden for providing myxoma virus DNA. The work was supported by a grant from NIAID to Y. Xiang through the Western Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research (NIH grant number U54 AI057156) and NIH grant R21AI065731.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andrade MA, Ponting CP, Gibson TJ, Bork P. Homology-based method for identification of protein repeats using statistical significance estimates. J Mol Biol. 2000;298(3):521–37. doi: 10.1006/jmbi.2000.3684. [DOI] [PubMed] [Google Scholar]
- Barrett JW, Shun Chang C, Wang G, Werden SJ, Shao Z, Barrett C, Gao X, Belsito TA, Villenevue D, McFadden G. Myxoma virus M063R is a host range gene essential for virus replication in rabbit cells. Virology. 2007;361(1):123–32. doi: 10.1016/j.virol.2006.11.015. [DOI] [PubMed] [Google Scholar]
- Bradley RR, Terajima M. Vaccinia virus K1L protein mediates host-range function in RK-13 cells via ankyrin repeat and may interact with a cellular GTPase-activating protein. Virus Res. 2005;114(1–2):104–12. doi: 10.1016/j.virusres.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Buller RM, Burnett J, Chen W, Kreider J. Replication of molluscum contagiosum virus. Virology. 1995;213(2):655–9. doi: 10.1006/viro.1995.0037. [DOI] [PubMed] [Google Scholar]
- Buller RM, Smith GL, Cremer K, Notkins AL, Moss B. Decreased virulence of recombinant vaccinia virus expression vectors is associated with a thymidine kinase-negative phenotype. Nature. 1985;317(6040):813–5. doi: 10.1038/317813a0. [DOI] [PubMed] [Google Scholar]
- Drillien R, Koehren F, Kirn A. Host range deletion mutant of vaccinia virus defective in human cells. Virology. 1981;111(2):488–99. doi: 10.1016/0042-6822(81)90351-2. [DOI] [PubMed] [Google Scholar]
- Earl PL, Moss B, Wyatt LS, Carroll MW. Generation of Recombinant Vaccinia Viruses. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, editors. Current protocols in molecular biology. John Wiley & Sons, Inc; 1998. pp. 16.17.1–16.17.19. [Google Scholar]
- Fischer T, Planz O, Stitz L, Rziha H-J. Novel Recombinant Parapoxvirus Vectors Induce Protective Humoral and Cellular Immunity against Lethal Herpesvirus Challenge Infection in Mice. J Virol %R. 2003;77(17):9312–9323. doi: 10.1128/JVI.77.17.9312-9323.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuerst TR, Niles EG, Studier FW, Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci U S A. 1986;83(21):8122–6. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillard S, Spehner D, Drillien R, Kirn A. Localization and sequence of a vaccinia virus gene required for multiplication in human cells. Proc Natl Acad Sci U S A. 1986;83(15):5573–7. doi: 10.1073/pnas.83.15.5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao JC, Chung CS, Drillien R, Chang W. The cowpox virus host range gene, CP77, affects phosphorylation of eIF2 [alpha] and vaccinia viral translation in apoptotic HeLa cells. Virology. 2004;329(1):199–212. doi: 10.1016/j.virol.2004.07.032. [DOI] [PubMed] [Google Scholar]
- Hsiao JC, Chao CC, Young MJ, Chang YT, Cho EC, Chang W. A poxvirus host range protein, CP77, binds to a cellular protein, HMG20A, and regulates its dissociation from the vaccinia virus genome in CHO-K1 cells. J Virol. 2006;80(15):7714–28. doi: 10.1128/JVI.00207-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ink BS, Gilbert CS, Evan GI. Delay of vaccinia virus-induced apoptosis in nonpermissive Chinese hamster ovary cells by the cowpox virus CHOhr and adenovirus E1B 19K genes. J Virol. 1995;69(2):661–8. doi: 10.1128/jvi.69.2.661-668.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones-Trower A, Garcia A, Meseda CA, He Y, Weiss C, Kumar A, Weir JP, Merchlinsky M. Identification and preliminary characterization of vaccinia virus (Dryvax) antigens recognized by vaccinia immune globulin. Virology. 2005;343(1):128–40. doi: 10.1016/j.virol.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Langland JO, Jacobs BL. Inhibition of PKR by vaccinia virus: role of the N- and C-terminal domains of E3L. Virology. 2004;324(2):419–29. doi: 10.1016/j.virol.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Lun X, Yang W, Alain T, Shi ZQ, Muzik H, Barrett JW, McFadden G, Bell J, Hamilton MG, Senger DL, Forsyth PA. Myxoma virus is a novel oncolytic virus with significant antitumor activity against experimental human gliomas. Cancer Res. 2005;65(21):9982–90. doi: 10.1158/0008-5472.CAN-05-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden G. POXVIRUS TROPISM. Nat Rev Micro. 2005;3(3):201–213. doi: 10.1038/nrmicro1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Xiang Y. Vaccinia virus K1L protein supports viral replication in human and rabbit cells through a cell-type-specific set of its ankyrin repeat residues that are distinct from its binding site for ACAP2. Virology. 2006;353(1):220–33. doi: 10.1016/j.virol.2006.05.032. [DOI] [PubMed] [Google Scholar]
- Moss B. Poxviridae: The Viruses and Their Replication. In: Knipe DM, Howley PM, editors. Fields Virology. 4. Vol. 2. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 2849–2883. [Google Scholar]
- Najera JL, Gomez CE, Domingo-Gil E, Gherardi MM, Esteban M. Cellular and biochemical differences between two attenuated poxvirus vaccine candidates (MVA and NYVAC) and role of the C7L gene. J Virol. 2006;80(12):6033–47. doi: 10.1128/JVI.02108-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkus ME, Goebel SJ, Davis SW, Johnson GP, Limbach K, Norton EK, Paoletti E. Vaccinia virus host range genes. Virology. 1990;179(1):276–286. doi: 10.1016/0042-6822(90)90296-4. [DOI] [PubMed] [Google Scholar]
- Ramsey-Ewing AL, Moss B. Complementation of a vaccinia virus host-range K1L gene deletion by the nonhomologous CP77 gene. Virology. 1996;222(1):75–86. doi: 10.1006/viro.1996.0399. [DOI] [PubMed] [Google Scholar]
- Sedgwick SG, Smerdon SJ. The ankyrin repeat: a diversity of interactions on a common structural framework. Trends Biochem Sci. 1999;24(8):311–6. doi: 10.1016/s0968-0004(99)01426-7. [DOI] [PubMed] [Google Scholar]
- Shisler JL, Jin XL. The vaccinia virus K1L gene product inhibits host NF-kappaB activation by preventing IkappaBalpha degradation. J Virol. 2004;78(7):3553–60. doi: 10.1128/JVI.78.7.3553-3560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somogyi P, Frazier J, Skinner MA. Fowlpox virus host range restriction: gene expression, DNA replication, and morphogenesis in nonpermissive mammalian cells. Virology. 1993;197(1):439–44. doi: 10.1006/viro.1993.1608. [DOI] [PubMed] [Google Scholar]
- Tartaglia J, Perkus ME, Taylor J, Norton EK, Audonnet JC, Cox WI, Davis SW, van der Hoeven J, Meignier B, Riviere M, et al. NYVAC: a highly attenuated strain of vaccinia virus. Virology. 1992;188(1):217–32. doi: 10.1016/0042-6822(92)90752-b. [DOI] [PubMed] [Google Scholar]