

Abstract
The repair of the multitude of single-base lesions formed daily in cells of all living organisms is accomplished primarily by the base excision repair pathway that initiates repair through a series of lesion-selective glycosylases. In this article, single-turnover kinetics have been measured on a series of oligonucleotide substrates containing both uracil and purine analogs for the Escherichia coli mispaired uracil glycosylase (MUG). The relative rates of glycosylase cleavage have been correlated with the free energy of helix formation and with the size and electronic inductive properties of a series of uracil 5-substituents. Data are presented that MUG can exploit the reduced thermodynamic stability of mispairs to distinguish U:A from U:G pairs. Discrimination against the removal of thymine results primarily from the electron-donating property of the thymine 5-methyl substituent, whereas the size of the methyl group relative to a hydrogen atom is a secondary factor. A series of parameters have been obtained that allow prediction of relative MUG cleavage rates that correlate well with observed relative rates that vary over 5 orders of magnitude for the series of base analogs examined. We propose that these parameters may be common among DNA glycosylases; however, specific glycosylases may focus more or less on each of the parameters identified. The presence of a series of glycosylases that focus on different lesion properties, all coexisting within the same cell, would provide a robust and partially redundant repair system necessary for the maintenance of the genome.
The DNA of all living organisms is constantly damaged by multiple pathways, leading to a complex array of lesions. Most of these endogenous lesions are single-base damage products that are generally repaired by the base excision repair pathway. The base excision repair pathway is initiated by one of a series of damage-specific glycosylases (1–4) that find and remove the damaged base by cleavage of the N-glycosidic bond. The resulting abasic site is then processed and the damaged segment is resynthesized by a group of interacting nucleases, polymerases, and ligases in a repair sequence common to most types of single-base damage.
Significant efforts are now underway in several laboratories to determine the mechanisms by which the lesions are initially located and distinguished from normal DNA bases. It is estimated that in the human genome, there are 104 to 105 lesions per cell per day from endogenous damage events (5, 6). However, these lesions are dispersed among 109 normal DNA bases, creating a substantial challenge for finding the damage. Furthermore, the fidelity of the glycosylase must exceed the fidelity of the DNA polymerases. If the glycosylase selectivity was 1/105, it would remove a normal DNA base each time it removed a damaged base, and if lower, could cause substantial collateral damage to the genome.
Among the DNA repair glycosylases is the uracil glycosylase superfamily, members of which are found in all organisms including bacteria and primates as well as viruses (7–10). Common among these glycosylases is their capacity to recognize and remove uracil from DNA. Uracil residues may be found in DNA from the misincorporation of dUTP during DNA synthesis or the deamination of cytosine residues in DNA. The most active of these glycosylases is uracil DNA glycosylase, (UNG),3 which removes uracil when paired with either adenine or guanine, as well as uracil in single-stranded DNA. UNG does not cleave the normal DNA bases thymine or cytosine. Discrimination against thymine has been explained by the presence of a tyrosine residue (Tyr66 in Escherichia coli, Tyr147 in human) that creates a steric opposition with the thymine methyl group (11). Discrimination against cytosine results in part from the formation of specific hydrogen bonds with the N3 hydrogen and O4 carbonyl of uracil. In humans, the UNG gene is located on chromosome 12q and is spliced into two forms; UNG2 is targeted to the nucleus, whereas UNG1 is targeted to the mitochondria (12, 13).
On chromosome 12 in humans, two additional members of the uracil glycosylase family are found: the thymine DNA glycosylase that can remove thymine when mispaired with guanine (14, 15) and SMUG1 (16, 17), a glycosylase that appears to selectively remove uracil from single-stranded DNA and has the further capacity to remove a series of oxidized thymine analogs (18, 19). The thymine DNA glycosylase homolog in E. coli is the mispaired uracil glycosylase (MUG) (20–25). MUG preferentially repairs uracil when mispaired with guanine, and has a very weak activity against mispaired thymine.
The substrate preferences of MUG have been explained on the basis of DNA protein contacts observed in the crystal structure (24). The preference of MUG for mispaired uracil has been ascribed to the formation of specific hydrogen bonds between amino acid residues of MUG and the “widowed guanine” remaining in the DNA helix following the extrusion of the target uracil residue. The strong selectivity for uracil over thymine by UNG has been attributed to a steric block created by an active site tyrosine residue (11). The strong selectivity of MUG for uracil over thymine similarly has been attributed to steric blocking of the thymine methyl group (24). However, in the case of MUG, the corresponding residue is glycine, which would not be expected to have a substantial steric blocking effect.
Previously, we investigated the substrate preferences of MUG using oligonucleotides containing a series of base analogs under steady-state enzyme reaction conditions (25). Upon the basis of results obtained with a series of purine analogs paired with the target uracil residue, we proposed that the reduced thermal stability of a duplex containing uracil in a mispair, as opposed to the formation of specific hydrogen bonds, more likely explained the preference of MUG for mispaired uracil. In similar studies with 5-substituted pyrimidine analogs, we observed that MUG had substantial activity against 5-bromouracil and 5-iodouracil, leading us to propose that the electronic-inductive properties of the target base were more important than steric size in establishing the apparent selectivity of MUG for uracil over thymine.
Subsequently, others investigating the kinetics of MUG activity established that MUG had a high affinity for the abasic site remaining in DNA following glycosylase removal of uracil (22). The high affinity for the abasic site would reduce the rate of MUG turnover, reducing the reliability of kinetic data obtained under steady-state conditions. In this article, we have reinvestigated the activity of MUG against a series of oligonucleotides containing purine and pyrimidine analogs. We have also obtained thermal melting and thermodynamic data on helix formation for a series of oligonucleotides containing this series of base analogs. We report here that the size and electronic-inductive properties of the pyrimidine 5-substituent, as well as helix stability can be used to reasonably explain the substrate preferences of MUG.
MATERIALS AND METHODS
Oligonucleotide Synthesis and Characterization—Oligonucleotides were prepared by solid phase synthesis methods as described previously (26, 27). Following synthesis and deprotection, oligonucleotides were purified with Poly-Pak II cartridges, and denaturing gel purified when necessary. The presence of modified bases was verified by gas chromatography/mass spectrometry following acid hydrolysis and conversion to the trimethylsilyl ethers.
Two sets of oligonucleotides were synthesized. A set of oligonucleotide 24-mers containing uracil with different 5-substituents (X) and purine analogs (P) was synthesized for the MUG activity assay (Fig. 1A). Another set of self-complementary 12-mers containing a uracil and a purine analog was synthesized for melting temperature (Tm) measurements in which the target uracil analog was placed within the same sequence context as in the 24-mer glycosylase assays (Fig. 1B). A 12-base sequence was selected for the thermodynamic studies because the predicted Tm would be within an appropriate range for UV melting studies. The self-complementary 12-mers were designed by keeping the two adjacent bases on each side of the uracil and purine analog base pair constant, and linking the two 5-base fragments in the 5′ → 3′ orientation. Proposed structures of the base pairs examined here are shown in Fig. 2.
FIGURE 1.

Sequences of the oligonucleotides examined for this study, where X = U, T, 5-fluorouracil, 5-chlorouracil, 5-bromouracil, or 5-iodouracil. In the purine series, P = A or G, hypoxanthine, purine, 2-aminoadenine, or 2-aminopurine. A, 24-mer oligonucleotide used for the enzyme kinetic experiments. B, 12-mer oligonucleotides used for the thermodynamic experiments.
FIGURE 2.

Proposed conformers of uracil base pairs with purine analogue base pairs in this study. R = H for uracil.
Determination of Enzyme Kinetic Parameters under Single Turnover Conditions—Oligonucleotide substrates were 5′-32P-end labeled by T4 polynucleotide kinase (New England Biolabs, Beverly, CA) with [γ-32P]ATP (MP Biomedicals, Costa Mesa, CA) under conditions recommended by the enzyme supplier and subsequently purified using G-50 Sephadex columns (Roche Applied Science). Labeled strands were annealed to a 2-fold excess of unlabeled complementary strands in 20 mm Tris-HCl, pH 8.0, 0.1 mg/ml bovine serum albumin, 1 mm EDTA, 1 mm EGTA, and 1 mm dithiothreitol, incubated at 95 °C for 5 min, and allowed to cool to room temperature slowly for duplex formation. MUG was obtained from Trevigen, Gaithersburg, MD.
To compare MUG activity between different duplexes, we measured the cleavage rates under single turnover conditions as described by O'Neill et al. (22). Reactions were performed at 25 °C with 1.4 nm substrate and 0.56 μm MUG in the standard reaction buffer (20 mm Tris-HCl, pH 8.0, 0.1 mg/ml bovine serum albumin, 1 mm EDTA, 1 mm EGTA, and 1 mm dithiothreitol). At selected time points 10-μl samples were removed and quenched with 5 μl of 0.1 m NaOH. Abasic sites were then cleaved by heating the samples at 95 °C for 30 min after addition of an equal volume of the Maxam-Gilbert loading buffer (98% formamide, 0.01 m EDTA, 1 mg/ml xylene cyanol, and 1 mg/ml bromophenol blue). Cleaved DNA fragments were separated from intact DNA by 20% denaturing polyacrylamide gels (8 m urea), and subsequently quantified using a PhosphorImager (Molecular Dynamics, Sunnyvale, CA) and ImageQuant software. The reaction rate constant, kobs, was determined by fitting time course data to a single exponential (y = a(1–e–bx)) using SigmaPlot 8.0, where a is the maximum level of product ratio and b is the kobs.
A rapid quench-flow apparatus (RQF-3, KinTek Corp., Austin, TX) was used for reactions requiring a short time course (53 ms to 200 s, U:G, U:HX, FU:G, and ClU:G). Reactions were performed in the standard conditions as described above except that the reaction volume was 35.5 μl, and 100 μl of 50 mm NaOH was used to quench reactions. The quenched reactions were heated at 95 °C for 20 min to cleave the abasic sites, and then dried under reduced pressure. DNA was redissolved in 14 μl of Maxam-Gilbert loading buffer and 1 μl of 10 pmol/μl of the uracil-containing oligonucleotide was added as a competitor. Samples were analyzed as described above.
Determination of Helix Melting Behavior—Samples containing self-complementary 12-mer oligonucleotides were prepared in buffer containing 0.1 m NaCl, 0.01 m sodium phosphate, and 0.1 mm EDTA, pH 7.0. Concentration dependent Tm measurements were conducted with total strand concentration (CT) between 2 and 75 μm in cuvettes with path lengths between 1 and 10 mm. Molar extinction coefficients of oligonucleotides were calculated (28) to determine single strand concentrations, and the molar extinction coefficients of 9.9, 12.5, 7.02, 1, and 7.7 × 103 m–1 cm–1, were used for U, HX, Pu, 2AP, and 2AA, respectively. Oligonucleotide melting temperatures (Tm) were determined using a Varian Cary 100 Bio UV-visible spectrophotometer (Varian, Walnut Creek, CA). Five temperature ramps were performed on each sample per run at 260 nm: 1) 15 °C to 90 °C at a rate of 0.5 °C/min, 2) 90 °C to 15 °C at a rate of 0.5 °C/min, 3) 15 °C to 90 °C at a rate of 0.5 °C/min, 4) 90 °C to 15 °C at a rate of 0.5 °C/min, and 5) 15 °C to 90 °C at a rate of 0.5 °C/min. The sample was held for 3 min when the temperature reached 90 °C and 10 min when it reached 15 °C and started the next cycle. Data were collected at 0.5 °C intervals while monitoring the temperature with a probe inserted into a cuvette containing only buffer. The Tm of each duplex was determined using Cary WinUV Thermal software (Varian) with a total of 3 to 4 independent Tm measurements (Table 1).
TABLE 1.
MUG reaction rate constants and physical properties of substrates
Observed Tm, ΔG, ΔH, ΔS, glycosylase cleavage rate constants and reported size and electronic-inductive properties for the substrates examined here. The thermodynamic parameters are obtained with a 12-mer oligodeoxynucleotide, whereas the kinetic parameters are obtained with a 24-mer.
| Base pair | Rate constant (kobs) | ΔG | σm | Size | ΔH | ΔS | Tm |
|---|---|---|---|---|---|---|---|
| s-1 | kcal mol-1 | Å | kcal mol-1 | cal mol-1 K-1 | °C | ||
| U:G | 3.90 ± 0.17 × 10-2 | -10.8 ± 0.4 | 0.00 | 2.26 | -83.2 ± 5.0 | -237 ± 15.5 | 49.1 ± 0.1 |
| U:HX | 3.40 ± 0.24 × 10-2 | -10.4 ± 0.5 | 0.00 | 2.26 | -75.6 ± 8.1 | -213 ± 25.5 | 49.0 ± 0.7 |
| U:Pu | 3.90 ± 0.50 × 10-3 | -12.1 ± 0.3 | 0.00 | 2.26 | -90.8 ± 2.9 | -257 ± 9.2 | 52.4 ± 0.3 |
| U:A | 1.60 ± 0.01 × 10-3 | -15.9 ± 0.5 | 0.00 | 2.26 | -103.7 ± 3.7 | -286 ± 10.6 | 64.4 ± 0.5 |
| U:2AA | 7.00 ± 1.00 × 10-4 | -15.3 ± 0.9 | 0.00 | 2.26 | -92.9 ± 7.5 | -253 ± 21.9 | 66.3 ± 0.5 |
| U:2AP | 2.80 ± 0.30 × 10-3 | -14.7 ± 0.3 | 0.00 | 2.26 | -99.4 ± 3.2 | -276 ± 9.5 | 61.1 ± 0.1 |
| FU:G | 1.86 ± 0.08 × 10-1 | -10.9 ± 0.4 | 0.35 | 2.65 | -85.1 ± 5.6 | -243 ± 17.1 | 48.9 ± 0.4 |
| CIU:G | 6.20 ± 0.53 × 10-2 | -11.1 ± 0.2 | 0.35 | 3.52 | -83.3 ± 3.1 | -236 ± 9.5 | 50.4 ± 0.4 |
| BrU:G | 2.40 ± 0.15 × 10-2 | -11.1 ± 0.2 | 0.38 | 3.82 | -83.7 ± 3.0 | -238 ± 9.2 | 50.0 ± 0.3 |
| IU:G | 3.40 ± 0.10 × 10-3 | -11.1 ± 0.4 | 0.35 | 4.23 | -85.7 ± 5.5 | -244 ± 17.1 | 49.9 ± 0.3 |
| T:G | 2.50 ± 0.20 × 10-6 | -11.3 ± 0.3 | -0.07 | 3.50 | -86.6 ± 3.2 | -246 ± 9.7 | 50.2 ± 0.1 |
Thermodynamic parameters were calculated in two ways: 1) averages from fits of individual melting curves at different concentrations using Van't Hoff calculation in the Cary WinUV Thermal software; 2) the 1/Tm versus ln CT plots fitted to the following equation (data shown as supplementary figures) for the self-complementary sequences examined here.
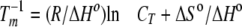 |
(Eq. 1) |
Both methods assume a two-state model and ΔCp = 0 for the transition equilibrium. The two-state approximation was assumed to be valid for sequences in which the ΔH values derived from the two methods agreed within 15% (29). The ΔH values derived from the two methods agree within 15%, indicating that the two-state approximation is valid for all other sequences employed in this study. The thermodynamic parameters from fits of melting curves are reported in Table 1.
RESULTS
The rates of MUG cleavage of oligonucleotides were determined from a gel-based electrophoresis assay as described above. To obtain data under single turnover conditions, an excess of MUG was used, and time points were obtained using a Kintec™ rapid kinetics instrument. Illustrative data are shown in Fig. 3.
FIGURE 3.

Kinetic study of MUG cleavage of 5-substituted uracil analogues paired with guanine illustrating the gel electrophoretic assay (left) and time-dependent product ratio (right). Single turnover reactions were performed at 25 °C with 1.4 nm substrate and 0.56 μm MUG in the standard reaction buffer. A rapid quench-flow apparatus was used for reactions conducted from 53 ms to 200 s. Upper, FU:G as a substrate; lower, U:G as a substrate.
Kinetic constants for the single-turnover reactions against the 11 substrates examined here are shown in Table 1. The trends observed are similar to those observed previously by us under steady-state conditions (25); uracil mispaired with guanine (U:G) is repaired faster than uracil paired with adenine (U:A). Cleavage is observed with the halogenated uracil analogs, with cleavage rates inversely proportional to substituent size. Cleavage against mispaired 5-fluorouracil is the fastest of the series, and in this study, we can observe and measure the rate of thymine cleavage.
Helix melting was measured, and the results are reported in Table 1 for each of the 11 substrates examined. The oligonucleotide substrates used in the thermal analyses are shorter than the oligonucleotides used in the enzyme cleavage assay so that the melting temperatures were within a range observable by this method. The sequence surrounding the target uracil, however, is identical for the series of oligonucleotides used in the enzyme cleavage assay and in the thermal denaturation assays. The oligonucleotides used in the thermal studies are symmetric and contain two substitutions per duplex.
The duplex containing the U:G mispair has a lower helix formation energy than the U:A duplex, a result in accord with previous studies of duplexes containing mispairs (29). Based upon the ΔG values, the U:HX base pair (Fig. 2) is the least stable of the base pairs examined. Both the U:G and U:HX base pairs are presumed to be in a wobble geometry (Fig. 2). The difference is the guanine 2-amino group that does not participate in base-base hydrogen bonding. The base pair formed with purine (nebularine) would likely have one hydrogen bond, whereas uracil paired with 2-aminopurine and 2-aminoadenine would form two and three hydrogen bonds, respectively (30–33). The results observed here are in accord with the proposal that differences in oligonucleotide melting temperatures for the series of oligonucleotides examined here can be attributed to differences in base-stacking interactions, as opposed to the number of hydrogen bonds formed between bases in opposing strands (34), and that base pair geometry (pseudo wobble versus pseudo Watson-Crick) modulates base stacking and melting behaviors.
Upon the basis of previous studies, we examined the impact of three parameters on the rates of glycosylase cleavage: 1) stability of the helix containing the target pyrimidine, 2) size of the pyrimidine 5-substituent, and 3) the electronic-inductive property of the 5-substituent of 5-substituted pyrimidines. To investigate the impact of helix stability, we compared the rates for the subset of duplexes containing uracil paired with a series of purines. In this series, the target pyrimidine, uracil, was constant allowing the isolation of the contribution of helix stability to the relative glycosylase rate. To examine the impact of substituent size, we compared the subset of pyrimidines including the 5-fluoro, 5-chloro, 5-bromo, and 5-iodo substituents paired with guanine. Within this series of halogens, substituent size increases through the series, however, the electronic-inductive property and helix stability are relatively constant. To estimate the impact of the electronic-inductive property, we compared a subset including uracil, thymine, and 5-fluorouracil, representing an electronically neutral substituent, an electron-donating substituent, and the smallest of the electron-withdrawing substituents.
The relationship between helix formation energy and MUG cleavage kinetics is shown in Fig. 4. In this series, the target pyrimidine, uracil, is constant, and the purine in the opposing strand is varied. A plot of the natural logarithm of the observed rate constant versus the free energy of helix formation reveals an inverse relationship. The trend observed is that the uracil residues from the more easily disrupted helices are most rapidly cleaved.
FIGURE 4.

Relationship between helix formation energy, ΔG, and glycosylase kinetics. The natural logarithm of the observed rate constant (ln kobs) of the MUG cleavage reaction is plotted versus ΔG (kcal/mol).
The slope of the line is 0.63, the intercept 3.02, and the correlation coefficient (r2) is 0.85. Upon the basis of the observed relationship (Fig. 4) the relationship between helix formation energy and MUG cleavage rate can be described by Equations 2 and 3.
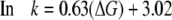 |
(Eq. 2) |
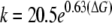 |
(Eq. 3) |
Upon the basis of Equation 3 and the data in Table 1, the expected rate for cleavage of uracil paired with guanine (U:G) would be 2.27 × 10–2 s–1. Using this value, the rate of cleavage of uracil paired with other bases, relative to the U:G mispair, krel, ΔG, can be determined as a function of the free energy of helix formation as shown in Equation 4.
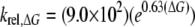 |
(Eq. 4) |
The influence of the 5-substituent size can be estimated by examining the cleavage rates of the 5-halouracils paired with guanine. Within this series, the free energy of helix formation and the electronic-inductive properties of the 5-subsituent denoted by the Hammett meta parameter (35), σm are similar. In Fig. 5, the observed rate constant for MUG cleavage is plotted versus the size of the 5-substituent. The size of the 5-substituent is estimated to be the sum of the van der Waals radius (36) and the length of the carbon-halogen bond (37).
FIGURE 5.

Relationship between substituent size and glycosylase cleavage rates. The observed rate constants (kobs) for the MUG cleavage reactions are plotted against the size of the halogen substituents at the C(5) position of the substituted uracil paired with guanine.
An inverse relationship (Fig. 5) is observed in that the larger the size of the 5-substituent, the slower the apparent rate of MUG cleavage. The slope of the line in Fig. 5 is –0.118, the intercept is 0.50, and the correlation coefficient is 0.97. The relationship between the expected cleavage rate and the size of the pyrimidine 5-substituent can be described by Equation 5.
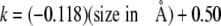 |
(Eq. 5) |
Upon the basis of Equation 5, the expected rate for the cleavage of a uracil analog with a 5-substituent the size of the hydrogen atom, and assuming the same electronic-inductive properties as the halogens would be 2.33 × 10–1 s–1. Therefore, the rate of cleavage of a pyrimidine analog, relative to the cleavage of uracil, can be determined as indicated in Equation 6.
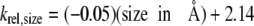 |
(Eq. 6) |
The predicted rate constant for glycosylase cleavage of uracil from the U:G mispair estimated from Equation 5 is 2.33 × 10–1 s–1, whereas the experimentally determined rate constant is 3.90 × 10–2 s–1. The value predicted from Equation 5 is ∼6-fold higher than the observed rate constant. However, Equation 5 was generated by comparing the measured rate constants for the halogenated derivatives. The difference between the observed rate constant for uracil cleavage and the rate constant predicted from Equation 5 suggests that the impact of substituting a hydrogen atom with a small, electron-withdrawing halogen would increase the rate constant by a factor of ∼6.
The influence of the electronic-inductive properties of the 5-substituent can be estimated independently by comparing the cleavage rates for U:G with that of T:G, the only one of the substituents that is electron donating to the ring, and therefore has a negative σm, and that of FU:G, the halogen with the size most similar to that of hydrogen. A plot of the observed MUG cleavage rate versus σm is shown in Fig. 6. A slope of 0.44, intercept of 0.035, and correlation coefficient of 0.99 are observed. The relationship between cleavage rate and substituent σm can be described by Equation 7.
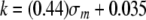 |
(Eq. 7) |
The value of the electronic-inductive property for a 5-substituent, denoted by σm, is determined relative to the hydrogen substituent, which is assigned the value of zero. Electron-donating substituents, such as a methyl group have negative values for σm, whereas electron withdrawing substituents like halogens, have positive values. As the Hammett parameter (σm) for hydrogen is zero, the relative rate of glycosylase cleavage can be written as Equation 8.
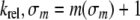 |
(Eq. 8) |
The slope (m) of the line described by Equation 8 was obtained by comparing the observed rates of cleavage of the uracil and thymine-containing oligonucleotides as indicated in Equation 9.
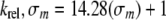 |
(Eq. 9) |
The relationship indicated by Equation 9 would predict that the relative rate constant for cleavage of a 5-halogenated uracil analog (σm = 0.35) would be ∼6-fold higher than that of uracil (σm = 0), if the size of the 5-substituents were the same. This independent prediction of the impact of a halogen substituent on the rate constant is the same as the estimate derived from Equation 5 and discussed above.
FIGURE 6.

Relationship between electronic-inductive properties and glycosylase cleavage rates. The observed rate constants (kobs) for the MUG cleavage reactions are plotted against the electronic inductive properties (σm) of the substituent at the C(5) position of the substituted uracil paired with guanine.
In the discussion above, we attempted to estimate independently the effects of helix stability, substituent size, and the electronic-inductive property by comparing subsets of analogs in which a specific factor could be isolated. Equations were generated to define the impact of each parameter on the relative rate of cleavage for a uracil analog. We observe that the expected relative rates increase with decreasing helix stability, decreasing 5-substituent size and increasing electron-withdrawing property. When the glycosylase encounters a target pyrimidine, all of these factors contribute simultaneously to the relative cleavage rate. If the effect of each of the parameters can be independently estimated, the observed relative rate should then be the product of the relative rates determined for each of the three parameters examined.
Upon the basis of the above discussion, the expected rate of cleavage of a 5-substituted uracil analog, in a given base pair and relative to U:G, should then be expressed as the product of the relative rates resulting from the effects of helix stability, substituent size, and substituent inductive property as shown in Equations 10 and 11.
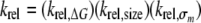 |
(Eq. 10) |
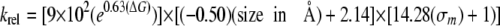 |
(Eq. 11) |
A plot of the expected rate of cleavage, relative to U:G, for the substrates examined here, is plotted versus the observed relative cleavage rates as shown in Fig. 7. A linear relationship is observed, with a slope of 0.95, intercept of 0.15, and correlation coefficient of 0.98.
FIGURE 7.

Relationship between expected relative rates and observed relative rates based on the helix formation energy of the oligonucleotide, size, and electronic inductive property of the uracil C(5) substituent plotted on a log-log scale. The relative rates are calculated with respect to the rate of cleavage of the U:G base pair.
DISCUSSION
In this article, we have re-examined the rate of MUG cleavage under single-turnover conditions for a series of oligonucleotide substrates with differing physical characteristics. The first property examined was the free energy of helix formation. The free energy of helix formation was systematically varied by placing the target uracil residue opposite a series of purine analogs. This series included the U:G pair, the presumed in vivo target of MUG, to which the others were subsequently compared. It is well established that oligonucleotides containing mispairs are less thermodynamically stable than those containing the corresponding correct base pairs, attributed primarily to reduced base-stacking interactions. The rationale for examining helix formation energy within the context of glycosylase substrate preference is that the glycosylases function by a “base flipping” mechanism (38, 39) in which the target base is extruded from the helix, requiring elimination of hydrogen-bonding and base-stacking interactions with the targeted base. Although we are not presuming that the energetics of helix thermal denaturation and glycosylase base flipping are identical, the examination of differences in helix denaturation within a homologous series allows isolation of the base-stacking and hydrogen-bonding interactions characteristic of a particular base pair.
Other laboratories have similarly proposed that the local helix “instability” resulting from DNA base damage or base mispair formation could be exploited by DNA repair enzymes that search for DNA damage and attempt to distinguish damaged and normal DNA (40–43). The data presented here are consistent with that concept. Previously, the preference of MUG for mispaired uracil was attributed to the formation of specific hydrogen bonds with the guanine remaining in the helix following uracil extrusion, specifically the N2-amino group and N1 proton (24). Our results showed that the replacement of guanine by hypoxanthine, which lacks the N2-amino group, had very little impact on the observed cleavage rates, suggesting that specific interactions with the 2-amino group do not strongly influence MUG base selection. Similarly, addition of a 2-amino group to the U:A base pair, with formation of the uracil-2-aminoadenine base pair (U:2AA), results in a decreased rather than increased cleavage rate. We therefore propose that the mechanism by which MUG distinguishes uracil paired with adenine from uracil mispaired with guanine is relative helix stability and not specific interactions with the “widowed” guanine. The scattering of the data from the line as shown in Fig. 4 could be attributed to a secondary effect of specific DNA-protein interactions or to energetic differences between helix thermal denaturation and glycosylase helix bending and base flipping.
In this article, we determined relative helix stability based upon experimentally determined oligonucleotide duplex melting temperatures. When measuring melting temperatures for an homologous series of duplex oligonucleotides, the impact of a specific base substitution can be estimated. Our results are in accord with other published studies suggesting that reduced helix stability could be exploited by DNA repair enzymes that search for damaged bases (25, 40–43). Helix stability could impact glycosylase cleavage rates in two ways. First, all of the known glycosylases act by a base-flipping mechanism whereby the target base is extruded from the helix into a glycosylase active site for further interrogation. Reduced helix stability for a given base would facilitate base flipping and thus enhance cleavage rates. Second, reduced helix stability for a given base would decrease the time the target base occupied an intrahelical position and increase the proportion of time spent in an extrahelical position. In accord with this second proposal, recent data presented by Stivers and co-workers (44) indicate that target selection by UNG is determined by base pair dynamics and not by active participation of the enzyme. The data presented in this article are consistent with both potential explanations, but cannot distinguish between them.
Glycosylase discrimination for substituted pyrimidines paired with guanine has been previously attributed to both size and electronic-inductive effects (24, 25). The discrimination of UNG against thymine has been ascribed to a steric effect imposed by a tyrosine residue in the pyrimidine pocket. Consistent with this suggestion, the substrate range of UNG is largely limited to uracil. Although 5-fluorouracil is cleaved, it is cleaved at a slower rate relative to uracil, and pyrimidines with larger 5-halogens are not substrates. In contrast, MUG does cleave halogenated pyrimidines, although size is a factor. As indicated in Fig. 5, an inverse linear relationship is observed between cleavage rates and substituent size.
Although the 5-methyl substituent of thymine and the 5-bromo-substituent of 5-bromouracil are similar in size, the observed cleavage rates differ by a factor of 104. The preference of MUG for 5-bromouracil over thymine was attributed previously to the contrasting electronic inductive properties of a methyl group and a bromine substituent (25). Linear free energy relationships have been demonstrated between σm, an index of inductive properties for 5-substituents on the pyrimidine ring system and the pKa of the N1 and N3 protons, both of which are meta to the 5-substituent (45). Shapiro and Kang (46) previously demonstrated a relationship between inductive properties of 5-substituents and nucleoside hydrolysis rates in aqueous solution. Both hydrolytic and enzymatic cleavage of the nucleotide glycosidic bond are proposed to proceed via increasing charge density at the C1′ position, which is facilitated by electron withdrawing substituents and inhibited by electron-donating substituents (45–49).
The substrates tested here have observed cleavage rates that range over 5 orders of magnitude. Substituent size, electronic-inductive properties, and helix stability are all parameters that can influence MUG selectivity, as measured by relative cleavage rates. The data presented in Fig. 7 suggest strongly that the parameters examined here can be assessed independently, yet conspire together to determine the relative cleavage rate for a given substrate. The selectivity of MUG for U:G over U:A can be attributed primarily to differences in helix formation energy, which we propose is proportional to the energetic cost of extruding the target uracil from the helix. The selectivity of MUG for U:G over T:G can be attributed to all three factors; the methyl group slightly increases helix stability, and the larger size of the methyl group causes steric problems once the pyrimidine is in the cleavage pocket. However, the primary factor that distinguishes the cleavage rate for U:G versus T:G is the inductive property of the methyl group. The electron-donating group destabilizes the transition state, slowing the chemical cleavage step.
The data presented here indicate that the cleavage preferences of MUG for the series examined can be attributed, primarily to: 1) differences in helix stability and the 2) size and 3) electronic-inductive properties of the 5-substituents. We suggest that these properties are critical for other glycosylases, although other glycosylases may depend more or less on these properties. Data presented by Drohat and co-workers (15) indicate that the inductive property is even more substantial with human TDG than MUG; FU is cleaved 78 times faster than U, which is an order of magnitude greater than with MUG reported here. In contrast, the discrimination demonstrated by UNG relies upon steric exclusion as opposed to electronic-inductive effects, and the propensity to cleave U:A, U:G, and uracil in single-stranded DNA with similar efficiency suggests that helix formation energy is less important for UNG selectivity.
The third of the three glycosylases on human chromosome 12 is SMUG1, the single-strand specific glycosylase. SMUG1 has the unusual capacity to cleave oxidized thymine analogs such as 5-hydroxymethyluracil and 5-formyluracil, as well as uracil, but not thymine (18, 19). It has been proposed that specific hydrogen bonding interactions and a displaceable water molecule allow this unusual selectivity. A thymine DNA glycosylase analog found in a thermophilic species (50) selectively acts on pyrimidine analogs mispaired only with guanine, possibly through the formation of specific hydrogen bonds as had been previously proposed for MUG (24).
In addition to serving as good size markers, the halopyrimidines are also biologically important in their own right. Fluorouracil (51, 52) is a common chemotherapy agent, and its cytotoxic activity is in part attributed to its incorporation into and glycosylase cleavage from DNA. Both 5-chlorouracil (53, 54) and 5-bromouracil (55, 56) can occur in DNA from reactive inflammatory species such as HOCl from neutrophils and HOBr from eosinophils. The repair of these lesions is likely to be biologically important as well. An understanding of the mechanisms of glycosylase selectivity is important for understanding the substrate overlap among glycosylases, and ultimately to understand the vulnerabilities of the human genome to DNA damage.
Supplementary Material
This work was supported in part by National Institutes of Health Grants GM50351 and CA112293. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S11.
Footnotes
The abbreviations used are: UNG, uracil DNA glycosylase; MUG, mispaired uracil DNA glycosylase; SMUG, single stranded uracil DNA glycosylase; Tm, melting temperature; U, uracil; A, adenine; T, thymine; FU, 5-fluorouracil; ClU, 5-chlorouracil; HX, hypoxanthine; Pu, purine; 2AA, 2-aminoadenine; 2AP, 2-aminopurine; σm, Hammett meta parameter; ΔG, free energy; ΔH, enthalpy; ΔS, entropy; krel, relative rate constant, relative to the rate of U:G cleavage.
References
- 1.Lindahl, T. (2000) Mutat. Res. 462 129–135 [DOI] [PubMed] [Google Scholar]
- 2.Krokan, H. E., Nilsen, H., Skorpen, F., Otterlei, M., and Slupphaug, G. (2000) FEBS Lett. 476 73–77 [DOI] [PubMed] [Google Scholar]
- 3.Sung, J. S., and Demple, B. (2006) FEBS J. 273 1620–1629 [DOI] [PubMed] [Google Scholar]
- 4.Wilson, D. M., and Bohr, V. A. (2007) DNA Repair 6 544–559 [DOI] [PubMed] [Google Scholar]
- 5.Saul, R. L., and Ames, B. N. (1986) Basic Life Sci. 38 529–535 [DOI] [PubMed] [Google Scholar]
- 6.Mullaart, E., Lohman, P. H., Berends, F., and Vijg, J. (1990) Mutat. Res. 237 189–210 [DOI] [PubMed] [Google Scholar]
- 7.Lindahl, T. (1980) Methods Enzymol. 65 284–290 [DOI] [PubMed] [Google Scholar]
- 8.Caradonna, S. J., and Cheng, Y. C. (1980) J. Biol. Chem. 255 2293–2300 [PubMed] [Google Scholar]
- 9.Mosbaugh, D. W., and Bennett, S. E. (1994) Prog. Nucleic Acids Res. Mol. Biol. 48 315–370 [DOI] [PubMed] [Google Scholar]
- 10.Pearl, L. H. (2000) Mutat. Res. 460 165–181 [DOI] [PubMed] [Google Scholar]
- 11.Saava, R., McAuley-Hecht, K., Brown, T., and Pearl, L. (1995) Nature 373 487–493 [DOI] [PubMed] [Google Scholar]
- 12.Nilsen, H., Otterlei, M., Haug, T., Solum, K., Nagelhus, T. A., Skorpen, F., and Krokan, H. E. (1997) Nucleic Acids Res. 25 750–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sousa, M. M., Krokan, H. E., and Slupphaug, G. (2007) Mol. Aspects Med. 28 276–306 [DOI] [PubMed] [Google Scholar]
- 14.Neddermann, P., and Jiricny, J. (1993) J. Biol. Chem. 268 21218–21224 [PubMed] [Google Scholar]
- 15.Bennett, M. T., Rodgers, M. T., Hebert, A. S., Ruslander, L. E., Eisele, L., and Drohat, A. C. (2006) J. Am. Chem. Soc. 128 12510–12519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nilsen, H., Haushalter, K. A., Robins, P., Barnes, D. E., Verdine, G. L., and Lindahl, T. (2001) EMBO J. 20 4278–4286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wibley, J. E., Walters, T. R., Haushalter, K., Verdine, G. L., and Pearl, L. H. (2003) Mol. Cell 11 1647–1659 [DOI] [PubMed] [Google Scholar]
- 18.Boorstein, R. J., Cummings, A., Marenstein, D. R., Chan, M. K., Ma, Y., Neubert, T. A., Brown, S. M., and Teebor, G. W. (2001) J. Biol. Chem. 276 41991–41997 [DOI] [PubMed] [Google Scholar]
- 19.Matsubara, M., Tanaka, T., Terato, H., Ohmae, E., Izumi, S., Katayanagi, K., and Ide, H. (2004) Nucleic Acids Res. 32 5291–5302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sung, J. S., and Mosbaugh, D. W. (2000) Biochemistry 39 10224–10235 [DOI] [PubMed] [Google Scholar]
- 21.Hang, B., Downing, G., Guliaev, A. B., and Singer, B. (2002) Biochemistry 41 2158–2165 [DOI] [PubMed] [Google Scholar]
- 22.O'Neill, R. J., Vorob'eva, O. V., Shahbakhti, H., Zmuda, E., Bhagwat, A. S., and Baldwin, G. S. (2003) J. Biol. Chem. 278 20526–20532 [DOI] [PubMed] [Google Scholar]
- 23.Lari, S. U., Chen, C. Y., Vertessy, B. G., Morre, J., and Bennett, S. E. (2006) DNA Repair 5 1407–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barrett, T. E., Scharer, O. D., Savva, R., Brown, T., Jiricny, J., Verdine, G. L., and Pearl, L. H. (1999) EMBO J. 18 6599–6609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu, P., Burdzy, A., and Sowers, L. C. (2002) Chem. Res. Toxicol. 15 1001–1009 [DOI] [PubMed] [Google Scholar]
- 26.Gait, M. J. (ed) (1983) Oligonucleotide Synthesis: A Practical Approach, Oxford University Press, Oxford, NY
- 27.Fujimoto, J., Nuesca, Z., Mazurek, M., and Sowers, L. C. (1996) Nucleic Acids Res. 24 754–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richard, E. G. (1975) in CRC Handbook of Biochemistry and Molecular Biology: Nucleic Acid (Fasman, G.D., ed) 3rd Ed., Vol. 1, p. 589, Cleveland, OH [Google Scholar]
- 29.Allawi, H. T., and SantaLucia, J. (1997) Biochemistry 36 10581–10594 [DOI] [PubMed] [Google Scholar]
- 30.Watkins, N. E., Jr., and SantaLucia, J., Jr. (2005) Nucleic Acids Res. 33 6258–6267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Eritja, R., Horowitz, D. M., Walker, P. A., Ziehler-Martin, P., Boosalis, M., Goodman, M. F., Itakura, K., and Kaplan, B. E. (1986) Nucleic Acids Res. 14 8135–8153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cheong, C., Tinoco, I., Jr., and Chollet, A. (1988) Nucleic Acids Res. 16 5115–5122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Law, S. M., Eritja, R., Goodman, M. F., and Breslauer, K. J. (1996) Biochemistry 35 12329–12337 [DOI] [PubMed] [Google Scholar]
- 34.Petruska, J., Sowers, L. C., and Goodman, M. F. (1986) Proc. Natl. Acad. Sci. U. S. A. 83 1559–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hansch, C., Leo, A., Unger, S. H., Kim, K. H., Nikaitani, D., and Lien, E. J. (1973) J. Med. Chem. 16 1207–1216 [DOI] [PubMed] [Google Scholar]
- 36.Sober, H. A. (1970) Handbook of Biochemistry, pp. J3, The Chemical Rubber Co., Cleveland, OH
- 37.Valinluck, V., Wu, W., Liu, P., Neidigh, J. W., and Sowers, L. C. (2006) Chem. Res. Toxicol. 19 556–562 [DOI] [PubMed] [Google Scholar]
- 38.Slupphaug, G., Mol, C. D., Kavli, B., Arvai, A. S., Krokan, H. E., and Tainer, J. A. (1996) Nature 384 87–92 [DOI] [PubMed] [Google Scholar]
- 39.Wong, I., Lundquist, A. J., Bernards, A. S., and Mosbaugh, D. W. (2002) J. Biol. Chem. 277 19424–19432 [DOI] [PubMed] [Google Scholar]
- 40.Sagi, J., Hang, B., and Singer, B. (1999) Chem. Res. Toxicol. 12 917–923 [DOI] [PubMed] [Google Scholar]
- 41.Valinluck, V., Liu, P., Burdzy, A., Ryu, J., and Sowers, L. C. (2002) Chem. Res. Toxicol. 15 1595–1601 [DOI] [PubMed] [Google Scholar]
- 42.Minetti, C. A., Remeta, D. P., Zharkov, D. O., Plum, G. E., Johnson, F., Grollman, A. P., and Breslauer, K. J. (2003) J. Mol. Biol. 328 1047–1060 [DOI] [PubMed] [Google Scholar]
- 43.Krosky, D. J., Schwarz, F. P., and Stivers, J. T. (2004) Biochemistry 43 4188–4195 [DOI] [PubMed] [Google Scholar]
- 44.Parker, J. B., Bianchet, M. A., Krosky, D. J., Freidman, J. I., Amzel, L. M., and Stivers, J. T. (2007) Nature 449 433–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jang, Y. H., Sowers, L. C., Cagin, T., and Goddard, W. A., III (2001) J. Phys. Chem. 105 274–280 [Google Scholar]
- 46.Shapiro, R., and Kang, S. (1969) Biochemistry 8 1806–1810 [DOI] [PubMed] [Google Scholar]
- 47.Sun, B., Latham, K. A., Dodson, M. L., and Lloyd, R. S. (1995) J. Biol. Chem. 270 19501–19508 [DOI] [PubMed] [Google Scholar]
- 48.Dinner, A. R., Blackburn, G. M., and Karplus, M. (2001) Nature 413 752–755 [DOI] [PubMed] [Google Scholar]
- 49.Bianchet, M. A., Seiple, L. A., Jiang, Y. L., Ichikawa, Y., Amzel, L. M., and Stivers, J. T. (2003) Biochemistry 42 12455–12460 [DOI] [PubMed] [Google Scholar]
- 50.Liu, P., Burdzy, A., and Sowers, L. C. (2003) DNA Repair 2 199–210 [DOI] [PubMed] [Google Scholar]
- 51.Parker, W. B., and Cheng, Y. C. (1990) Pharmacol. Ther. 48 381–395 [DOI] [PubMed] [Google Scholar]
- 52.Seiple, L., Jaruga, P., Dizdaroglu, M., and Stivers, J. T. (2006) Nucleic Acids Res. 34 140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Whiteman, M., Jenner, A., and Halliwell, B. (1997) Chem. Res. Toxicol. 10 1240–1246 [DOI] [PubMed] [Google Scholar]
- 54.Jiang, Q., Blount, B. C., and Ames, B. N. (2003) J. Biol. Chem. 278 32834–32840 [DOI] [PubMed] [Google Scholar]
- 55.Henderson, J. P., Byun, J., Williams, M. V., Mueller, D. M., McCormick, M. L., and Heinecke, J. W. (2001) J. Biol. Chem. 276 7867–7875 [DOI] [PubMed] [Google Scholar]
- 56.Henderson, J. P., Byun, J., Takeshita, J., and Heinecke, J. W. (2003) J. Biol. Chem. 278 23522–23528 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.