

Abstract
Multiple osteochondromas (MO) is characterised by development of two or more cartilage capped bony outgrowths (osteochondromas) of the long bones. The prevalence is estimated at 1:50,000, and it seems to be higher in males (male-to-female ratio 1.5:1). Osteochondromas develop and increase in size in the first decade of life, ceasing to grow when the growth plates close at puberty. They are pedunculated or sessile (broad base) and can vary widely in size. The number of osteochondromas may vary significantly within and between families, the mean number of locations is 15–18. The majority are asymptomatic and located in bones that develop from cartilage, especially the long bones of the extremities, predominantly around the knee. The facial bones are not affected. Osteochondromas may cause pain, functional problems and deformities, especially of the forearm, that may be reason for surgical removal. The most important complication is malignant transformation of osteochondroma towards secondary peripheral chondrosarcoma, which is estimated to occur in 0.5–5%. MO is an autosomal dominant disorder and is genetically heterogeneous. In almost 90% of MO patients germline mutations in the tumour suppressor genes EXT1 or EXT2 are found. The EXT genes encode glycosyltransferases, catalyzing heparan sulphate polymerization. The diagnosis is based on radiological and clinical documentation, supplemented with, if available, histological evaluation of osteochondromas. If the exact mutation is known antenatal diagnosis is technically possible. MO should be distinguished from metachondromatosis, dysplasia epiphysealis hemimelica and Ollier disease. Osteochondromas are benign lesions and do not affect life expectancy. Management includes removal of osteochondromas when they give complaints. Removed osteochondromas should be examined for malignant transformation towards secondary peripheral chondrosarcoma. Patients should be well instructed and regular follow-up for early detection of malignancy seems justified. For secondary peripheral chondrosarcoma, en-bloc resection of the lesion and its pseudocapsule with tumour-free margins, preferably in a bone tumour referral centre, should be performed.
Disease name and synonyms
Multiple Osteochondromas (MO) MIM 133700
Hereditary Multiple Exostoses (HME), Multiple Hereditary Exostoses (MHE), EXT, diaphyseal aclasis, (multiple hereditary) osteochondromatosis, multiple cartilaginous exostoses
Definition and diagnostic criteria
Osteochondroma (osteocartilaginous exostosis) is a cartilage capped bony projection arising on the external surface of bone containing a marrow cavity that is continuous with that of the underlying bone [1]. A diagnosis of MO can be made when radiologically at least two osteochondromas of the juxta-epiphyseal region of long bones are observed. In the majority of patients a positive family history and/or mutation in one of the EXT genes can be detected [2,3].
Epidemiology
The prevalence of MO is estimated at 1:50,000 persons within the general population [4] and seems to be higher in males (male-to-female ratio 1.5:1) [2,5]. This is probably due to the fact that females tend to have a milder phenotype and are therefore more easily overlooked [2]. The solitary (sporadic) form of osteochondroma is approximately six times more common than the occurrence within the context of MO. Approximately 62% of the patients with multiple osteochondromas have a positive family history [2].
Clinical description
Osteochondromas develop and increase in size in the first decade of life, ceasing to grow when the growth plates close at puberty. They are pedunculated or sessile (broad base) and can vary widely in size. The majority are asymptomatic and located in bones that develop from cartilage, especially the long bones of the extremities, predominantly around the knee (Figures 1 and 2A). The facial bones are not affected. The number of osteochondromas may vary significantly within and between families, the mean number of locations is 15–18 [6]. In addition, in MO patients a variety of orthopaedic deformities can be found like deformities of the forearm (shortening of the ulna with secondary bowing of radius) (39–60%) [4,6,7] (Figure 2C), inequality in limb length (10–50%) [4,7], varus or valgus angulation of the knee (8–33%) [4,7], deformity of the ankle (2–54%) [4,7] and disproportionate short stature (37–44%) [2,5,6].
Figure 1.
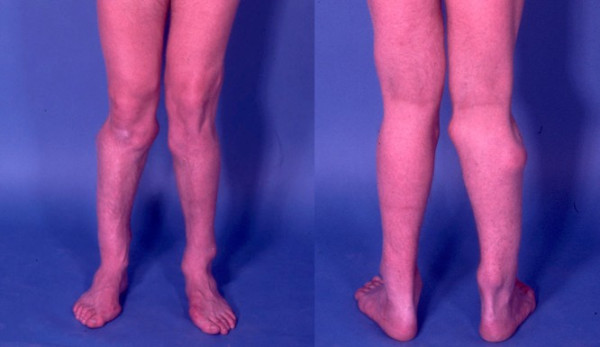
Photograph of the legs of a 26 year old male showing multiple lumps leading to deformity.
Figure 2.

Examples of radiographs demonstrating multiple osteochondromas around the knee (A) and at the pelvis and proximal femur (B), while (C) demonstrates the deformity of the forearm (shortening of the ulna with secondary bowing of radius) that is found in 39–60% of the patients.
Other complications of the osteochondromas include osseous and cosmetic deformities, bursa formation, arthritis (14%) [5] and impingement on adjacent tendons, nerves (22.6%) [5], vessels (11.3%) [5] or spinal cord (0.6%) [5,8]. MO patients may have abnormal scar formation [9]. Osteochondromas bear the risk for fracture of the bony stalk during physical exercise. This is estimated to occur in approximately 5% of osteochondromas [10] and may be reason for surgical removal.
The majority of MO patients experiences pain [11,12], approximately half of which concerns generalised pain [11]. Therefore, the number of MO individuals having pain has been underestimated and pain seems a problem that must be addressed when caring for MO patients. The occurrence of pain was associated with MO related complications and surgery [11].
The most important complication of MO is malignant transformation of an osteochondroma, which is estimated to occur in 0.5–5% of patients [2,4,5,13,14]. Clinical signs of malignant transformation include an increase in size and pain [6]. Malignant transformation of osteochondroma leads to a secondary peripheral chondrosarcoma in 94% of the cases [15]. The suspicion of secondary chondrosarcoma is indicated by growth of the tumour after puberty, the presence of pain, or a thickness over 1 cm of the cartilaginous cap in adults.
Aetiology
Two genes, EXT1 and EXT2 located respectively at 8q24 and 11p11-p12, have been isolated to cause MO [16-19]. Additional linkage to chromosome 19p has been found, suggesting the existence of an EXT3-gene [20]. However, the gene has never been identified. Moreover, the increased sensitivity of mutation detection and the use of new techniques screening for larger deletions, such as MLPA, have dramatically decreased the proportion of MO patients without an EXT1 or EXT2 mutation to <15% [21-23]. These data question the existence of an EXT3-gene at 19p.
The EXT1 gene is composed of 11 exons and has a coding region of 2238 bp [17-24]. The EXT2-gene contains 16 exons [18,19] and its cDNA defines a single open reading frame of 2154 bp. EXT1 and EXT2 are highly similar, especially in the carboxy terminal region [18,19].
The EXT1 gene was reported to show linkage in 44%–66% of the MO families [25,26], whereas EXT2 would be involved in 27% [26]. Germline mutations of EXT1 and EXT2 in MO patients have been studied extensively in Caucasian as well as Asian populations [27]. In EXT1, mutations are more or less randomly distributed over the first 6 exons, while the last 5 exons, containing the conserved carboxyterminal region, contain significantly less mutations [27]. Similarly, in EXT2 most mutations are found in the first eight exons. No mutational hotspots are found. Approximately 80% of the mutations are either non-sense, frameshift, or splice-site mutations leading to premature termination of EXT proteins [25,28-32]. The majority of missense mutations also lead to defective EXT protein function [33]. Mutations in EXT1 seem associated with a more severe phenotype as compared to EXT2 [34-37].
It has long been thought that osteochondromas are the result of skeletal dysplasia. It is now however generally accepted that osteochondromas are neoplastic, since genetic changes are found in the cartilage cap [1,38-42]. The EXT-genes are tumour suppressor genes. Loss of the remaining EXT1 wildtype allele has been demonstrated in the cartilage cap of osteochondromas from MO patients [39]. However, in a considerable proportion of MO patients loss of the remaining wildtype allele could not be detected so far [43]. In seven out of eight solitary osteochondromas, homozygous deletions of EXT1 are found [38] further supporting the two-hit model. Moreover, the deletions were confined to the cartilage cap. Thus, the cartilage cap is the clonal neoplastic element, while the stalk is reactive [38].
Both EXT1 and EXT2 mRNA is ubiquitously expressed [17-19]. A high level of expression of Ext1 and Ext2 mRNA has been found in developing limb buds of mouse embryos [44,45] and expression was demonstrated to be confined to the proliferating and prehypertrophic chondrocytes of the growth plate [46]. In osteochondromas and peripheral chondrosarcomas the expression of EXT1 and/or EXT2 is decreased, corresponding to the mutation status [47].
The gene products, exostosin-1 (EXT1) and exostosin-2 (EXT2), are endoplasmic reticulum localized type II transmembrane glycoproteins which form a Golgi-localised hetero-oligomeric complex that catalyzes heparan sulphate (HS) polymerization [48-51]. Heparan sulphate proteoglycans (HSPG) are large macromolecules composed of heparan sulphate glycosaminoglycan chains linked to a protein core. Four important HSPG families are syndecan, glypican, perlecan and isoforms of CD44 bearing variable exon 3 (CD44v3). In osteochondromas in which EXT expression is decreased due to mutation or deletion, the heparan sulphate proteoglycans seem to accumulate in the cytoplasm of the cell, instead of being transported to be expressed at the cell surface [47].
EXT and HSPGs are required for high-affinity binding of fibroblast growth factor to its receptor and for the diffusion of the morphogens Hedgehog (Hh, human homologues Indian (IHH) and Sonic Hedgehog (SHH) [52-54], decapentaplegic (dpp, human homologues TGF-beta and BMP) and wingless (wng, human homologue Wnt) [55,56]. These three pathways are important during development and are specifically active in the growth plate during endochondral bone formation. During normal growth, IHh and PTHLH are involved in a delicate paracrine feedback loop regulating proliferation and differentiation of the chondrocytes of the growth plate (Figure 3). In osteochondroma, IHH signalling is still active and is probably cell autonomous [57,58]. PTHLH signalling, which is downstream of IHH and is responsible for chondrocyte proliferation, is absent in osteochondroma, while being upregulated upon malignant transformation of osteochondroma [59,60]. Wnt signalling and TGF-beta signalling are also active in the majority of osteochondromas [57]. The exact role of EXT in orchestrating these pathways leading to osteochondroma formation in MO patients needs to be further elucidated.
Figure 3.
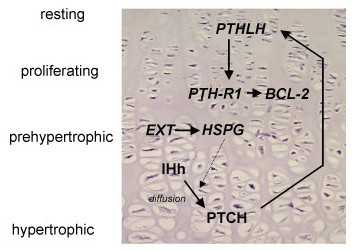
Growth plate signaling in the normal growth plate. Indian Hedgehog protein (IHh) is expressed in the prehypertrophic cells, and diffuses over a variable distance to its receptor Patched (PTCH). Subsequently, increased secretion of ParaThyroid Hormone Like Hormone (PTHLH) is induced at the apical perichondrium via an incompletely understood mechanism. PTHLH then diffuses to its receptor, whose expression is restricted to the late proliferating chondrocytes, inhibiting their further differentiation, resulting in less IHh producing cells, which closes the feedback loop. Thus, PTHLH regulates the pace of chondrocyte differentiation by delaying the progression of chondrocytes towards the hypertrophic zone, allowing longitudinal bone growth. Defective or absent EXT proteins leading to altered or absent HSPG expression at the cell surface may affect this negative feedback loop by disturbing the diffusion of IHh, produced at the pre-hypertrophic chondrocytes, towards its receptor Ptc.
Diagnostic methods
When a patient is suspected to have MO, the full radiological documentation, histology (if available), patient history and family history have to be carefully reviewed. Given the specific radiological and histological expertise needed, and the rarity of the disorder and of those in the differential diagnosis, it is recommended that this review is performed by specialists in the field, for instance through a national bone tumour registry consisting of clinicians, radiologists and pathologists. If this review is indicative for MO, the peripheral blood of the patient may be screened for germline mutations in EXT1 or EXT2 [61]. In case of a positive family history in which MO is clearly established in relatives, the diagnosis of MO can be clinically made and mutation analysis is not essential. With the currently used methods it is possible to detect point mutations or gross deletions in almost 90% of MO patients [21-23,61-63].
To evaluate possible malignant transformation in case of complaints or growth of the lesion after puberty, the size of the cartilaginous cap can be well established with T2-weighted magnetic resonance (MR) imaging [64]. A cartilage cap >1.5 cm should be regarded with caution. The role of 18 Fluoro-deoxyglucose positron emission tomography (18FDG PET) needs to be further established [65].
Differential diagnosis
Dysplasia Epiphysealis Hemimelica (DEH, Trevor's disease, tarso-epiphysial aclasis) and metachondromatosis (MC) are considered in the differential diagnosis of solitary and hereditary osteochondromas. Despite their similarities, they were shown to be separate entities [66] and the EXT downstream pathway is not involved [67].
DEH is a developmental disorder with cartilaginous overgrowth of a portion of one or more epiphyses [68]. It predominantly affects the lower extremity on one side of the body. It is usually restricted to either the medial (most frequent) or lateral side of the limb (hemimelic). Similar to osteochondroma, DEH is usually diagnosed prior to the age of 15 years, more often in boys than in girls, and growth of these lesions end at puberty as the growth plates close [68,69]. In contrast to MO, malignant transformation has not been reported so far [68] and there does not appear to be any genetic transmission [69-71].
MC is a rare disorder exhibiting, synchronous, both multiple osteochondromas and enchondromas in children. It has an autosomal dominant mode of inheritance [72-74] but the disorder has not been mapped in the human genome so far. MC related osteochondromas characteristically occur in the hands and feet, predominantly the digits and toes, and point toward the adjacent growth plate, while in MO the osteochondromas are mainly located in the long or other tubular bones and point away from the epiphysis [72]. Differentiation from MO is of great clinical significance because in patients with MC the lesions do not result in shortening or deformity of affected bones as in MO, and may spontaneously decrease in size or resolve completely, both clinically and radiologically [72,74].
Moreover, MO should be distinguished from enchondromatosis (Ollier disease and Maffucci syndrome), in which multiple cartilage tumours are found in the medulla of bone, with a predilection for the short tubular bones and a unilateral predominance [75].
Upon histopathological examination of osteochondroma after surgical removal malignancy should be considered. Malignant transformation in the cartilage cap of osteochondroma leads to a secondary peripheral chondrosarcoma. Occasionally, osteosarcomas and spindle cell sarcomas develop in the stalk of the osteochondroma [15,76-80]. Extremely rare is the occurrence of dedifferentiated peripheral chondrosarcoma, in which a low-grade chondrosarcoma that developed within an osteochondroma "dedifferentiates" into a high grade sarcoma [81,82].
Genetic counselling
MO is an autosomal dominant disorder. Affected individuals have 50% risk of transmitting the disorder to their offspring. MO has nearly 100% penetrance. If the exact mutation is known antenatal diagnosis is technically possible.
Management including treatment
Osteochondromas are only removed when they cause pain, when they give functional complaints for instance due to compression on nerves or vessels, or for cosmetic reasons.
Surgical treatment of forearm deformities remains controversial. In a retrospective series 23 MO patients corrective osteotomy and/or lengthening of forearm bones was not beneficial [83]. Moreover, one should consider the possible recurrence of ulnar shortening within 1.5 years when operating skeletally immature patients [83,84]. The most beneficial procedure was excision of the osteochondromas. The simple removal of an osteochondroma can improve forearm rotation and correct deformity [83], especially if there is an isolated tumour of the distal part of the ulna.
If the diagnosis of MO is established and all tumours are identified, patients should be well instructed to seek earlier medical attention if their condition changes, for instance if there is pain or growth of a known lesion [61]. It is important to realise that no new osteochondromas develop after puberty. Moreover, regular follow-up to discover potential malignant transformation at an early stage to enable adequate treatment should be considered. The risk of malignant transformation of osteochondroma towards secondary peripheral chondrosarcoma is estimated at 1–5% [2,4,5,13,14,34]. After skeletal maturation a base-line bone scan is recommended [61]. Furthermore, baseline plain radiographs of areas that can not be manually examined, like the chest, pelvis and scapula can be performed [61]. After the base-line documentation one should consider screening patients regularly, for instance every year or every other year. There are as yet no studies available that have proven efficacy of screening. If lesions change over time, further examination, using magnetic resonance (MR) imaging including contrast enhanced MR sequences, is indicated [61].
In case of malignancy, en-bloc resection of the lesion and its pseudocapsule with tumour-free margins, preferably in a bone tumour referral centre, should be performed, resulting in excellent long term clinical and local results. The most common location is however the pelvis where the large cartilage cap can be difficult to excise. In a series of 61 patients with grade I or II secondary peripheral chondrosarcoma of the pelvis published by Donati et al., a 3% local recurrence rate was found after wide resection, in contrast with 23% after inadequate excision [85].
Prognosis
Osteochondromas are benign lesions and do not affect life expectancy. The risk of malignant transformation is 1–5%. The prognosis for secondary peripheral chondrosarcoma is depending on histological grade: 10 year survival rates are 83% for grade I chondrosarcomas compared to 29% for grade III chondrosarcomas [86].
Unresolved questions
• How can the enormous difference in disease severity within and between families be explained?
• What drives malignant transformation of osteochondroma and can this be prevented?
• What is the role of EXT in normal cartilage growth and differentiation and in osteochondroma formation?
Acknowledgments
Acknowledgements
The author would like to thank Prof. Dr. A.H.M. Taminiau, Department of Orthopaedic Surgery, Leiden University Medical Center and Dr. S.J. Ham, Department of Orthopaedic Surgery, Onze Lieve Vrouwe Gasthuis Amsterdam for providing figures 1 and 2.
References
- Khurana J, Abdul-Karim F, Bovée JVMG. Osteochondroma. In: Fletcher CDM, Unni KK and Mertens F, editor. World Health Organization classification of tumours Pathology and genetics of tumours of soft tissue and bone. Lyon, IARC Press; 2002. pp. 234–236. [Google Scholar]
- Legeai-Mallet L, Munnich A, Maroteaux P, Le Merrer M. Incomplete penetrance and expressivity skewing in hereditary multiple exostoses. Clin Genet. 1997;52:12–16. doi: 10.1111/j.1399-0004.1997.tb02508.x. [DOI] [PubMed] [Google Scholar]
- Bovée JVMG, Hogendoorn PCW. Multiple osteochondromas. In: Fletcher CDM, Unni KK and Mertens F, editor. World Health Organization classification of tumours Pathology and genetics of tumours of soft tissue and bone. Lyon, IARC Press; 2002. pp. 360–362. [Google Scholar]
- Schmale GA, Conrad EU, Raskind WH. The natural history of hereditary multiple exostoses. J Bone Joint Surg [Am] 1994;76:986–992. doi: 10.2106/00004623-199407000-00005. [DOI] [PubMed] [Google Scholar]
- Wicklund LC, Pauli RM, Johnston D, Hecht JT. Natural history study of hereditary multiple exostoses. Am J Med Genet. 1995;55:43–46. doi: 10.1002/ajmg.1320550113. [DOI] [PubMed] [Google Scholar]
- Hennekam RC. Hereditary multiple exostoses. J Med Genet. 1991;28:262–266. doi: 10.1136/jmg.28.4.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro F, Simon S, Glimcher MJ. Hereditary multiple exostoses. Anthropometric, roentgenographic, and clinical aspects. J Bone Joint Surg Am. 1979;61:815–824. [PubMed] [Google Scholar]
- Vanhoenacker FM, Van Hul W, Wuyts W, Willems PJ, De Schepper AM. Hereditary multiple exostoses: from genetics to clinical syndrome and complications. Eur J Radiol. 2001;40:208–217. doi: 10.1016/S0720-048X(01)00401-6. [DOI] [PubMed] [Google Scholar]
- Hosalkar H, Greenberg J, Gaugler RL, Garg S, Dormans JP. Abnormal scarring with keloid formation after osteochondroma excision in children with multiple hereditary exostoses. J Pediatr Orthop. 2007;27:333–337. doi: 10.1097/BPO.0b013e3180326732. [DOI] [PubMed] [Google Scholar]
- Carpintero P, Leon F, Zafra M, Montero M, Berral FJ. Fractures of osteochondroma during physical exercise. Am J Sports Med. 2003;31:1003–1006. doi: 10.1177/03635465030310060101. [DOI] [PubMed] [Google Scholar]
- Darilek S, Wicklund C, Novy D, Scott A, Gambello M, Johnston D, Hecht J. Hereditary multiple exostosis and pain. J Pediatr Orthop. 2005;25:369–376. doi: 10.1097/01.bpo.0000150813.18673.ad. [DOI] [PubMed] [Google Scholar]
- Bottner F, Rodl R, Kordish I, Winklemann W, Gosheger G, Lindner N. Surgical treatment of symptomatic osteochondroma. A three- to eight-year follow-up study. J Bone Joint Surg Br. 2003;85:1161–1165. doi: 10.1302/0301-620X.85B8.14059. [DOI] [PubMed] [Google Scholar]
- Gordon SL, Buchanan JR, Ladda RL. Hereditary multiple exostoses: report of a kindred. J Med Genet. 1981;18:428–430. doi: 10.1136/jmg.18.6.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson HA. Multiple hereditary osteochondromata. Clin Orthop. 1989;239:222–230. [PubMed] [Google Scholar]
- Willms R, Hartwig CH, Böhm P, Sell S. Malignant transformation of a multiple cartilaginous exostosis - a case report. Int Orthop. 1997;21:133–136. doi: 10.1007/s002640050136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook A, Raskind W, Blanton SH, Pauli RM, Gregg RG, Francomano CA, Puffenberger E, Conrad EU, Schmale G, Schellenberg G, Wijsman E, Hecht JT, Wells D, Wagner MJ. Genetic heterogeneity in families with hereditary multiple exostoses. Am J Hum Genet. 1993;53:71–79. [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Ludecke HJ, Lindow S, Horton WA, Lee B, Wagner MJ, Horsthemke B, Wells DE. Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1) Nature Genet. 1995;11:137–143. doi: 10.1038/ng1095-137. [DOI] [PubMed] [Google Scholar]
- Wuyts W, Van Hul W, Wauters J, Nemtsova M, Reyniers E, Van Hul E, De Boulle K, De Vries BBA, Hendrickx J, Herrygers I, Bossuyt P, Balemans W, Fransen E, Vits L, Coucke P, Nowak NJ, Shows TB, Mallet L, Van den Ouweland AMW, McGaughran J, Halley DJJ, Willems P. Positional cloning of a gene involved in hereditary multiple exostoses. Hum Mol Genet. 1996;5:1547–1557. doi: 10.1093/hmg/5.10.1547. [DOI] [PubMed] [Google Scholar]
- Stickens D, Clines G, Burbee D, Ramos P, Thomas S, Hogue D, Hecht JT, Lovett M, Evans GA. The EXT2 multiple exostoses gene defines a family of putative tumour suppressor genes. Nature Genet. 1996;14:25–32. doi: 10.1038/ng0996-25. [DOI] [PubMed] [Google Scholar]
- Le Merrer M, Legeai-Mallet L, Jeannin PM, Horsthemke B, Schinzel A, Plauchu H, Toutain A, Achard F, Munnich A, Maroteaux P. A gene for hereditary multiple exostoses maps to chromosome 19p. Hum Mol Genet. 1994;3:717–722. doi: 10.1093/hmg/3.5.717. [DOI] [PubMed] [Google Scholar]
- Signori E, Massi E, Matera MG, Poscente M, Gravina C, Falcone G, Rosa MA, Rinaldi M, Wuyts W, Seripa D, Dallapiccola B, Fazio VM. A combined analytical approach reveals novel EXT1/2 gene mutations in a large cohort of Italian multiple osteochondromas patients. Genes Chromosomes Cancer. 2007;46:470–477. doi: 10.1002/gcc.20429. [DOI] [PubMed] [Google Scholar]
- Vink GR, White SJ, Gabelic S, Hogendoorn PC, Breuning MH, Bakker E. Mutation screening of EXT1 and EXT2 by direct sequence analysis and MLPA in patients with multiple osteochondromas: splice site mutations and exonic deletions account for more than half of the mutations. Eur J Hum Genet. 2004;13:470–474. doi: 10.1038/sj.ejhg.5201343. [DOI] [PubMed] [Google Scholar]
- White SJ, Vink GR, Kriek M, Wuyts W, Schouten J, Bakker B, Breuning MH, den Dunnen JT. Two-color multiplex ligation-dependent probe amplification: detecting genomic rearrangements in hereditary multiple exostoses. Hum Mutat. 2004;24:86–92. doi: 10.1002/humu.20054. [DOI] [PubMed] [Google Scholar]
- Ludecke HJ, Ahn J, Lin X, Hill A, Wagner MJ, Schomburg L, Horsthemke B, Wells DE. Genomic organization and promotor structure of the human EXT1 gene. Genomics. 1997;40:351–354. doi: 10.1006/geno.1996.4577. [DOI] [PubMed] [Google Scholar]
- Raskind WH, Conrad EU, III, Matsushita M, Wijsman EM, Wells DE, Chapman N, Sandell LJ, Wagner M, Houck J. Evaluation of locus heterogeneity and EXT1 mutations in 34 families with hereditary multiple exostoses. Hum Mutat. 1998;11:231–239. doi: 10.1002/(SICI)1098-1004(1998)11:3<231::AID-HUMU8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Legeai-Mallet L, Margaritte-Jeannin P, Lemdani M, Le Merrer M, Plauchu H, Maroteaux P, Munnich A, Clerget-Darpoux F. An extension of the admixture test for the study of genetic heterogeneity in hereditary multiple exostoses. Hum Genet. 1997;99:298–302. doi: 10.1007/s004390050361. [DOI] [PubMed] [Google Scholar]
- Wuyts W, Van Hul W. Molecular basis of multiple exostoses: mutations in the EXT1 and EXT2 genes. Hum Mutat. 2000;15:220–227. doi: 10.1002/(SICI)1098-1004(200003)15:3<220::AID-HUMU2>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Seki H, Kubota T, Ikegawa S, Haga N, Fujioka F, Ohzeki S, Wakui K, Yoshikawa H, Takaoka K, Fukushima Y. Mutation frequencies of EXT1 and EXT2 in 43 Japanese families with hereditary multiple exostoses. Am J Med Genet. 2001;99:59–62. doi: 10.1002/1096-8628(20010215)99:1<59::AID-AJMG1115>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Wuyts W, Van Hul W, De Boulle K, Hendrickx J, Bakker E, Vanhoenacker F, Mollica F, Ludecke HJ, Sitki Sayli B, Pazzaglia UE, Mortier G, Hamel B, Conrad EU, Matsushita M, Raskind WH, Willems PJ. Mutations in the EXT1 and EXT2 genes in hereditary multiple exostoses. Am J Hum Genet. 1998;62:346–354. doi: 10.1086/301726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Xia J, Jiang H, Zhou J, Li H, Wang D, Pan Q, Long Z, Fan C, Deng HX. Mutation analysis of hereditary multiple exostoses in the Chinese. Hum Genet. 1999;105:45–50. doi: 10.1007/s004390051062. [DOI] [PubMed] [Google Scholar]
- Park KJ, Shin KH, Ku JL, Cho TJ, Lee SH, Choi IH, Phillipe C, Monaco AP, Porter DE, Park JG. Germline mutations in the EXT1 and EXT2 genes in Korean patients with hereditary multiple exostoses. J Hum Genet. 1999;44:230–234. doi: 10.1007/s100380050149. [DOI] [PubMed] [Google Scholar]
- Francannet C, Cohen-Tanugi A, Le Merrer M, Munnich A, Bonaventure J, Legeai-Mallet L. Genotype-phenotype correlation in hereditary multiple exostoses. J Med Genet. 2001;38:430–434. doi: 10.1136/jmg.38.7.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung PK, McCormick C, Crawford BE, Esko JD, Tufaro F, Duncan G. Etiological point mutations in the hereditary multiple exostoses gene EXT1: a functional analysis of heparan sulfate polymerase activity. Am J Hum Genet. 2001;69:55–66. doi: 10.1086/321278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DE, Lonie L, Fraser M, Dobson-Stone C, Porter JR, Monaco AP, Simpson AH. Severity of disease and risk of malignant change in hereditary multiple exostoses. A genotype-phenotype study. J Bone Joint Surg Br. 2004;86:1041–1046. doi: 10.1302/0301-620X.86B7.14815. [DOI] [PubMed] [Google Scholar]
- Alvarez C, Tredwell S, De VM, Hayden M. The genotype-phenotype correlation of hereditary multiple exostoses. Clin Genet. 2006;70:122–130. doi: 10.1111/j.1399-0004.2006.00653.x. [DOI] [PubMed] [Google Scholar]
- Alvarez CM, De Vera MA, Heslip TR, Casey B. Evaluation of the anatomic burden of patients with hereditary multiple exostoses. Clin Orthop Relat Res. 2007;462:73–79. doi: 10.1097/BLO.0b013e3181334b51. [DOI] [PubMed] [Google Scholar]
- Jager M, Westhoff B, Portier S, Leube B, Hardt K, Royer-Pokora B, Gossheger G, Krauspe R. Clinical outcome and genotype in patients with hereditary multiple exostoses. J Orthop Res. 2007;25:1541–1551. doi: 10.1002/jor.20479. [DOI] [PubMed] [Google Scholar]
- Hameetman L, Szuhai K, Yavas A, Knijnenburg J, van Duin M, Van Dekken H, Taminiau AH, Cleton-Jansen AM, Bovee JV, Hogendoorn PC. The role of EXT1 in nonhereditary osteochondroma: identification of homozygous deletions. J Natl Cancer Inst. 2007;99:396–406. doi: 10.1093/jnci/djk067. [DOI] [PubMed] [Google Scholar]
- Bovée JVMG, Cleton-Jansen AM, Wuyts W, Caethoven G, Taminiau AHM, Bakker E, Van Hul W, Cornelisse CJ, Hogendoorn PCW. EXT-mutation analysis and loss of heterozygosity in sporadic and hereditary osteochondromas and secondary chondrosarcomas. Am J Hum Genet. 1999;65:689–698. doi: 10.1086/302532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridge JA, Nelson M, Orndal C, Bhatia P, Neff JR. Clonal karyotypic abnormalities of the hereditary multiple exostoses chromosomal loci 8q24.1 (EXT1) and 11p11-12 (EXT2) in patients with sporadic and hereditary osteochondromas. Cancer. 1998;82:1657–1663. doi: 10.1002/(SICI)1097-0142(19980501)82:9<1657::AID-CNCR10>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Mertens F, Rydholm A, Kreicbergs A, Willen H, Jonsson K, Heim S, Mitelman F, Mandahl N. Loss of chromosome band 8q24 in sporadic osteocartilaginous exostoses. Genes Chromosomes Cancer. 1994;9:8–12. doi: 10.1002/gcc.2870090103. [DOI] [PubMed] [Google Scholar]
- Feely MG, Boehm AK, Bridge RS, Krallman PM, Neff JR, Nelson M, Bridge JA. Cytogenetic and molecular cytogenetic evidence of recurrent 8q24.1 loss in osteochondroma. Cancer Genet Cytogenet. 2002;137:102–107. doi: 10.1016/S0165-4608(02)00557-5. [DOI] [PubMed] [Google Scholar]
- Hall CR, Cole WG, Haynes R, Hecht JT. Reevaluation of a genetic model for the development of exostosis in hereditary multiple exostosis. Am J Med Genet. 2002;112:1–5. doi: 10.1002/ajmg.10635. [DOI] [PubMed] [Google Scholar]
- Lin X, Gan L, Klein WH, Wells DE. Expression and functional analysis of mouse EXT1, a homolog of the human multiple exostoses type 1 gene. Biochem Biophys Res Commun. 1998;248:738–743. doi: 10.1006/bbrc.1998.9050. [DOI] [PubMed] [Google Scholar]
- Stickens D, Evans GA. Isolation and characterization of the murine homolog of the human EXT2 multiple exostoses gene. Biochem Mol Med. 1997;61:16–21. doi: 10.1006/bmme.1997.2588. [DOI] [PubMed] [Google Scholar]
- Stickens D, Brown D, Evans GA. EXT genes are differentially expressed in bone and cartilage during mouse embryogenesis. Dev Dyn. 2000;218:452–464. doi: 10.1002/1097-0177(200007)218:3<452::AID-DVDY1000>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Hameetman L, David G, Yavas A, White SJ, Taminiau AHM, Cleton-Jansen AM, Hogendoorn PCW, Bovée JVMG. Decreased EXT expression and intracellular accumulation of HSPG in osteochondromas and peripheral chondrosarcomas. J Pathol. 2007;211:399–409. doi: 10.1002/path.2127. [DOI] [PubMed] [Google Scholar]
- Lind T, Tufaro F, McCormick C, Lindahl U, Lidholt K. The putative tumor suppressors EXT1 and EXT2 are glycosyltransferases required for the biosynthesis of heparan sulfate. J Biol Chem. 1998;273:26265–26268. doi: 10.1074/jbc.273.41.26265. [DOI] [PubMed] [Google Scholar]
- McCormick C, Leduc Y, Martindale D, Mattison K, Esford LE, Dyer AP, Tufaro F. The putative tumour suppressor EXT1 alters the expression of cell-surface heparan sulfate. Nature Genet. 1998;19:158–161. doi: 10.1038/514. [DOI] [PubMed] [Google Scholar]
- Simmons AD, Musy MM, Lopes CS, Hwang LY, Yang YP, Lovett M. A direct interaction between EXT proteins and glycosyltransferases is defective in hereditary multiple exostoses. Hum Mol Genet. 1999;8:2155–2164. doi: 10.1093/hmg/8.12.2155. [DOI] [PubMed] [Google Scholar]
- McCormick C, Duncan G, Goutsos KT, Tufaro F. The putative tumor suppressors EXT1 and EXT2 form a stable complex that accumulates in the golgi apparatus and catalyzes the synthesis of heparan sulfate. Proc Natl Acad Sci USA. 2000;97:668–673. doi: 10.1073/pnas.97.2.668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellaiche Y, The I, Perrimon N. Tout-velu is a drosophila homologue of the putative tumour suppressor EXT1 and is needed for Hh diffusion. Nature. 1998;394:85–88. doi: 10.1038/27932. [DOI] [PubMed] [Google Scholar]
- The I, Bellaiche Y, Perrimon N. Hedgehog movement is regulated through tout velu -dependant synthesis of a heparan sulfate proteoglycan. Mol Cell. 1999;4:633–639. doi: 10.1016/S1097-2765(00)80214-2. [DOI] [PubMed] [Google Scholar]
- Toyoda H, Kinoshita-Toyoda A, Selleck SB. Structural analysis of glycosaminoglycans in drosophila and caenorhabditis elegans and demonstration that tout-velu, a drosophila gene related to EXT tumor suppressors, affects heparan sulfate in vivo. J Biol Chem. 2000;275:2269–2275. doi: 10.1074/jbc.275.4.2269. [DOI] [PubMed] [Google Scholar]
- Han C, Belenkaya TY, Khodoun M, Tauchi M, Lin X, Lin X. Distinct and collaborative roles of Drosophila EXT family proteins in morphogen signalling and gradient formation. Development. 2004;131:1563–1575. doi: 10.1242/dev.01051. [DOI] [PubMed] [Google Scholar]
- Takei Y, Ozawa Y, Sato M, Watanabe A, Tabata T. Three Drosophila EXT genes shape morphogen gradients through synthesis of heparan sulfate proteoglycans. Development. 2004;131:73–82. doi: 10.1242/dev.00913. [DOI] [PubMed] [Google Scholar]
- Hameetman L, Rozeman LB, Lombaerts M, Oosting J, Taminiau AHM, Cleton-Jansen AM, Bovée JVMG, Hogendoorn PCW. Peripheral chondrosarcoma progression is accompanied by decreased Indian Hedgehog (IHH) signalling. J Pathol. 2006;209:501–511. doi: 10.1002/path.2008. [DOI] [PubMed] [Google Scholar]
- Benoist-Lasselin C, de Margerie E, Gibbs L, Cormier S, Silve C, Nicolas G, Lemerrer M, Mallet JF, Munnich A, Bonaventure J, Zylberberg L, Legeai-Mallet L. Defective chondrocyte proliferation and differentiation in osteochondromas of MHE patients. Bone. 2006;39:17–26. doi: 10.1016/j.bone.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Bovée JVMG, Van den Broek LJCM, Cleton-Jansen AM, Hogendoorn PCW. Up-regulation of PTHrP and Bcl-2 expression characterizes the progression of osteochondroma towards peripheral chondrosarcoma and is a late event in central chondrosarcoma. Lab Invest. 2000;80:1925–1933. doi: 10.1038/labinvest.3780202. [DOI] [PubMed] [Google Scholar]
- Hameetman L, Kok P, Eilers PHC, Cleton-Jansen AM, Hogendoorn PCW, Bovée JVMG. The use of Bcl-2 and PTHLH immunohistochemistry in the diagnosis of peripheral chondrosarcoma in a clinicopathological setting. Virchows Arch. 2005;446:430–437. doi: 10.1007/s00428-005-1208-4. [DOI] [PubMed] [Google Scholar]
- Hameetman L, Bovée JVMG, Taminiau AHM, Kroon HM, Hogendoorn PCW. Multiple Osteochondromas: Clinicopathological and Genetic Spectrum and Suggestions for Clinical Management. Hereditary Cancer in Clinical Practice. 2004;2:161–173. doi: 10.1186/1897-4287-2-4-161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuyts W, Radersma R, Storm K, Vits L. An optimized DHPLC protocol for molecular testing of the EXT1 and EXT2 genes in hereditary multiple osteochondromas. Clin Genet. 2005;68:542–547. doi: 10.1111/j.1399-0004.2005.00538.x. [DOI] [PubMed] [Google Scholar]
- Lonie L, Porter DE, Fraser M, Cole T, Wise C, Yates L, Wakeling E, Blair E, Morava E, Monaco AP, Ragoussis J. Determination of the mutation spectrum of the EXT1/EXT2 genes in British Caucasian patients with multiple osteochondromas, and exclusion of six candidate genes in EXT negative cases. Hum Mutat. 2006;27:1160. doi: 10.1002/humu.9467. [DOI] [PubMed] [Google Scholar]
- Geirnaerdt MJ, Hogendoorn PCW, Bloem JL, Taminiau AHM, Van der Woude HJ. Cartilaginous tumors: fast contrast-enhanced MR imaging. Radiology. 2000;214:539–546. doi: 10.1148/radiology.214.2.r00fe12539. [DOI] [PubMed] [Google Scholar]
- Feldman F, Vanheertum R, Saxena C. 18Fluoro-deoxyglucose positron emission tomography evaluation of benign versus malignant osteochondromas: preliminary observations. J Comput Assist Tomogr. 2006;30:858–864. doi: 10.1097/01.rct.0000228160.86096.ca. [DOI] [PubMed] [Google Scholar]
- Glick R, Khaldi L, Ptaszynski K, Steiner GC. Dysplasia epiphysealis hemimelica (Trevor disease): a rare developmental disorder of bone mimicking osteochondroma of long bones. Hum Pathol. 2007;38:1265–1272. doi: 10.1016/j.humpath.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Bovée JVMG, Hameetman L, Kroon HM, Aigner T, Hogendoorn PCW. EXT-related pathways are not involved in pathogenesisof Dysplasia Epiphysealis Hemimelica and Metachondromatosis. J Pathol. 2006;209:411–419. doi: 10.1002/path.1985. [DOI] [PubMed] [Google Scholar]
- Murphey MD, Choi JJ, Kransdorf MJ, Flemming DJ, Gannon FH. Imaging of osteochondroma: variants and complications with radiologic-pathologic correlation. RadioGraphics. 2000;20:1407–1434. doi: 10.1148/radiographics.20.5.g00se171407. [DOI] [PubMed] [Google Scholar]
- Silverman FN. Dysplasia epiphysealis hemimelica. Semin Roentgenol. 1989;24:246–258. doi: 10.1016/0037-198X(89)90022-9. [DOI] [PubMed] [Google Scholar]
- Ippolito E, Tudisco C. Dysplasia epiphysealis hemimelica. Clinical, histological and histochemical features. Ital J Orthop Traumatol. 1983;9:101–107. [PubMed] [Google Scholar]
- Kuo RS, Bellemore MC, Monsell FP, Frawley K, Kozlowski K. Dysplasia epiphysealis hemimelica: clinical features and management. J Pediatr Orthop. 1998;18:543–548. doi: 10.1097/00004694-199807000-00028. [DOI] [PubMed] [Google Scholar]
- Bassett GS, Cowell HR. Metachondromatosis. Report of four cases. J Bone Joint Surg Am. 1985;67:811–814. [PubMed] [Google Scholar]
- Herman TE, Chines A, McAlister WH, Gottesman GS, Eddy MC, Whyte MP. Metachondromatosis: report of a family with facial features mildly resembling trichorhinophalangeal syndrome. Pediatr Radiol. 1997;27:436–441. doi: 10.1007/s002470050164. [DOI] [PubMed] [Google Scholar]
- Kennedy LA. Metachondromatosis. Radiology. 1983;148:117–118. doi: 10.1148/radiology.148.1.6602353. [DOI] [PubMed] [Google Scholar]
- Mertens F, Unni KK. Enchondromatosis: Ollier disease and Maffucci syndrome. In: Fletcher CDM, Unni KK and Mertens F, editor. World Health Organization Classification of Tumours Pathology and genetics of tumours of soft tissue and bone. Lyon, IARC Press; 2002. pp. 356–357. [Google Scholar]
- van Lerberghe E, Van Damme B, van Holsbeeck M, Burssens A, Hoogmartens M. Case report 626: Osteosarcoma arising in a solitary osteochondroma of the femur. Skeletal Radiol. 1990;19:594–597. doi: 10.1007/BF00241283. [DOI] [PubMed] [Google Scholar]
- Lamovec J, Spiler M, Jevtic V. Osteosarcoma arising in a solitary osteochondroma of the fibula. Arch Pathol Lab Med. 1999;123:832–834. doi: 10.5858/1999-123-0832-OAIASO. [DOI] [PubMed] [Google Scholar]
- Matsuno T, Ichioka Y, Yagi T, Ishii S. Spindle-cell sarcoma in patients who have osteochondromatosis. A report of two cases. J Bone Joint Surg [Am] 1988;70:137–141. [PubMed] [Google Scholar]
- Bovée JVMG, Sakkers RJ, Geirnaerdt MJ, Taminiau AH, Hogendoorn PCW. Intermediate grade osteosarcoma and chondrosarcoma arising in an osteochondroma. A case report of a patient with hereditary multiple exostoses. J Clin Pathol. 2002;55:226–229. doi: 10.1136/jcp.55.3.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya H, Morikawa S, Tomita K. Osteosarcoma arising from a multiple exostoses lesion: case report. Jpn J Clin Oncol. 1990;20:296–298. [PubMed] [Google Scholar]
- Bertoni F, Present D, Bacchini P, Picci P, Pignatti G, Gherlinzoni F, Campanacci M. Dedifferentiated peripheral chondrosarcomas, a report of seven cases. Cancer. 1989;63:2054–2059. doi: 10.1002/1097-0142(19890515)63:10<2054::AID-CNCR2820631030>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Staals EL, Bacchini P, Mercuri M, Bertoni F. Dedifferentiated chondrosarcomas arising in preexisting osteochondromas. J Bone Joint Surg Am. 2007;89:987–993. doi: 10.2106/JBJS.F.00288. [DOI] [PubMed] [Google Scholar]
- Akita S, Murase T, Yonenobu K, Shimada K, Masada K, Yoshikawa H. Long-term results of surgery for forearm deformities in patients with multiple cartilaginous exostoses. J Bone Joint Surg Am. 2007;89:1993–1999. doi: 10.2106/JBJS.F.01336. [DOI] [PubMed] [Google Scholar]
- Matsubara H, Tsuchiya H, Sakurakichi K, Yamashiro T, Watanabe K, Tomita K. Correction and lengthening for deformities of the forearm in multiple cartilaginous exostoses. J Orthop Sci. 2006;11:459–466. doi: 10.1007/s00776-006-1047-4. [DOI] [PubMed] [Google Scholar]
- Donati D, El Ghoneimy A, Bertoni F, Di Bella C, Mercuri M. Surgical treatment and outcome of conventional pelvic chondrosarcoma. Journal of Bone and Joint Surgery-British Volume. 2005;87:1527–1530. doi: 10.1302/0301-620X.87B11.16621. [DOI] [PubMed] [Google Scholar]
- Evans HL, Ayala AG, Romsdahl MM. Prognostic factors in chondrosarcoma of bone. A clinicopathologic analysis with emphasis on histologic grading. Cancer. 1977;40:818–831. doi: 10.1002/1097-0142(197708)40:2<818::AID-CNCR2820400234>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]