

Abstract
RAD51 is an important component of double-stranded DNA–repair mechanisms that interacts with both BRCA1 and BRCA2. A single-nucleotide polymorphism (SNP) in the 5′ untranslated region (UTR) of RAD51, 135G→C, has been suggested as a possible modifier of breast cancer risk in BRCA1 and BRCA2 mutation carriers. We pooled genotype data for 8,512 female mutation carriers from 19 studies for the RAD51 135G→C SNP. We found evidence of an increased breast cancer risk in CC homozygotes (hazard ratio [HR] 1.92 [95% confidence interval {CI} 1.25–2.94) but not in heterozygotes (HR 0.95 [95% CI 0.83–1.07]; P=.002, by heterogeneity test with 2 degrees of freedom [df]). When BRCA1 and BRCA2 mutation carriers were analyzed separately, the increased risk was statistically significant only among BRCA2 mutation carriers, in whom we observed HRs of 1.17 (95% CI 0.91–1.51) among heterozygotes and 3.18 (95% CI 1.39–7.27) among rare homozygotes (P=.0007, by heterogeneity test with 2 df). In addition, we determined that the 135G→C variant affects RAD51 splicing within the 5′ UTR. Thus, 135G→C may modify the risk of breast cancer in BRCA2 mutation carriers by altering the expression of RAD51. RAD51 is the first gene to be reliably identified as a modifier of risk among BRCA1/2 mutation carriers.
Germline mutations in BRCA1 (MIM 113705) and BRCA2 (MIM 600185) confer high risks of breast and ovarian cancer. In a meta-analysis of mutation-positive families identified through population-based studies of breast and ovarian cancer cases, the cumulative risks of breast cancer by age 70 years were estimated to be 65% and 45% for BRCA1 and BRCA2 mutation carriers, respectively.1 However, these and other population-based estimates of penetrance have, in general, been lower than estimates based on families with multiple affected individuals.2–4 Moreover, the breast cancer risk has been found to vary by the age at diagnosis and the type of cancer in the index patient.1,5 Such observations are consistent with the hypothesis that breast cancer risk in mutation carriers is modified by other genetic or environmental factors that cluster in families.
RAD51 is the homolog of bacterial RecA, which is required for recombinational repair of double-strand DNA breaks.6,7 Both BRCA1 and BRCA2 interact with RAD51,8,9 and the Rad51-knockout mouse phenotype resembles the Brca1- and Brca2-knockout phenotypes.10 To examine the effect of RAD51 SNPs on cancer risk in BRCA1 and BRCA2 mutation carriers, Wang et al.11 searched for common sequence variants by resequencing the RAD51 gene. No SNPs in the coding region were identified, but two SNPs (135G→C [rs1801320] and 172G→T [rs1801321]) were discovered in the 5′ UTR of RAD51. The latter SNP was found to have no effect on cancer risk, but carriers of the 135C allele were reported to have an increased risk of breast cancer among the subset of BRCA2 carriers (n=216; odds ratio [OR] 3.2 [95% CI 1.4–40]).11 Two additional studies of the 135G→C SNP also found an association with cancer risk in BRCA2 carriers. A study of Israeli Ashkenazi Jewish carriers from 141 BRCA1 families and 64 BRCA2 families found a significant association of the C allele with cancer risk (breast or ovarian) in BRCA2 carriers (hazard ratio [HR] of 4.0 [95% CI 1.3–9.2]), largely because of its effect on breast cancer risk.12 Kadouri et al.13 also found an increased risk of breast cancer (HR 2.09 [95% CI 1.04–4.18]) for BRCA2 carriers and reported that the median age at onset of breast cancer in BRCA2 carriers with the RAD51 C allele was 7 years younger than that in RAD51 wild-type carriers.
In contrast, Jakubowska et al.14 evaluated this RAD51 SNP in a small study of 83 discordant pairs (affected with breast cancer and unaffected) of female carriers of the BRCA1 founder mutation 5382insC and observed a significantly reduced risk of breast cancer among RAD51 135C allele carriers (OR 0.23 [95% CI 0.07–0.62]). In a recent report by the same group,15 the effect was only marginally significant among the 5382insC carriers (P=.046) and became more significant when carriers of the two other BRCA1 Polish founder mutations (4153delA and 300T→G) were included in the analysis (OR 0.58 [95% CI 0.38–0.91]; P=.018).
Studies of genetic modifiers of BRCA1/2 have been hampered by small sample size, such that the power to detect even moderate effects on cancer risk has been limited. To address this problem, we established CIMBA to conduct collaborative analyses of genetic polymorphisms, involving many thousands of samples, as modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers.16 In this report, we evaluate the association of the RAD51 135G→C polymorphism (rs1801320) with breast cancer risk in BRCA1 and BRCA2 female mutation carriers.
Material and Methods
Study Population
Eligible study subjects were women aged ⩾18 years who carry a pathogenic mutation in BRCA1 or BRCA2. Information on study subjects was submitted from 19 participating studies from 13 countries. These women participated in clinical and research studies at the host institutions under institutional review board–approved protocols. Data collected included year of birth, mutation description, family membership, ethnicity, country of residence, age at last follow-up, ages at diagnosis of breast and ovarian cancer, and information on bilateral prophylactic mastectomy. Mutations were included in the analysis if they were pathogenic according to generally recognized criteria (from the Breast Cancer Information Core)—that is, (i) mutations generating a premature termination codon as a result of nonsense substitution, frameshift due to small deletion or insertion, aberrant splicing, or large genomic rearrangement (excluding truncating variants in exon 27 of BRCA2); (ii) mutations resulting in loss of expression due to deletion of promoter and transcription start site; (iii) large in-frame deletions spanning one or more exons caused by aberrant splicing or large genomic rearrangement; and (iv) missense mutations classified as pathogenic by use of the algorithms of Goldgar et al.17 and Chenevix-Trench et al.18
The functional consequences of pathogenic BRCA1/2 mutations may depend on their type.19–21 To examine whether the effect of the RAD51 135G→C on breast cancer risk is different for carriers of different types of BRCA1/2 mutations, we grouped the mutations into two categories. Class 1 corresponds to loss-of-function mutations expected to result in reduced transcript or protein level because of mRNA nonsense-mediated decay (NMD) and/or degradation or instability of truncated proteins,20–22 translation reinitiation but no production of stable protein,23 or the absence of expression because of deletion of transcription regulatory regions. Class 2 consists of mutations likely to generate potentially stable mutant protein that might have dominant negative action, partially preserved normal function, or loss of function. Class 2 mutations are missense substitutions and truncating mutations with premature stop codon occurring in the last exon.
Genotyping
Most centers genotyped the 135G→C SNP in the 5′ UTR of RAD51 by the 5′ nuclease assay (TaqMan) on an ABI 7900HT Sequence Detection System (Applied Biosystems). PCR primers were forward primer 5′-GCTGGGAACTGCAACTCATCT-3′ and reverse primer 5′-GCAGCGCTCCTCTCTCCAGC-3′. Probes were VIC-5′-CAACGCCCGTGGCTTACGCT-3′ and FAM-5′-CCCCAACGCCCCTGGCTTAC-3′. The annealing temperature was 60°C. The Interdisciplinary Health Research International Team on Breast Cancer Susceptibility (INHERIT BRCAs) and Iceland Landspitali-University Hospital (ILUH) samples and most of the samples from the Ontario Cancer Genetics Network (OCGN) were genotyped by direct sequencing. INHERIT BRCAs and OCGN used forward primer 5′-GTTTGGCGGGAATTCTGAAAGCCG-3′ and reverse primer 5′-GTTCTAAAGACTGAGGTCCACTTG-3′. ILUH used forward primer 5′-TGGGAACTGCAACTCATCTGG-3′ and reverse primer 5′-GCGCTCCTCTCTCCAGCAG-3′. GCHBOC used the iCycler technology (Bio-Rad Laboratories) with forward primer 5′-GGGCAAGCGAGTAGAGAAGTG-3′ and reverse primer 5′-CGCGCTCCGACTTCAC-3′. Probes were VIC-5′-CAACGCCCCTGGCTT-3′ and FAM-5′-ACGCCCGTGGCTT-3′. The International Hereditary Cancer Centre (IHCC), Helsinki Breast Cancer Study (HEBCS), Deutsche Krebsforschungszentrum (DKFZ), and National Israeli Cancer Center Control (NICCC) used RFLP-PCR analysis, and Memorial Sloan-Kettering Cancer Center (MSKCC) used pyrosequencing (details available on request). All centers included at least 2% of samples in duplicate, no template controls on every plate, and a random mixture of samples of affected and unaffected mutation carriers on each plate. The minimum accepted call rate was 95%. Call rates for each study were in the range 96%–100% (mean 98.5%). There was one discordant result (0.1%) among the 1,008 duplicates genotyped. For each study, the genotype frequencies among unrelated carriers were consistent with the expected frequencies under the assumption of Hardy-Weinberg equilibrium.
All reported CC homozygotes were confirmed by sequencing with use of forward primer 5′-GAGGGCAGTCTGTAAAACTC-3′ and reverse primer 5′-AACTGCCGCTGAGCACTGGA-3′. One putative homozygote was shown to be a GC heterozygote by sequencing; for another, there was no DNA available for sequencing, so it was excluded from the analysis.
A total of 8,606 mutation carriers with an observed RAD51 135G→C genotype were eligible for inclusion in the study. In some instances, more than one study had enrolled carriers from the same family. We therefore investigated for possible overlap between studies by comparing the year of birth, mutation, and the reported ages, to identify potential duplicate individuals. When a potential duplicate was identified, we contacted the relevant centers for further information about these individuals, without revealing their identity. When potential overlap was identified, centers were contacted to determine precisely the extent of true overlap in subjects and families appearing more than once in the data set. To avoid having families extending over several centers, we excluded the smallest version of the family. In total, 94 carriers with a submitted genotype were excluded: 85 appeared twice in the data set, and 9 were excluded because they were part of the family being studied at another center. The RAD51 135G→C genotypes between the two submissions of the 85 carriers who appeared twice were identical (one was a CC homozygote). A total of 8,512 unique BRCA1 and BRCA2 mutation carriers remained for analysis (table 1).
Table 1. .
Description of Studies and Number of Carriers Included in the Analysis
| No. of Carriers (%) with RAD51 135G→C Genotype |
|||||||
| Study | Country of Residence | Ascertainment Basis | GG | GC | CC | Total | Genotyping Platform |
| CNIO | Spain | Clinic | 188 (82.8) | 37 (16.3) | 2 (.9) | 227 | TaqMan |
| DKFZ Heidelberg | Germany, Pakistan, Colombia | Clinic | 151 (87.3) | 21 (12.1) | 1 (.6) | 173 | RFLP confirmed by sequencing |
| EMBRACE | United Kingdom, Ireland | Clinic | 659 (87.2) | 91 (12.0) | 6 (.8) | 756 | TaqMan |
| GCHBOC | Germany | Clinic | 630 (86.8) | 92 (12.7) | 4 (.5) | 726 | Bio-Rad iCycler |
| GEMO | France, United States, Greece | Clinic | 1,098 (84.3) | 197 (15.1) | 8 (.6) | 1,303 | TaqMan |
| HEBCS | Finland | Clinic | 160 (86.0) | 25 (13.4) | 1 (.5) | 186 | RFLP confirmed by sequencing |
| INHERIT BRCAs | Canada | Clinic | 132 (86.8) | 18 (11.8) | 2 (1.3) | 152 | Sequencing |
| KConFab | Australia, New Zealand | Clinic | 631 (88.0) | 84 (11.7) | 2 (.3) | 717 | TaqMan |
| Leiden University Medical Centre | Netherlands | Clinic | 242 (93.4) | 16 (6.2) | 1 (.4) | 259 | TaqMan |
| MAGIC | United States, Austria, Canada | Clinic | 888 (85.4) | 142 (13.6) | 10 (1.0) | 1,040 | TaqMan |
| MAYO Clinic | United States | Clinic | 92 (82.1) | 18 (16.1) | 2 (1.8) | 112 | TaqMan |
| MSKCC | United States, Brazil, Canada, Philippines | Clinic | 406 (90.0) | 43 (9.5) | 2 (.4) | 451 | Pyrosequencing |
| Mod-Squad | Czech Republic | Clinic/population | 160 (83.3) | 30 (15.6) | 2 (1.0) | 192 | TaqMan |
| NCI | United States | Clinic/community | 205 (86.9) | 30 (12.7) | 1 (.4) | 236 | TaqMan, sequencing |
| NICCC | Israel | Clinic/population | 308 (93.6) | 21 (6.4) | 0 (0) | 329 | RFLP confirmed by sequencing |
| OCGN | Canada | Clinic/population | 261 (86.4) | 39 (12.9) | 2 (.7) | 302 | TaqMan, sequencing |
| IHCC | Poland | Clinic | 661 (77.1) | 186 (21.7) | 10 (1.2) | 857 | PCR-RFLP, sequencing |
| ILUH | Iceland | Clinic/population | 101 (95.3) | 5 (4.7) | 0 (0) | 106 | Sequencing |
| Sheeba Medical Centre | Israel | Clinic | 350 (90.2) |
37 (9.5) |
1 (.3) |
388 |
TaqMan |
| All | … | … | 7,323 (86.0) | 1,132 (13.3) | 57 (.7) | 8,512 | … |
Statistical Analysis
Individuals were classified according to their age at diagnosis of breast cancer or their age at last follow-up. For this purpose, individuals were censored at the age at the first of the following events: breast cancer diagnosis (n=4,443), ovarian cancer diagnosis (n=798), bilateral prophylactic mastectomy (n=176), or last observation (n=3,095). For the purpose of our analysis, only carriers censored at breast cancer diagnosis were assumed to be affected. To investigate whether our results were sensitive to the inclusion of prevalent cancer cases, we also performed analyses after excluding patients with breast or ovarian cancer diagnosed >5 years before their age at last follow-up. The 5-year cutoff was selected to maintain a sample size that would still have reasonable power to investigate the effects under consideration, while excluding long-term survivors. Moreover, prognostic data suggest that 5-year survival after breast cancer is ∼95%, so that, within this time frame, survival bias would be minimal.24 The same cutoff has been used in studies investigating environmental modifiers of risk.25 For this analysis, we excluded subjects for whom an age at interview was not provided. The IHCC study was also excluded because only a censoring age was provided. This left a total of 5,198 mutation carriers for the sensitivity analysis.
To examine whether our results are modified by consideration of information on bilateral prophylactic oophorectomy (BPO), we also performed analyses whereby carriers were censored at age at BPO.26
The analysis of associations in BRCA1 and BRCA2 mutation carriers is complicated by the fact that mutation carriers are not randomly sampled with respect to their disease phenotype. Many carriers are sampled through genetic clinics, and it is likely that affected individuals are oversampled. In such cases, standard methods of analysis, like Cox regression, do not give valid estimates of the HRs.27 To correct for this potential bias, we analyzed the data within a retrospective likelihood framework, by modeling the likelihood of the observed RAD51 135G→C genotypes and disease phenotypes conditional on the disease phenotypes28 (appendix A). In this model, the breast cancer incidence was assumed to depend on the underlying RAD51 135G→C genotype through a Cox proportional hazards model: λi(t)=λ0(t)exp(βi), where exp(βi) is the HR for genotype i and λ0(t) is the breast cancer incidence rate in the baseline category. We estimated the log-HRs for genotypes GC and CC, using the GG homozygotes as the baseline category. The baseline age-specific incidence rates were chosen such that the overall breast cancer incidence rates, averaged over all genotypic categories, agreed with external estimates of BRCA1 and BRCA2 incidence rates. This process is described in detail elsewhere.29 For this purpose, we used the calendar-specific, cohort-specific (for cohorts based on birth year: before 1920, 1920–1929, 1930–1939, 1940–1949, and 1950 and after), and age-specific incidence rates derived using combined data from the meta-analysis of the families of BRCA1/2 carriers identified through population-based studies of breast and ovarian cancer1 and data from three population-based studies of breast cancer31–35 (A.C.A., unpublished data). These analyses were performed using the pedigree analysis software MENDEL.30 We also fitted models in which the log-relative hazards were allowed to vary with age. Significance tests for the null hypothesis that the log-HRs are equal to 0 were also performed using a score test statistic based on the retrospective likelihood described above36 (also see appendix A). Between-study heterogeneity was examined by comparing the log-likelihood of models with study-specific log-HRs (logL1) against the log-likelihood of models in which the same log-HR was assumed to apply to all studies (logL2). These likelihood-ratio tests are approximate because of the small numbers of carriers in each study and the fact that some parameter estimates converge to boundaries.
All analyses used mutation-, calendar-, and cohort-specific breast cancer incidence rates and were stratified by study, country of residence, and reported ethnicity. Analyses were performed for BRCA1 and BRCA2 mutation carriers combined and separately. Fourteen compound BRCA1 and BRCA2 mutation carriers were included in all analyses. In all instances, a robust variance approach was used to allow for the dependence between related carriers.37,38
RNA Extraction and RT-PCR
Total RNA was extracted from lymphoblastoid cell lines (in INHERIT BRCAs and GEMO studies) with the RNeasy mini kit (QIAGEN) according to the manufacturer’s instruction, with a digestion step by use of DNase I (QIAGEN). A total of 5 μg of RNA was reverse transcribed with SuperScript II Rnase H-Reverse Transcriptase (Invitrogen Life Technologies) by use of random primers (Promega). To explore the alternative splicing within the 5′ UTR of RAD51, RT-PCR was performed in the presence of 1.5% dimethyl sulfoxide (DMSO) by use of a forward primer located in the 5′ UTR (5′-AGACCGAGCCCTAAGGAGAG-3′) and a reverse primer located at the exon 2–exon 3 junction (5′-CCACACTGCTCTAACCGTGA-3′) (fig. 1). This RT-PCR was also performed in human mammary epithelial cells (CC-2551 [Lonza]) and human breast cancer cell lines MCF-7 (HTB-22 [ATCC]), MDA-MB-157 (HTB-24 [ATCC]), ZR-75-1 (CRL-1500 [ATCC]), and T47D (HTB-133 [ATCC]). To test whether the RAD51 5′ UTR isoform 2 was associated with specific alternative splicing of coding exons, we performed RT-PCR by using a forward primer specific to isoform 2 (5′-GAAGTGGAGCTAATGGCAATG-3′) and a reverse primer in exon 7 (5′-CTGGTGGTCTGTGTTGAACG-3′). The amplified fragments were sequenced using BDT V1.1 (Applied Biosystems) on the ABI PRISM 3100 (Applied Biosystems). These experiments and the quantitative real-time PCR measurements were performed in the Cancer Genomics Laboratory, Centre Hospitalier Universitaire (CHU) de Quebec & Laval University, and in the Unité Mixte de Génétique Constitutionnelle des Cancers Fréquents, Hospices Civils de Lyon–Centre Léon Bérard, Lyon.
Figure 1. .

RAD51 135G→C variant and alternative splicing within the 5′ UTR. A, Schematic representation and sequence of 5′ RAD51 exons. Exons are represented by hatched boxes (in 5′ UTR) and unblackened boxes (in coding region). Major splicing patterns are shown by blue connecting lines above (for isoform 1) and below (for isoform 2) the gene scheme. ATG is the translation initiation codon. The nucleotide sequence of the full-length 5′ UTR is in blue, the 5′ UTR sequence alternatively spliced as part of intron 1 is in italics, and the canonical motif of the alternative 5′ splice site within the 5′ UTR is underlined. B, Results of the RT-PCR performed with the primers shown in panel A in lymphoblastoid cell lines from carriers of three genotypes of the RAD51 135G→C variant. A predominant RAD51 transcript with the longest 5′ UTR (isoform 1, with full-length 5′ UTR of length 257 nt [GenBank accession number NM_002875]) and a less abundant transcript, with the shortest 5′ UTR (isoform 2, with truncated 5′ UTR of length 153 nt [GenBank accession number AK131299]), as well as several minor RAD51 transcript isoforms with intermediate 5′ UTR lengths characterized by sequencing, were detected.
Quantitative Real-Time PCR Experiments
Experiment 1
First-strand cDNA synthesis was performed using 1–5 μg of total RNA with Superscript III Rnase H-Reverse Transcriptase (Invitrogen Life Technologies) and oligo-dT18. The resulting products were purified with Qiaquick PCR purification kits (QIAGEN). cDNA corresponding to 40–300 ng of total RNA was used to perform fluorescent-based real-time PCR quantification by use of LightCycler FastStart DNA MasterPlus SYBR Green I (Roche) on the LightCycler Realtime PCR apparatus (Roche) as described by the manufacturer. The amplification of RAD51 isoform 1 required the addition of 4% DMSO in the reaction mix for PCR. To ensure that a specific fluorescence signal was read, PCRs were brought for 3 s to a temperature a few degrees below the melting temperature of DNA fragments. The fluorescence signal was then registered at this temperature at the end of each cycle (for isoform 1, 84°C; for isoform 2, 78°C). A melting curve was performed at the end of each run to assess nonspecific signal. The primer pairs used for the specific amplification of RAD51 isoforms were as follows: for RAD51 isoform 1, forward primer 5′-AAGCGAGTAGAGAAGTGGAGCGTA-3′ and reverse primer 5′-ACTGCTCTAACCGTGAAATGGG-3′; for RAD51 isoform 2, forward primer 5′-AGAGAAGTGGAGCTAATGGCAATG-3′ and reverse primer 5′-ACTGCTCTAACCGTGAAATGGG-3′. The specificity of isoform 1 primers was also verified by performing PCR with the purified isoform 2 amplicon as a template, and no amplified product was detected. The housekeeping gene glucose-6-phosphate dehydrogenase (G6PD [MIM 305900]) (forward primer 5′-GATGTCCCCTGTCCCACCAACTCTG-3′; reverse primer 5′-GCAGGGCATTGAGGTTGGGAG-3′) was used for normalization. Standard curves were established using serial dilutions of known cDNA amounts for each RAD51 isoform and G6PD, and the expression was quantified as described elsewhere.39 All experiments were done in duplicate.
Experiment 2
A total of 2 μg of total RNA was reverse transcribed using the First-Strand cDNA Synthesis Kit and pd(N)6 random primers (Amersham Biosciences). Quantification of RAD51 isoform 1 and isoform 2 expression by real-time PCR was performed using LightCycler FastStart DNA MasterPLUS SYBR Green I (Roche) on the LightCycler 2.0 instrument (Roche) as described by the manufacturer. Primer pairs used for specific amplification of RAD51 isoforms were as follows: for RAD51 isoform 1, forward primer 5′-GGCCTGCTGGAGAGAGGA-3′ and reverse primer 5′-CCACACTGCTCTAACCGTGA-3′; for RAD51 isoform 2, forward primer 5′-GAAGTGGAGCTAATGGCAATG-3′ and reverse primer 5′-CCACACTGCTCTAACCGTGA-3′. A melting curve was performed at the end of each run to assess nonspecific signal. The levels of the reference housekeeping genes glyceraldehyde-3-phosphatase dehydrogenase (GAPDH [MIM 138400]) (forward primer 5′-AGCCACATCGCTCAGACAC-3′ and reverse primer 5′-GCCCAATACGACCAAATCC-3′) and β-actin (ACTB [MIM 102630]) (forward primer 5′-ATTGGCAATGAGCGGTTC-3′ and reverse primer 5′-GGATGCCACAGGACTCCAT-3′) were quantified using TaqMan probes UPL#60 and UPL#11, respectively, and LightCycler Taqman Master (Roche) as described by the manufacturer. All experiments were done in duplicate and were normalized to the geometric mean of the level of these reference genes. Relative standard curves determining the PCR efficiencies of the RAD51 isoforms, GAPDH, and ACTB were established using cDNA serial dilutions. Efficiency-corrected and calibrator-normalized calculations were performed using the LightCycler Relative Quantification Software (Roche). The nonparametric Kruskal-Wallis test was used to test for differences in the distribution of expression levels between genotypes implemented in STATA (version 8.2 for Unix [Statacorp]).
Results
A total of 8,512 BRCA1 and BRCA2 mutation carriers were used in the analysis (tables 1 and 2). The overall RAD51 genotype frequencies (GC 13.3%; CC 0.7%) were similar to those reported in population-based studies in the United Kingdom and Australia,40,41 but there was some variation in the frequencies between the CIMBA centers. Carriers of the RAD51 135C allele were least common in the Icelandic (ILUH) study and were most frequent among Polish BRCA1 mutation carriers. The RAD51 genotype frequencies were similar across the larger studies of GEMO, Modifiers and Genetics in Cancer (MAGIC), GCHBOC, EMBRACE, and kConFab.
Table 2. .
Patient Characteristics[Note]
|
BRCA1 Mutation Carriersa |
BRCA2 Mutation Carriersa |
||||
| Characteristic | All Patients | Unaffected | With Breast Cancer | Unaffected | With Breast Cancer |
| No. of carriers | 8,512 | 2,902 | 2,876 | 1,174 | 1,574 |
| Length of follow-up (person-years) | 363,476 | 122,953 | 118,711 | 52,575 | 69,825 |
| Mean ± SD age at censure (years) | 42.9±11.2 | 42.6±12.5 | 41.4±9.3 | 45.1±13.1 | 44.5±10.1 |
| Year of birth: | |||||
| Before 1920 | 91 (1.1) | 19 (.7) | 29 (1.0) | 17 (1.5) | 26 (1.7) |
| 1920–1929 | 334 (3.9) | 74 (2.6) | 115 (4.0) | 43 (3.7) | 102 (6.5) |
| 1930–1939 | 829 (9.7) | 214 (7.4) | 291 (10.1) | 105 (8.9) | 221 (14.0) |
| 1940–1949 | 1,704 (20.0) | 422 (14.5) | 701 (24.4) | 193 (16.4) | 389 (24.7) |
| 1950–1959 | 2,590 (30.4) | 807 (27.8) | 985 (34.3) | 299 (25.5) | 504 (32.0) |
| 1960 or later | 2,964 (34.8) | 1,366 (47.1) | 755 (26.3) | 517 (44.0) | 332 (21.1) |
| Ethnicity: | |||||
| White | 6,935 (81.5) | 2,344 (80.8) | 2,351 (81.8) | 916 (78.0) | 1,334 (84.8) |
| Ashkenazi Jewish | 1,329 (15.6) | 473 (16.3) | 425 (14.8) | 229 (19.5) | 206 (13.1) |
| African American | 54 (.6) | 17 (.6) | 21 (.7) | 9 (.8) | 7 (.4) |
| Asian | 57 (.7) | 16 (.6) | 26 (.9) | 2 (.2) | 13 (.8) |
| Hispanic | 79 (.9) | 29 (1.0) | 28 (1.0) | 12 (1.0) | 10 (.6) |
| Other | 58 (.7) | 23 (.8) | 25 (.9) | 6 (.5) | 4 (.3) |
| Parity: | |||||
| Nulliparous | 693 (8.1) | 285 (9.8) | 190 (6.6) | 123 (10.5) | 95 (6.0) |
| 1 child | 652 (7.7) | 190 (6.6) | 249 (8.7) | 89 (7.6) | 124 (7.9) |
| 2 children | 1,564 (18.4) | 486 (16.8) | 544 (18.9) | 222 (18.9) | 314 (20.0) |
| 3 children | 885 (10.4) | 257 (8.9) | 303 (10.5) | 141 (12.0) | 185 (11.8) |
| ⩾4 children | 517 (6.1) | 168 (5.8) | 154 (5.4) | 86 (7.3) | 110 (7.0) |
| Data missing | 4,201 (49.4) | 1,516 (52.2) | 1,436 (49.9) | 513 (43.7) | 746 (47.4) |
| Oophorectomy: | |||||
| None | 4,148 (48.7) | 1,275 (43.9) | 1,457 (50.6) | 621 (52.9) | 803 (51.0) |
| BPO | 452 (5.3) | 243 (8.4) | 62 (2.2) | 105 (8.9) | 42 (2.7) |
| Data missing | 3,912 (46.0) | 1,384 (47.7) | 1,357 (47.2) | 448 (38.2) | 729 (46.3) |
Note.— Data are no. (%) of carriers, unless otherwise indicated.
Includes 14 compound BRCA1 and BRCA2 mutation carriers.
The genotype frequencies by mutation and disease status are shown in table 3, along with the estimated HRs. There was evidence of association between the RAD51 135G→C genotype and breast cancer risk among BRCA1 and BRCA2 mutation carriers combined (P=.002, by 2-df test). The estimated breast cancer HR was 0.95 (95% CI 0.83–1.07; P=.292) in GC individuals and 1.92 (95% CI 1.25–2.94; P=.0008) in CC individuals. The estimated HR for the CC homozygotes was higher among BRCA2 mutation carriers (HR 3.18 [95% CI 1.39–7.27]; P=.0004) than among BRCA1 mutation carriers (HR 1.59 [95% CI 0.96–2.63]), although the difference in HR was not statistically significant. There were no statistically significant differences between the estimated HRs for patients aged <40 years and those aged ⩾40 years, for either gene (results not shown). Likelihood-ratio tests suggest some evidence of heterogeneity in the HR estimates among centers for BRCA2 (χ2=41.6, df=24) but not for BRCA1 (χ2=32.7, df=32).
Table 3. .
RAD51 135G→C Genotype Distribution and Estimated HRs in the Total Sample of BRCA1 and BRCA2 Mutation Carriers[Note]
| No. (%) of Carriers |
||||
| Gene and Genotype or Test |
Unaffected | With Breast Cancer | HR (95% CI) | Pa |
| BRCA1/2: | ||||
| GG | 3,485 (85.6) | 3,838 (86.4) | 1.00 | … |
| GC | 565 (13.9) | 567 (12.7) | .95 (.83–1.07) | .292 |
| CC | 19 (.5) | 38 (.9) | 1.92 (1.25–2.94) | .0008 |
| 2-df Test | … | … | … | .002 |
| Trend test | … | … | … | .801 |
| BRCA1: | ||||
| GG | 2,456 (84.6) | 2,475 (86.0) | 1.00 | … |
| GC | 429 (14.8) | 376 (13.1) | .86 (.77–1.02) | .095 |
| CC | 17 (.6) | 25 (.9) | 1.59 (.96–2.63) | .067 |
| 2-df Test | … | … | … | .046 |
| Trend test | … | … | … | .386 |
| BRCA2: | ||||
| GG | 1,036 (88.2) | 1,370 (87.0) | 1.00 | … |
| GC | 136 (11.6) | 191 (12.1) | 1.17 (.91–1.51) | .123 |
| CC | 2 (.2) | 13 (.8) | 3.18 (1.39–7.27) | .0004 |
| 2-df Test | … | … | … | .0007 |
| Trend test | … | … | … | .007 |
Note.— Compound BRCA1 and BRCA2 carriers were included in all analyses.
P value by the score test.
To investigate whether our results were sensitive to the inclusion of patients with cancer diagnosed a long time before their recruitment into the various studies (i.e., “prevalent” cancer cases) who may introduce a survival bias, we repeated our analysis after excluding patients with breast or ovarian cancer diagnosed >5 years before their last follow-up. The estimated HRs for CC individuals were very similar to those obtained with the complete data set (BRCA1/2 combined HR 2.36 [95% CI 1.31–4.26]; BRCA1 HR 1.98 [95% CI 0.88–4.46]; BRCA2 HR 3.37 [95% CI 1.33–8.55]).
It is well established that BPO significantly reduces the risk of breast cancer among BRCA1 and BRCA2 mutation carriers.26,42 We therefore examined whether our results are modified by considering the information on BPO, by censoring carriers at BPO in two ways: mutation carriers with missing information on BPO either were included in the analysis under the assumption that they had not undergone BPO or were excluded from the analysis. When all carriers were used in the analysis, the results were very similar to those of the primary analysis (data not shown). When subjects with missing information on BPO were excluded, the HR among BRCA2 mutation carriers with the GC genotype was estimated to be 1.35 (95% CI 0.97–1.89) and, among BRCA2 carriers with the CC genotype, was estimated to be 5.67 (95% CI 1.64–19.63). Thus, the estimated HRs were comparable to those in the overall analysis, although the effect in the combined sample of BRCA1 and BRCA2 mutation carriers was no longer statistically significant.
Although there are limited experimental data on the functional consequence of BRCA1 and BRCA2 mutations, current evidence suggests that most of the premature stop-codon mutations result in loss of function caused by reduced transcript and protein levels due to NMD and/or instability of the truncated proteins (class 1 mutations).20–23 On the other hand, missense or in-frame deletion/insertion mutations, as well as truncating mutations that are not subject to NMD, might generate stable mutant protein that has consequences other than loss of function (class 2 mutations). It is possible that the BRCA1/2 mutations in these two categories may interact in different ways with the potential cancer risk modifier effect of RAD51 135G→C. Of the BRCA1 mutation carriers, 64% carried class 1 mutations, 33% carried class 2 mutations, and 3% carried mutations of unpredictable consequence at transcript or protein level. The majority (95%) of the BRCA2 mutation carriers carried class 1 mutations. There was some indication that the RAD51 135G→C genotype association was greater in carriers of class 1 BRCA1 mutations, with an estimated HR among CC homozygotes of 1.97 (95% CI 1.05–3.70; P=.041), and had no effect in carriers of class 2 mutations (HR 1.11 [95% CI 0.42–2.93]) (table 4). However, the difference in the HRs by mutation type was not statisically significant. The corresponding HR estimates among the GC heterozygotes did not differ significantly from unity (0.83 [95% CI 0.68–1.00] and 0.93 [95% CI 1.05–3.70] for carriers of class 1 and class 2 BRCA1 mutations, respectively).
Table 4. .
RAD51 135G→C Genotype Distribution and Estimated HRs by BRCA1 and BRCA2 Mutation Class[Note]
| No. (%) of Carriers |
||||
| Mutation Class and Genotype or Test |
Unaffected | With Breast Cancer | HR (95% CI) | Pa |
| BRCA1 class 1: | ||||
| GG | 1,639 (85.8) | 1,562 (87.5) | 1.00 | … |
| GC | 262 (13.7) | 208 (11.6) | .83 (.68–1.00) | .058 |
| CC | 9 (.5) | 16 (.9) | 1.97 (1.05–3.70) | .041 |
| 2-df Test | … | … | … | .021 |
| Trend test | … | … | … | .284 |
| BRCA1 class 2: | ||||
| GG | 753 (81.9) | 825 (83.9) | 1.00 | … |
| GC | 158 (17.2) | 150 (15.3) | .93 (.74–1.17) | .567 |
| CC | 8 (.9) | 8 (.8) | 1.11 (.42–2.93) | .780 |
| 2-df Test | … | … | … | .817 |
| Trend test | … | … | … | .525 |
| BRCA2 class 1: | ||||
| GG | 995 (87.8) | 1,289 (87.0) | 1.00 | … |
| GC | 136 (12.0) | 179 (12.1) | 1.14 (.88–1.48) | .123 |
| CC | 2 (.2) | 13 (.9) | 3.24 (1.41–7.45) | .0004 |
| 2-df Test | … | … | … | .0007 |
| Trend test | … | … | … | .007 |
Note.— See main text for definition of mutation classes.
P value by the score test.
To evaluate whether the observed association between the RAD51 135G→C genotype and breast cancer risk was an indirect effect due to another (untested) variant in the RAD51 region, we used the HapMap data for CEPH individuals from Utah data to identify SNPs in strong linkage disequilibrium with 135G→C (International HapMap Project). Fifty SNPs had an r2>0.50 with 135G→C, in a region of 285 kb. Of these, 44 were perfectly correlated with 135G→C (r2=1.0) and would not be distinguishable by genetic epidemiological studies in European populations. Only 2 of the 50 SNPs, however, were located within RAD51 (r2=1.0 and 0.51). SNP rs2304579 (r2=1.0) is in the RAD51 intron 2 (IVS2+110A→G), and SNP rs4144242 (r2=0.51) is in intron 5 (IVS5-4016G→A). By use of software tools that assess splice sites and splice-enhancer motifs, these deep intronic SNPs were not predicted to be of functional significance (SpliceSiteFinder, Splice Site Prediction, and ESE Finder). Three of the other proxy SNPs for RAD51 135G→C are located within the neighboring cancer susceptibility candidate 5 gene (CASC5 [MIM 609173]). SNP rs10518696 is in an intron, and SNPs rs8034726 and rs16970854 are in the promoter region. CASC5 (also known as “AF15Q14” and “D40”) is a component of the hMis12 kinetochore complex essential for chromosome segregation43 and is fused to the MLL gene as a result of translocations in some cases of acute lymphoblastic and myeloblastic leukemias.44–46 However, there is no known interaction between CASC5 and BRCA1 or BRCA2, so we consider RAD51 to be the more plausible modifier of risk.
The molecular mechanism by which the RAD51 135G→C SNP results in increased breast cancer risk in BRCA2 mutation carriers is unclear. Alternative splicing of the human RAD51 gene, involving coding and noncoding exons, has been reported in genomic databases (including GenBank). Some mRNAs had 5′ UTRs shorter than the 5′ UTR of the full-length RAD51 transcript (GenBank accession number NM_002875), with 135G→C located 1 nt next to the canonical motif of the alternative 5′ splice site within the 5′ UTR (fig. 1A).
To investigate whether 135G→C is involved in the regulation of the pattern of the 5′ UTR alternatively spliced RAD51 transcripts, we examined their relative abundance in lymphoblastoid cell lines established from 5 CC homozygotes, 20 GC heterozygotes, and 20 GG homozygotes by performing RT-PCR, encompassing the 5′ UTR, exon 1, and exon 2, including the beginning of the coding region (fig. 1A and 1B). Several RAD51 transcripts with varying length for the 5′ UTR were detected. The two predominant RAD51 transcripts had the longest and the shortest 5′ UTRs: isoform 1 (full-length 5′ UTR of 257 nt [GenBank accession number NM_002875]) and isoform 2 (truncated 5′ UTR of 153 nt [GenBank accession number AK131299]), generated by an alternative splicing within the 5′ UTR, which removes the 104-nt 5′ UTR sequence recognized as part of intron 1. These RT-PCR experiments suggested that the level of the isoform 2 transcript varied between cell lines with different 135G→C genotypes and was found to be particularly low in CC homozygotes (fig. 1B). To investigate this observation further, we used quantitative real-time PCR to compare the expression of the RAD51 isoforms 1 and 2 across three 135G→C genotypes. The quantitative real-time PCR quantification was performed in two laboratories with overlapping sets of samples, by use of different housekeeping genes for normalizing the RAD51 expression data. The results between the two experiments were very similar and are shown in detail in figure 2. There was no statistically significant difference in the expression levels for isoform 1 among the three genotypes (experiment 1, P=.92; experiment 2, P=.44), but there were significant differences among the genotypes in the expression of isoform 2 (experiment 1, P=.008; experiment 2, P=.001). Pairwise genotype comparisons of the mean expression levels of isoform 2 revealed 3–7-fold differences between GG and CC homozygotes (experiment 1, P=.007; experiment 2, P=.002) and 2–4-fold differences between GC heterozygotes and CC homozygotes (experiment 1, P=.012; experiment 2, P=.002). There were no statistically significant differences between the expression levels of isoform 2 between GG homozygotes and GC heterozygotes (experiment 1, P=.137; experiment 2, P=.087).
Figure 2. .

Relative levels of the RAD51 isoforms 1 and 2 transcripts by genotype, measured by quantitative RT-PCR in lymphoblastoid cell lines established from individuals with different RAD51 135G→C genotypes. The relative expression level of each RAD51 isoform across the three 135G/C variant genotypes was normalized by the geometric mean of the expression level of the reference housekeeping genes (in experiment 1, G6PD; in experiment 2, GAPDH and ACTB) and are given in arbitrary units relative to the mean level for the GG genotype. Two replicates were performed for each experiment. The nonparametric Kruskal-Wallis tests were performed to investigate differences in the distributions of the isoform levels across the genotypes.
To test whether the RAD51 5′ UTR isoform 2 is associated with specific alternative splicing of coding exons and may, therefore, give rise to a variant RAD51 protein, we performed RT-PCR, using the forward primer specific to isoform 2 and the reverse primer in exon 7 (see the “Material and Methods” section), generating an amplicon covering the RAD51 exons reported to be alternatively spliced. However, no isoform 2 transcripts with alternative splicing of coding exons were detected.
To find out whether alternative RAD51 splicing that produces isoforms 1 and 2 transcripts is present in mammary gland and is not limited to lymphoblastoid cell lines, we studied human mammary epithelial cells and four human breast cancer cell lines: MCF-7, MDA-MB-157, ZR-75-1, and T47D. Both isoforms 1 and 2 were detected in the breast epithelium and cancer samples tested (data not shown).
Discussion
In this study, we combined data from 19 studies to investigate the effect of the RAD51 135G→C SNP on breast cancer risk among BRCA1 and BRCA2 female mutation carriers and found evidence that this polymorphism is associated with breast cancer risk among BRCA2 mutation carriers (P=.007, by trend test; P=.0007, by 2-df test). We found no evidence of a higher risk in GC heterozygotes, and the sample size was sufficient to rule out any substantial risk in this group (HR 1.17 [95% CI 0.91–1.51]). However, we found strong evidence of an increased risk in CC homozygotes (HR 3.18 [95% CI 1.39–7.27]). The association with breast cancer risk was weaker or nonexistent in BRCA1 carriers, but larger studies will be needed to determine whether there is any association in this group.
Since CC homozygotes are rare, previous studies would have been too small to detect this effect. Only a study of this size, made possible through the CIMBA collaboration, allows such effects to be detected reliably. The association with risk in BRCA2 carriers was essentially unaltered by restriction to incident cases and by adjustment for prophylactic oophorectomy.
There was some evidence of heterogeneity in the HRs between the studies for BRCA2 mutation carriers. However, this may be partly because of the very small number of rare homozygotes in each study, which results in imprecise study-specific estimates. Studies with larger numbers of BRCA2 mutation carriers are required to clarify this further.
Three previously published studies found evidence that carriers of the 135C allele among BRCA2 mutation carriers were at a significantly increased risk of breast cancer,11–13 with the evidence largely coming from the heterozygotes. Although the data from one of these studies are included in the current combined CIMBA analysis11 and although partial overlap may exist with the other two studies, which we cannot establish with certainty, we did not find significant evidence of an increased risk among the heterozygotes.
Two additional studies investigated the effect of the RAD51 135G→C polymorphism among Polish BRCA1 mutation carriers and found a significant protective effect among carriers of the 135C allele.14,15 These data were included in the CIMBA analysis (as a subset of the IHCC study), but the combined analysis does not support this finding. This discrepancy may reflect differences in the eligibility criteria for participation in each study, differences in the study size, or differences in the statistical methods used. In the present analysis, the IHCC study–specific HR estimates were not significantly different from 1 for either the heterozygotes or the rare homozygotes. Interestingly, 93% of the carriers in the IHCC group carry BRCA1 class 2 mutations, for which no effect was detected in our analysis.
The effect of the RAD51 135G→C SNP in unselected series of breast cancer cases has been investigated in a small number of studies. Kuschel et al.40 found no significant association with breast cancer among heterozygote carriers, whereas the OR estimate for rare homozygotes was 2.5 (95% CI 0.6–10.9). However, preliminary analyses of a larger sample from the same population found an OR of 0.97 (95% CI 0.45–2.07) among the CC homozygotes (K. Pooley, personal communication). Another population-based study from Australia also found no evidence of association with breast cancer risk.41 These observations raise the possibility that the association between RAD51 135G→C and breast cancer risk is specific to BRCA2 carriers.
BRCA2 mediates the homologous recombination activity of RAD51 through binding to this DNA recombinase and is essential for the repair of DNA double-strand breaks.6,47 The effect on breast cancer risk observed in our study suggests an interaction between the functional outcomes of the RAD51 135C allele and BRCA2-inactivating mutations. Because of the strong linkage disequilibrium in this region, genetic studies cannot distinguish whether the association is being driven by RAD51 135G→C itself or by other polymorphisms in the region (including CASC5), but RAD51 135G→C is the most plausible causative variant. Prior characterization of RAD51 suggested that it contains a 747-nt basic promoter region that includes the 135G→C polymorphic site.48 Furthermore, expression constructs containing the 135C allele had higher activity than that of the wild type.
Our study suggests an effect of the RAD51 135G→C variant on the RAD51 alternative splicing within the 5′ UTR. The level of the RAD51 alternative isoform 2 transcript is significantly decreased in the lymphoblastoid cell lines of 135CC homozygotes, compared with other genotypes. Interestingly, the part of the 5′ UTR sequence that is lacking in isoform 2 has a particularly high GC content (77%) and is predicted to create highly stable stem-loop secondary structures (RNA and DNA Folding Applications).49 Such structures are known to negatively regulate the translation potential, by inhibiting binding or scanning of the translation machinery.50,51 Therefore, isoform 2 would be expected to have high translation efficiency. These findings suggest that the RAD51 135C allele may cause an overall lower abundance of RAD51 protein, thereby providing an insight into the molecular mechanism through which this RAD51 variant may affect cancer risk. Further studies will be needed to characterize the consequences of this SNP at the protein level, particularly in breast tissue.
The identification of RAD51 as a genetic modifier may have implications for the clinical management of BRCA2 mutation carriers. On the basis of the estimated HR in CC homozygotes, the estimated absolute risk of breast cancer in BRCA2 carriers by age 70 years in the most recent birth cohort would be 90% in CC homozygotes, compared with 51% in GG homozygotes. This difference may be sufficiently large to alter management for those rare patients who are RAD51 CC homozygotes.
Acknowledgments
The MAGIC Consortium centers and individuals are as follows: Baylor–Charles A. Sammons Cancer Center: Joanne L. Blum, Becky Althaus, and Gaby Ethington; Baylor College of Medicine: Claire Noll and Sharon Plon; Beth Israel Deaconess Medical Center: Nadine Tung; City of Hope National Medical Center: Veronica Lagos and Jeffery Weitzel; Creighton University: Carrie Snyder, Henry T. Lynch, and Patrice Watson; Dana Farber Cancer Institute: Jamie Elkins, Kathryn Stoeckert, and Judy E. Garber; Duke University: Sydnee Crankshaw and Joellen Schildkraut; Evanston Northwestern Healthcare Center for Medical Genetics: Suzanne O’Neil, Sue Nelson, and Wendy Rubinstein; Fox Chase Cancer Center: Mary B. Daly and Andrew Godwin; Georgetown University: Camille Jasper and Claudine Isaacs; Jonsson Comprehensive Cancer Center at the University of California, Los Angeles: Joyce Seldon and Patricia A. Ganz; Mayo Clinic College of Medicine: Linda Wadum and Fergus Couch; University of Chicago: Shelly Cummings amd Olufunmilayo Olopade; University of California–Irvine: Susan L. Neuhausen and Linda Steele; University of Pennsylvania: Susan Domchek, K. Nathanson, Tara Friebel, and Timothy Rebbeck; University of Texas, Southwestern: Gail Tomlinson; University of Vienna: Theresa Wagner; and Women’s College Hospital: Steven A. Narod.
EMBRACE collaborating centers and individuals are as follows: Coordinating Centre, Cambridge, United Kingdom: Susan Peock, Margaret Cook, and Cassandra Engel; North of Scotland Regional Genetics Service, Aberdeen: Neva Haites and Helen Gregory; Northern Ireland Regional Genetics Service, Belfast: Patrick Morrison; West Midlands Regional Clinical Genetics Service, Birmingham, United Kingdom: Trevor Cole and Carole McKeown; South West Regional Genetics Service, Bristol: Alan Donaldson; East Anglian Regional Genetics Service, Cambridge: Joan Paterson; Medical Genetics Services for Wales, Cardiff: Mark Rogers, Alexandra Murray, and Jonathon Gray; St. James’s Hospital, Dublin: Peter Daly; National Centre for Medical Genetics, Dublin: David Barton; South East of Scotland Regional Genetics Service, Edinburgh: Mary Porteous and Michael Steel; Peninsula Clinical Genetics Service, Exeter: Carole Brewer and Julia Rankin; West of Scotland Regional Genetics Service, Glasgow: Rosemarie Davidson and Victoria Murday; South East Thames Regional Genetics Service, Guys Hospital, London: Louise Izatt and Gabriella Pichert; North West Thames Regional Genetics Service, Harrow: Huw Dorkins; Leicestershire Clinical Genetics Service, Leicester: Richard Trembath; Yorkshire Regional Genetics Service, Leeds: Tim Bishop and Carol Chu; Merseyside and Cheshire Clinical Genetics Service, Liverpool: Ian Ellis; Manchester Regional Genetics Service, Manchester: Gareth Evans, Fiona Lalloo, and Andrew Shenton; North East Thames Regional Genetics Service, London: James Mackay and Anne Robinson; Nottingham Centre for Medical Genetics, Nottingham: Susan Ritchie; Northern Clinical Genetics Service, Newcastle: Fiona Douglas and John Burn; Oxford Regional Genetics Service, Oxford: Lucy Side and Sarah Durell; Department of Cancer Genetics, Royal Marsden Hospital: Ros Eeles; Sheffield Clinical Genetics Service, Sheffield: Jackie Cook and Oliver Quarrell; South West Thames Regional Genetics Service, London: Shirley Hodgson; and Wessex Clinical Genetics Service, Southampton: Diana Eccles and Anneke Lucassen.
GEMO study collaborators are as follows: Institut Gustave Roussy, Villejuif, France: Agnès Chompret, Brigitte Bressac-de-Paillerets, Véronique Byrde, Corinne Capoulade, and Gilbert Lenoir; Centre Jean Perrin, Clermont-Ferrand, France: Yves-Jean Bignon and Nancy Uhrhammer; Institut Curie, Paris: Dominique Stoppa-Lyonnet, Marion Gauthier-Villars, Muriel Belotti, and Antoine de Pauw; Hospices Civils de Lyon/Centre Léon Bérard, Lyon, France: Olga Sinilnikova, Laure Barjhoux, Mélanie Léone, and Sophie Giraud; Centre Léon Bérard, Lyon, France: Christine Lasset and Valérie Bonadona; Centre François Baclesse, Caen, France: Agnès Hardouin and Pascaline Berthet; Institut Paoli Calmettes, Marseille, France: Hagay Sobol, Violaine Bourdon, and François Eisinger; Hopital Pitié-Salpétrière, Paris: Florence Coulet, Chrystelle Colas, and Florent Soubrier; CHU de Arnaud-de-Villeneuve, Montpellier, France: Isabelle Coupier; Centre Oscar Lambret, Lille, France: Jean-Philippe Peyrat, Joëlle Fournier, Philippe Vennin, and Claude Adenis; Centre René Huguenin, Saint-Cloud, France: Catherine Nogues; INSERM-U735, Centre René Huguenin, Saint-Cloud, France: Rosette Lidereau; Centre Paul Strauss, Strasbourg, France: Danièle Muller and Jean-Pierre Fricker; Institut Bergonié, Bordeaux, France: Michel Longy; Institut Claudius Regaud, Toulouse, France: Christine Toulas, Rosine Guimbaud, Laurence Gladieff, and Viviane Feillel; CHU, Grenoble, France: Dominique Leroux, Hélène Dreyfus, and Christine Rebischung; CHU, Dijon, France: Laurence Olivier-Faivre; CHU, Saint-Étienne, France: Fabienne Prieur; Centre Antoine Lacassagne, Nice, France: Marc Frénay; Centre National de la Recherche Scientifique (CNRS) Unité Mixte de Recherche (UMR) 5201, Lyon, France: Sylvie Mazoyer; Creighton University, Omaha: Henry T. Lynch; National Center for Scientific Research Demokritos, Athens: Drakoulis Yannoukakos.
INHERIT BRCAs collaborating centers and individuals are as follows: CHU de Quebec & Laval University, Quebec: Jacques Simard, Francine Durocher, Rachel Laframboise, and Marie Plante; Molecular Diagnostic Laboratory, Alberta Children’s Hospital, Calgary: Peter Bridge and Jilian Parboosingh; Hôpital du Saint-Sacrement, Quebec: Jocelyne Chiquette; and Hôpital du Sacré-Cour de Montréal, Montreal: Bernard Lesprance and Roxanne Pichette.
A.C.A., C.B., M.C., S.P., the CIMBA data management, and EMBRACE are funded by Cancer Research UK. D.F.E. is a Principal Research Fellow of Cancer Research UK. The GEMO study was supported by the Programme Hospitalier de Recherche Clinique grant AOR01082, by the Programme Incitatif et Coopératif Génétique et Biologie de Cancer du Sein, Institut Curie, and by the Association “Le cancer du sein, parlons-en!” Award. This publication was supported in part by revenue from Nebraska cigarette taxes awarded to Creighton University by the Nebraska Department of Health and Human Services. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the State of Nebraska or the Nebraska Department of Health and Human Services. Support was also received from National Institutes of Health (NIH) grants 5UO1 CA86389 (to H.T.L.), R01-CA083855 (to S.L.N.), R01-CA74415 (to S.L.N.), R01-CA102776 (to T.R.R.), and R01-CA083855 (to T.R.R.). Sample collection and participation for J.W. was supported in part by a General Clinical Research Center grant from the NIH (MO1 RR00043) awarded to the City of Hope National Medical Center, Duarte, California. The EMBRACE team thanks C. Luccarini for technical assistance with the DNA samples. The GEMO collaborators thank Benjamin Bouchet and Gaël Grelier for their assistance with real-time PCR analysis. The GCHBOC is supported by German Cancer Aid grant 107054 and Center for Molecular Medicine Cologne grant TV 93 (to R.K.S.). GCHBOC acknowledges the contributions of Christian Sutter, Institute of Human Genetics, University of Heidelberg; Juergen Horst, Institute of Human Genetics, University of Muenster; Dieter Schaefer, Institute of Human Genetics, University of Frankfurt; Wera Hofmann, Division of Tumorgenetics, Max Delbrück Center for Molecular Medicine, Berlin; Karin Kast, Department of Gynecology and Obstetrics, University of Dresden; and Dorothea Gadzicki, Department of Human Genetics, Medical University, Hannover. kConFab thanks Heather Thorne, Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow Up Study (funded by National Health and Medical Research Council [NHMRC] grants 145684 and 288704), for their contributions to this resource, and the many families who contribute to kConFab. kConFab is supported by grants from the National Breast Cancer Foundation, NHMRC; the Queensland Cancer Fund; the Cancer Councils of New South Wales, Victoria, Tasmania, and South Australia; and the Cancer Foundation of Western Australia. The kConFab genotyping was supported by an NHMRC Programme grant (to G.C.T.). G.C.T. is an NHMRC Senior Principal Research Fellow, and A.B.S. is an NHMRC Career Development Award recipient. This research was supported in part by funding from the Intramural Research Program of the NIH, National Cancer Institute (NCI), Division of Cancer Epidemiology and Genetics, and the Center for Cancer Research, and by Clinical Genetic Branch’s support services contract NO2-CP-11019-50 with Westat. We acknowledge the long-term clinical support of Jennifer Loud, Ruthann Giusti, and Ron Kase and his staff, as well as Lutecia H. Mateus Pereira and Marbin A. Pineda for laboratory assistance. This work was supported by Cancer Care Ontario and Request For Application CA-95-003 as part of the NCI Breast Cancer Family Registries (to I.L.A.). This work was supported by the Canadian Institutes of Health Research for the INHERIT BRCAs program, the CURE Foundation, and the Fonds de la recherche en Santé du Quebec/Reseau de Medecine Genetique Appliquee. J.S. holds the Canada Research Chair in Oncogenetics. K.O. is supported by funding from Komen Foundation grant BCTR0601361, and T.K. holds a Frankel Fellowship. This work was partially funded by grants from the Israeli Cancer Association and the Sackler School of Medicine, Tel-Aviv University, Tel-Aviv (to E.F.). M.P.G.V. was supported by Dutch Cancer Society grant UL-2001-2471. We thank Hans Vasen, Hanne Meijers-Heijboer, and Christi van Asperen, for their ascertainment and blood sampling of breast cancer families, and Dieneke Biemans, for providing follow-up data. The Spanish Consortium for the Study of Genetic Modifiers of BRCA1 and BRCA2 (Spanish National Cancer Centre [CNIO]) was supported by the “Mutua Madrileña” and “Genoma España” foundations. We acknowledge Roger Milne, Rosario Alonso, Guillermo Pita, Jesús López, and Miguel Urioste for their assistance. HEBCS was supported by Academy of Finland grant 110663, the Finnish Cancer Society, the Helsinki University Central Hospital Research Fund, and the Sigrid Juselius Fund. We thank Drs. Kristiina Aittomäki, Carl Blomqvist, and Kirsimari Aaltonen, as well as Anitta Tamminen, for their kind help. For Modifier Study of Quantitative Effects on Disease (Mod-Squad), C.I.S. is partially supported by Susan G. Komen Foundation Basic, Clinical, and Translational Research grant BCTR0402923, Research Project of the Ministry of Education, Youth, and Sports of the Czech Republic grant MSM0021620808 (to M.Z., Zdenek Kleibl, and Petr Pohlreich). We acknowledge the contributions of Petr Pohlreich and Zdenek Kleibl (Department of Biochemistry and Experimental Oncology, First Faculty of Medicine, Charles University, Prague, Czech Republic) and Lenka Foretova, Machakova Eva, and Lukesova Miroslava (Department of Cancer Epidemiology and Genetics, Masaryk Memorial Cancer Institute, Brno, Czech Republic) with the support of Ministry of Health grant CR-MZ0 MOU 2005. We thank Diana Torres and Muhammad U. Rashid for providing DNA samples and supplying data. We thank Antje Seidel-Renkert and Michael Gilbert for expert technical assistance. This work was supported in part by grants from the Breast Cancer Research Foundation, U.S. Army Medical Research and Materiel Command grant W81XWH-04-1-0588, and Mayo Clinic Breast Cancer Specialized Programs of Research Excellence grant P50-CA116201 (to F.J.C.). The Mayo Clinic study thanks Noralane Lindor and Linda Wadum for their contributions. ILUH collaborators are Rosa B. Barkardottir, Gudrun Johannesdottir, Bjarni A. Agnarsson, and Oskar T. Johannsson. ILUH was funded by the Science Fund of Landspitali-University Hospital and by the Memorial Fund of Bergthora Magnusdottir and Jakob Bjarnason.
Author affiliations.—Cancer Research UK, Genetic Epidemiology Unit, Department of Public Health and Primary Care (A.C.A.; S.P.; M.C.; D.F.E.), and Cancer Research UK, Human Cancer Genetics Group, Department of Oncology (C.B.), University of Cambridge, Cambridge, United Kingdom; Unité Mixte de Génétique Constitutionnelle des Cancers Fréquents, Hospices Civils de Lyon–Centre Léon Bérard (O.M.S.; M.L.; L.B.), Laboratoire de Génétique Moléculaire, Signalisation et Cancer UMR5201 CNRS, Université Claude Bernard (O.M.S.; L.B.), Université de Lyon, Université Lyon 1, CNRS UMR5558, Laboratoire de Biometrie et Biologie Evolutive (C.L.; V.B.), and Centre Léon Bérard (C.L.; V.B.), Lyon, France; Cancer Genomics Laboratory, Oncology and Molecular Endocrinology Research Center, CHU de Quebec and Laval University, Quebec (J.S.; M.D.; F.D.); Department of Epidemiology, University of California–Irvine, Irvine (S.L.N.); Clinical Genetics Branch, Division of Cancer Epidemiology and Genetics (J.P.S.; M.H.G.; S.A.P.), and Laboratory of Population Genetics (J.P.S.) and Cancer Genetics Branch (S.A.P.), Center for Cancer Research, US NCI, Bethesda; INSERM U509, Service de Génétique Oncologique, Institut Curie, Paris (D.S-.L.; I.C.; M.B.); Department of Clinical Medicine, Trinity College (D.J.H.), Department of Medical Oncology, St. James’s Hospital and Trinity College (P.A.D.), and Cancer Genetics Program, St. James’s Hospital (P.D.), Dublin; INSERM UMR484, Centre Jean Perrin, Clermont–Ferrand, France (Y.-J.B.); Center for Clinical Epidemiology and Biostatistics (T.R.R.), Abramson Cancer Center (T.R.R.; S.M.D.; K.L.N.), and Department of Medicine (S.M.D.; K.L.N.), The University of Pennsylvania School of Medicine, and Fox Chase Cancer Center (A.G.), Philadelphia; University of Vienna, Vienna (T.W.); Creighton University, Omaha (H.T.L.); Dana Farber Cancer Institute, Boston (J.E.G.); City of Hope National Medical Center, Duarte, CA (J.W.); Women’s College Hospital (S.A.N.), Fred A. Litwin Center for Cancer Genetics, Samuel Lunenfeld Research Institute, Mount Sinai Hospital (I.L.A.; E.I.; H.O.), and OCGN, Cancer Care Ontario (I.L.A.), Toronto; Department of Pediatrics, University of Texas, Southwestern, Dallas (G.T.); Department of Medicine, University of Chicago, Chicago (O.I.O.); Lombardi Cancer Center, Georgetown University, Washington, DC (C.I.); IHCC, Department of Genetics and Pathology, Pomeranian Medical University, Szczecin, Poland (A.J.; J.L.; J.G.; B.G.; T.B.; T.H.); Medical Genetics Services for Wales, University Hospital of Wales, Cardiff (A. Murray; M.R.); North West Thames Regional Genetics Service, Northwick Park Hospital, Harrow, United Kingdom (H. Dorkins); Division of Molecular Gyneco-Oncology, Department of Obstetrics Gynaecology, University of Cologne, Cologne, Germany (R.K.S.; B.V.); Institute for Medical Informatics, Statistics and Epidemiology, University of Leipzig, Leipzig, Germany (C.E.); Department of Gynecology and Obstetrics, Technical University, Munich (A. Meindl); Department of Gynecology and Obstetrics, University of Schleswig-Holstein, Kiel, Germany (N.A.); Molecular Genetics Laboratory, Department of Gynecology and Obstetrics, University of Düsseldorf, Düsseldorf (D.N.); Department of Gynecology and Obstetrics, University of Ulm, Ulm, Germany (H. Deissler); Queensland Institute of Medical Research (A.B.S.; X.C.; N.W.; G.C.-T.) and Institute of Molecular Biosciences, University of Queensland (N.C.), Brisbane, Australia; Peter MacCallum Cancer Centre, Melbourne (kConFab); Clinical Genetics Service, MSKCC, New York (T.K.; K.O.); The Susanne Levy Gertner Oncogenetics Unit and the Oncology Division, Sheba Medical Center, Tel-Hashomer, Israel (E.F.; B.K.; Y.L.; G.G.); CHS NICCC at Carmel Medical Center and B. Rappaport Faculty of Medicine, Technion, Haifa, Israel (G.R.; F.L.; L.R.); Department of Human Genetics, Leiden University Medical Center, Leiden, The Netherlands (P.D.; M.P.G.V.); Human Cancer Genetics Programme, CNIO, Madrid (A.O.; J.B.); Department of Biochemistry and Experimental Oncology, First Faculty of Medicine, Charles University Prague, Prague (M.Z.); Department of Laboratory Medicine and Experimental Pathology (C.I.S.) and Department of Laboratory Medicine and Pathology (F.J.C.), Mayo Clinic College of Medicine, Rochester, MN; Helsinki University Central Hospital, Department of Obstetrics and Gynecology, Helsinki (O.K.; H.N.); Deutsches Krebsforschungszentrum Heidelberg, Heidelberg, Germany (U.H.); and Department of Pathology, Landspitali-University Hospital, Reykjavik (A.A.).
Appendix A : Retrospective Likelihood Approach and Score Test
Analytical Framework
We assume that the study includes n carriers who receive a diagnosis of breast cancer at ages t1,t2,…,tn and m-n unaffected carriers censored at ages tn+1,…,tm and that we observe genotype vectors z1,z2,…,zm (or, in general, other covariates) for these individuals. We assume that the breast cancer incidence rate for individual i depends on the underlying genotype through a Cox proportional hazards model: λi(t)=λ0(t)exp(βtzi). Our aim is to estimate and perform significance tests on the log-risk ratios β. Note that the baseline incidence rate λ0(t) is not known but can be inferred from the overall incidence rates μ(t) obtained, for example, from external sources.
Retrospective Likelihood
The conditional likelihood of the genotypes (assumed to be random effects) given the disease phenotypes can be written as
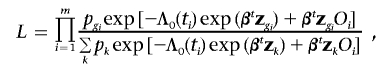 |
where the summation in the denominator is over all genotypes, pk is the frequency of genotype k, and Oi is 0 (zero) if individual i is unaffected and 1 if affected. Λ0(t) is the baseline cumulative incidence rate 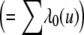 and is unknown but can be obtained by constraining the overall incidence to agree with the assumed incidence rates for BRCA1 and BRCA2 mutation carriers as described elsewhere.29 This is a true likelihood and can be maximized jointly over p and β by use of the software MENDEL.30 Stratified data can be dealt with by considering the stratified conditional likelihood
and is unknown but can be obtained by constraining the overall incidence to agree with the assumed incidence rates for BRCA1 and BRCA2 mutation carriers as described elsewhere.29 This is a true likelihood and can be maximized jointly over p and β by use of the software MENDEL.30 Stratified data can be dealt with by considering the stratified conditional likelihood
 |
where pkj is the frequency of the kth genotype within stratum j (j=1,…,S) and the subscript ji indicates the observed data for individual i in the jth stratum. This likelihood can be maximized jointly over β and the stratum specific genotype frequencies.
Score Test
By use of the likelihood, it is also possible to derive the score test statistic for testing the null hypothesis H0: β=0. Let U represent the score vector. Following the notation in the previous section, it can be shown that, under the null hypothesis, the kth element of U has the form
 |
where zki is value of the covariate for the ith individual and 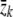 is the mean of the kth covariate over all individuals. Under the null hypothesis, the statistic UtV-1U has a χ2 distribution with ν-1 df, where V is the information matrix under the null hypothesis and ν is the dimension of the covariate vector z. A stratified version of the test can also be derived. This test is fully efficient when the effect size is small and λ0(t) agrees with the true incidence rates but is valid even if these assumptions are not met. The score test is similar in concept to the log-rank test used in standard survival analysis.32 The difference is that, in the log-rank test, the expected number of events is computed using observed data, whereas, in this test, the expected number is based on the assumed carrier incidence rates.
is the mean of the kth covariate over all individuals. Under the null hypothesis, the statistic UtV-1U has a χ2 distribution with ν-1 df, where V is the information matrix under the null hypothesis and ν is the dimension of the covariate vector z. A stratified version of the test can also be derived. This test is fully efficient when the effect size is small and λ0(t) agrees with the true incidence rates but is valid even if these assumptions are not met. The score test is similar in concept to the log-rank test used in standard survival analysis.32 The difference is that, in the log-rank test, the expected number of events is computed using observed data, whereas, in this test, the expected number is based on the assumed carrier incidence rates.
Web Resources
Accession numbers and URLs for data presented herein are as follows:
- Breast Cancer Information Core, http://research.nhgri.nih.gov/projects/bic/
- ESE Finder, http://rulai.cshl.edu/tools/ESE/
- GenBank, http://www.ncbi.nlm.nih.gov/Genbank/ (for RAD51 transcripts [accession numbers NM_002875 and AK131299])
- International HapMap Project, http://www.hapmap.org/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for BRCA1, BRCA2, G6PD, GAPDH, ACTB, and CASC5)
- RNA and DNA Folding Applications, http://www.bioinfo.rpi.edu/applications/mfold/
- SpliceSiteFinder, http://violin.genet.sickkids.on.ca/~ali/splicesitefinder.html
- Splice Site Prediction, http://www.fruitfly.org/seq_tools/splice.html
References
- 1.Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL, Loman N, Olsson H, Johannsson O, Borg A, et al (2003) Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 72:1117–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE, Breast Cancer Linkage Consortium (1994) Risks of cancer in BRCA1-mutation carriers. Lancet 343:692–695 10.1016/S0140-6736(94)91578-4 [DOI] [PubMed] [Google Scholar]
- 3.Ford D, Easton DF, Stratton M, Narod S, Goldgar D, Devilee P, Bishop DT, Weber B, Lenoir G, Chang-Claude J, et al (1998) Genetic heterogeneity and penetrance analysis of the BRCA1 and BRCA2 genes in breast cancer families. Am J Hum Genet 62:676–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, Timmerman MM, Brody LC, Tucker MA (1997) The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med 336:1401–1408 10.1056/NEJM199705153362001 [DOI] [PubMed] [Google Scholar]
- 5.Simchoni S, Friedman E, Kaufman B, Gershoni-Baruch R, Orr-Urtreger A, Kedar-Barnes I, Shiri-Sverdlov R, Dagan E, Tsabari S, Shohat M, et al (2006) Familial clustering of site-specific cancer risks associated with BRCA1 and BRCA2 mutations in the Ashkenazi Jewish population. Proc Natl Acad Sci USA 103:3770–3774 10.1073/pnas.0511301103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pellegrini L, Venkitaraman A (2004) Emerging functions of BRCA2 in DNA recombination. Trends Biochem Sci 29:310–316 10.1016/j.tibs.2004.04.009 [DOI] [PubMed] [Google Scholar]
- 7.Karran P (2000) DNA double strand break repair in mammalian cells. Curr Opin Genet Dev 10:144–150 10.1016/S0959-437X(00)00069-1 [DOI] [PubMed] [Google Scholar]
- 8.Scully R, Chen J, Plug A, Xiao Y, Weaver D, Feunteun J, Ashley T, Livingston DM (1997) Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell 88:265–275 10.1016/S0092-8674(00)81847-4 [DOI] [PubMed] [Google Scholar]
- 9.Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL (1997) RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem 272:31941–31944 10.1074/jbc.272.51.31941 [DOI] [PubMed] [Google Scholar]
- 10.Shu Z, Smith S, Wang L, Rice MC, Kmiec EB (1999) Disruption of muREC2/RAD51L1 in mice results in early embryonic lethality which can be partially rescued in a p53−/− background. Mol Cell Biol 19:8686–8693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang WW, Spurdle AB, Kolachana P, Bove B, Modan B, Ebbers SM, Suthers G, Tucker MA, Kaufman DJ, Doody MM, et al (2001) A single nucleotide polymorphism in the 5′ untranslated region of RAD51 and risk of cancer among BRCA1/2 mutation carriers. Cancer Epidemiol Biomarkers Prev 10:955–960 [PubMed] [Google Scholar]
- 12.Levy-Lahad E, Lahad A, Eisenberg S, Dagan E, Paperna T, Kasinetz L, Catane R, Kaufman B, Beller U, Renbaum P, et al (2001) A single nucleotide polymorphism in the RAD51 gene modifies cancer risk in BRCA2 but not BRCA1 carriers. Proc Natl Acad Sci USA 98:3232–3236 10.1073/pnas.051624098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kadouri L, Kote-Jarai Z, Hubert A, Durocher F, Abeliovich D, Glaser B, Hamburger T, Eeles RA, Peretz T (2004) A single-nucleotide polymorphism in the RAD51 gene modifies breast cancer risk in BRCA2 carriers, but not in BRCA1 carriers or noncarriers. Br J Cancer 90:2002–2005 10.1038/sj.bjc.6601837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jakubowska A, Narod SA, Goldgar DE, Mierzejewski M, Masojc B, Nej K, Huzarska J, Byrski T, Gorski B, Lubinski J (2003) Breast cancer risk reduction associated with the RAD51 polymorphism among carriers of the BRCA1 5382insC mutation in Poland. Cancer Epidemiol Biomarkers Prev 12:457–459 [PubMed] [Google Scholar]
- 15.Jakubowska A, Gronwald J, Menkiszak J, Gorski B, Huzarski T, Byrski T, Edler L, Lubinski J, Scott RJ, Hamann U (2007) The RAD51 135 G>C polymorphism modifies breast cancer and ovarian cancer risk in Polish BRCA1 mutation carriers. Cancer Epidemiol Biomarkers Prev 16:270–275 10.1158/1055-9965.EPI-06-0562 [DOI] [PubMed] [Google Scholar]
- 16.Chenevix-Trench G, Milne RL, Antoniou AC, Couch FJ, Easton DF, Goldgar DE (2007) An international initiative to identify genetic modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: the Consortium of Investigators of Modifiers of BRCA1 and BRCA2 (CIMBA). Breast Cancer Res 9:104 10.1186/bcr1670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldgar DE, Easton DF, Deffenbaugh AM, Monteiro AN, Tavtigian SV, Couch FJ (2004) Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet 75:535–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chenevix-Trench G, Healey S, Lakhani S, Waring P, Cummings M, Brinkworth R, Deffenbaugh AM, Burbidge LA, Pruss D, Judkins T, et al (2006) Genetic and histopathologic evaluation of BRCA1 and BRCA2 DNA sequence variants of unknown clinical significance. Cancer Res 66:2019–2027 10.1158/0008-5472.CAN-05-3546 [DOI] [PubMed] [Google Scholar]
- 19.Liu HX, Cartegni L, Zhang MQ, Krainer AR (2001) A mechanism for exon skipping caused by nonsense or missense mutations in BRCA1 and other genes. Nat Genet 27:55–58 [DOI] [PubMed] [Google Scholar]
- 20.Perrin-Vidoz L, Sinilnikova OM, Stoppa-Lyonnet D, Lenoir GM, Mazoyer S (2002) The nonsense-mediated mRNA decay pathway triggers degradation of most BRCA1 mRNAs bearing premature termination codons. Hum Mol Genet 11:2805–2814 10.1093/hmg/11.23.2805 [DOI] [PubMed] [Google Scholar]
- 21.Ware MD, DeSilva D, Sinilnikova OM, Stoppa-Lyonnet D, Tavtigian SV, Mazoyer S (2006) Does nonsense-mediated mRNA decay explain the ovarian cancer cluster region of the BRCA2 gene? Oncogene 25:323–328 [DOI] [PubMed] [Google Scholar]
- 22.Mikaelsdottir EK, Valgeirsdottir S, Eyfjord JE, Rafnar T (2004) The Icelandic founder mutation BRCA2 999del5: analysis of expression. Breast Cancer Res 6:R284-R290 10.1186/bcr785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buisson M, Anczukow O, Zetoune AB, Ware MD, Mazoyer S (2006) The 185delAG mutation (c.68_69delAG) in the BRCA1 gene triggers translation reinitiation at a downstream AUG codon. Hum Mutat 27:1024–1029 10.1002/humu.20384 [DOI] [PubMed] [Google Scholar]
- 24.Bonadona V, Dussart-Moser S, Voirin N, Sinilnikova OM, Mignotte H, Mathevet P, Bremond A, Treilleux I, Martin A, Romestaing P, et al (2007) Prognosis of early-onset breast cancer based on BRCA1/2 mutation status in a French population-based cohort and review. Breast Cancer Res Treat 101:233–245 10.1007/s10549-006-9288-7 [DOI] [PubMed] [Google Scholar]
- 25.Andrieu N, Goldgar D, Easton D, Rookus MA, Brohet R, Antoniou AC, Peock S, Evans DG, Eccles D, Douglas F, et al (2006) Pregnancies, breastfeeding and breast cancer risk in the International BRCA1/2 Carrier Cohort Study (IBCCS). J Natl Cancer Inst 98:535–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rebbeck TR, Lynch HT, Neuhausen SL, Narod SA, Van’t Veer L, Garber JE, Evans G, Isaacs C, Daly MB, Matloff E, et al (2002) Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med 346:1616–1622 10.1056/NEJMoa012158 [DOI] [PubMed] [Google Scholar]
- 27.Antoniou AC, Goldgar DE, Andrieu N, Chang-Claude J, Brohet R, Rookus MA, Easton DF (2005) A weighted cohort approach for analysing factors modifying disease risks in carriers of high-risk susceptibility genes. Genet Epidemiol 29:1–11 10.1002/gepi.20074 [DOI] [PubMed] [Google Scholar]
- 28.Kadouri L, Kote-Jarai Z, Easton DF, Hubert A, Hamoudi R, Glaser B, Abeliovich D, Peretz T, Eeles RA (2004) Polyglutamine repeat length in the AIB1 gene modifies breast cancer susceptibility in BRCA1 carriers. Int J Cancer 108:399–403 10.1002/ijc.11531 [DOI] [PubMed] [Google Scholar]
- 29.Antoniou AC, Pharoah PD, McMullan G, Day NE, Ponder BA, Easton D (2001) Evidence for further breast cancer susceptibility genes in addition to BRCA1 and BRCA2 in a population-based study. Genet Epidemiol 21:1–18 10.1002/gepi.1014 [DOI] [PubMed] [Google Scholar]
- 30.Lange K, Weeks D, Boehnke M (1988) Programs for pedigree analysis: MENDEL, FISHER, and dGENE. Genet Epidemiol 5:471–472 10.1002/gepi.1370050611 [DOI] [PubMed] [Google Scholar]
- 31.Anglian Breast Cancer Study Group (2000) Prevalence and penetrance of BRCA1 and BRCA2 mutations in a population-based series of breast cancer cases. Br J Cancer 83:1301–1308 10.1054/bjoc.2000.1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cox DR, Oakes D (1984) Analysis of survival data. Chapman and Hall, London [Google Scholar]
- 33.Lalloo F, Varley J, Ellis D, Moran A, O’Dair L, Pharoah P, Evans DG (2003) Prediction of pathogenic mutations in patients with early-onset breast cancer by family history. Lancet 361:1101–1102 10.1016/S0140-6736(03)12856-5 [DOI] [PubMed] [Google Scholar]
- 34.Peto J, Collins N, Barfoot R, Seal S, Warren W, Rahman N, Easton DF, Evans C, Deacon J, Stratton MR (1999) Prevalence of BRCA1 and BRCA2 gene mutations in patients with early-onset breast cancer. J Natl Cancer Inst 91:943–949 10.1093/jnci/91.11.943 [DOI] [PubMed] [Google Scholar]
- 35.Antoniou AC, Pharoah PP, Smith P, Easton DF (2004) The BOADICEA model of genetic susceptibility to breast and ovarian cancer. Br J Cancer 91:1580–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kadouri L, Easton DF, Edwards S, Hubert A, Kote-Jarai Z, Glaser B, Durocher F, Abeliovich D, Peretz T, Eeles RA (2001) CAG and GGC repeat polymorphisms in the androgen receptor gene and breast cancer susceptibility in BRCA1/2 carriers and non-carriers. Br J Cancer 85:36–40 10.1054/bjoc.2001.1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huber PJ (1967) The behavior of maximum likelihood estimates under non-standard conditions. Proc Fifth Berkley Symp Math Stat Probab 1:221–233 [Google Scholar]
- 38.Boos DD (1992) On generalised score tests. Am Stat 46:327–333 10.2307/2685328 [DOI] [Google Scholar]
- 39.Luu-The V, Paquet N, Calvo E, Cumps J (2005) Improved real-time RT-PCR method for high-throughput measurements using second derivative calculation and double correction. Biotechniques 38:287–293 [DOI] [PubMed] [Google Scholar]
- 40.Kuschel B, Auranen A, McBride S, Novik KL, Antoniou A, Lipscombe JM, Day NE, Easton DF, Ponder BA, Pharoah PD, et al (2002) Variants in DNA double-strand break repair genes and breast cancer susceptibility. Hum Mol Genet 11:1399–1407 10.1093/hmg/11.12.1399 [DOI] [PubMed] [Google Scholar]
- 41.Webb PM, Hopper JL, Newman B, Chen X, Kelemen L, Giles GG, Southey MC, Chenevix-Trench G, Spurdle AB (2005) Double-strand break repair gene polymorphisms and risk of breast or ovarian cancer. Cancer Epidemiol Biomarkers Prev 14:319–323 10.1158/1055-9965.EPI-04-0335 [DOI] [PubMed] [Google Scholar]
- 42.Kramer JL, Velazquez IA, Chen BE, Rosenberg PS, Struewing JP, Greene MH (2005) Prophylactic oophorectomy reduces breast cancer penetrance during prospective, long-term follow-up of BRCA1 mutation carriers. J Clin Oncol 23:8629–8635 10.1200/JCO.2005.02.9199 [DOI] [PubMed] [Google Scholar]
- 43.Obuse C, Iwasaki O, Kiyomitsu T, Goshima G, Toyoda Y, Yanagida M (2004) A conserved Mis12 centromere complex is linked to heterochromatic HP1 and outer kinetochore protein Zwint-1. Nat Cell Biol 6:1135–1141 10.1038/ncb1187 [DOI] [PubMed] [Google Scholar]
- 44.Kuefer MU, Chinwalla V, Zeleznik L, Behm FG, Naeve CW, Rakestraw KM, Mukatira ST, Raimondi SC, Morris SW (2003) Characterization of the MLL partner gene AF15q14 involved in t(11;15)(q23;q14). Oncogene 22:1418–1424 10.1038/sj.onc.1206272 [DOI] [PubMed] [Google Scholar]
- 45.Hayette S, Tigaud I, Vanier A, Martel S, Corbo L, Charrin C, Beillard E, Deleage G, Magaud JP, Rimokh R (2000) AF15q14, a novel partner gene fused to the MLL gene in an acute myeloid leukaemia with a t(11;15)(q23;q14). Oncogene 19:4446–4450 10.1038/sj.onc.1203789 [DOI] [PubMed] [Google Scholar]
- 46.Chinwalla V, Chien A, Odero M, Neilly MB, Zeleznik L, Rowley JD (2003) A t(11;15) fuses MLL to two different genes, AF15q14 and a novel gene MPFYVE on chromosome 15. Oncogene 22:1400–1410 10.1038/sj.onc.1206273 [DOI] [PubMed] [Google Scholar]
- 47.Shivji MK, Davies OR, Savill JM, Bates DL, Pellegrini L, Venkitaraman AR (2006) A region of human BRCA2 containing multiple BRC repeats promotes RAD51-mediated strand exchange. Nucleic Acids Res 34:4000–4011 10.1093/nar/gkl505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hasselbach L, Haase S, Fischer D, Kolberg HC, Sturzbecher HW (2005) Characterisation of the promoter region of the human DNA-repair gene Rad51. Eur J Gynaecol Oncol 26:589–598 [PubMed] [Google Scholar]
- 49.Zuker M (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415 10.1093/nar/gkg595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Davuluri RV, Suzuki Y, Sugano S, Zhang MQ (2000) CART classification of human 5′ UTR sequences. Genome Res 10:1807–1816 10.1101/gr.GR-1460R [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hughes TA (2006) Regulation of gene expression by alternative untranslated regions. Trends Genet 22:119–122 10.1016/j.tig.2006.01.001 [DOI] [PubMed] [Google Scholar]