

Abstract
Considerable attention has focused on the health-promoting effects of red wine and its nonflavonoid polyphenol compound resveratrol. However, the underlying molecular mechanisms and molecular target(s) of red wine or other potentially active ingredients in red wine remain unknown. Here we report that red wine extract (RWE) or the red wine flavonoid, quercetin, inhibited 12-O-tetradecanoylphorbol-13-acetate (TPA)-induced transformation of JB6 promotion sensitive mouse skin epidermal (JB6 P+) cells. The activation of activator protein (AP)-1 and nuclear factor (NF)-κB induced by TPA was dose-dependently inhibited by RWE or quercetin treatment. Western blot and kinase assay data revealed that RWE or quercetin inhibited MEK1 and Raf1 kinase activities and subsequently attenuated TPA-induced phosphorylation of ERK/p90RSK. Although either RWE or quercetin suppressed Raf1 kinase activity, they were more effective in inhibiting MEK1 activity. Importantly, quercetin exerted stronger inhibitory effects than PD098059, a well-known pharmacological inhibitor of MEK. Resveratrol did not affect either MEK1 or Raf1 kinase activity. Pull-down assays revealed that RWE or quercetin (but not resveratrol) bound with either MEK1 or Raf1. RWE or quercetin also dose-dependently suppressed JB6 P+ cell transformation induced by epidermal growth factor or H-Ras, both of which are known to be involved in the activation of MEK/ERK signaling. Docking data suggested that quercetin, but not resveratrol, formed a hydrogen bond with the backbone amide group of Ser212, which is the key interaction for stabilizing the inactive conformation of the activation loop of MEK1.
Keywords: Red wine, quercetin, MEK1, Raf1, neoplastic transformation
Introduction
Considerable attention has been focused on the health benefits of red wine, which are associated with its high content of a variety of polyphenols (1). Red wine can also reportedly prevent various types of cancers (2). Even though red wine contains different types of polyphenols, multiple lines of evidence indicate the health-promoting effects of resveratrol (3,5,4′-trihydroxy-trans-stilbene), which is a nonflavonoid compound present in red wine. Previous studies reported that resveratrol exerted antitumor effects through the activation of p53-mediated apoptosis (3), inhibited cyclooxygenase-2 expression and two-stage skin cancer induced by 12-O-tetradecanoylphorbol-13-acetate (TPA) in mouse skin (4). However, the concentration of resveratrol required for achieving its beneficial effects is unlikely to be attained through a moderate consumption of red wine, due to its low content of resveratrol. The resveratrol content in red wine [0.6–6.8 mg/L in French red wines (5)] is about 30 times lower than the flavonol content, with one of the major flavonols in red wine being quercetin (3,3′,4′,5,7-pentahydroxyflavone) (6). Quercetin has been suggested as a potent anticarcinogenic flavonol. In 9,10-dimethyl-1,2-benzanthracene-initiated and TPA-promoted two-stage mouse skin cancer models, quercetin exerted the strongest anticarcinogenic effects (7). Thus, the identification of the actual active components responsible for the chemopreventive effects of red wine and molecular mechanism(s) of action is needed.
Activator protein (AP)-1 and nuclear factor (NF)-κB act as pivotal transcription factors involving neoplastic transformation and development of cancer (8–11), and are regulated by upstream kinases, including mitogen-activated protein kinases (MAPKs), signaling pathways. MAPK signaling pathways are commonly upregulated in various cancer cell types, and these pathways are known to be involved in cell proliferation and survival (12). Among the components of the MAPK pathways, the MAPK kinase kinase (Raf)/ MAPK kinase (MEK)/extracellular signal-regulated kinase (ERK) cascade has been the focus of cancer chemotherapy because of its relevance in carcinogenesis. A variety of tumor promoters including TPA and epidermal growth factor (EGF) are known to induce neoplastic transformation through activation of Raf/MEK/ERK pathway in various cell lines (8, 13, 14). The Raf/MEK/ERK pathway has also been identified as a key downstream effecter of Ras, which is a frequently mutated oncogene in 30% or more of human cancers (15, 16). This pathway plays a critical role in linking extracellular signals associated with Ras activation to nuclear transcription events (17). Because aberrant activation of ERK has been demonstrated in various types of tumors (18, 19), the targeted downregulation of ERK through the inhibition of upstream kinases such as Raf or MEK is an effective method for intervening in carcinogenesis.
The JB6 mouse epidermal cell system, including promotion-sensitive (P+) and promotion-resistant (P−) components, is regarded as an appropriate model for studying tumor-promoter-induced carcinogenic processes at the molecular level. The present study aimed to elucidate the mechanism of the antitumorigenic effects of red wine and to identify potentially effective compounds by investigating the possible inhibitory effects of red wine extract (RWE), quercetin, or resveratrol on TPA-induced neoplastic transformation of JB6 P+ cells. Here we report that RWE or quercetin, but not resveratrol, is a potent inhibitor of MEK1 activity. The inhibition of MEK1 suppressed downstream ERK phosphorylation and activation of AP-1 and NF-κB, which subsequently inhibited neoplastic transformation. RWE or quercetin also inhibited Raf1 activity, but this was less substantial than the inhibition of MEK1.
Materials and Methods
Chemicals
Quercetin, resveratrol, EGF, and TPA were obtained from Sigma Chemical (St. Louis, MO); Eagle’s minimum essential medium (MEM), basal medium Eagle (BME), gentamicin, and L-glutamine were purchased from GIBCO BRL (Carlsbad, CA); and fetal bovine serum (FBS) was purchased from Gemini Bio-Products (Calabasas, CA). PD098059 and GW5047 were obtained from Calbiochem (San Diego, CA). The antibodies against phosphorylated MEK (Ser217/221), phosphorylated ERK (Thr202/Tyr204), total ERK, phosphorylated p90RSK (Thr359/Ser363), p90 ribosomal s6 kinase (p90RSK), phosphorylated c-Jun N-terminal kinase (JNK, Thr183/Tyr185), and total JNK were purchased from Cell Signal Biotechnology (Beverly, MA). The antibodies against total MEK and Raf1 were from Santa Cruz Biotechnology (Santa Cruz, CA). The MEK1 and Raf1 kinase assay kits were obtained from Upstate Biotechnology (Lake Placid, NY). CNBr-Sepharose 4B, glutathione-Sepharose 4B, [γ-32P]ATP, and the chemiluminescence detection kit were purchased from Amersham Pharmacia Biotech (Piscataway, NJ), and the protein assay kit was obtained from Bio-Rad Laboratories (Hercules, CA). G418 and the luciferase assay substrate were purchased from Promega (Madison, WI).
Preparation of RWE
RWE was kindly provided by Dr. Schini-Kerth (Universite Louis Pasteur, France). The procedure used to prepare RWE has been described previously (20). In brief, phenolic compounds were adsorbed onto a preparative column, the alcohol was desorbed, the alcoholic eluent was gently evaporated, and finally the concentrated residue was lyophilized and finely sprayed to obtain RWE dry powder.
Cell culture
The JB6 P+ mouse epidermal (JB6 P+) and H-Ras-transformed JB6 P+ (H-Ras JB6 P+) cell lines were cultured in monolayers at 37°C in a 5% CO2 incubator in MEM containing 5% FBS, 2 mM L-glutamine, and 25 µg/ml gentamicin. The JB6 mouse epidermal cell line was stably transfected with an AP-1 or NF-κB luciferase reporter plasmid, and maintained in MEM supplemented with 5% FBS containing 200 µg/ml G418.
Anchorage-independent cell transformation assay
The effects of RWE and red wine compounds on TPA- or EGF-induced cell transformation were investigated in JB6 P+ cells. Cells (8×103/ml) were exposed to TPA or EGF with or without RWE and red wine compounds in 1 ml of 0.33% BME agar containing 10% FBS or in 3.5 ml of 0.5% BME agar containing 10% FBS. The effects of RWE and red wine compounds on H-Ras-induced cell transformation were investigated in H-Ras transformed JB6 cells. The H-Ras cells (8×103/ml) were incubated with or without RWE or red wine compounds in 1 ml of 0.33% BME agar containing 10% FBS or in 3.5 ml of 0.5% BME agar containing 10% FBS. The cultures were maintained at 37°C in a 5% CO2 incubator for 14 days, after which the cell colonies were counted under a microscope with the aid of the Image-Pro Plus software program (v.4, Media Cybernetics, Silver Spring, MD) (21).
Luciferase assay for AP-1 and NF-κB transactivation
AP-1 or NF-κB luciferase reporter JB6 P+ cells (8×103), suspended in 100 µl of 5% FBS/MEM, were added to each well of a 96-well plate and incubated at 37°C in a humidified atmosphere of 5% CO2. When cells reached 80–90% confluence, they were starved by culturing in 0.1% FBS MEM for an additional 24 h. The cells were then treated for 1 h with RWE (0–40 µg/ml), quercetin (0–40 µM), or resveratrol (0–40 µM), and then exposed to 20 ng/ml TPA for 24 h. After treatment, cells were disrupted with 100 µl of lysis buffer [0.1 M potassium phosphate buffer (pH 7.8), 1% Triton X-100, 1 mM dithiothreitol (DTT), and 2 mM EDTA], and the luciferase activity was measured using a luminometer (Luminoskan Ascent, Thermo Electron, Helsinki, Finland).
Western blot analysis
After the cells (1.5×106) were cultured in a 10-cm dish for 48 h, they were starved in serum-free medium for an additional 24 h. The cells were then treated with RWE (0–20 µg/ml) or quercetin (0–20 µM) for 1 h before they were exposed to 20 ng/ml TPA for an additional 30 min. The harvested cells were disrupted and the supernatant fractions were boiled for 5 min. The protein concentration was determined using a dye-binding protein assay kit as described in the manufacturer’s manual. Lysate protein (20 µg) was subjected to 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis and electrophoretically transferred to a PVDF membrane. After blotting, the membrane was incubated with the specific primary antibody at 4°C overnight. Protein bands were visualized by a chemiluminescence detection kit after hybridization with an HRP-conjugated secondary antibody.
In vitro MEK1 and Raf1 assays
The in vitro kinase assays were performed in accordance with the instructions provided by Upstate Biotechnology. In brief, every reaction contained 20 µl of assay dilution buffer [20 mM MOPS (pH 7.2), 25 mM β-glycerol phosphate, 5 mM EGTA, 1 mM sodium orthovanadate, and 1 mM DTT] and a magnesium-ATP cocktail buffer. For MEK1, 1 µg of the inactive ERK2 substrate peptide was also included. For Raf1, 0.4 µg of inactive MEK1 and 1 µg of inactive ERK2 were included. A 4-µl aliquot was removed from the reaction mixture, containing 20 µg of MBP substrate peptide and 10 µl of diluted [γ-32P]ATP solution, and incubated at 30°C for 30 min. This mixture was incubated for 10 min at 30°C, and then 25-µl aliquots were transferred onto p81 filter paper and washed three times with 0.75% phosphoric acid for 5 min per wash and once with acetone for 2 min. The radioactive incorporation was determined using a scintillation counter (LS6500, Beckman Coulter, Fullerton, CA). Each experiment was performed three times.
Ex vivo MEK1 and Raf1 immunoprecipitation and kinase assay
JB6 P+ cells were cultured to 80% confluence and then serum-starved in 0.1% FBS/MEM for 24 h at 37°C. Cells were either treated or not treated with RWE, quercetin, or resveratrol for 1 h, then treated with 20 ng/ml TPA for 30 min, disrupted with lysis buffer [20 mM Tris-HCl (pH 7.4), 1 mM EDTA, 150 mM NaCl, 1 mM EGTA, 1% Triton X-100, 1 mM β-glycerophosphate, 1 mg/ml leupeptin, 1 mM Na3VO4, and 1 mM phenylmethylsulfonyl fluoride (PMSF)], and finally centrifuged at 14,000 rpm for 10 min in a microcentrifuge. The lysates, each containing 500 µg of protein, were used for immunoprecipitation with an antibody against MEK1 or Raf1 and then incubated at 4°C overnight. Protein A/G Plus agarose beads were then added and the mixture was continuously rotated for an additional 3 h at 4°C. The beads were washed three times with kinase buffer [20 mM MOPS (pH 7.2), 25 mM β-glycerol phosphate, 5 mM EGTA, 1 mM sodium orthovanadate, and 1 mM DTT], and then resuspended in 20 µl of 1×kinase buffer supplemented with 1 µg of inactive ERK2 (for MEK1) or with 0.4 µg of inactive MEK1 and 1 µg of inactive ERK2 (for Raf1) and incubated for an additional 30 min at 30°C. Then MBP (20 µg) and 10 µl of diluted [γ32P]ATP solution were added and the mixture was incubated for 10 min at 30°C. A 20-µl aliquot was transferred onto p81 filter paper and washed three times with 0.75% phosphoric acid for 5 min per wash and once with acetone for 2 min. The radioactive incorporation was determined using a scintillation counter. Each experiment was performed three times.
In vitro and ex vivo pull-down assays
Recombinant MEK1 (2 µg) (or Raf1) or a JB6 P+ cellular supernatant fraction (500 µg protein) was incubated with the RWE- or quercetin-Sepharose 4B (or Sepharose 4B as control) beads (100 µl, 50% slurry) in reaction buffer [50 mM Tris-HCl, (pH 7.5), 5 mM EDTA, 150 mM NaCl, 1 mM DTT, 0.01% Nonidet P-40, 2 µg/ml bovine serum albumin, 0.02 mM PMSF, and 1× protease inhibitor mixture]. After incubation with gentle rocking overnight at 4°C, the beads were washed five times with buffer [50 mM Tris-HCl (pH 7.5), 5 mM EDTA, 150 mM NaCl, 1 mM DTT, 0.01% Nonidet P-40, and 0.02 mM PMSF], and proteins bound to the beads were analyzed by immunoblotting.
Molecular modeling
Insight II (Accelrys, San Diego, CA) was used for the docking study and structure analysis with the crystal coordinates of MEK1 (accession code 1S9J), which are available in the Protein Data Bank (http://www.rcsb.org/pdb/).
Statistical analysis
When necessary, data were expressed as means ± S.D. values, and the ANOVA was used for multiple statistical comparisons. A probability value of p < 0.05 was used as the criterion for statistical significance.
Results
RWE inhibits TPA-induced neoplastic transformation of JB6 P+ cells
To investigate whether red wine exerts health-promoting effects by intervening in carcinogenesis processes, we first examined the effect of RWE on neoplastic transformation. Results indicated that treatment with RWE markedly inhibited TPA-promoted neoplastic transformation of JB6 P+ cells in a dose-dependent manner (Fig. 1A). Based on the numbers of cell colonies, RWE at only 5 µg/ml suppressed TPA-induced JB6 P+ cell transformation by 59%, and at or above 20 µg/ml almost completely prevented transformation (Fig. 1B).
Figure 1.
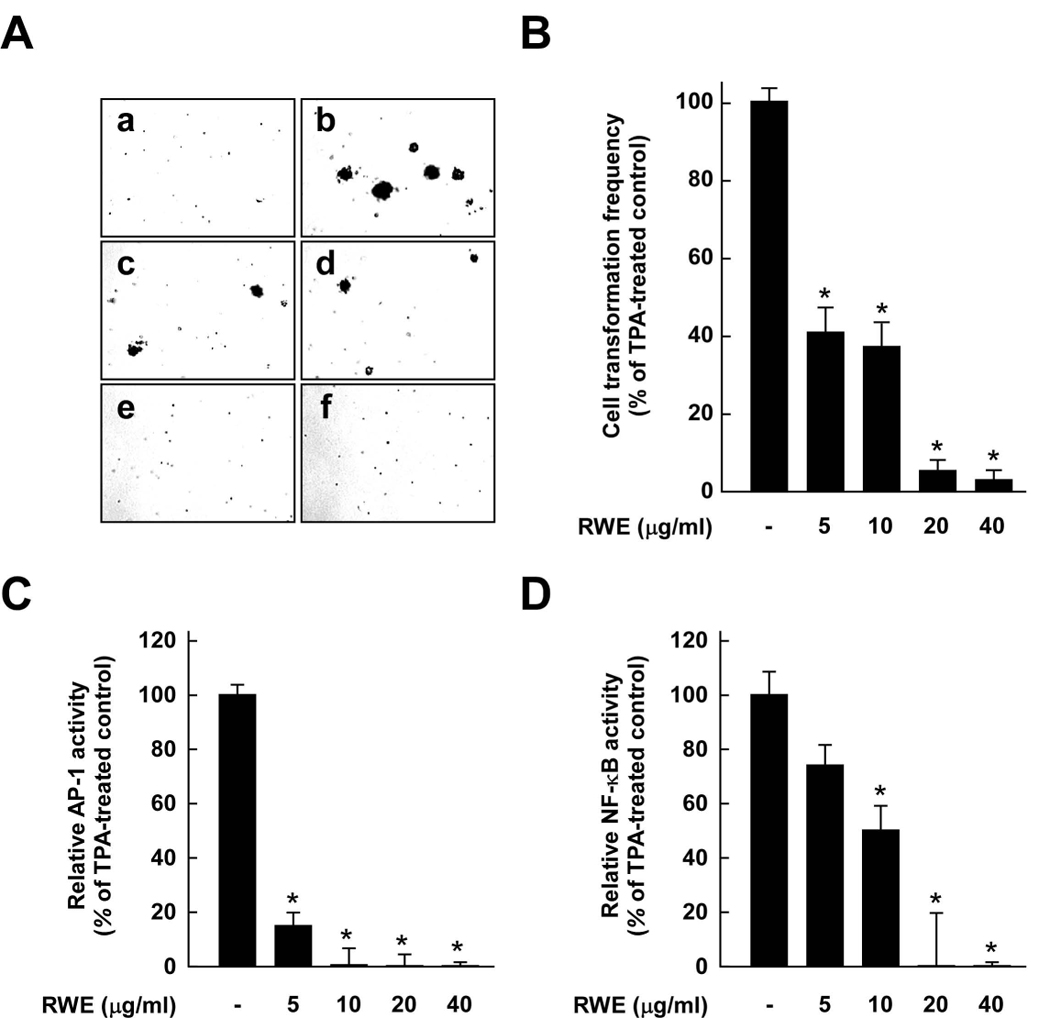
Effects of red wine extract (RWE) on TPA-induced neoplastic transformation, and AP-1 and NF-κB transactivation in JB6 P+ cells. A, RWE inhibited TPA-induced JB6 P+ cell transformation. JB6 P+ cells were treated as described in the Materials and Methods and colonies were counted 14 days later: untreated control (a), TPA alone (b), TPA and 5 µg/ml RWE (c), TPA and 10 µg/ml RWE (d), TPA and 20 µg/ml RWE (e), and TPA and 40 µg/ml RWE (f). B, Cell colonies were counted under a microscope with the aid of Image-Pro Plus software (v.4). The effects of RWE on neoplastic transformation of JB6 P+ cells are presented as the percent inhibition of cell transformation compared with cells treated only with TPA in soft agar. Data are presented as means ± S.D. of the percent inhibition as determined from three separate experiments. C and D, RWE inhibited TPA-induced AP-1 (C) or NF-κB (D) activation. The JB6 P+ cells, which were stably transfected with AP-1 or NF-κB luciferase reporter plasmids, were pretreated with RWE for 1 h at the indicated concentrations (5, 10, 20, or 40 µg/ml) followed by exposure to 20 ng/ml TPA for 24 h. The relative activity was measured by the luciferase assay as described in the Materials and Methods. Data are presented as means ± S.D. values of AP-1 and NF-κB luciferase activities calculated from three independent experiments. For B–D, the asterisk (*) indicates significant differences between groups treated with TPA and RWE together and the group treated with TPA alone (p < 0.05).
RWE inhibits TPA-induced AP-1 and NF-κB transactivation in JB6 P+ cells
AP-1 and NF-κB are major transcription factors involved in TPA-induced neoplastic transformation of JB6 P+ cells (22–24). To investigate whether RWE downregulates cell transformation through the inhibition of these transcription factors, we measured AP-1 and NF-κB transactivation by using JB6 P+ cells stably transfected with an AP-1 or NF-κB luciferase reporter plasmid. RWE inhibited TPA-induced transactivation of either AP-1 or NF-κB in a dose-dependent manner, and treatment with RWE at a low concentration inhibited AP-1 more effectively compared to NF-κB (Fig. 1C and D, respectively).
RWE suppresses TPA-induced phosphorylation of MEK, ERK and p90RSK in JB6 P+ cells
Among the kinases belonging to the MAPK family, ERK has been reported to be the most important in TPA-induced JB6 P+ cell transformation (9, 24, 25). We next examined whether RWE downregulates ERK and JNK pathways stimulated by TPA in JB6 P+ cells. RWE at 5 µg/ml inhibited TPA-induced phosphorylation of MEK, but more strongly suppressed phosphorylation of the MEK downstream kinases ERK and p90RSK (Fig. 2A). RWE, at 20 µg/ml, slightly inhibited TPA-induced JNK phosphorylation, but to a much lower degree than ERK (data not shown). These results suggested that the inhibition of the MEK/ERK/p90RSK signal pathway by RWE leads to the suppression of AP-1 and NF-κB resulting in decreased neoplastic transformation.
Figure 2.
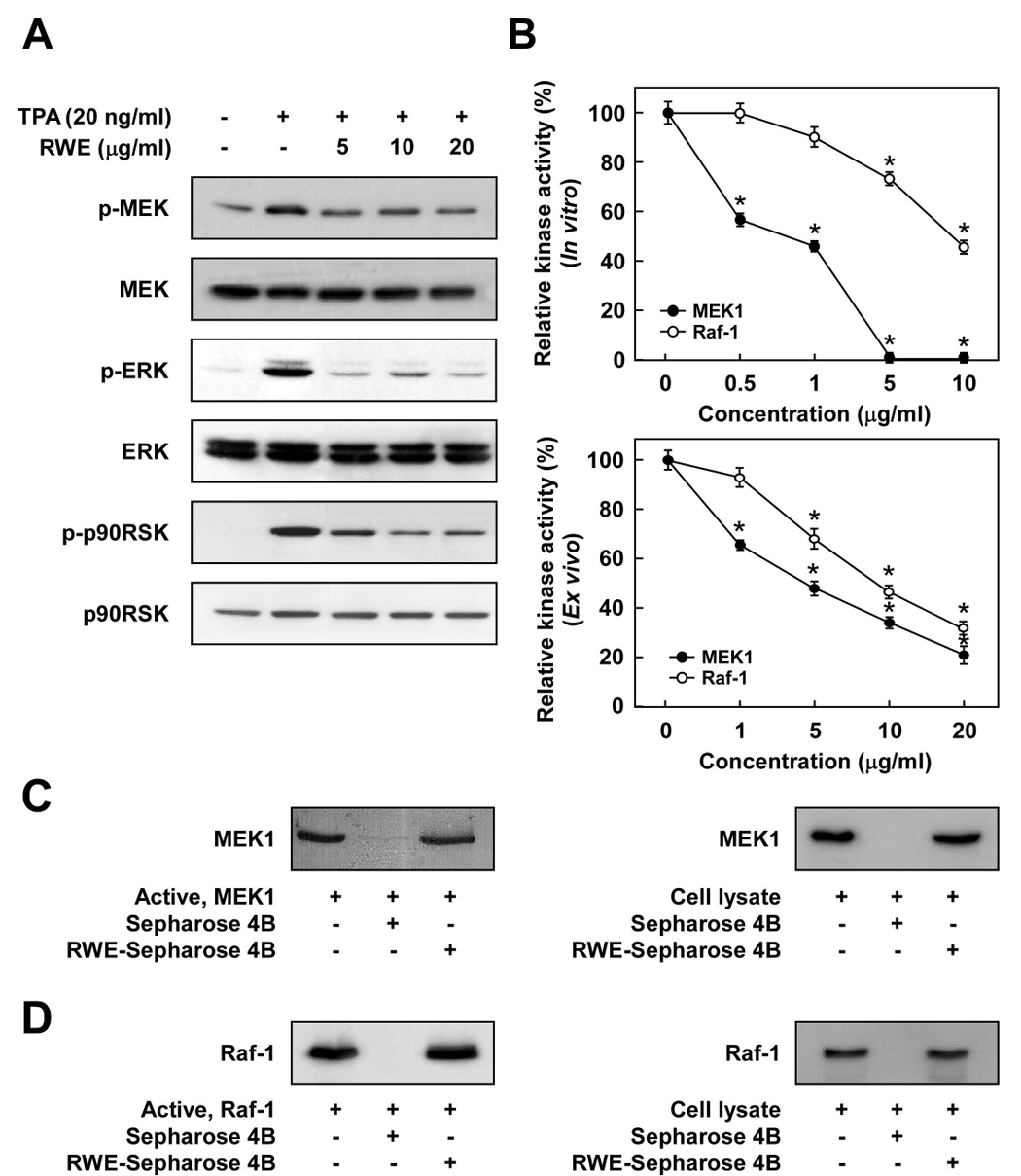
The Raf1/MEK/ERK/p90RSK signaling cascade is downregulated by RWE through the direct inhibition of both Raf1 and MEK1 activities. A, RWE inhibited TPA-induced phosphorylation of MEK, ERK, and p90RSK in JB6 P+ cells. JB6 P+ cells were treated with RWE (5, 10 or 20 µg/ml) for 1 h before being stimulated by 20 ng/ml TPA for an additional 15 min. The cells were lysed and the levels of phosphorylated and total MEK, ERK, and p90RSK proteins were determined by Western blot analysis as described in the Materials and Methods using specific antibodies against the respective phosphorylated and total proteins. Data are representative of three independent experiments that gave similar results. B, RWE inhibited MEK1 activity more than Raf1 activity both in vitro (upper panel) and ex vivo (lower panel). An in vitro MEK1 or Raf1 kinase assay was performed as described in the Materials and Methods, and the kinase activity is expressed as the percent inhibition relative to the activity of untreated MEK1 or Raf1 control. For the ex vivo MEK1 or Raf1 kinase assay, cells were pretreated with RWE at the indicated concentrations (1, 5, 10, or 20 µg/ml) for 1 h and then stimulated with 20 ng/ml TPA for 30 min. Cells were harvested, and immunoprecipitation and an ex vivo MEK1 or Raf1 kinase assay was performed. The kinase activity is expressed as the percent inhibition relative to cells treated with TPA only. The average 32P count was determined from three separate experiments, and the data are presented as means ± S.D. values. For in vitro kinase assays, the asterisk (*) indicates a significant decrease (p < 0.05) in kinase activity between the groups treated with active MEK1 (or Raf1) and RWE together and the group treated with active MEK1 (or Raf1) alone. For ex vivo kinase assays, the asterisk (*) indicates a significant decrease in kinase activity between cells treated with TPA and RWE together and the cells treated with TPA alone (p < 0.05). C, RWE specifically binds with MEK1 both in vitro and ex vivo. The MEK1–RWE binding in vitro was confirmed by immunoblotting using an antibody against MEK1 (left panel): lane 1 (input control), MEK1 protein standard; lane 2 (control), Sepharose 4B was used to pull down MEK1 as described in the Materials and Methods; and lane 3, RWE-Sepharose 4B affinity beads were used to pull down MEK1. The MEK1–RWE binding ex vivo was confirmed by immunoblotting using an antibody against MEK1 (right panel): lane 1 (input control), whole-cell lysates from JB6 P+ cells; lane 2 (control), a lysate of JB6 P+ cells precipitated with Sepharose 4B beads; and lane 3, whole-cell lysates from JB6 P+ cells precipitated by RWE-Sepharose 4B affinity beads. D, RWE specifically binds with Raf1 both in vitro and ex vivo. The Raf1–RWE binding in vitro was confirmed by immunoblotting using an antibody against Raf1 (left panel): lane 1 (input control), Raf1 protein standard; lane 2 (control), Sepharose 4B used to pull down Raf1 as described in the Materials and Methods; and lane 3, RWE-Sepharose 4B affinity beads were used to pull down Raf1. The Raf1–RWE binding ex vivo was confirmed by immunoblotting using an antibody against Raf1 (right panel): lane 1 (input control), whole-cell lysates from JB6 P+ cells; lane 2 (control), a lysate of JB6 P+ cells precipitated with Sepharose 4B beads; and lane 3, whole-cell lysates from JB6 P+ cells precipitated by RWE-Sepharose 4B affinity beads. Each experiment was performed two times and representative blots are shown.
RWE inhibits MEK1 activity more strongly than Raf1 activity
We next investigated the effects of RWE on the kinase activity of MEK1 and Raf1. Kinase assay data revealed that RWE inhibited MEK1 activity more strongly than Raf1 activity (Fig. 2B). RWE at 0.5 µg/ml blocked active MEK1 activity by 53.2%, while it had no effect on active Raf1 activity. In vitro MEK1 activity was completely inhibited by RWE at or above 5 µg/ml, whereas treatment with 5 µg/ml RWE reduced Raf1 activity by only 24.9% (Fig. 2B, upper panel). Consistent with results from an in vitro kinase assay, an ex vivo kinase assay also revealed that RWE inhibited TPA-induced MEK1 activity in JB6 P+ cells more than Raf1 activity. RWE at 1 µg/ml blocked TPA-induced MEK1 activity by about 34.6%, whereas no significant inhibition against TPA-induced Raf1 activity was detected. However, at the highest concentration, RWE effectively suppressed either MEK1 or Raf1 activity stimulated by TPA (Fig. 2B, lower panel). These results indicated that the inhibition of cell transformation by RWE was mainly caused by the suppression of MEK1 activity and to a lesser extent by inhibition of Raf1 and its downstream signaling pathways.
RWE directly binds with either MEK1 or Raf1
To determine whether the inhibition of MEK1 and Raf1 kinase activities by RWE was caused by a direct interaction, we next performed pull-down assays. Using an in vitro pull-down assay, MEK1 was found in the RWE-Sepharose 4B beads (Fig. 2C, left panel), but not in Sepharose 4B beads alone (Fig. 2C, left panel). We also observed ex vivo binding between RWE and MEK1 in JB6 P+ cell lysates (Fig. 2C, right panel). Because RWE could also inhibit Raf1 activity, we performed in vitro pull-down assays to determine whether RWE directly interacts with Raf1. Raf1 was found in the RWE-Sepharose 4B beads (Fig. 2D, left panel), but not in Sepharose 4B beads alone (Fig. 2D, left panel). As for MEK, we also observed ex vivo binding of RWE and Raf1 in JB6 P+ cell lysates (Fig. 2D, right panel). These results suggested that the inhibition of MEK1 or Raf1 activities by RWE occurs through direct binding of RWE with either protein.
Quercetin inhibits MEK1 activity more strongly than Raf1 activity, whereas resveratrol has no inhibitory effect on either kinase
To elucidate the active components of RWE contributing to its chemopreventive effects, we next examined the effects of quercetin or resveratrol (Fig. 3A) on MEK1 and Raf1 kinase activities. Similar to RWE, quercetin inhibited MEK1 activity more effectively compared to inhibition of Raf1 activity, while resveratrol had no inhibitory effect on either kinase (Fig. 3B). MEK1 kinase assay revealed that quercetin at 5 µM almost completely blocked MEK1 activity, and the effect was greater than the inhibition by PD098059, a specific MEK inhibitor. Furthermore, resveratrol at up to 20 µM exerted no effect on MEK1 activity (Fig. 3B, left panel). In contrast, quercetin inhibited Raf1 activity by only 23.8% at 10 µM, and resveratrol at up to 10 µM exerted no effect (Fig. 3B, right panel). GW5047, a well-known inhibitor of Raf1, was used as a positive control.
Figure 3.
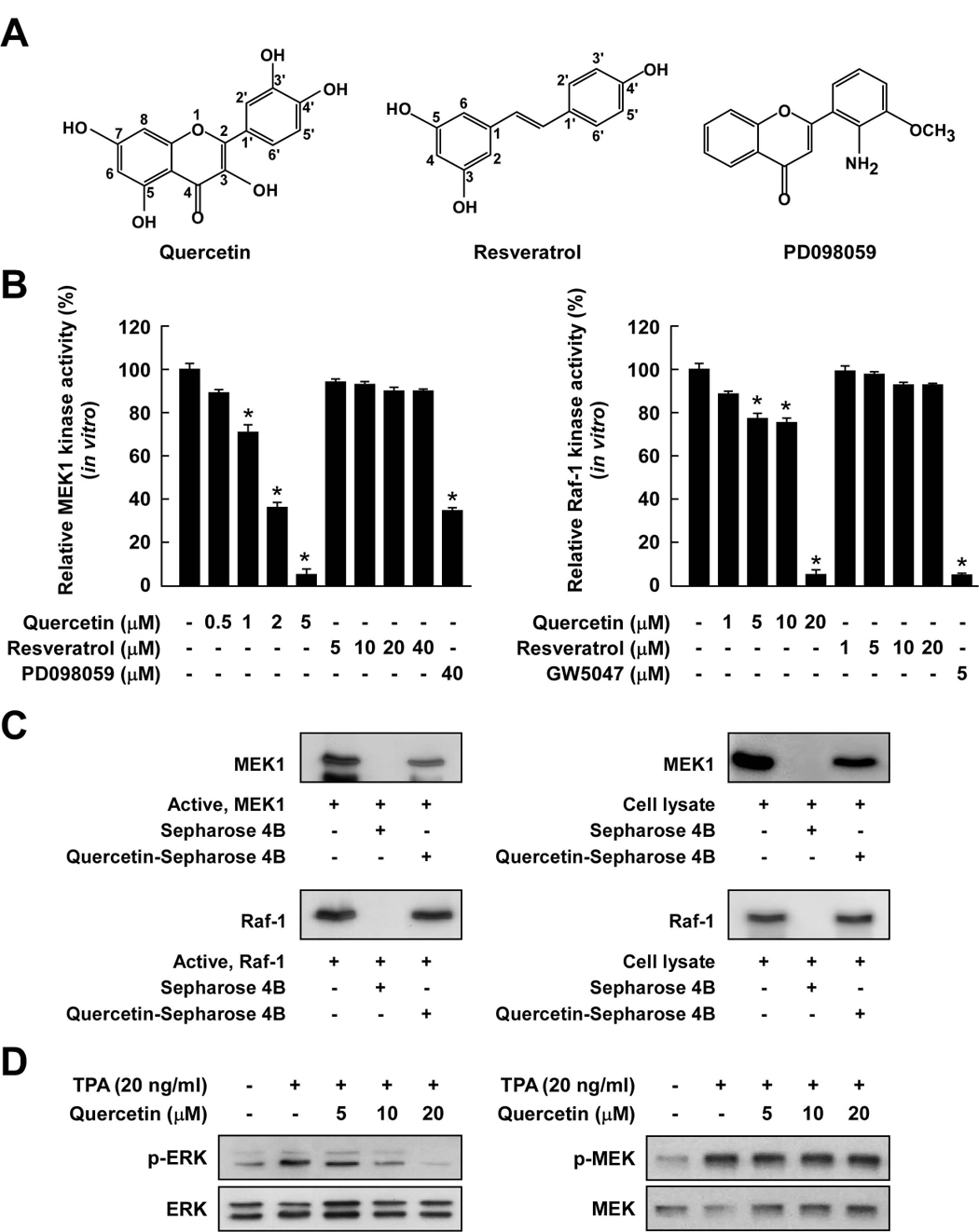
Comparison of the inhibitory effects of quercetin or resveratrol against activation of Raf/MEK/ERK signaling cascades. A, Chemical structures of quercetin, resveratrol, and PD098059. B, Quercetin inhibited MEK1 activity more strongly than Raf1 activity, whereas resveratrol did not inhibit either kinase activity. An in vitro MEK1 (left panel) or Raf1 (right panel) kinase assay was performed as described in the Materials and Methods, and the kinase activity is expressed as the percent inhibition relative to the activity of untreated MEK1 or Raf1 control. The asterisk (*) indicates a significant decrease (p < 0.05) in kinase activity between the groups treated with active MEK1 or Raf1 and quercetin (or resveratrol or PD098059 or GW5074) together and the group treated with active MEK1 or Raf1 alone. C, Quercetin specifically binds with either MEK1 or Raf1. The in vitro MEK1 (or Raf1)–quercetin binding was confirmed by immunoblotting using an antibody against MEK1 (left-upper panel) or Raf1 (left-lower panel): lane 1 (input control), MEK1 or Raf1 protein standard; lane 2 (control), Sepharose 4B was used to pull down MEK1 or Raf1; and lane 3, MEK1 or Raf1 was pulled down using quercetin-Sepharose 4B affinity beads. The ex vivo MEK1 (or Raf1)–quercetin binding was confirmed by immunoblotting using an antibody against MEK1 (right-upper panel) or Raf1 (right-lower panel): lane 1 (input control), whole-cell lysates from JB6 P+ cells; lane 2 (control), a lysate of JB6 P+ cells precipitated with Sepharose 4B beads; and lane 3, whole-cell lysates from JB6 P+ cells precipitated by quercetin-Sepharose 4B affinity beads. Each experiment was performed two times and representative blots are shown. D, Quercetin inhibited TPA-induced ERK phosphorylation but had no effect on MEK phosphorylation in JB6 P+ cells. JB6 P+ cells were treated with quercetin at the indicated concentrations (5, 10, or 20 µM) for 1 h before being stimulated by 20 ng/ml TPA for an additional 15 min. The cells were lysed and the levels of phosphorylated and total ERK and MEK proteins were determined by Western blot analysis as described in the Materials and Methods using specific antibodies against the respective phosphorylated and total proteins. Data are representative of three independent experiments that gave similar results.
To further determine whether quercetin directly binds with MEK1 or Raf1, we performed in vitro and ex vivo pull-down assays. MEK1 was found in the quercetin-Sepharose 4B beads, but not in Sepharose 4B beads alone (Fig. 3C, left-upper panel). We also observed en vivo binding of quercetin and MEK1 in JB6 P+ cell lysates (Fig. 3C, right-upper panel). In addition, Raf1 was found in the quercetin-Sepharose 4B beads, but not in Sepharose 4B beads alone (Fig. 3C, left-lower panel). We also observed ex vivo binding of quercetin and Raf1 in JB6 P+ cell lysates (Fig. 3C, right lower panel).
Western blot analysis confirmed that quercetin inhibited TPA-induced ERK phosphorylation, whereas quercetin had no effect on TPA-induced MEK phosphorylation in JB6 P+ cells (Fig. 3D). This is consistent with the previous result showing that quercetin blocked MEK1 activity more effectively than Raf1 activity. In addition, quercetin (at up to 20 µM) had no effect on TPA-induced phosphorylation of JNK in JB6 P+ cells (data not shown). These results indicated that quercetin interacts with MEK1 and Raf1 protein kinases, and results in downregulation of their kinase activities. Of note, MEK is a more important target molecule of quercetin for the suppression of TPA-induced JB6 P+ cell transformation.
Quercetin inhibits TPA-induced neoplastic transformation and transactivation of AP-1 and NF-κB in JB6 P+ cells, whereas resveratrol had no effect
To confirm that inhibition of the Raf/MEK/ERK signaling pathway by quercetin leads to the suppression of neoplastic transformation, we next examined quercetin’s effects on TPA-induced JB6 P+ cell transformation and transactivation of AP-1 and NF-κB. Treatment with quercetin markedly inhibited TPA-promoted neoplastic transformation of JB6 P+ cells in a dose-dependent manner (Fig. 4A). Based on the numbers of cell colonies, quercetin at 40 µM almost completely suppressed TPA-induced neoplastic transformation, while resveratrol at up to 40 µM had no effect. To determine whether the blocking of transformation by quercetin involves the inhibition of AP-1 or NF-κB activity, we measured AP-1 and NF-κB transactivation. Quercetin suppressed TPA-induced transactivation of either AP-1 or NF-κB in a dose-dependent manner (Fig. 4B and C). Even though resveratrol at 40 µM slightly inhibited TPA-induced AP-1 or NF-κB activity, this inhibition might have been insufficient to suppress TPA-induced transformation. PD098059 and GW5047 were used as positive controls, and the inhibitory effect of quercetin was greater than that of PD098059 for treatment at the same concentration.
Figure 4.
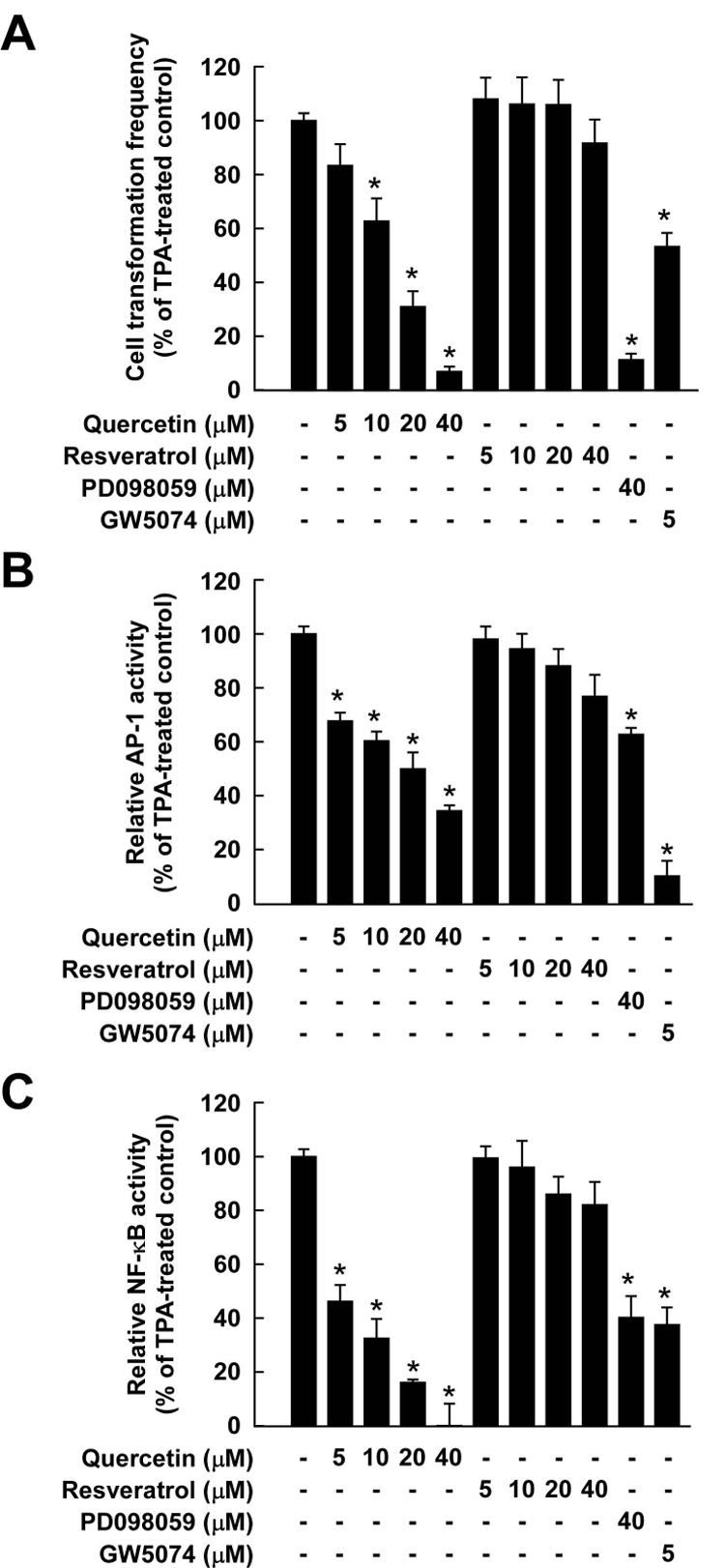
Comparison of inhibitory effects of quercetin or resveratrol against TPA-induced neoplastic transformation, and AP-1 and NF-κB transactivation in JB6 P+ cells. A, Quercetin was more effective than resveratrol at inhibiting TPA-induced JB6 P+ cell transformation. JB6 P+ cells were treated as described in the Materials and Methods, and colonies were counted 14 days later under a microscope with the aid of Image-Pro Plus software (v.4). The effects of quercetin or resveratrol on neoplastic transformation of JB6 P+ cells are presented as the percent inhibition of cell transformation compared with cells treated with only TPA in soft agar. Data are presented as means ± S.D. values of percent inhibition as determined from three separate experiments. The asterisk (*) indicates a significant difference (p < 0.05) between groups treated with TPA and quercetin (or resveratrol or PD098059 or GW5074) together and the group treated with TPA alone. B and C, TPA-induced activation of AP-1 (B) or NF-κB was inhibited more strongly by quercetin than by resveratrol. The JB6 P+ cells, which were stably transfected with AP-1 or NFκB luciferase reporter plasmids, were pretreated with quercetin (or resveratrol or PD098059 or GW5074) for 1 h followed by exposure to 20 ng/ml TPA for 24 h. The relative activity was measured by the luciferase assay as described in the Materials and Methods. Data are presented as means and S.D. values of AP-1 and NF-κB luciferase activities calculated from three independent experiments. The asterisks (*) indicate significant differences (p < 0.05) between groups treated with TPA and quercetin (or resveratrol or PD098059 or GW5074) together and the group treated with TPA alone (* indicates p < 0.05).
RWE or quercetin inhibits H-Ras- or EGF-induced neoplastic transformation of JB6 P+ cells, whereas resveratrol has no effect
Because H-Ras or EGF acts as a potent activator of the Raf/MEK/ERK signaling pathway leading to neoplastic transformation in JB6 P+ cells (9, 26, 27), we next examined whether RWE, quercetin, or resveratrol could inhibit H-Ras-or EGF-induced JB6 P+ cell transformation. RWE at 20 µg/ml suppressed H-Ras- or EGF-induced neoplastic transformation by 87.1% and 91.2%, respectively (Fig. 5A and B, respectively). Based on the number of cell colonies, quercetin at 20 µM blocked H-Ras- or EGF-induced cell transformation by 94.7% or 66.31%, respectively (Fig. 5C and D, respectively), and this ability was significantly better than or similar to that of PD098059 or GW5074 at the same concentration, respectively. Consistent with other results, resveratrol at up to 20 µM had no significant effect on H-Ras- or EGF-induced neoplastic transformation of JB6 P+ cells (Fig. 5C and D). Taken together, these findings support the idea that RWE or quercetin suppresses cell transformation mainly by targeting the Raf1/MEK/ERK signaling cascades.
Figure 5.
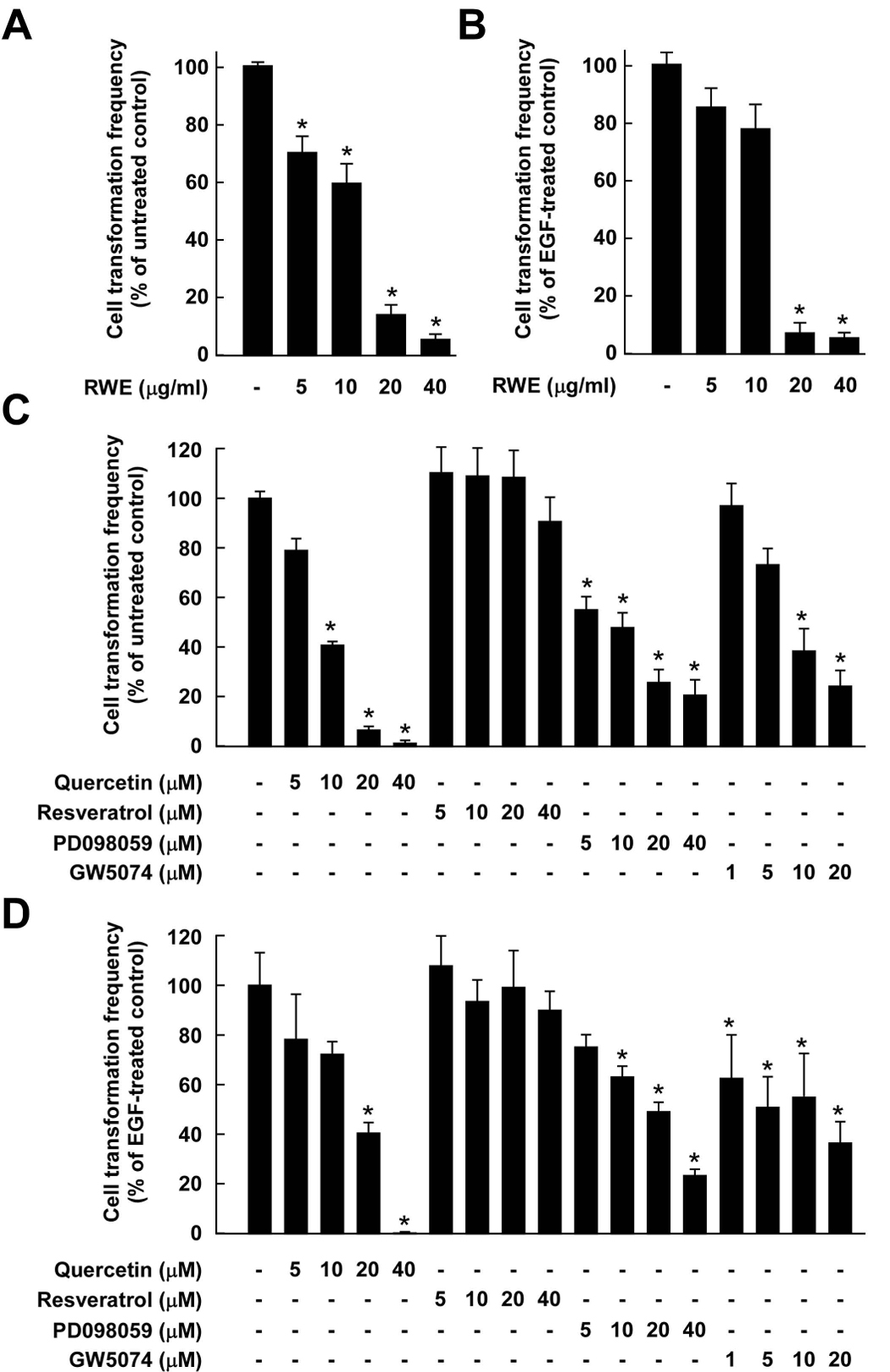
Effects of RWE, quercetin, or resveratrol on H-Ras- or EGF-induced cell transformation. Cell transformation was induced by H-Ras or EGF as described in the Materials and Methods and colonies were counted 14 days later under a microscope with the aid of Image-Pro Plus software (v.4). The effects of RWE, quercetin, or resveratrol on cell transformation of JB6 P+ cells are presented as the percent inhibition of cell transformation compared with H-Ras-or EGF-stimulated cells in soft agar. Data are presented as means ± S.D. values of the percent inhibition as determined from three independent experiments. A, RWE inhibited H-Ras-induced cell transformation of JB6 P+ cells. The asterisk (*) indicates a significant difference (p < 0.05) between groups treated with RWE and the untreated control group. B, RWE inhibited EGF-induced cell transformation of JB6 P+ cells. The asterisk (*) indicates a significant difference (p < 0.05) between groups treated with EGF and RWE together and the group treated with EGF alone. C, Quercetin (but not resveratrol) inhibited H-Ras-induced cell transformation of JB6 P+ cells. The asterisk (*) indicates a significant difference (p < 0.05) between groups treated with quercetin (or resveratrol or PD098059 or GW5074) and the untreated control group (p < 0.05). D, Quercetin (but not resveratrol) inhibited EGF-induced cell transformation of JB6 P+ cells. The asterisk (*) indicates a significant difference (p < 0.05) between groups treated with EGF and quercetin (or resveratrol or PD098059 or GW5074) together and the group treated with EGF alone.
Discussion
Although red wine has been reported to exert anticarcinogenic effects, including growth inhibition of some cancer cells (28, 29), the underlying mechanisms and molecular targets remain unclear. In the present study, we found that RWE inhibited TPA-induced neoplastic transformation of JB6 P+ cells. Previous studies have focused on the critical role of AP-1 in regulating transformation of JB6 P+ cells, because blocking of AP-1 activity by phytochemicals is linked to the suppression of JB6 P+ cell transformation (11, 25, 30). However, other studies have shown that NF-κB activation is also required for TPA-induced neoplastic transformation of JB6 P+ cells (22, 23). Interestingly, a close interaction of c-Fos or c-Jun (AP-1 subunits) with p65 (an NF-κB subunit) has been reported (22). These findings suggest that inhibiting both or either transcription factor effectively suppresses neoplastic transformation. Our results demonstrated that RWE inhibited TPA-induced activation of both AP-1 and NF-κB in JB6 P+ cells.
Transcription factors such as AP-1 and NF-κB are regulated mainly by MAPKs (31, 32). Previous studies have demonstrated the importance of the involvement of ERK in JB6 P+ cell transformation. In contrast to P+ cells, P− cells do not respond to tumor promoters, and this was shown to be due to low levels of phosphorylated and total ERK proteins (9). The ERK signaling pathway involves Raf, MEK, ERK, and p90RSK proteins (33, 34). In this study, RWE inhibited TPA-induced phosphorylation of MEK, ERK, and p90RSK in JB6 P+ cells, and this inhibition of the ERK pathway led to the suppression of neoplastic transformation through the inhibition of AP-1 and NF-κB. RWE was more effective at inhibiting MEK1 activity than Raf1 activity, which is consistent with the above results. A strong inhibition of MEK1 activity resulted in RWE being very effective at suppressing phosphorylation of ERK, due to this being a downstream effecter kinase of MEK. In addition to the ERK pathway, JNK has been reported to involve AP-1 activation and neoplastic transformation in JB6 P+ cells (35). Other studies have found that JNK2-deficient mice failed to induce skin tumorigenesis in response to TPA, which also supports the important role of JNK in skin tumorigenesis (36). This prompted us to examine the effect of RWE on TPA-induced JNK phosphorylation in JB6 P+ cells. RWE at only 20 µM slightly inhibited TPA-induced JNK phosphorylation. These results indicated that the inhibition of the ERK pathway by RWE might be responsible for RWE’s strong inhibition of neoplastic transformation. Further, in vitro and ex vivo pull-down assays revealed that RWE bound with either MEK1 or Raf1, which may be contribute to the observed reduced kinase activities of MEK1 and Raf1.
The identification of the actual active components responsible for the chemopreventive effects of red wine and elucidation of the molecular mechanism(s) of action are needed. The present study compared the effects of resveratrol and quercetin and results indicated that quercetin inhibited MEK1 and Raf1 activities, whereas resveratrol at up to 20 µM had no effect. Quercetin inhibited MEK1 activity more efficiently than Raf1 activity. Of note, quercetin was more effective than PD098059 for inhibiting MEK1 activity. Quercetin inhibited MEK1 and Raf1 activities through direct binding with each protein and blocking of MEK1 activity inhibited TPA-induced ERK phosphorylation in JB6 P+ cells. Interestingly, however, the TPA-induced MEK phosphorylation was not suppressed by quercetin even though quercetin slightly blocked Raf1 activity. These results suggested the presence of a negative feedback loop in MEK regulation. Other investigators have found that inhibition of MEK activity by PD184352 elevated MEK phosphorylation because the binding of PD184352 prevents the catalytic activity of MEK, but still allows the phosphorylation of Ser218 and Ser222 (37). Therefore, because quercetin strongly inhibited MEK1 kinase activity, phosphorylation of MEK would not change in spite of the slight inhibition of Raf1 activity by quercetin. Quercetin, but not resveratrol, inhibited TPA-induced AP-1 and NF-κB activation by suppressing the MEK/ERK, but not the JNK, pathway, subsequently leading to the suppression of neoplastic transformation. Further investigation revealed that RWE or quercetin suppressed, not only TPA-induced, but also H-Ras- or EGF-induced neoplastic transformation, and these results support the finding that RWE or quercetin inhibits the MEK/ERK pathway, regardless of the types of inducers stimulating this pathway.
In contrast to other protein kinase inhibitors, MEK1 inhibitors including PD098059, U0126, PD184352, and PD318088 do not compete with ATP, which accounts for their high selectivity (38). PD184352 is another MEK1-specific inhibitor used in clinical trials, and it binds in a unique inhibitor binding pocket that is adjacent to the ATP-binding site (39). In contrast to the highly homologous ATP-binding site, this unique binding pocket of MEK contains distinctive sequences that are not shared with other kinases. The binding of PD184352 with MEK1 results in a stabilized inactive conformation and a deformation of catalytic sites (39). To investigate the MEK1 inhibition mechanism of quercetin, we carried out a modeling study (Fig. 6). Interestingly, quercetin could be docked to the pocket separate from but adjacent to the ATP binding site similar to that observed for PD318088 (Fig. 6A) in the crystal structure of the MEK1-PD318088 complex (39). The predicted binding mode of quercetin is also similar to that of PD318088. The hydroxyl group at the 7 position can make a hydrogen bond with the backbone carbonyl group of Val127 in the ATP noncompetitive binding site. In addition, several van der Waals interactions exist with the hydrophobic surface formed by Ile99, Ile141, Phe209, and Leu118. The C ring interacts with the residues in the activation loop of the inactive MEK1. Val211 and Leu215 form van der Waals interactions with the C ring of the inhibitor. The hydroxyl group at the 3′ position of the C ring can make a critical hydrogen bond with the backbone amide group of Ser212. These interactions of quercetin with the activation loop would lock MEK1 into a catalytically inactive species by stabilizing the inactive conformation of the activation loop. The low MEK1 inhibitory activity of resveratrol relative to quercetin also can be explained with the docking study (Fig. 6B). Assuming that the binding mode of resveratrol would be similar to that of quercetin, the 3′ position of resveratrol would be placed on the 3′ position of quercetin. However, the lack of the hydroxyl group at the 3′ position of resveratrol would result in the failure of the formation of the hydrogen bond between resveratrol and the backbone amide group of Ser212. Compared to quercetin, kaempferol (3, 4′, 5, 7-tetrahydroxyflavon), a compound structurally related to quercetin, has a lesser inhibitory effect on MEK1 (data not shown). The only structural difference between kaempherol and quercetin is the lack of the hydroxyl group at the 3′ position. Therefore, kaempferol cannot inhibit MEK1 effectively for the same reason as resveratrol (Fig. 6B).
Figure 6.
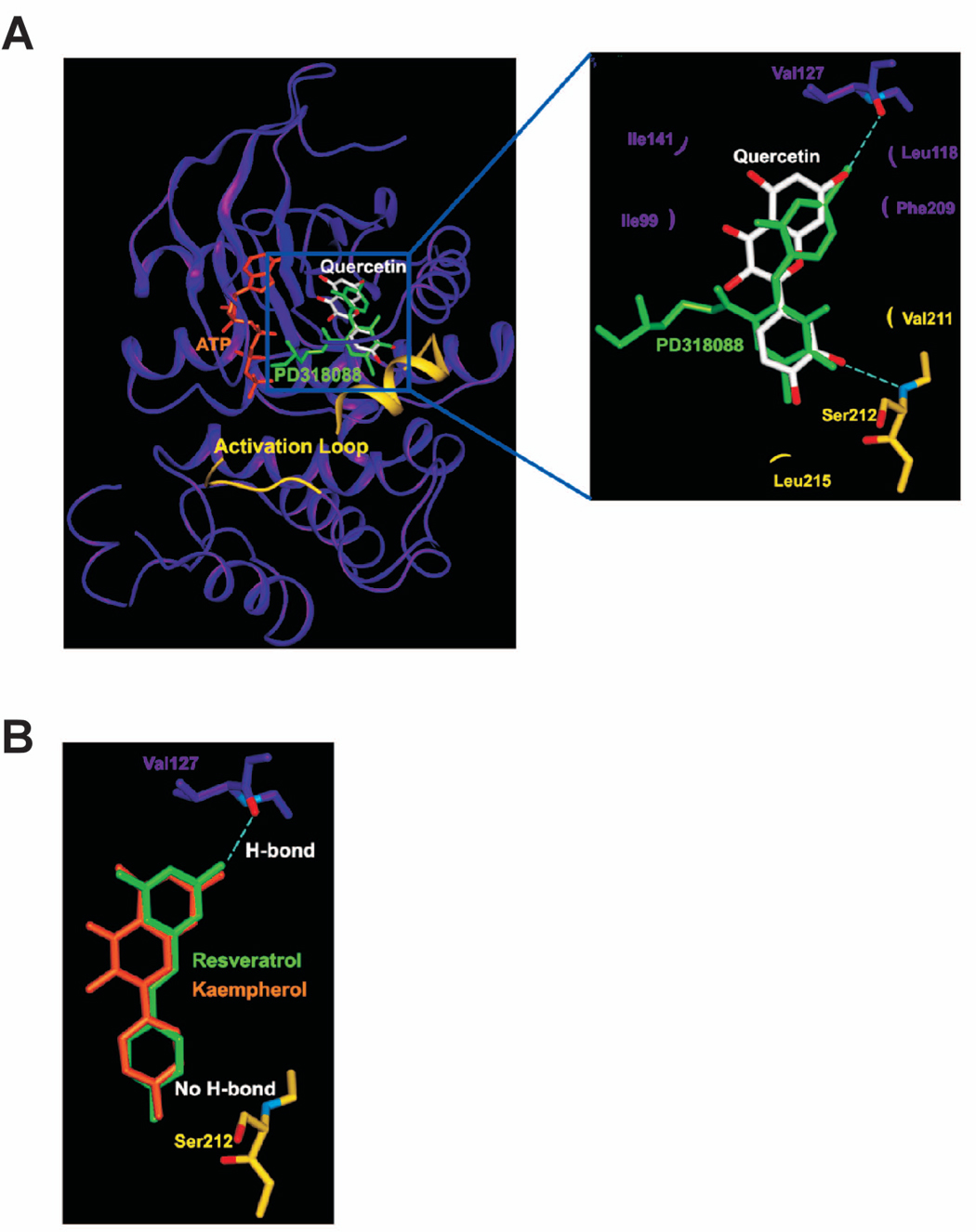
Modeling study of the MEK1 binding of quercetin, resveratrol, or kaempherol. A. Hypothetical model of MEK1-quercetin complex. Quercetin (white color) binds to the pocket adjacent to the ATP (orange) binding site. PD318088 (green) is superimposed on the model structure of MEK1-quercetin complex for comparison. The partially disordered activation loop is colored yellow. The residues involved in the interactions with quercetin are indicated. The hydrogen bonds are depicted as dashed lines. B. Hypothetical model of MEK1 in complex with resveratrol (green) or kaempherol (orange). Although each of these compounds can retain the hydrogen bond with Val127 and the van der Waals interactions involved in the binding of quercetin to MEK1, neither compound can form a hydrogen bond with the activation loop of MEK1 due to the lack of a hydrogen bond acceptor at the 3′ position of their respective ring adjacent to the activation loop.
In summary, RWE or quercetin inhibited tumor promoter-induced neoplastic transformation of JB6 P+ cells. This inhibition was mediated mainly through the blocking of the Raf/MEK/ERK/p90RSK pathway and subsequent suppression of AP-1 and NF-κB activity. RWE or quercetin binds with MEK1 or Raf1 but inhibits MEK1 activity more strongly than Raf1 activity. Together these results suggested that MEK1 is the most potent molecular target of RWE or quercetin for suppressing neoplastic transformation, and that the chemopreventive effects of RWE are more likely to be attributable to quercetin than to resveratrol.
Acknowledgements
We thank Andria Hansen for secretarial assistance.
Grant support: This work is supported in part by The Hormel Foundation, NIH grants CA27502, CA120388, CA111536, CA88961, and CA81064, and by a grant from the BioGreen21 Program (nos. 20070301-034-027 and 20070301-034-042), Rural Development Administration, Republic of Korea.
References
- 1.German JB, Walzem RL. The health benefits of wine. Annu Rev Nutr. 2000;20:561–593. doi: 10.1146/annurev.nutr.20.1.561. [DOI] [PubMed] [Google Scholar]
- 2.Damianaki A, Bakogeorgou E, Kampa M, et al. Potent inhibitory action of red wine polyphenols on human breast cancer cells. J Cell Biochem. 2000;78:429–441. doi: 10.1002/1097-4644(20000901)78:3<429::aid-jcb8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 3.She QB, Bode AM, Ma WY, Chen NY, Dong Z. Resveratrol-induced activation of p53 and apoptosis is mediated by extracellular-signal-regulated protein kinases and p38 kinase. Cancer Res. 2001;61:1604–1610. [PubMed] [Google Scholar]
- 4.Kundu JK, Chun K-S, Kim SO, Surh Y-J. Resveratrol inhibits phorbol ester-induced cyclooxygenase-2 expression in mouse Skin : MAPKs and AP-1 as potential molecular targets. BioFactors. 2004;21:33–39. doi: 10.1002/biof.552210108. [DOI] [PubMed] [Google Scholar]
- 5.Ribeiro de Lima MT, Waffo-Teguo P, Teissedre PL, et al. Determination of stilbenes (trans-astringin, cis- and trans-piceid, and cis- and trans-resveratrol) in Portuguese wines. J Agric Food Chem. 1999;47:2666–2670. doi: 10.1021/jf9900884. [DOI] [PubMed] [Google Scholar]
- 6.Waterhouse AL. Wine phenolics. Ann N Y Acad Sci. 2002;957:21–36. doi: 10.1111/j.1749-6632.2002.tb02903.x. [DOI] [PubMed] [Google Scholar]
- 7.Soleas GJ, Grass L, Josephy PD, Goldberg DM, Diamandis EP. A comparison of the anticarcinogenic properties of four red wine polyphenols. Clin Biochem. 2006;39:492–497. doi: 10.1016/s0009-9120(02)00275-8. [DOI] [PubMed] [Google Scholar]
- 8.Dong Z, Birrer MJ, Watts RG, Matrisian LM, Colburn NH. Blocking of tumor promoter-induced AP-1 activity inhibits induced transformation in JB6 mouse epidermal cells. Proc Natl Acad Sci USA. 1994;91:609–613. doi: 10.1073/pnas.91.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huang C, Ma WY, Young MR, Colburn N, Dong Z. Shortage of mitogen-activated protein kinase is responsible for resistance to AP-1 transactivation and transformation in mouse JB6 cells. Proc Natl Acad Sci USA. 1998;95:156–161. doi: 10.1073/pnas.95.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young MR, Li JJ, Rincon M, et al. Transgenic mice demonstrate AP-1 (activator protein-1) transactivation is required for tumor promotion. Proc Natl Acad Sci U S A. 1999;96:9827–9832. doi: 10.1073/pnas.96.17.9827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu G, Bode A, Ma WY, Sang S, Ho CT, Dong Z. Two novel glycosides from the fruits of Morinda citrifolia (noni) inhibit AP-1 transactivation and cell transformation in the mouse epidermal JB6 cell line. Cancer Res. 2001;61:5749–5756. [PubMed] [Google Scholar]
- 12.Sebolt-Leopold JS, Herrera R. Targeting the mitogen-activated protein kinase cascade to treat cancer. Nat Rev Cancer. 2004;4:937–947. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- 13.Nomura M, Ichimatsu D, Moritani S, et al. Inhibition of epidermal growth factor-induced cell transformation and Akt activation by caffeine. Mol Carcinog. 2005;44:67–76. doi: 10.1002/mc.20120. [DOI] [PubMed] [Google Scholar]
- 14.Suzukawa K, Weber TJ, Colburn NH. AP-1, NF-kappa-B, and ERK activation thresholds for promotion of neoplastic transformation in the mouse epidermal JB6 model. Environ Health Perspect. 2002;110:865–870. doi: 10.1289/ehp.02110865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thompson N, Lyons J. Recent progress in targeting the Raf/MEK/ERK pathway with inhibitors in cancer drug discovery. Curr Opin Pharmacol. 2005;5:350–356. doi: 10.1016/j.coph.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 16.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 17.Schreck R, Rapp UR. Raf kinases: oncogenesis and drug discovery. Int J Cancer. 2006;119:2261–2271. doi: 10.1002/ijc.22144. [DOI] [PubMed] [Google Scholar]
- 18.Gioeli D, Mandell JW, Petroni GR, Frierson HF, Jr, Weber MJ. Activation of mitogen-activated protein kinase associated with prostate cancer progression. Cancer Res. 1999;59:279–284. [PubMed] [Google Scholar]
- 19.Oka H, Chatani Y, Hoshino R, et al. Constitutive activation of mitogen-activated protein (MAP) kinases in human renal cell carcinoma. Cancer Res. 1995;55:4182–4187. [PubMed] [Google Scholar]
- 20.Oak MH, Chataigneau M, Keravis T, et al. Red wine polyphenolic compounds inhibit vascular endothelial growth factor expression in vascular smooth muscle cells by preventing the activation of the p38 mitogen-activated protein kinase pathway. Arterioscler Thromb Vasc Biol. 2003;23:1001–1007. doi: 10.1161/01.ATV.0000070101.70534.38. [DOI] [PubMed] [Google Scholar]
- 21.Colburn NH, Wendel EJ, Abruzzo G. Dissociation of mitogenesis and late-stage promotion of tumor cell phenotype by phorbol esters: mitogen-resistant variants are sensitive to promotion. Proc Natl Acad Sci USA. 1981;78:6912–6916. doi: 10.1073/pnas.78.11.6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li JJ, Westergaard C, Ghosh P, Colburn NH. Inhibitors of both nuclear factor-kappaB and activator protein-1 activation block the neoplastic transformation response. Cancer Res. 1997;57:3569–3576. [PubMed] [Google Scholar]
- 23.Nomura M, Ma W, Chen N, Bode AM, Dong Z. Inhibition of 12-O-tetradecanoylphorbol-13-acetate-induced NF-kappaB activation by tea polyphenols, (-)-epigallocatechin gallate and theaflavins. Carcinogenesis. 2000;21:1885–1890. doi: 10.1093/carcin/21.10.1885. [DOI] [PubMed] [Google Scholar]
- 24.Wang SY, Feng R, Lu Y, Bowman L, Ding M. Inhibitory effect on activator protein-1, nuclear factor-kappaB, and cell transformation by extracts of strawberries (Fragaria x ananassa Duch.) J Agric Food Chem. 2005;53:4187–4193. doi: 10.1021/jf0478049. [DOI] [PubMed] [Google Scholar]
- 25.Hou DX, Kai K, Li JJ, et al. Anthocyanidins inhibit activator protein 1 activity and cell transformation: structure-activity relationship and molecular mechanisms. Carcinogenesis. 2004;25:29–36. doi: 10.1093/carcin/bgg184. [DOI] [PubMed] [Google Scholar]
- 26.Chung JY, Huang C, Meng X, Dong Z, Yang CS. Inhibition of activator protein 1 activity and cell growth by purified green tea and black tea polyphenols in H-ras-transformed cells: structure-activity relationship and mechanisms involved. Cancer Res. 1999;59:4610–4617. [PubMed] [Google Scholar]
- 27.Chung JY, Park JO, Phyu H, Dong Z, Yang CS. Mechanisms of inhibition of the Ras-MAP kinase signaling pathway in 30.7b Ras 12 cells by tea polyphenols (-)-epigallocatechin-3-gallate and theaflavin-3,3′-digallate. FASEB J. 2001;15:2022–2024. doi: 10.1096/fj.01-0031fje. [DOI] [PubMed] [Google Scholar]
- 28.Hakimuddin F, Paliyath G, Meckling K. Treatment of mcf-7 breast cancer cells with a red grape wine polyphenol fraction results in disruption of calcium homeostasis and cell cycle arrest causing selective cytotoxicity. J Agric Food Chem. 2006;54:7912–7923. doi: 10.1021/jf060834m. [DOI] [PubMed] [Google Scholar]
- 29.Briviba K, Pan L, Rechkemmer G. Red wine polyphenols inhibit the growth of colon carcinoma cells and modulate the activation pattern of mitogen-activated protein kinases. J Nutr. 2002;132:2814–2818. doi: 10.1093/jn/132.9.2814. [DOI] [PubMed] [Google Scholar]
- 30.Ding M, Lu Y, Bowman L, et al. Inhibition of AP-1 and neoplastic transformation by fresh apple peel extract. J Biol Chem. 2004;279:11670–11676. doi: 10.1074/jbc.M311465200. [DOI] [PubMed] [Google Scholar]
- 31.Karin M, Liu Z, Zandi E. AP-1 function and regulation. Curr Opin Cell Biol. 1997;9:240–246. doi: 10.1016/s0955-0674(97)80068-3. [DOI] [PubMed] [Google Scholar]
- 32.Schulze-Osthoff K, Ferrari D, Riehemann K, Wesselborg S. Regulation of NF-kappa B activation by MAP kinase cascades. Immunobiology. 1997;198:35–49. doi: 10.1016/s0171-2985(97)80025-3. [DOI] [PubMed] [Google Scholar]
- 33.Mansour SJ, Matten WT, Hermann AS, et al. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science. 1994;265:966–970. doi: 10.1126/science.8052857. [DOI] [PubMed] [Google Scholar]
- 34.Sebolt-Leopold JS, Dudley DT, Herrera R, et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat Med. 1999;5:810–816. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- 35.Dong Z, Ma WY, Huang C, Yang CS. Inhibition of tumor promoter-induced activator protein1 activation and cell transformation by tea polyphenols, (-)-epigallocatechin gallate, and theaflavins. Cancer Res. 1997;57:4414–4419. [PubMed] [Google Scholar]
- 36.Chen N, Nomura M, She QB, et al. Suppression of skin tumorigenesis in c-Jun NH(2)-terminal kinase-2-deficient mice. Cancer Res. 2001;61:3908–3912. [PubMed] [Google Scholar]
- 37.Delaney AM, Printen JA, Chen H, Fauman EB, Dudley DT. Identification of a novel mitogen-activated protein kinase kinase activation domain recognized by the inhibitor PD 184352. Mol Cell Biol. 2002;22:7593–7602. doi: 10.1128/MCB.22.21.7593-7602.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Favata MF, Horiuchi KY, Manos EJ, et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J Biol Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 39.Ohren JF, Chen H, Pavlovsky A, et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nat Struct Mol Biol. 2004;11:1192–1197. doi: 10.1038/nsmb859. [DOI] [PubMed] [Google Scholar]