

Abstract
Intracellular signal transduction proteins typically utilize multiple interaction domains for proper targeting, and thus a broad diversity of distinct signaling complexes may be assembled. Considering the coordination of only two such domains, as in tandem Src homology 2 (SH2) domain constructs, gives rise to a kinetic scheme that is not adequately described by simple models used routinely to interpret in vitro binding measurements. To analyze the interactions between tandem SH2 domains and bisphosphorylated peptides, we formulated detailed kinetic models and applied them to the phosphoinositide 3-kinase p85 regulatory subunit/platelet-derived growth factor β-receptor system. Data for this system from different in vitro assay platforms, including surface plasmon resonance, competition binding, and isothermal titration calorimetry, were reconciled to estimate the magnitude of the cooperativity characterizing the sequential binding of the high and low affinity SH2 domains (C-SH2 and N-SH2, respectively). Compared with values based on an effective volume approximation, the estimated cooperativity is 3 orders of magnitude lower, indicative of significant structural constraints. Homodimerization of full-length p85 was found to be an alternative mechanism for high avidity binding to phosphorylated platelet-derived growth factor receptors, which would render the N-SH2 domain dispensable for receptor binding.
Intracellular signal transduction networks, under the control of activated cell surface receptors, govern cell functional behaviors such as proliferation, migration, differentiation, and programmed cell death (1). Proper communication between signaling proteins is generally contingent upon noncovalent, intermolecular interactions, mediated by well conserved protein domains. A key feature of these domains is their modular nature, which has facilitated the extensive characterization of their binding affinities and specificities in vitro, as well as the construction of “synthetic” signaling proteins with prescribed function (2). The prototypical and best characterized interaction domains in signaling are the Src homology 2 (SH2)3 domains, which direct interactions of proteins with receptor tyrosine kinases and other tyrosine-phosphorylated proteins (3). Receptors of the receptor tyrosine kinase family, which engage growth factor ligands such as platelet-derived growth factor (PDGF), are activated through ligand binding, receptor oligomerization, and autophosphorylation on multiple intracellular residues, which then serve as a scaffold for recruitment of proteins containing SH2 and analogous domains (4, 5).
Signaling proteins typically contain three or more modular interaction domains of various types, and therefore the diversity of interactions that might take place in the cell is staggering (6). Further complicating the problem is the avidity effect, which tends to promote the cooperative association of different domains with binding partners in the same multimolecular complex or subcellular compartment. Other mechanisms of binding cooperativity might also depend on the modification of signaling proteins at multiple sites (7). This context-dependent diversity of interactions is a prime example of what has been called combinatorial complexity (8). Although kinetic modeling has emerged as a powerful tool in the analysis of signal transduction networks (9–11), the very large number of potential state variables that can arise even for combinations of a handful of proteins has prohibited detailed modeling of signaling interactions. The recent development of rule-based modeling tools (12) has enabled modeling of more complex systems; in previous work, we used this approach to analyze the function of the protein-tyrosine phosphatase Shp2 (13), demonstrating the application of rule-based modeling at the level of modular protein domains.
In this paper, we present mathematical models and analysis focused on the interactions between tandem SH2 domains derived from signal transduction proteins and peptides or proteins bearing two phosphotyrosine-binding sites. Such interactions have been characterized in vitro by a variety of biochemical methods (14–18), but the various types of complexes that can form between multivalent binding partners cannot be resolved, making the measurements potentially difficult to interpret. Although dual SH2 domains are found in a number of signaling proteins, including isoforms of phospholipase C, the aforementioned Shp2, and the nonreceptor tyrosine kinases Syk and ZAP70, we focus in particular on the interactions between the p85 regulatory subunit of phosphoinositide 3-kinase (PI3K) and sequences derived from the PDGF β-receptor. PI3Ks are lipid kinases that are strongly activated by PDGF receptors and by many other cell surface receptors, and they play pivotal roles in cell migration, survival, and proliferation pathways (19, 20). The interactions of the p85 SH2 domains are critical for targeting and allosteric activation of the enzyme in cells (21–24).
Analysis of the models reconciles various published in vitro p85/phosphopeptide binding studies that have utilized different assay platforms, namely surface plasmon resonance (SPR) and other solid phase binding assays, competition binding, and isothermal titration calorimetry (ITC). Thus, the consensus magnitude of the cooperativity parameter characterizing the sequential association of the two SH2 domains was evaluated and found to be orders of magnitude lower than expected based on search volume considerations. We address the implications of this apparent structural constraint in the context of PI3K recruitment and activation in cells.
EXPERIMENTAL PROCEDURES
General Modeling Considerations and Implementation—Our kinetic models are executed in the second generation version of the rule-based modeling software, BioNetGen (25). BioNetGen 2 uses a programming syntax that was described in detail in the supplemental material of Barua et al. (13). Graph theoretic methods are used to automatically generate a complete set of kinetic equations (ordinary differential equations in time) based on a set of user-specified rules. In this modeling framework, molecules and complexes thereof are called species, and distinct domains/motifs within the molecules are called components. Other nomenclature specific to the models presented here is as follows. The phosphopeptide has two components, Y1 and Y2, which represent phosphorylated Tyr751 and Tyr740 of the human PDGF β-receptor, respectively. The tandem SH2 construct also has two components, C-SH2 and N-SH2, corresponding to the C-terminal and more N-terminal SH2 domains of p85, respectively. The components are easily silenced in the model by removing their corresponding rules to accommodate peptides with a single phosphorylation site or p85 constructs with only one of the SH2 domains. Each of the four combinations of interactions between phosphotyrosine and SH2 components is assigned a second order association rate constant kon and a first order dissociation rate constant koff, which characterize the reversible binding of two species to form one (Fig. 1A). At equilibrium, it is only the ratio of these rate constants that matters, with KD = koff/kon given in units of molar concentration; incidentally, we used the same realistic value of kon = 1 μm–1 s–1 for all interactions and models, and koff values were specified according to the corresponding KD.
FIGURE 1.

Rule-based model of tandem SH2 binding to bisphosphorylated peptide. A, rules for bimolecular complex formation and associated rate constants. The dashed lines indicate that the remainder of each species is unknown, potentially subject to context-dependent rules. B, ring closure transitions and associated rate constants. The cooperativity factor χ has units of concentration and applies to all such transitions. C, all 10 of the distinct tandem SH2·phosphopeptide complexes containing one peptide molecule, as in the case where the peptide is immobilized at low density. Type I complexes contain one tandem SH2 domain molecule that is singly bound, Type II complexes contain one tandem SH2 domain that is doubly bound forming a ring, and Type III complexes contain two singly bound tandem SH2 domain molecules. D, classification of chain and ring structures containing more than one peptide molecule. Type IV complexes are 1:2 chains, whereas Type V complexes are chains with 2:2 or higher stoichiometry. Type VI complexes are ring structures with 2:2 or higher stoichiometry. E, examples of complex structures that can form when dimerization of full-length protein, such as PI3K p85, are considered.
Tandem SH2 domains, such as in the p85 regulatory subunit of PI3K, engage cognate bisphosphorylated peptides and proteins in a cooperative manner, with binding of one SH2 domain facilitating the binding of the other through a ring closure transition (Fig. 1B). These interactions are characterized by a first order association rate constant that is the product of the corresponding kon and a cooperativity parameter χ, which is the effective concentration of each free binding site within the same molecular complex (13, 26), assumed to be the same value for all ring complexes. If such a site were able to freely search a characteristic volume of 100 nm3 (within a 3-nm radius), that concentration would be ∼20 mm. A more conservative estimate would account for the flexibility of the peptide and other structural constraints within the complex (27), and hence we varied χ between 1 μm and 1 mm and evaluated its effect on the overall binding avidity and other aspects of complex formation. As the value of χ is increased, the ring closure interactions become increasingly favorable, and the overall binding avidity of the complex is enhanced. The reverse, ring opening rate constant is given by the corresponding koff. The assumption that only the forward rate constant is modified affects the binding kinetics but not the equilibrium.
The kinetic equations were integrated numerically for sufficient time to achieve steady state (104 s, typically). All of the model codes are available upon request.
Model 1: Immobilized Phosphopeptide—In the simplest model, the bisphosphorylated peptide is immobilized to a surface or solid matrix, and the tandem SH2 construct binds from solution. It is assumed that the immobilized peptide is present at a sufficiently low density, such that bound complexes are comprised of only one peptide and either one or two tandem SH2 molecules. The peptide is present at an arbitrarily low concentration (10 pm was used) so that the tandem SH2 domain is far in excess, with its free concentration approximately equal to the total. Each of the peptide phosphorylation sites (Y1 or Y2), if unoccupied, may reversibly bind tandem SH2 from solution (both SH2 domains must be unoccupied) via C-SH2 or N-SH2; these four combinations constitute separate rules (Fig. 1A). A peptide·SH2 complex with Y1 or Y2 unoccupied may engage in reversible ring closure transitions (four separate rules shown in Fig. 1B). As a result, there are 12 distinct species in this model, the two unbound molecules and 10 distinct peptide·SH2 complexes; the complexes are classified as Type I, II, or III depending on their structure (Fig. 1C).
Model 2: Immobilized Phosphopeptide with Competition—This model is the same as the previous except that the system also includes soluble, bisphosphorylated peptide as a competitive inhibitor with respect to tandem SH2 binding to the surface, which allows several types of extended structures to form (Fig. 1D). Although complexes may contain only one immobilized peptide molecule, any species containing an unoccupied SH2 domain can combine with any other having an unoccupied competitor peptide site, and thus molecular chains with more than one bisphosphorylated peptide molecule may be formed. Chains comprised of one tandem SH2 and two peptide molecules are classified as Type IV complexes, and chains comprised of four or more molecules are classified as Type V complexes. Ring structures containing four or more molecules can also form; these are classified as Type VI complexes. To simplify matters, the immobilized peptide is only monophosphorylated (on Y1, corresponding to Tyr(P)751 of PDGF β-receptor), matching the conditions of the published experiments (17). Thus, there are only two rules for C-SH2 or N-SH2 binding to the surface, four rules for binding of two species containing unoccupied SH2 and competitor peptide sites, and four rules for unimolecular ring closure involving unoccupied SH2 and competitor peptide sites. In BioNetGen 2, it is possible to set the maximum number of each molecule type in the generated species. Thus, cross-linking of immobilized sites was prohibited here by setting the maximum number of immobilized peptide molecules in a complex to 1, and the potentially infinite sizes of the chain and ring structures were truncated at a maximum number of N molecules each of the tandem SH2 and bisphosphorylated competitor peptide per complex. Values of n = 2, 3, and 4 were used and found to give nearly identical results. These models vary in complexity as N is increased, yielding 68, 272, and 1,075 distinct species, respectively.
Model 3: Solution Phase Binding—In this model, both the tandem SH2 construct and bisphosphorylated peptide are in solution, as in ITC measurements. The binding rules are the same as in the immobilized phosphopeptide with competition model, except that the immobilized peptide is absent. Thus, for the same value of N as described for Model 2, there are correspondingly fewer distinct species in Model 3 (37, 145, and 629 species for n = 2, 3, and 4, respectively). As with Model 2, these values of N produced nearly identical results.
Model 4: Immobilized Phosphopeptide with p85 Dimerization—This model is a modification of Model 1, in which p85 has an additional domain that mediates p85 dimerization (Fig. 1E), with six additional rules. Two of these are for dimerization, one for when at least one of the p85 molecules binds from solution and another for when both p85 molecules are bound to the same peptide; in the latter case, the χ value for dimerization, χdimer, is distinguished from that of 1:1 ring formation (Type II complex), called χSH2. To satisfy the principle of detailed balance, χdimer also applies to the ring closure of peptide-p85-p85 chains via either of the unoccupied SH2 domains in the second p85 molecule. The network for this model is comprised of 35 distinct species.
RESULTS
Cooperativity of Tandem SH2/Phosphopeptide Binding as a Key Determinant of Complex Avidity, Stoichiometry, and Equilibration Time—The simplest model is one in which the phosphopeptide is immobilized, such that complexes contain only one peptide molecule (Model 1) (Fig. 2). This scenario simulates SPR and other solid phase binding assays and is analogous to p85 recruitment to the plasma membrane. For simplicity, the two phosphotyrosine sites are assumed here to be equivalent, and the C-SH2 and N-SH2 domains are assigned single-site KD values characteristic of PI3K p85 (50 nm and 1.5 μm, respectively) (16–18, 28). Each SH2 domain by itself exhibits the expected hyperbolic binding isotherm, with half-maximal binding at a SH2 concentration equal to its KD and a stoichiometry of 2:1 SH2 molecules/peptide at saturation. By comparison, the binding isotherm of the tandem construct is altered relative to that of the higher affinity C-SH2, depending on the value of χ. As expected, the change is dramatic when χ = ∼10 μm or greater, exceeding the KD of the low affinity N-SH2 domain (Fig. 2A). At tandem SH2 concentrations below the C-SH2 KD, overall binding is enhanced because of the cooperativity of the SH2 domains in forming stable, Type II ring structures (Fig. 1C), the effective KD for these structures being given by Equation 1.
FIGURE 2.

Binding properties of tandem SH2 constructs to immobilized, bisphosphorylated peptides. Calculations were performed using Model 1, assuming SH2 domain KD values characteristic of PI3K p85. Constant parameter values were kon,C1 = kon,C2 = kon,N1 = kon,N2 = 1 μm–1 s–1, KD,C1 = KD,C2 = 50 nm, KD,N1 = KD,N2 = 1.5 μm. A, equilibrium binding isotherms. The value of χ was varied as indicated. B, structure types of complexes formed with χ = 100 μm (refer to Fig. 1C). C, tandem SH2 binding as a function of time (χ = 100 μm), with t = 100, 200, 500, and 1,000 s.
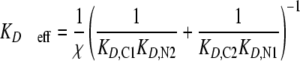 |
(Eq. 1) |
Effective KD values for p85 tandem SH2 binding to the Tyr(P)740/Tyr(P)751 bisphosphorylated peptide have been reported to lie in the vicinity of 1 nm (16, 18); for the single-site KD values assumed here, an order of magnitude estimate of χ = ∼30 μm is obtained. A somewhat lower estimate (χ = ∼10 μm) is obtained if KD,C1 and KD,C2 are allowed to adopt different values spanning the range of 10–100 nm.
In contrast, at tandem SH2 concentrations above the KD of C-SH2, overall binding is diminished because the ring structure reduces the overall stoichiometry of SH2 binding. Indeed, as the value of χ is increased, there is an apparent saturation of binding at 1:1 stoichiometry, and increasingly higher tandem SH2 concentrations are needed to shift the equilibrium from Type II rings to Type III chain structures with 2:1 stoichiometry (Fig. 2B).
Another consequence of cooperative tandem SH2 binding is slower binding kinetics (Fig. 2C). For a simple receptor/ligand system with 1:1 binding stoichiometry, it is well known that the characteristic time constant for approaching equilibrium is the inverse of koff(1 + [L]/KD), where koff is the dissociation rate constant, and [L] is the free ligand concentration (29). Formation of the Type II ring structure effectively increases the dwell time of the tandem SH2 molecule on the peptide, thus reducing the overall off rate and slowing the approach to steady state. Indeed, with the highest values of χ the t½ for approaching steady state at low concentrations is greater than 5 min, compared with t½ = ln 2/koff = 14 s for C-SH2 alone.
Analysis of Tandem SH2/Phosphopeptide Interactions in Competition Binding Experiments Establishes a Lower Limit on the Cooperativity Parameter χ—To further characterize the cooperativity of tandem SH2/phosphopeptide binding, we analyzed the data of Harpur and colleagues (17), who assessed the ability of Tyr(P)740, Tyr(P)751, and Tyr(P)740/Tyr(P)751 peptides to inhibit the binding of various p85 constructs (C-SH2, tandem SH2, as well as full-length) to a SPR chip bearing Tyr(P)751; this experiment is recapitulated in our Model 2 (Fig. 3). In the relatively simple case of C-SH2 and monophosphorylated peptide as the competitor, the fractional occupancy of the immobilized peptide sites, assumed to be small in number compared with the SH2 molecules, is given by Equations 2, 3, 4, 5.
 |
(Eq. 2) |
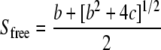 |
(Eq. 3) |
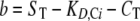 |
(Eq. 4) |
 |
(Eq. 5) |
Equations 2, 3, 4, 5 show that the free SH2 concentration, Sfree, depends on the total concentrations of both SH2 (ST) and the peptide competitor (CT) and the KD of C-SH2 binding to the competitor site (i = 1 for Tyr(P)751, i = 2 for Tyr(P)740). It was assumed that ST was chosen to yield ≈50% surface occupancy in the absence of competitor. Hence, good agreement with the C-SH2 inhibition data were found with ST = KD,C1 = 10 nm and KD,C2 = 75 nm (Fig. 3A), and those parameter values were kept the same in Fig. 3B, described below.
FIGURE 3.

Evaluation of competition binding experiments. The calculations were performed using Model 2 (see “Experimental Procedures”). A, inhibition of monovalent p85 C-SH2 domain binding to Tyr(P)751 of PDGF β-receptor by different competitor peptides as indicated. Fig. 6A of Ref. 17 was recapitulated with ST = KD,C1 = 10 nm and KD,C2 = 75 nm, where ST is the total concentration of C-SH2. B, inhibition of p85 tandem SH2 construct by different competitor peptides as indicated. In the case of the bisphosphorylated competitor, the value of χ is varied as indicated. Fig. 6 (B and C) of Ref. 17 compares favorably with these results when χ > 10 μm.
The experiments also showed that whereas each of the monophosphorylated competitor peptides inhibits C-SH2 and tandem SH2 binding with roughly the same potency, the bisphosphorylated competitor peptide is more potent toward tandem SH2 and full-length p85 binding, indicative of the cooperative formation of ring structures; the inhibition curve also exhibited a much steeper dose response (17). Those results are matched nicely by the Model 2 calculations when the value of χ is much greater than the N-SH2 affinity (χ = ∼10 μm or greater; Fig. 3B). Interestingly, the shape of the inhibition curve is not attributable to the multivalent nature of the competitor binding but rather to the near stoichiometric avidity of the interaction. When the binding avidity is arbitrarily high, the fractional occupancy of immobilized peptide (Equations 2, 3, 4, 5) is closely approximated by taking Sfree ≈ ST – CT, or Sfree ≈ 0 when the competitor is in excess (CT > ST), which produces the characteristic steepness of the inhibition curve. A dramatic reduction of ST, to a value well below the effective KD of ring formation (Equation 1), eliminates this feature (results not shown); however, doing so would reduce the fractional occupancy on the surface in the absence of competitor, perhaps to an unacceptably low level for SPR detection.
Analysis of Tandem SH2/Phosphopeptide Interactions in ITC Measurements Establishes an Upper Limit on the Cooperativity Parameter χ—ITC experiments provide information about molecular interactions through measurements of heat liberated upon serial injections of one solution into another (30). O'Brien and colleagues (18) performed such experiments with full-length p85, injecting increasing amounts of bisphosphorylated Tyr(P)740/Tyr(P)751 peptide into the calorimeter; the net energy change required to maintain the system at constant temperature with each injection was plotted as a function of the increasing molar ratio of peptide/p85. Two distinct changes in the heat/injection were observed, one starting at a molar ratio of ≈0.5 and another, more dramatic reduction induced at a molar ratio of ≈1.0; at a molar ratio of 2.0, the heat released was near 0, indicating saturation of the SH2 domains. Based on those molar ratios, a conceptual model was proposed in which the predominant complex at lower peptide concentrations is the 2:1 chain (Type III complex), whereas a 1:1 complex (depicted as a Type II ring) dominates for molar ratios approaching 1.0 (18).
Our calculated results (Model 3), which allow us to resolve the various types of complexes, shed additional light on those conclusions and provide further evidence for the magnitude of χ (Fig. 4). Based on a concentration of 10 μm p85 in the calorimeter initially and given that 1.5 nmol of peptide was introduced per injection, achieving a molar ratio of 2.0 after 16 injections of 15 μl each (18), the total concentrations of p85 and peptide after each injection were determined. Thus, the total peptide concentration increases from 1.2 μm after the first injection up to 16.7 μm at the end; the p85 is diluted in the process, with a final concentration of 8.3 μm. Based on those concentrations, and using the same default KD values from Fig. 2, we determined the net changes in the amounts of complexes after each injection. Changes in these amounts are related to changes in enthalpy and thus the amount of energy required to maintain constant temperature after each injection. For the sake of simplicity, we adopt a thermodynamic model in which the enthalpy change (ΔH) is a weighted sum of the numbers of bonds formed with C-SH2 and N-SH2 (nCSH2 and nNSH2, respectively), regardless of the structures of the complexes formed.
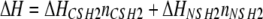 |
(Eq. 6) |
This is equivalent to assuming that the induced proximity effect that distinguishes ring closure from chain extension equilibria is attributed to a difference in conformational entropy.
FIGURE 4.

Evaluation of ITC experiments. The calculations were performed using the solution phase binding model (Model 3). In ITC experiments performed by O'Brien et al. (18), 16 aliquots of bisphosphorylated Tyr(P)751/Tyr(P)740 peptide were added sequentially to a fixed amount of p85 in solution, eventually reaching a molar ratio of 2.0 peptide molecules/p85. Total p85 and peptide concentrations were determined as described in the main text, and single-site KD values are as assumed in Fig. 2. The value of χ is given above each set of panels. A, net change in the numbers of C-SH2 and N-SH2 bonds formed with each injection of peptide. B, hypothetical enthalpy change with each injection of peptide, with ΔHCSH2 = –60 kJ/mol for C-SH2 bonds, and various ratios of ΔHNSH2/ΔHCSH2; the curves in gray are with ΔHNSH2/ΔHCSH2 = 0.2, 0.4, 0.6, and 0.8.
At lower values of χ (1 and 10 μm), the calculated numbers of high affinity C-SH2 bonds formed with each injection show the characteristic plateau at low molar ratios, thereafter yielding to formation of N-SH2 interactions, whereas for higher values of χ (100 μm and 1 mm), the plateau is absent (Fig. 4A). Using Equation 6 to calculate the heat release/injection and varying the ratio of specific enthalpies (ΔHNSH2/ΔHCSH2), only χ = ∼10–30 μm correctly recapitulates the experimentally observed hump in the heat/injection at molar ratios between 0.5 and 1.0 (Fig. 4B and supplemental Fig. S1). Allowing the individual KD parameters to adopt various values within the reported ranges yielded similar results (supplemental Fig. S1).
Further analysis of the complexes formed revealed that when the molar ratio is between 0.5 and 1, χ = 1 μm produces a mixture of extended chain structures, whereas χ = 10 μm leads predominantly to the formation of ring structures. In both cases, there is a shift to 1:2 (Type IV) chains as the molar ratio is increased above 1.0. In contrast, with χ = 100 μm, the shift from Type III chains to Type II rings proceeds steadily for molar ratios up to 1.0, and with χ = 1 mm, the Type II ring structure dominates throughout the hypothetical ITC run (supplemental Fig. S2). Taking the results of this and the previous sections together, it is suggested that the order of magnitude value of χ, characterizing the cooperativity of both SH2 domains of PI3K p85 engaging bisphosphorylated peptides derived from PDGF β-receptor, is 10 μm.
To What Extent Can Dimerization of p85 Stabilize p85 Binding to Bisphosphorylated Peptide?—It has been shown that purified PI3K p85 dimerizes in vitro via a Src homology 3 (SH3) domain/proline-rich sequence interaction, estimated to be of micromolar affinity, perhaps aided by a second, lower affinity interaction (17, 31, 32). These domains are not present in truncated, p85-derived tandem SH2 constructs, but in the context of full-length p85, we were curious as to how p85 dimerization might affect p85 interactions with the bisphosphorylated Tyr(P)740/Tyr(P)751 peptide (Fig. 5). In the corresponding model, Model 4, p85 dimerization is treated as a single interaction with KD = 1 μm in solution. As in Model 1, the phosphopeptide is assumed to be immobilized at low density. Here, the structural constraints governing the formation of Type II rings are distinguished from those governing ring formation via dimerization of p85 molecules attached to the same peptide chain, characterized by distinct values of χ (χSH2 and χdimer, respectively). The principle of detailed balance dictates that χdimer also applies to the cyclization of ring structures via one of the two unoccupied SH2 domains of a p85 molecule already dimerized with another, peptide-bound p85 molecule.
FIGURE 5.

Effect of p85 dimerization on binding to immobilized, bisphosphorylated peptides. The calculations were performed using the immobilized phosphopeptide with p85 dimerization model (Model 4). Self-association of the dimerization domain in solution is characterized by a dissociation constant KD,dimer = 1 μm, and formation of Type II rings (Fig. 1C) is distinguished from other ring closure transitions by assignment of distinct χ values, χSH2 and χdimer, respectively (as explained in the text). KD values for the C-SH2 and N-SH2 domains are the same as in Figs. 2 and 4. A, equilibrium binding isotherm of full-length p85, relative to C-SH2 or N-SH2 alone, with χSH2 = 10 μm and χdimer varied as indicated. B, same as A, but with N-SH2 deleted from p85; the value of χdimer is varied as indicated.
Assuming a value of χSH2 = 10 μm, consistent with the analysis in the previous sections, the calculations show that p85 dimerization can improve binding avidity at low nanomolar concentrations, but only when rings involving dimers are not subject to significant constraints; χdimer must be in the millimolar range (Fig. 5A). With low values of χdimer, comparable with χSH2, the binding avidity is not substantially enhanced beyond what is achieved through Type II ring formation (compare with Fig. 2A). Ring structures with dimerized p85 molecules are found in proportion to the free p85 concentration squared, which is manifested in the steepness of the binding isotherm at low p85 concentrations. At p85 concentrations that are far in excess of the dimerization KD, complexes with stoichiometry approaching 4:1 (two p85 dimers/peptide) are found. This model was also adapted to examine the binding of a p85 variant with the N-SH2 domain deleted (Fig. 5B). Here, the only ring structure that can form is the 2:1 complex with the p85 molecules dimerized. Comparing the isotherm with that of wild-type p85 in Fig. 5A, it is apparent that such rings are the predominant structure at low concentrations of p85 if χdimer is sufficiently high. Under those conditions, the N-SH2 domain is dispensable for binding to the bisphosphorylated motif.
DISCUSSION
In vitro measurements using purified components are predicated on the notion that they are indicative of interactions in cells, and they afford obvious advantages. However, when an interaction involves more than one discrete step, and especially when complexes of varying stoichiometry can form, the interpretation of the measurements can be challenging and perhaps misleading. Reconciling data obtained using different assay designs and platforms only adds to that challenge; here, we used kinetic, rule-based models to accomplish this goal. Interactions between the tandem SH2 domains of PI3K p85 regulatory subunit and its bisphosphorylated binding site in PDGF β-receptor were analyzed in detail, and the cooperativity of the SH2 domains in forming a high avidity ring complex was evaluated in terms of the concentration factor, χ. Analysis of SPR and ITC measurements, which differ with respect to peptide configuration (immobilized versus soluble) and species concentrations (nanomolar versus micromolar), yielded a consistent order of magnitude estimate of χ = ∼10 μm. Significantly lower values do not yield the effective KD values reported for tandem SH2 binding to Tyr(P)740/Tyr(P)751 (16, 18), nor do they give the extent of inhibition observed in competition binding assays (17). Significantly higher values promote ring formation even when one of the components is in micromolar excess, in clear disagreement with ITC measurements (18).
The estimate of χ obtained for p85 tandem SH2 binding is 3 orders of magnitude lower than the value anticipated based on simple search volume considerations, indicating significant structural constraints. Consistent with this conclusion, a worm-like chain model of peptide binding shows that consideration of the peptide flexibility alone can yield χ values in the low micromolar range (27). However, in experiments in which the length of the peptide spacer sequence between Tyr(P) sites was varied, peptide stimulation of PI3K kinase activity in vitro was apparently able to tolerate a reduction of the spacing to 6 residues (33); based on this assertion, the worm-like chain model produces a significantly higher estimate of χ = ∼10 mm (27). It seems clear that factors other than peptide flexibility, such as the conformational dynamics of the tandem SH2 construct (34) and the nature of the peptide residues flanking the Tyr(P) sites (35) and other peptide residues, must contribute to the structural constraints of the interaction.
Although still sufficient to enhance the binding of the tandem SH2 construct, the cooperativity of bisphosphorylated peptide recognition is deemed to be relatively weak, which has a number of implications for PI3K interactions with PDGF receptors in cells. Absent from experiments with receptor-derived peptides are the activities of the receptor tyrosine kinase and nonreceptor tyrosine kinases that associate with activated receptors. That is significant because p85 is tyrosine-phosphorylated in cells stimulated with PDGF, on a site that engages the N-SH2 domain (36, 37). Although it is presently unclear whether or not that interaction is intramolecular (which might lend further insights into the conformational dynamics of the p85 SH2 domains), what is clear is that the role of the interaction is to relieve the autoinhibition of PI3K catalytic activity. In our previous analysis of Shp2, which is regulated by its N-SH2 domain in a related fashion, it was shown that Shp2 phosphorylation and intramolecular N-SH2 binding gives rise to a receptor binding avidity that lies between two extremes; one of these is the case in which phosphorylation does not occur, and the other is the case where the N-SH2 is completely buffered from receptor binding (13). The modest value of χ for p85/receptor binding might represent a compromise between a need for selective recognition of activated PDGF receptors, as PI3K is recruited from the cytosol, and a need for displacement of N-SH2 from the receptor after p85 is phosphorylated.
If the above is true, then the implication is that the N-SH2 domain does not contribute to PDGF receptor binding in cells to the same extent as it does to binding of bisphosphorylated peptide in vitro. Indeed, it has been shown that removing the N-SH2 domain of p85 does not alter its binding to PDGF receptors, but intriguingly, neither does mutation of the phosphorylation site (36, 37), suggesting that the N-SH2 domain is dispensable for receptor binding. PI3K and PDGF receptor bind extraordinarily tightly (38), and PI3K signaling stimulated by PDGF is saturated at much lower concentrations than is PDGF receptor phosphorylation (39, 40), suggesting that interactions other than C-SH2 binding to the receptor are required to stabilize the complex. Our model calculations show that p85 dimerization, whether by SH3 domain/proline-rich sequence or N-SH2/phosphotyrosine interactions, could carry out this function, in a manner that renders the N-SH2 domain dispensable. In the context of PDGF receptor binding in cells, it is important to consider also the dimerization of PDGF receptors. This configuration might contribute parallel binding sites for the C-SH2 domains of two dimerized p85 molecules, such that the complex is less structurally constrained than in the case of binding to a single peptide or receptor molecule. Of course, interactions of the SH3 and proline-rich motifs with other molecules (31), not to mention those of the catalytic subunit with substrate and possibly other binding partners, could also contribute to the stability of PI3K recruitment in cells.
Supplementary Material
This work was supported in part by National Institutes of Health Grant R01-GM067739 and the Cell Migration Consortium under NIGMS, National Institutes of Health Grant U54-GM064346. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
Footnotes
The abbreviations used are: SH, Src homology; PDGF, platelet-derived growth factor; PI3K, phosphoinositide 3-kinase; SPR, surface plasmon resonance; ITC, isothermal titration calorimetry.
References
- 1.Hunter, T. (2000) Cell 100 113–127 [DOI] [PubMed] [Google Scholar]
- 2.Bhattacharyya, R. P., Remenyi, A., Yeh, B. J., and Lim, W. A. (2006) Annu. Rev. Biochem. 75 655–680 [DOI] [PubMed] [Google Scholar]
- 3.Songyang, Z., and Cantley, L. C. (1995) Trends Biochem. Sci. 20 470–475 [DOI] [PubMed] [Google Scholar]
- 4.van der Geer, P., Hunter, T., and Lindberg, R. A. (1994) Annu. Rev. Cell Biol. 10 251–337 [DOI] [PubMed] [Google Scholar]
- 5.Schlessinger, J. (2000) Cell 103 211–225 [DOI] [PubMed] [Google Scholar]
- 6.Pawson, T. (2004) Cell 116 191–203 [DOI] [PubMed] [Google Scholar]
- 7.Lenz, P., and Swain, P. S. (2006) Curr. Biol. 16 2150–2155 [DOI] [PubMed] [Google Scholar]
- 8.Hlavacek, W. S., Faeder, J. R., Blinov, M. L., Perelson, A. S., and Goldstein, B. (2003) Biotechnol. Bioeng. 84 783–794 [DOI] [PubMed] [Google Scholar]
- 9.Eungdamrong, N. J., and Iyengar, R. (2004) Trends Cell Biol. 14 661–669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kholodenko, B. N. (2006) Nat. Rev. Mol. Cell Biol. 7 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haugh, J. M., and Weiger, M. C. (2007) in Chemical Biology: From Small Molecules to Systems Biology and Drug Design (Schreiber, S., Kapoor, T., and Wess, G., eds) Vol. 3, pp. 1061–1081, Wiley-VCH, Weinheim, Germany [Google Scholar]
- 12.Hlavacek, W. S., Faeder, J. R., Blinov, M. L., Posner, R. G., Hucka, M., and Fontana, W. (2006) Science's STKE 344 re6. [DOI] [PubMed] [Google Scholar]
- 13.Barua, D., Faeder, J. R., and Haugh, J. M. (2007) Biophys. J. 92 2290–2300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pluskey, S., Wandless, T. J., Walsh, C. T., and Shoelson, S. E. (1995) J. Biol. Chem. 270 2897–2900 [DOI] [PubMed] [Google Scholar]
- 15.Eck, M. J., Pluskey, S., Trub, T., Harrison, S. C., and Shoelson, S. E. (1996) Nature 379 277–280 [DOI] [PubMed] [Google Scholar]
- 16.Ottinger, E. A., Botfield, M. C., and Shoelson, S. E. (1998) J. Biol. Chem. 273 729–735 [DOI] [PubMed] [Google Scholar]
- 17.Harpur, A. G., Layton, M. J., Das, P., Bottomley, M. J., Panayotou, G., Driscoll, P. C., and Waterfield, M. D. (1999) J. Biol. Chem. 274 12323–12332 [DOI] [PubMed] [Google Scholar]
- 18.O'Brien, R., Rugman, P., Renzoni, D., Layton, M., Handa, R., Hilyard, K., Waterfield, M. D., Driscoll, P. C., and Ladbury, J. E. (2000) Protein Sci. 9 570–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vanhaesebroeck, B., and Waterfield, M. D. (1999) Exp. Cell Res. 253 239–254 [DOI] [PubMed] [Google Scholar]
- 20.Rameh, L. E., and Cantley, L. C. (1999) J. Biol. Chem. 274 8347–8350 [DOI] [PubMed] [Google Scholar]
- 21.McGlade, C. J., Ellis, C., Reedijk, M., Anderson, D., Mbamalu, G., A. D., R., Panayotou, G., End, P., Bernstein, A., Kazlauskas, A., Waterfield, M. D., and Pawson, T. (1992) Mol. Cell Biol. 12 991–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Panayotou, G., Bax, B., Gout, I., Federwisch, M., Wroblowski, B., Dhand, R., Fry, M. J., Blundell, T. L., Wollmer, A., and Waterfield, M. D. (1992) EMBO J. 11 4261–4272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carpenter, C. L., Auger, K. R., Chanudhuri, M., Yoakim, M., Schaffhausen, B., Shoelson, S. E., and Cantley, L. C. (1993) J. Biol. Chem. 268 9478–9483 [PubMed] [Google Scholar]
- 24.Shoelson, S. E., Sivaraja, M., Williams, K. P., Hu, P., Schlessinger, J., and Weiss, M. A. (1993) EMBO J. 12 795–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blinov, M. L., Faeder, J. R., Goldstein, B., and Hlavacek, W. S. (2004) Bioinformatics 20 3289–3291 [DOI] [PubMed] [Google Scholar]
- 26.Haugh, J. M., Schneider, I. C., and Lewis, J. M. (2004) J. Theor. Biol. 230 119–132 [DOI] [PubMed] [Google Scholar]
- 27.Zhou, H.-X. (2003) J. Mol. Biol. 329 1–8 [DOI] [PubMed] [Google Scholar]
- 28.Piccione, E., Case, R. D., Domchek, S. M., Hu, P., Chaudhuri, M., Backer, J. M., Schlessinger, J., and Shoelson, S. E. (1993) Biochemistry 32 3197–3202 [DOI] [PubMed] [Google Scholar]
- 29.Lauffenburger, D. A., and Linderman, J. L. (1993) Receptors: Models for Binding, Trafficking, and Signaling, pp. 19–22, Oxford University Press, New York
- 30.Ladbury, J. E., and Chowdhry, B. Z. (1996) Chem. Biol. 3 791–801 [DOI] [PubMed] [Google Scholar]
- 31.Kapeller, R., Prasad, K. V. S., Janssen, O., Hou, W., Schaffhausen, B. S., Rudd, C. E., and Cantley, L. C. (1994) J. Biol. Chem. 269 1927–1933 [PubMed] [Google Scholar]
- 32.Layton, M. J., Harpur, A. G., Panayotou, G., Bastiaens, P. I. H., and Waterfield, M. D. (1998) J. Biol. Chem. 273 33379–33385 [DOI] [PubMed] [Google Scholar]
- 33.Herbst, J. J., Andrews, G., Contillo, L., Lamphere, L., Gardner, J., Lienhard, G. E., and Gibbs, E. M. (1994) Biochemistry 33 9376–9381 [DOI] [PubMed] [Google Scholar]
- 34.Siegal, G., Davis, B., Kristensen, S. M., Sankar, A., Linacre, J., Stein, R. C., Panayotou, G., Waterfield, M. D., and Driscoll, P. C. (1998) J. Mol. Biol. 276 461–478 [DOI] [PubMed] [Google Scholar]
- 35.Songyang, Z., Shoelson, S. E., Chaudhuri, M., Gish, G., Pawson, T., Haser, W. G., King, F., Roberts, T., Ratnofsky, S., Lechleider, R. J., Neel, B., Birge, R. B., Fajardo, J. E., Chou, M. M., Hanafusa, H., Schaffhausen, B., and Cantley, L. C. (1993) Cell 72 767–778 [DOI] [PubMed] [Google Scholar]
- 36.Kavanaugh, W. M., Klippel, A., Escobedo, J. A., and Williams, L. T. (1992) Mol. Cell Biol. 12 3415–3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.von Willebrand, M., Williams, S., Saxena, M., Gilman, J., Tailor, P., Jascur, T., Amarante-Mendes, G. P., Green, D. R., and Mustelin, T. (1998) J. Biol. Chem. 273 3994–4000 [DOI] [PubMed] [Google Scholar]
- 38.Kazlauskas, A., and Cooper, J. A. (1990) EMBO J. 9 3279–3286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park, C. S., Schneider, I. C., and Haugh, J. M. (2003) J. Biol. Chem. 278 37064–37072 [DOI] [PubMed] [Google Scholar]
- 40.Kaur, H., Park, C. S., Lewis, J. M., and Haugh, J. M. (2006) Biochem. J. 393 235–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.