

Abstract
Labetalol, a combined α1, β1, and β2 adrenoceptor-blocking drug, has been shown to have analgesic properties in vivo. To determine the underlying mechanisms, we examined its effects on GABAA receptor-mediated spontaneous inhibitory postsynaptic currents (sIPSCs) and spontaneous firings of rat ventrolateral periaqueductal gray (PAG) neurons, either mechanically dissociated, or in acute brain slices. These PAG neurons mediate opioid-mediated analgesia and pain transmission and are under tonic control of GABAergic interneurons. An increase in GABAergic transmission to these neurons yields an inhibitory hyperpolarized state and may interrupt pain signal transmission. Using patch clamp techniques, we found that labetalol reversibly increases the frequency of sIPSCs without changing their mean amplitude. This indicates that labetalol enhances GABAergic synaptic transmission by a presynaptic mechanism. Metoprolol, a specific β1-adrenoceptor antagonist, also reversibly enhanced sIPSC frequency. In the presence of metoprolol, labetalol-induced increase in sIPSC frequency was significantly attenuated or even abolished. These results suggest that labetalol shares the same pathway as metoprolol in enhancing GABAergic transmission via an inhibition of presynaptic β1-adrenoceptors. We further showed that labetalol reversibly reduced the firing rate of PAG neurons. This reduction was significantly attenuated in the presence of bicuculline, a selective antagonist of GABAA receptors. These data indicate that labetalol-induced inhibition of PAG cell firing is attributable to its potentiation of GABAergic transmission. Based on these data, we postulate that labetalol-induced analgesia is at least in part ascribed to its antagonistic effects on presynaptic β1- adrenoceptors.
1. Introduction
Labetalol is a potent antihypertensive agent. It exhibits selective α1- and nonselective β1- and β2-adrenergic antagonist effects (MacCarthy and Bloomfield, 1983). In an in vivo animal study (Khanna et al., 1978), using hot plate tests, it was found that intraperitoneal administration of labetalol enhanced the analgesic effects of morphine. These results imply the involvement of superaspinal mechanisms (Franklin and Abbott, 1989). Moreover, labetalol has been used clinically to relieve pathological pain in humans (Margaria et al., 1983). Although decades have passed, the mechanisms underlying labetalol-induced analgesia remain poorly understood.
The midbrain periaqueductal gray (PAG) is a major site of opioid and analgesic actions in the central nervous system (Vaughan et al., 1996). It has been proposed that opioids produce antinociception in the PAG by directly inhibiting tonically active GABAergic interneurons, thereby disinhibiting the ventrolateral PAG output neurons which project to the rostral ventromedial medulla (RVM). Microinjection of morphine in the PAG increases activity of RVM Off-cells and decreases that of RVM On-cells. The activity of the former one increases, whereas that of the later stops, just prior to the initiation of nociceptive response (Heinricher et al., 1987).
Norepinephrine is a neurotransmitter known to be involved in the ascending and descending pain transmission pathway (Cousins and Mather, 1984). Several lines of evidence point to the possibility that norepinephrine functions within the PAG 1) Both catecholamine-synthesizing enzymes: tyrosine hydroxylase (Pearson et al., 1983) and the norepinerphrine-synthesizing phenylethanolamine N-methyltransferase (Kopp et al., 1979) are expressed in the PAG. 2) Release-based studies have shown high concentrations of norepinephrine in the PAG (Behbehani, 1995). 3) Epinephrine causes prolonged changes in the basal firing rate of PAG cells (Jiang et al., 1992). 4) The existence of subtypes of adrenoceptors, including the α1- (Mitchell et al., 2003), α2- (Mitchell et al., 2003; Peng et al., 1996; Vaughan et al., 1996) and β-adrenoceptors (Behbehani, 1995) has been found in the PAG.
As a major inhibitory neurotransmitter in the central nervous system, γ-aminobutyric acid (GABA) regulates the excitability of neurons, including those involved in the relay of pain signals (Jasmin et al., 2004). Modulation of GABAergic synaptic transmission by norepinephrine was found in various preparations. These include: the anterior cerebral cortex, the thalamus and the hypothalamus (Kamisaki et al., 1992; Chong et al., 2004), the cerebellum (Cheun and Yeh, 1996; Saitow et al., 2000), the spinal cord (Baba et al., 2000), the substantia nigra (Cathala et al., 2002), the mesencephalic red nucleus (Ciranna et al., 2004), and the bed nucleus of the stria terminals (Dumont and Williams, 2004). However, it is unknown whether norepinephrine is also involved in the modulation of GABAergic synaptic transmission in the PAG.
In the present study, we hypothesized that labetalol disrupts adrenergic modulation of GABAergic function and thus increases inhibitory input to ventrolateral PAG neurons. Consequently, this would depress the activity of PAG cells and pain transmission.
2. Results
2.1. Spontaneous GABAergic IPSCs (sIPSCs) recorded in mechanically dissociated PAG neurons
In the presence of 50 μM 5-amino-phosphonovalerate (APV), a competitive antagonist to glutamate N-methyl D-aspartate (NMDA) receptors and 20 μM 6,7-dinitroquinoxaline-2,3-dione (DNQX), an antagonist to α-amino-3-hydroxy-5-methylisoxazole-4- propionic acid (AMPA) receptors, high rates of spontaneous postsynaptic current (sPSCs) were recorded at the holding potential (VH) of -50 mV, in the majority of the neurons mechanically dissociated from the ventrolateral region of PAG area (Fig. 1). Most of these sPSCs were stable for more than 40 min. Application of 10 μM bicuculline (BIC), a selective GABAA receptor antagonist, completely and reversibly eliminated these sPSCs, indicating that they are GABAAR-mediated spontaneous inhibitory postsynaptic currents (sIPSCs) (Fig. 2A, B).
Fig. 1.

Photomicrograph of rat PAG neurons. Photos in the left column were obtained by a Nikon E600FN upright microscope with the aid of near-infrared CCD camera. A, PAG within a rat midbrain slice (350 μm thick) observed at low power objective (4X). The Arrow indicates aqueduct and the circles indicate the ventrolateral region. B, PAG neurons in the ventrolateral region were obtained under differential interference contrast illumination (40 X, water immersion objective). In the right column, photos of a fusiform (C) and a pyramidal (D) PAG neuron mechanically dissociated from the midbrain slice were obtained by a Leica DMIRB invert microscope (40X objective). The much-reduced dendritic arbors of such neurons facilitate the space clamp. These nerve-bouton preparations often preserve some functional synaptic boutons.
Fig.2.
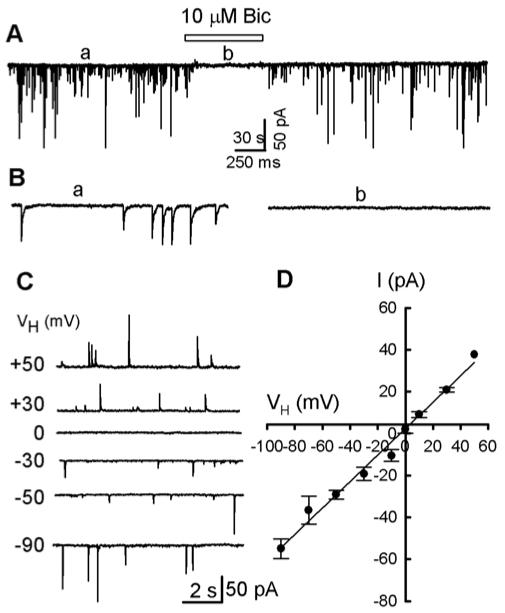
GABAA receptor mediated spontaneous IPSCs in mechanically dissociated PAG cells. A, 10 μM bicuculline (BIC) completely and reversibly blocked all postsynaptic currents (PSCs) recorded at a holding potential (VH) of -50 mV, using a conventional whole cell technique. B, Parts of trace A on an expanded time scale. C, 10 s traces of sPSCs were recorded from a mechanically dissociated PAG cell at various VH levels. D, I-V plot of mean (± SEM) data from 4 neurons and a successfully fitted line (the intercept at abscissa is 2.3 mV).
To determine the reversal potential of the sIPSCs, we measured their mean of peak amplitude (of 30 - 60 s) at various VH levels from -90 to + 50 mV with an increment of 20 mV. In Fig. 2C, typical traces (10 s) are shown at various VH levels. Fig. 2D summarizes the current-voltage relations of the peak amplitude of sIPSCs for four neurons. The reversal potential of sIPSCs was 3.6 mV. This is very close to the theoretical Cl- equilibrium potential (ECl), -1.2 mV, calculated by the Nernst equation, for extracellular and intracellular (the pipette solution) Cl- concentrations of 151 and 145 mM, respectively. These results confirm that sIPSCs, recorded in this preparation, were mediated by channels selectively permeable for Cl-.
2.2. Effects of labetalol on sIPSC frequency of PAG neurons
The ventrolateral PAG output neurons are known to be under the tonic control of GABAergic inhibitory synapses (Behbehani, 1995). To determine whether labetalol modulates GABAergic synaptic transmission of the PAG, we examined the effects of labetalol on sIPSCs of mechanically dissociated PAG neurons. The application of 1 nM labetalol (LAB) profoundly enhanced sIPSC frequency (Fig. 3A, C; by 41 ± 8%; from 1.42 ± 0.42 Hz in control to 1.88 ± 0.48 Hz in labetalol (n = 9 neurons, p < 0.001)). The increase in sIPSC frequency is further illustrated in Fig. 3B, on the left side, by the leftward shift of cumulative plots of the intervals between successive sIPSCs (interevent interval). The corresponding plot, in Fig. 3B, on the right hand side, illustrates the lack of change in their amplitude (14 ± 8%; p = 0.89; n = 9).
Fig. 3.
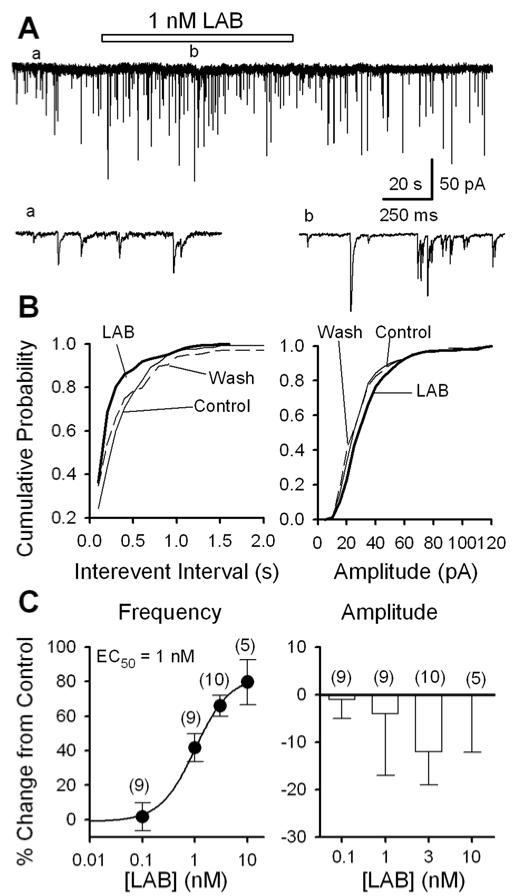
Labetalol raises the frequency of sIPSCs of PAG cells: data from mechanically dissociated neurons. A, GABAergic sIPSCs recorded before (a), during (b) and after the application of 1 nM labetalol (LAB) in a PAG cell; accelerated trace in (a) and (b) are shown. B, For the same data as A, cumulative probability plots of sIPSC interevent intervals (left: K - S test, p = 0.008, labetalol vs. control) and amplitudes (right: K - S test, p = 0.89, labetalol vs. control). C, Concentration-dependence of labetalol induced changes of the frequency (left panel, n = 5 - 10; with an EC50 of 1 nM and maximal effect of 83%) and amplitude (right panel, n = 5 - 10, p > 0.2) of sIPSCs. For all figures, the numbers in brackets are the number of neurons examined.
Fig. 3C illustrates the concentration-dependence of labetalol-induced changes of sIPSC frequency (left panel) and amplitude (right panel). The dose-response curve of labetalol-induced facilitation of sIPSC frequency was successfully fitted to a Logistic equation as defined in the Methods section, and an EC50 of 1 nM was obtained. At a concentration of 10 nM, labetalol increased the sIPSC frequency by 83%. In contrast, labetalol (0.1 - 10 nM) did not significantly alter the mean sIPSC amplitude (Fig. 3C, right panel).
These data also suggest that there is some free catecholamine transmitter in this preparation. This possibility is further supported by the effect of prazosin (see below Fig. 4). Future study identifying the source of this catecholamine is warranted.
Fig. 4.
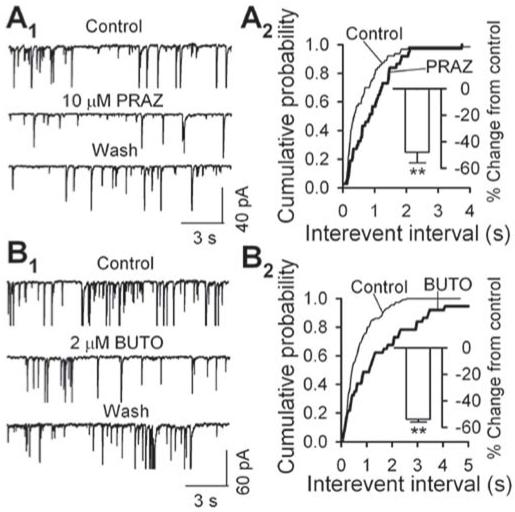
Adrenoceptor antagonists reduce sIPSC frequency: data from mechanically dissociated neurons. A1 - B1, sample traces of sIPSCs, showing that sIPSC frequency decreased in the presence of prazosin (PRAZ, 10 μM, A1), an antagonist of α1-adrenoceptor or butoxamine (BUTO, 2 μM, B1), an antagonist of β2- adrenoceptor. A2 - B2, cumulative probability plots of interevent intervals between successive sIPSCs before and during the application of PRAZ (A2) or BUTO (B2). The insets summarized PRAZ (A2) or BUTO (B2) induced inhibition of sIPSC frequency from 5 neurons. ** p < 0.01, A2, control vs. PRAZ, B2, control vs. BUTO.
2.3. Antagonist to β1-, but not α1- and β2- adrenoceptor, mimics the effects of labetalol on sIPSC frequency
Since there is tonic adrenergic innervation in our preparation, and labetalol is a combined α1-, β1-, and β2-adrenoceptor-blocking drug, in the following experiments we attempted to determine which subtype(s) of adrenoceptors is responsible for labetalol-induced facilitation of sIPSCs in mechanically dissociated PAG neurons.
Prazosin (PRAZ, 10 μM, Fig. 4A1), an antagonist to α1-adrenoceptor, and butoxamine (BUTO, 2 μM, Fig. 4B1), a β2-adrenoceptor, both prominently decreased sIPSC frequency. It was further illustrated in Fig. 4 A2 and B2 that both of them induced significant rightward shift of cumulative probability plots of interevent intervals. On average, PRAZ and BUTO respectively suppressed sIPSC frequency by 48 ± 8% (from 1.24 ± 0.23 Hz in control to 0.70 ± 0.21 Hz in PRAZ, n = 5, p = 0.002, inset in Fig. 4A2) and 54 ± 2% (from 0.93 ± 0.14 Hz in control to 0.43 ± 0.08 Hz in BUTO, n = 5, p = 0.001, inset in Fig. 4B2).
Metoprolol (MET, 100 nM), a β1-adrenoceptor antagonist, reversibly raised the frequency of sIPSCs (Fig. 5A, B). This effect was further illustrated in Fig. 5C, in which 100 nM metoprolol induced a significant leftward shift of the cumulative probability plot of interevent intervals (K - S test, p < 0.001). On average, 100 nM metoprolol enhanced sIPSC frequency by 42 ± 6% (from 1.61 ± 0.39 Hz in control to 2.24 ± 0.56 Hz in metoprolol, n = 9, p = 0.002, inset in Fig. 5C), however, it did not change sIPSC amplitude (103 ± 4% of control, from 17.3 ± 2.1 in control to 17.7 ± 2.3 in metoprolol, n = 9, p = 0.28, data not illustrated).
Fig. 5.
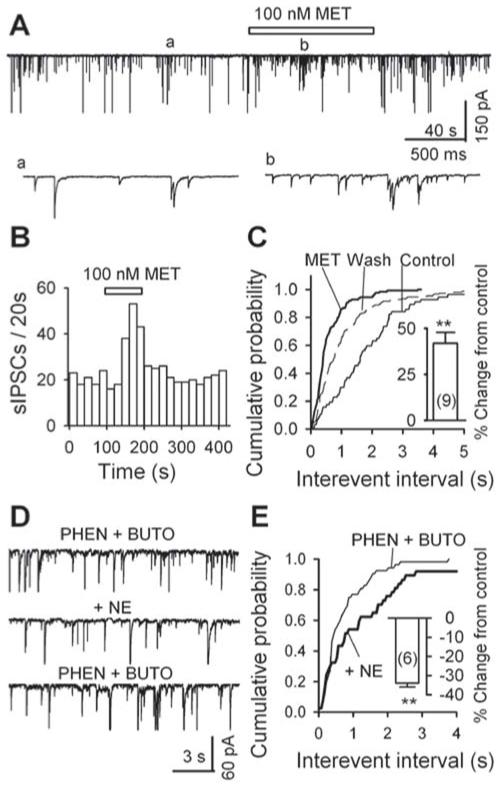
Metoprolol (MET) raises the frequency of sIPSCs: data from mechanically dissociated neurons. A, GABAergic sIPSCs recorded in a PAG cell before (a), during (b) and after the application of 100 nM MET; accelerated trace in (a) and (b). B, For the same data as in A, the time course of the enhancement of sIPSC frequency by 100 nM MET. C, Cumulative probability plots of interevent interval (left: K - S test, p < 0.001, metoprolol vs. control) and amplitude (right inset, K - S test, p = 0.037, metoprolol vs. control) of sIPSCs. Metoprolol (100 nM) significantly increased the frequency of sIPSCs (left inset: n = 9, p = 0.003). D, Norepinephrine (NE, 10 μM) decreased sIPSC frequency in the presence of phentolamine (PHEN, 10 μM), an α-adrenoceptor antagonist, and butoxamine (BUTO, 2 μM), a β2- adrenoceptor antagonist. E, cumulative probability plots of interevent interval of sIPSCs before (PHEN + BUTO) and during (+ NE) the application of 10 μM NE. Inset, the summary from 6 neurons, of the effect of norepinephrine on sIPSC frequency in the presence of phentolamine PHEN and butoxamine. ** p < 0.01, PHEN + BUTO vs. PHEN + BUTO + NE.
Metoprolol-induced enhancement of sIPSC frequency suggested that the activation of β1-adrenoceptor would decrease sIPSC frequency. To test this possibility, and due to the lack of specific β1-adrenoceptor agonist, we examined the effect of norepinephrine (NE), an agonist of α- and β-adrenoceptors, on sIPSC frequency in the presence of phentolamine (PHEN), an α-adrenoceptor antagonist, and butoxamine (BUTO), a β2-adrenoceptor antagonist. The application of 10 μM phentolamine and 2 μM butoxamine decreased sIPSC frequency by 33 ± 5% (from 1.25 ± 0.30 Hz in control to 0.96 ± 0.30 Hz in PHEN + BUTO, n = 6, p = 0.005, data not illustrated). At the newly established baseline, 10 μM norepinephrine further decreased sIPSC frequency (Fig. 5D) and induced a rightward shift of cumulative probability plot of interevent intervals (K-S test: p < 0.01, Fig. 5E). On average, 10 μM norepinephrine induced a decrease of 34 ± 2% in sIPSC frequency (0.66 ± 0.2 Hz in PHEN + BUTO + NE vs. 0.96 ± 0.30 Hz in PHEN + BUTO, n = 6, p = 0.0005, inset in Fig. 5E). This result supports our hypothesis that activation of β1-adrenoceptor decreases sIPSC frequency.
2.4. Labetalol enhances sIPSC frequency via an inhibition of β1- adrenoceptors
The above experiments imply that among the targeted adrenoceptors of labetalol, β1- adrenoceptor is most likely responsible for labetalol-induced facilitation of sIPSC frequency. To further test this possibility, we examined the effects of labetalol on sIPSCs recorded from mechanically dissociated neurons in the absence or the presence of metoprolol. The rationale for this series of experiments is that if labetalol and metoprolol acting on the same pathway in facilitating sIPSCs, they will compete with each other when they are applied together.
At a submaximal concentration (3 nM), labetalol prominently and reversibly increased sIPSC frequency (Fig. 6A-B) by 69 ± 17% (from 1.61 ± 0.74 Hz in control, to 2.74 ± 0.96 Hz in labetalol, n = 5 neurons, p < 0.001, Fig. 6D). Furthermore, labetalol induced a significant leftward shift of the cumulative probability plot of interevent intervals (K - S test, p = 0.005, Fig. 6C, left panel). Repeated application of labetalol increased sIPSC frequencies to a similar extent (data not shown). As expected, metoprolol (100 nM MET) also increased sIPSC frequency (Fig. 6A). After the establishment of a stable baseline in the presence of 100 nM metoprolol, 3 nM labetalol increased sIPSC frequency (Fig. 6A-B) by 18 ± 2% (from 1.83 ± 0.89 Hz in metoprolol to 2.18 ± 0.98 Hz in LAB + MET, n = 5 neurons, p < 0.01, Fig. 6D) and induced a slight leftward shift of cumulative probability plot of interevent intervals (Fig. 6C, right panel). This effect (18 ± 2%) is significantly weaker than 69 ± 17% by labetalol alone (Fig. 6D, n = 5 neurons, p = 0.02).
Fig. 6.
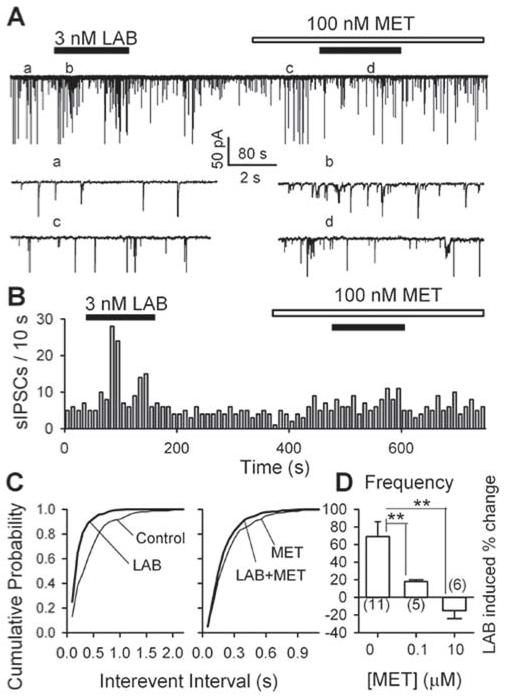
β1- adrenoceptors mediate labetalol-induced enhancement of sIPSC frequency: data from mechanically dissociated PAG neurons. A, GABAergic sIPSCs recorded before, during and after the application of 3 nM labetalol in the absence and presence of 100 nM metoprolol (MET); some accelerated traces in different conditions are shown. B, For the same data, the time course of 3 nM labetalol induced changes of sIPSC frequency in the absence and presence of 100 nM metoprolol. C, Cumulative probability plots of interevent intervals before and during the application of labetalol at the baseline of control (left panel: K - S test, p = 0.005, labetalol vs. control) and the new baseline established in the presence of 100 nM metoprolol (right panel, K - S test, p = 0.62, labetalol + metoprolol vs. metoprolol); D, 3 nM labetalol induced enhancement of sIPSC frequency was significantly diminished in the presence of 0.1 and 10 μM metoprolol. ** p < 0.01, compared with that in the absence of metoprolol (MET, left column, 0 μM).
In the other 6 experiments, 3 nM labetalol reversibly increased sIPSC frequency (by 52 ± 10%; from 0.82 ± 0.25 Hz in control to 1.13 ± 0.30Hz in labetalol, n = 6 neurons, p < 0.001, data not illustrated). 10 μM metoprolol significantly increased sIPSC frequency (by 91 ± 19%, from 0.61 ± 0.19 Hz to 1.23 ± 0.37 Hz, n = 6 neurons, p <0.001, data not illustrated). After a stable baseline was established in the presence of 10 μM metoprolol, the application of 3 nM labetalol did not induce a significant change in sIPSC frequency (a reduction of 15 ± 9%, from 1.23 ± 0.37 Hz in metoprolol to 1.02 ± 0.41 Hz in LAB + MET, n = 6 neurons, p > 0.05, Fig. 6D). This effect differs significantly from that in the absence of metoprolol (52 ± 10%, p < 0.05, n = 6 neurons; Fig. 6D).
These results suggest that labetalol and metoprolol compete for the same receptors. These results also suggest that β1- adrenoceptors mediate labetalol-induced facilitation of GABAergic transmission in the PAG cells.
2.5. Labetalol inhibits the activity of PAG neurons in midbrain slices
Next, we examined the effects of labetalol on the activity of neurons in ventrolateral PAG in midbrain slices. Spontaneous action potential firing was recorded from current- clamped neurons (at 0 pA). As illustrated in Fig. 7A, 10 nM labetalol prominently and reversibly inhibited the spontaneous action potential firings. Spontaneous firing was noted to vary prior to labetalol administration. Therefore, labetalol-induced changes in firing rates are usually expressed as percentage change from control.
Fig. 7.

Labetalol inhibited the activity of PAG cells in brain slices. The spontaneous firing was recorded from PAG cells in brain slices under current clamp conditions. A, left panel showed the time course of 10 nM labetalol induced inhibition of spontaneous firing of a PAG neuron. Sample traces of spontaneous firing recorded before (a) and during (b) the application of 10 nM labetalol are shown in the right. Dotted lines indicate the resting membrane potentials. B, Concentration-dependence of LAB-induced changes in spontaneous firing rate (left panel) and in membrane potentials (right panel). ** p < 0.01. labetalol vs. control. C, the time course of 10 nM labetalol induced-inhibition of spontaneous action potential firing recorded from a PAG neuron, in the absence (a, b, c) and the presence (d, e) of 10 μM bicuculline (BIC). D. 10 s episodes in the absence (c) and presence of 10 μM bicuculline (BIC) before (d) and during (e) the application of 10 nM labetalol are shown. E. Summary of 1 and 10 nM labetalol induced reduction of spontaneous firing rate in the absence (Control, white bar) and the presence of 10 μM bicuculline (BIC, gray bar). ** p < 0.01, bicuculline vs. control.
In 5 experiments, 10 nM labetalol inhibited the spontaneous firing rate by 63 ± 4% (from 3.07 ± 0.62 Hz in control to 1.19 ± 0.31 Hz in labetalol, p < 0.001, Fig. 7B, left panel). The inhibition depended on the concentration of labetalol (Fig. 7B, left panel). 1 nM and 100 nM labetalol respectively inhibited the spontaneous firing rate by 36 ± 5% (from 3.64 ± 0.91 Hz in control to 2.51 ± 0.93 in 1 nM labetalol, n = 5, p = 0.01) and 56 ± 7% (from 4.93 ± 0.38 Hz in control to 2.19 ± 0.33 Hz in 100 nM labetalol, n = 4, p < 0.001).
The above experiments demonstrate that labetalol enhanced GABAergic sIPSCs (Fig. 3) and inhibited PAG neuron firing (Fig. 7A). To determine whether there is a correlation between these two effects of labetalol, we compared the action of labetalol on the firing in the absence and the presence of bicuculline (BIC), a specific GABAA receptor antagonist. As illustrated in Fig. 7C and D, 10 μM bicuculline alone prominently facilitated the firing of the PAG neurons. This result indicates that these cells were under the tonic inhibition of GABAergic transmission in our experimental conditions. On average, 10 μM bicuculline increased the firing rate by 60 ± 19% (from 1.81 ± 0.22 Hz in control to 2.78 ± 0.34 Hz in BIC, n = 6, p = 0.003). After a stable baseline was established, the application of 1 and 10 nM labetalol respectively inhibited the firing rate by 16 ± 4% (from 2.58 ± 0.31 Hz in BIC to 2.12 ± 0.13 Hz in 1 nM labetalol + BIC, n = 5, p = 0.001) and 30 ± 6% (from 3.34 ± 0.47 Hz in BIC, 2.43 ± 0.43 Hz in 10 nM labetalol + BIC, n = 5, p = 0.000) (Fig. 7E). These are significantly less than the changes in the absence of BIC (16 ± 4% in BIC vs. 36 ± 5% without BIC, n = 5, p < 0.001; 30 ± 6% in BIC, vs. 63 ± 4% without BIC, n = 5, p < 0.001, Fig. 7E). These results suggest that the enhancement of sIPSCs at least partially contributes to labetalol-induced inhibition of the activity of PAG neurons.
Moreover, as illustrated in Fig. 8B (right panel), 1, 10 and 100 nM LAB significantly hyperpolarized the membrane: by 1.9 ± 0.5 mV (from 46.9 ± 0.7 mV in control to 48.8 ± 1.0 mV in 1 nM LAB, n = 5, p = 0.01), by 4.4 ± 0.9 mV (from 49.1 ± 0.6 mV in control to 53.5 ± 1.1 mV in 10 nM LAB, n = 5, p = 0.004), and by 3.4 ± 0.2 mV (from 47.9 ± 1.7 mV in control to 51.2 ± 1.7 mV in 100 nM LAB, n = 5, p < 0.001), respectively.
3. Discussion
In the present study, using patch clamp recording from PAG neurons either isolated mechanically or in brain slices, we demonstrated that labetalol enhanced GABA release, via inhibition of β1-adrenoceptors. We then showed that labetalol reduced the spontaneous firings of PAG neurons, which may be attributable to its enhancement of GABA release. As PAG neurons play a critical role in pain transmission, our data might provide an explanation, for the cellular and molecular mechanisms, underlying labetalol-induced analgesic effects observed in vivo.
3.1. Several subtypes of adrenoceptors modulate GABA release onto PAG neurons
As described in the beginning of this report, adrenergic modulation of GABA release has been found in several brain regions (Kamisaki et al., 1992; Cathala et al., 2002; Ciranna et al., 2004; Dumont and Williams, 2004). Although the existence of α1-, α2-, and β-adrenoceptors in the PAG has been found in several laboratories (Behbehani, 1995; Peng et al., 1996; Vaughan et al., 1996; Mitchell et al., 2003), their roles in GABA release have not been reported. In the present study, we showed that an antagonist of the β1-adrenoceptor (metoprolol) enhanced, while those of α1- (prazosin) and β2- (butoxamine) adrenoceptor decreased GABAergic sIPSC frequency. The spontaneous postsynaptic currents are events that represent the release of presynaptic vesicles. A change in the frequency usually represents a presynaptic action (Li et al., 1998). Our results suggest that α1-, β1-, and β2-adrenoceptors exist on GABAergic terminals which made synapses onto the PAG cells, and that activation of these presynaptic α1- and β2-adrenoceptors increases GABA release onto PAG neurons. This is consistent with previous reports on other brain regions (Baba et al., 2000; Chong et al., 2004; Dumont and Williams, 2004). However, our data indicate that activation of β1-adrenoceptor suppressed GABAergic synaptic transmission to PAG neurons. This was further confirmed by norepinephrine-induced inhibition of sIPSC frequency in the presence of α- and β2-adrenoceptor antagonists.
3.2. Labetalol enhances GABAergic synaptic transmission by a presynaptic mechanism
In the present study, on mechanically dissociated PAG neurons, labetalol enhanced sIPSC frequency without changing their mean amplitude. As stated above, when a modulator produces a change in the frequency, but not the amplitude of sIPSCs, its action is at the presynaptic site (Li et al., 1998). Our results suggest that labetalol enhanced sIPSCs by a presynaptic mechanism, increasing GABA release.
3.3. Selective β1- adrenoceptor blockade mimicked labetalol - induced facilitation of GABA release
Labetalol has antagonistic effects on α1-, β1- and β2-adrenoceptors. We observed that a β1-adrenoceptor antagonist (metoprolol) facilitated sIPSC frequency, while the antagonists of α1- and β2-adrenoceptors decreased sIPSC frequency. Furthermore, 100 nM metoprolol diminished, and 10 μM metoprolol abolished, labetalol-induced enhancement of GABA release. These results indicated that labetalol shares the same pathway as metoprolol in enhancing GABA release via blockade of β1-adrenoceptors. Our observation is also consistent with previous studies that showed that the potency of labetalol for β-adrenergic blockade is fivefold to tenfold of that for α-adrenergic blockade (see Blakeley and Summers, 1977; Drew et al., 1978; Gold et al., 1982).
3.4. Labetalol - induced enhancement of GABAergic inhibition of PAG neurons might contribute to its analgesic effect
We showed that PAG neurons were under the tonic control of GABAergic neurons, as evidenced by the spontaneous GABAergic IPSCs recorded from the PAG neurons. Furthermore, application of bicuculline, a GABAA receptor antagonist, eliminated all sIPSCs and markedly enhanced the frequency of spontaneous firings of PAG neurons. We additionally showed that labetalol alone inhibited spontaneous firing in PAG neurons. Interestingly, the application of bicuculline did significantly attenuate (by > 50%) labetalol-induced inhibition of the firing of PAG neuron. This suggests that the enhancement of GABA release, at least partially, contributes to labetalol-induced inhibition of cell firing, although there could be some other underlying mechanisms, which warrant further investigations.
Noradrenergic receptors modulate the physiological response to a painful stimulation. Previous studies have focused more on α-adrenoceptors than on β-adrenoceptors. The clinical influence, of β-blockers upon anesthesia and postoperative pain, has been examined in few previous studies. Stanley et al. (1982) showed a reduction, in sufentanil requirements for unconsciousness, in patients receiving chronic propranolol treatment before coronary-artery bypass surgery. The use of β-antagonists has been found to reduce anesthetic requirements during anesthesia (Johansen et al., 1997), to reduce inhalation anesthetic MAC (minimal alveoli concentration), and to improve postoperative recovery (Zaugg et al., 2002). In a recent clinical study of patients undergoing hysterectomy, Chia and colleagues (2004) found that the use of the perioperative β- antagonist, esmolol, administration during anesthesia, reduced the intraoperative use of inhalation anesthetics and fentanyl. Esmolol use also decreased sympathetically mediated hemodynamic responses. Furthermore, it reduced morphine consumption for the first 3 postoperative days.
It has been suggested that perioperative β- antagonist administration is an alternative to remifentanil, a short-acting μ-opioid receptor agonist, in maintaining stable intraoperative haemodynamics (Coloma et al., 2001). However, the specific mechanism, by which β-adrenoceptor blockers potentiate the analgesic effects of an opioid, or an inhalation anesthetic, remains controversial (Chia et al., 2004). Our data may provide an interpretation for these observations that both opioid agents and β-adrenoceptor blockers inhibit PAG neuron activity and interrupt pain transmission.
It is important to emphasize the limitations to the approach we have taken. First, we only used norepinephrine in the presence of α- and β2-receptor blockade to enhance IPSC frequency as evidence of β1 receptor involvement. Future study using a more specific β1 agonist to verify the current result is warranted. However, as metoprolol mimics the effect of labetalol on sIPSCs, this result enables us to demonstrate the involvement of β1 receptors in labetalol-induced facilitation of GABAergic transmission. Second, the principle cellular action of β agonists acting through guanine nucleotide binding (G) proteins Gs is to stimulate cAMP formation. Increased cAMP formation in the PAG and elsewhere profoundly (many-fold) enhances probability of GABA release events. The opposite was found in the current study. Future study is necessary to investigate the biochemical mechanism of enhanced GABA release by labetalol, to determine whether labetalol affects the protein kinase A pathways, etc.
In conclusion, we have demonstrated that functional α1-, β1- and β2- adrenoceptors exist on GABA-releasing terminals which make synapses onto PAG neurons. These presynaptic adrenoceptors are tonically activated. It is suggested that labetalol inhibits presynaptic β1-adrenoceptors, leading to an increased release of GABA and subsequent an inhibition of PAG neuronal activity. This may contribute to the analgesic effects of labetalol observed in vivo. Future studies are warranted to determine whether other β1-adrenoceptor antagonists also have an effect similar to that of labetalol shown in current investigation.
4. Experimental procedure
4.1. Slice preparation and mechanical dissociation
The care and use of animals, and the experimental protocol, were approved by the Institutional Animal Care and Use Committee of the University of Medicine and Dentistry of New Jersey. The experiments were done on PAG neurons from 2- to 4-wk old Sprague Dawley rats. The midbrain slices were prepared as described previously (Ye et al., 2006). In brief, rats were decapitated, and the brain was quickly excised and coronally sliced (300 μm) with a VF-200 Slicer (Precisionary Instruments, Greenville, NC).
This was done in ice-cold glycerol-based artificial cerebrospinal fluid (GACSF) - containing (in mM) 250 glycerol, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 25 NaHCO3, and 11 glucose, and saturated with 95%O2/5%CO2 (carbogen) (Ye et al., 2006). Midbrain slices were then kept in carbogen-saturated regular ACSF, in which the glycerol was replaced by 125 mM NaCl, at room temperature (22 - 24 °C) for at least 1 hr before use.
PAG neurons with functional terminals attached were isolated from the ventrolateral region of PAG area of midbrain slices using an enzyme-free mechanical dissociation procedure, as previously described (Akaike and Moorhouse, 2003; Ye et al., 2004). Briefly, midbrain slices were kept in carbogen-saturated ACSF at room temperature (22-24 °C) for at least one hour before mechanical dissociation. The slice was then transferred to a 35 mm culture dish (Falcon, Rutherford, NJ) filled with a standard external solution containing (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 Glucose (320 mOsm, pH set to 7.3 with Tris base). Under an inverted microscope (Nikon, Tokyo, Japan), PAG was identified. A heavily fire-polished glass micropipette was placed on the surface of the ventrolateral region of the PAG. This tip of the pipette was vibrated horizontally, at ∼20 Hz for 2 - 5 min by a homemade device. The slice was then removed. The liberated neurons were left to settle onto the base of the dish within 20 min. The fusiform or pyramidal neurons (Fig. 1C, D), with diameters greater than 15 μm were selected for electrophysiological recording.
The experimental procedure for recording from brain slices was similar to what has been described recently (Xiao et al., 2007; Ye et al., 2004). Briefly, slices (two per animal) were transferred in a 0.4 ml recording chamber and stabilized with a U-shaped stainless steel net. Cells were visualized using an upright microscope with near infrared illumination (E600FN, Nikon, Japan) (Fig. 1B). Throughout the experiments, the bath was continually superfused with carbogen-saturated ACSF at a rate of 2 - 3 ml/min.
4.2. Electrophysiological Recording
Whole-cell currents were recorded with Axopatch 200B and 700A amplifiers (Molecular Devices Co., Union City, CA, USA), via Digidata 1320A and 1322A analog-to-digital converters (Molecular Devices Co.), and pClamp 9 software (Molecular Devices Co). Data were sampled at 5 kHz and filtered at 1 kHz. Whole-cell recordings were made 5 - 7 min after the patched membrane was ruptured. This was in consideration of the equilibration of the gradients between the pipette solution and the cytoplasm.
The patch electrodes had a resistance of 3 - 5 MΩ when filled with pipette solutions containing (in mM): 140 KCl, 2 MgCl2, 4 EGTA, 0.4 CaCl2, 10 HEPES, 2 Mg-ATP, and 0.1 GTP (for voltage-clamp experiments), or 135 potassium gluconate, 5 KCl, 2 MgCl2, 10 HEPES, 2 Mg-ATP, and 0.2 GTP (for current clamp experiments). The pH was adjusted to 7.2 with Tris-base, and the osmolarity was adjusted to 280 - 300 mOsm with sucrose.
4.3. Chemicals and applications
Most of the chemicals, including bicuculline (BIC), adenosine 5′-triphosphate (ATP), guanine 5′-triphosphate (GTP), DL-2-amino-5-phosphono-valeric acid (DL-APV), 6,7-dinitroquinoxaline-2, 3-dione (DNQX), prazosin, and butoxamine were purchased from Sigma (St. Louis, MO). Labetalol, metoprolol, phentolamine, and norepinephrine were from Bedford Laboratories (Bedford, OH). Chemicals were applied, to dissociated neurons, with a Y-tube perfusion system. With this system, the external solution, surrounding the neurons could be exchanged within 40 ms. In experiments on brain slices, chemicals were prepared in the final concentration in ACSF, and were applied via bath perfusion. The fact that 10 μM bicuculline blocked spontaneous inhibitory postsynaptic currents (sIPSCs) within 90 sec is an indication of the effective bath exchange time (Ye et al., 2004).
4.4. Data analysis
The sIPSCs were counted and analyzed using Clampfit 9.2 software (Molecular Devices Corp, Sunnyvale, CA, USA). The sIPSCs were screened automatically using a template with an amplitude threshold of 5 pA and then visually accepted or rejected based upon their 10-90% rise and 90-37% decay times. The majority (above 95%), of those sIPSCs, which were visually accepted, were screened using a suitable template. The frequency and amplitude of all events, during and after drug applications, were normalized to the mean values obtained during the control period. The inter-event intervals and amplitudes of sIPSCs obtained from the same neuron were examined by constructing cumulative probability distributions, and these distributions under different experimental conditions were compared using the Kolmogorov-Smirnov (K-S) test with Clampfit 9.2. For other plots, the frequency and amplitude of all events over a 1-2 min during the peak of a drug response were normalized to the average values of those during the initial control period (5-10 min). A concentration response curve was fitted with a Logistic equation: y = A2 + (A1-A2)/(1 + (x/x0)p), where y is the drug-elicited percentage change, of the frequency of sIPSCs, compared to control. A1 is the minimum effect and A2 is the maximum effect. X0 and p denote the half-effective concentration (EC50) and Hill coefficients, respectively. Differences in amplitude and frequency were tested by Student’s paired two-tailed t-test using their normalized values. Numerical values were provided as the mean ± standard error of the mean (SEM). Values of p < 0.05 were considered significant.
Acknowledgment
This work is made possible by the NIH grants AA01595 and AT001182 to JHY.
Nonstandard abbreviations
- BIC
bicuculline
- BUTO
butoxamine
- carbogen
95% O2/5%CO2
- GABA
γ-aminobutyric acid
- IPSC
inhibitory postsynaptic current
- sIPSC
spontaneous inhibitory postsynaptic current
- LAB
Labetalol
- MET
metoprolol
- NE
nor-epinephrine
- PAG
periaqueductal gray
- PHEN
phentolamine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Akaike N, Moorhouse AJ. Techniques: Applications of the nerve-bouton preparation in neuropharmacology. Trends Pharmacol. Sci. 2003;24:44–47. doi: 10.1016/s0165-6147(02)00010-x. [DOI] [PubMed] [Google Scholar]
- Baba H, Shimoji K, Yoshimura M. Norepinephrine facilitates inhibitory transmission in substantia gelatinosa of adult rat spinal cord (part 1): effects on axon terminals of GABAergic and glycinergic neurons. Anesthesiology. 2000;92:473–484. doi: 10.1097/00000542-200002000-00030. [DOI] [PubMed] [Google Scholar]
- Behbehani MM. Functional characteristics of the midbrain periaqueductal gray. Prog. Neurobiol. 1995;46:575–605. doi: 10.1016/0301-0082(95)00009-k. [DOI] [PubMed] [Google Scholar]
- Blakeley AG, Summers RJ. The effects of labetalol (AH 5158) on adrenergic transmission in the cat spleen. Br J Pharmacol. 1977;59(4):643–50. doi: 10.1111/j.1476-5381.1977.tb07733.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathala L, Guyon A, Eugene D, Paupardin-Tritsch D. Alpha2- adrenoceptor activation increases a cationic conductance and spontaneous GABAergic synaptic activity in dopaminergic neurones of the rat substantia nigra. Neuroscience. 2002;115:1059–1065. doi: 10.1016/s0306-4522(02)00542-0. [DOI] [PubMed] [Google Scholar]
- Cheun JE, Yeh HH. Noradrenergic potentiation of cerebellar Purkinje cell responses to GABA: cyclic AMP as intracellular intermediary. Neuroscience. 1996;74:835–844. doi: 10.1016/0306-4522(96)00130-3. [DOI] [PubMed] [Google Scholar]
- Chia YY, Chan MH, Ko NH, Liu K. Role of beta-blockade in anaesthesia and postoperative pain management after hysterectomy. Br. J. Anaesth. 2004;93:799–805. doi: 10.1093/bja/aeh268. [DOI] [PubMed] [Google Scholar]
- Chong W, Li LH, Lee K, Lee MH, Park JB, Ryu PD. Subtypes of alpha1- and alpha2- adrenoceptors mediating noradrenergic modulation of spontaneous inhibitory postsynaptic currents in the hypothalamic paraventricular nucleus. J. Neuroendocrinol. 2004;16:450–457. doi: 10.1111/j.1365-2826.2004.01180.x. [DOI] [PubMed] [Google Scholar]
- Ciranna L, Licata F, Li Volsi G, Santangelo F. Alpha 2- and beta-adrenoceptors differentially modulate GABAA- and GABAB-mediated inhibition of red nucleus neuronal firing. Exp. Neurol. 2004;185:297–304. doi: 10.1016/j.expneurol.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Coloma M, Chiu JW, White PF, Armbruster SC. The use of esmolol as an alternative to remifentanil during desflurane anesthesia for fast-track outpatient gynecologic laparoscopic surgery. Anesth. Analg. 2001;92:352–357. doi: 10.1097/00000539-200102000-00014. [DOI] [PubMed] [Google Scholar]
- Cousins MJ, Mather LE. Intrathecal and epidural administration of opioids. Anesthesiology. 1984;61:276–310. [PubMed] [Google Scholar]
- Drew GM, Hilditch A, Levy GP. Effect of labetalol on the uptake of [3H]-(-)-noradrenaline into the isolated vas deferens of the rat. Br J Pharmacol. 1978;63:471–474. doi: 10.1111/j.1476-5381.1978.tb07799.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont EC, Williams JT. Noradrenaline triggers GABAA inhibition of bed nucleus of the stria terminalis neurons projecting to the ventral tegmental area. J. Neurosci. 2004;24:8198–8204. doi: 10.1523/JNEUROSCI.0425-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Abbott FV. Techniques for assessing the effects of drugs on nociceptive responses. In: Boulton AA, Baker GB, Greenshaw AJ, editors. Neuromethods: psychopharmacology. Humana Press; New Jersey: 1989. pp. 145–215. [Google Scholar]
- Gold EH, Chang W, Cohen M, Baum T, Ehrreich S, Johnson G, Prioli N, Sybertz EJ. Synthesis and comparison of some cardiovascular properties of the stereoisomers of labetalol. J. Med. Chem. 1982;25:1363–1370. doi: 10.1021/jm00353a017. [DOI] [PubMed] [Google Scholar]
- Heinricher M, Cheng Z, Fields HL. Evidence for two classes of nociceptive modulating neurons in the periaqueductal gray. J. Neurosci. 1987;7:271–278. doi: 10.1523/JNEUROSCI.07-01-00271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasmin L, Wu MV, Ohara PT. GABA puts a stop to pain. Curr. Drug Targets CNS Neurol. Disord. 2004;3:487–505. doi: 10.2174/1568007043336716. [DOI] [PubMed] [Google Scholar]
- Jiang M, Chandler SD, Ennis M, Shipley MT, Behbehani MM. Actions of epinephrine on neurons in the rat midbrain periaqueductal gray maintained in vitro. Brain Res. Bull. 1992;29:871–877. doi: 10.1016/0361-9230(92)90158-t. [DOI] [PubMed] [Google Scholar]
- Johansen JW, Flaishon R, Sebel PS. Esmolol reduces anesthetic requirement for skin incision during propofol/nitrous oxide/morphine anesthesia. Anesthesiology. 1997;86:364–371. doi: 10.1097/00000542-199702000-00011. [DOI] [PubMed] [Google Scholar]
- Kamisaki Y, Hamahashi T, Hamada T, Maeda K, Itoh T. Presynaptic inhibition by clonidine of neurotransmitter amino acid release in various brain regions. Eur. J. Pharmacol. 1992;217:57–63. doi: 10.1016/0014-2999(92)90511-2. [DOI] [PubMed] [Google Scholar]
- Khanna NK, Dadhich AP, Vyas DS. Modification of morphine analgesia by Labetalol. Indian J. Exp. Biol. 1978;16:1091–1092. [PubMed] [Google Scholar]
- Kopp N, Denoroy L, Renaud L, Puzol JF, Tabib A. Tommas A: Distribution of adrenalin- synthesizing enzyme activity in the human brain. J. Neurol. Sci. 1979;41:397–409. doi: 10.1016/0022-510x(79)90098-4. [DOI] [PubMed] [Google Scholar]
- Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J. Neurosci. 1998;18:10231–10240. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCarthy EP, Bloomfield SS. Labetalol: a review of its pharmacology, pharmacokinetics, clinical uses and adverse effects. Pharmacotherapy. 1983;3:193–219. doi: 10.1002/j.1875-9114.1983.tb03252.x. [DOI] [PubMed] [Google Scholar]
- Margaria E, Gagliardi M, Palieri L, Treves S, Fanzago E. Analgesic effect of peridural labetalol in the treatment of cancer pain. Int. J. Clin. Pharmacol. Therap. Toxicol. 1983;21:47–50. [PubMed] [Google Scholar]
- Mitchell VA, Christie MJ, Vaughan CW. Developmental changes in the alpha-adrenergic responses of rat periaqueductal grey neurons. Neuroreport. 2003;14:1637–1639. doi: 10.1097/00001756-200308260-00019. [DOI] [PubMed] [Google Scholar]
- Pearson J, Goldstein M, Markey K, Brandeis L. Human brainstem catecholamine neuronal anatomy as indicated by immunicytochemistry with antibodies to tyrosine hydroxylase. Neuroscience. 1983;8:3–32. doi: 10.1016/0306-4522(83)90023-4. [DOI] [PubMed] [Google Scholar]
- Peng YB, Lin Q, Willis WD. Involvement of alpha-2 adrenoceptors in the periaqueductal gray- induced inhibition of dorsal horn cell activity in rats. J. Pharmacol. Exp. Ther. 1996;278:125–135. [PubMed] [Google Scholar]
- Saitow F, Satake S, Yamada J, Konishi S. Beta-adrenergic receptor-mediated presynaptic facilitation of inhibitory GABAergic transmission at cerebellar interneuron-Purkinje cell synapses. J. Neurophysiol. 2000;84:2016–2025. doi: 10.1152/jn.2000.84.4.2016. [DOI] [PubMed] [Google Scholar]
- Stanley TH, de Lange S, Boscoe MJ, de Bruijn N. The influence of chronic preoperative propranolol therapy on cardiovascular dynamics and narcotic requirements during operation in patients with coronary artery disease. Can. Anaesth. Soc. J. 1982;29:319–324. doi: 10.1007/BF03007519. [DOI] [PubMed] [Google Scholar]
- Tseng KY, O’Donnell P. Dopamine- glutamate interactions controlling prefrontal cortical pyramidal cell excitability involve multiple signaling mechanisms. J. Neurosci. 2004;24:5131–5139. doi: 10.1523/JNEUROSCI.1021-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Bandler R, Christie MJ. Differential responses of lateral and ventrolateral rat periaqueductal grey neurones to noradrenaline in vitro. J. Physiol. 1996;490:373–381. doi: 10.1113/jphysiol.1996.sp021151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Xiao C, Ye JH. Taurine activates excitatory non- synaptic glycine receptors on dopamine neurons in ventral tegmental area of young rats. J. Physiol. 2005;565:503–516. doi: 10.1113/jphysiol.2005.085423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Zhou C, Li K, Ye JH. Presynaptic GABAA receptors facilitate GABAergic transmission to dopaminergic neurons in the ventral tegmental area of young rats. J Physiol. 2007;580(Pt3):731–743. doi: 10.1113/jphysiol.2006.124099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye JH, Wang F, Krnjevic K, Wang W, Xiong Z. Presynaptic glycine receptors on GABAergic terminals facilitate discharge of dopaminergic neurons in ventral tegmental area. J. Neurosci. 2004;24:8961–8974. doi: 10.1523/JNEUROSCI.2016-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye JH, Zhang J, Xiao C, Kong JQ. Patch-clamp studies in the CNS illustrate a simple new method for obtaining viable neurons in rat brain slices: Glycerol replacement of NaCl protects CNS neurons. J. Neurosci Methods. 2006;158:251–259. doi: 10.1016/j.jneumeth.2006.06.006. [DOI] [PubMed] [Google Scholar]
- Zaugg M, Schaub MC, Pasch T, Spahn DR. Modulation of β- adrenergic receptor subtype activities in perioperative medicine: mechanisms and sites of action. Br. J. Anaesthesia. 2002;88:101–123. doi: 10.1093/bja/88.1.101. [DOI] [PubMed] [Google Scholar]