

Abstract
Human cardiac fibroblasts are protected from oxidative stress triggered by inflammation after myocardial injury (Li, P. F., Dietz, R., and von Harsdorf, R. (1999) FEBS Lett. 448, 206–210) by expressing potent antioxidant defenses such as superoxide dismutases, catalases, glutathione-peroxidases, and peroxiredoxins. Recently the transcription factor FOXO3A has been shown to increase resistance to oxidative stress by up-regulation of mitochondrial superoxide dismutase and peroxisomal catalase (Kops, G. J., Dansen, T. B., Polderman, P. E., Saarloos, I., Wirtz, K. W., Coffer, P. J., Huang, T. T., Bos, J. L., Medema, R. H., and Burgering, B. M. (2002) Nature 419, 316–321; Nemoto, S., and Finkel, T. (2002) Science 295, 2450–2452). We hypothesized that FOXO3A also regulates the expression of Prx III, the mitochondrial peroxiredoxin, in human cardiac fibroblasts. We found that depletion of FOXO3A leads to a dramatic reduction of Prx III mRNA and protein in serum-deprived human cardiac fibroblasts. These data suggest that endogenous FOXO3A is necessary for base-line expression of Prx III. Next, we identified two putative FOXO3A DNA binding sites in Prx III promoter at –267 and –244 nucleotides relative to the start codon. We demonstrated that both sequences are required for binding of endogenous FOXO3A to the Prx III promoter by performing electromobility shift assays and chromatin immunoprecipitation assays. Inhibition of endogenous FOXO3A by insulin growth factor 1 prevented binding of FOXO3A to Prx III promoter. In contrast, overexpression of FOXO3A increased Prx III promoter activity. Furthermore, depletion of Prx III was associated with enhanced apoptosis and oxidative stress after serum deprivation. We conclude that FOXO3A mediates Prx III expression, and this may play a critical role in the resistance to oxidative stress in cardiac fibroblasts.
Peroxiredoxins (Prx)2 are antioxidant enzymes that modulate the cellular response to oxidative stress, in particular to H2O2 (4, 5). Multiple mammalian peroxiredoxins (I through VI) often coexist in the same cell in various intracellular locations and function as scavengers of cellular H2O2 released after stimulation with growth factors during proliferation, apoptosis, or oxidative stress (6, 7). Peroxiredoxins inactivate H2O2 by oxidizing a key cysteine residue in their catalytic center, which is subsequently reduced by thioredoxins, ultimately reduced by electrons from NADPH provided by cellular thioredoxin reductases (5, 6, 8, 9). All six peroxiredoxins are present in human cardiac fibroblasts, and Prx III is localized exclusively in mitochondria because of a mitochondrial targeting sequence (10). Prx III was originally identified in differentiating murine erythroleukemia cells and subsequently shown to reduce H2O2 using electrons from GSH via thioredoxin (7). Prx III is up regulated upon exposure to H2O2 or inhibitors of mitochondrial electron chain transporters (11), demonstrating that Prx III is important for cellular resistance to mitochondrial-derived oxidative stress. Prx III can also be induced by oxidative stress following serum stimulation in breast cancer cells or the oncogenic transcription factor c-myc in rat fibroblasts (12).
Serum deprivation also induces oxidative stress, which is at least partially derived from mitochondria (2). A key transcription factor that coordinates resistance to oxidative stress during serum withdrawal is the forkhead box transcription factor FOXO3A (a.k.a. FKHLR1). FOXO3A is part of the FOXO family of transcription factors that upon exposure to oxidative stress, translocate to nucleus and activate transcription by specifically binding to the consensus sequence TTGTTTAC in the promoters of target genes (13). Thus, oxidative stress triggered by serum deprivation causes FOXO3A to bind to gene promoters and activate transcription of the two essential antioxidant enzymes mitochondrial superoxide dismutase (MnSOD) and catalase, which scavenge superoxide and hydrogen peroxide, respectively (2, 14).
In the current study, we show that FOXO3A also regulates expression of Prx III in serum-deprived human cardiac fibroblasts and we identify specific DNA-binding elements in the Prx III promoter. These data provide new insights into the mechanisms of cellular resistance to oxidative stress in human fibroblasts.
EXPERIMENTAL PROCEDURES
Cell Culture—Primary human cardiac fibroblast cells (catalog number ACBRI 5118) were purchased at passage two from Cell Systems (Kirkland, WA) and grown in FGM-2 media (Cambrex, Baltimore, MD). All cells were harvested at passage 3–6 and used at 50–70% confluence for transfection experiments. Human embryonic kidney-293 (HEK-293) cells were bought from ATCC (Manassas, Virginia) and grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum.
Antibodies—FOXO3A antibodies were purchased from Cell Signaling (Western blotting) and from Santa Cruz Biotechnology (chromatin immunoprecipitation assay). Prx I and III antibodies were purchased from Lab Frontier (Seoul, Korea), and FLAG antibody was purchased from Sigma. The 32P radioactive isotope (10 μCi/μl) was purchased from GE Healthcare.
Plasmid Constructs—The immediate Prx III promoter (–554 to +1) was amplified using genomic DNA as template and the primer pair, 5′-CTT CAA GTA CTA TGC TCA CCA C-3′ and 5′-GCA GCC GAG ATC TTC AGT GCA-3′. The DNA fragment obtained by PCR was digested with ScaI and BglII restriction endonucleases, purified, and ligated in pGL3-Basic vector (Promega, Madison, WI), previously digested with SmaI and BglII, yielding pGL3-Basic-Prx III promoter plasmid.
A 100-bp double stranded DNA fragment spanning from –300 to –200 within PRX III promoter, which spans exactly the two putative FOXO3A DNA binding elements (FDBE), having compatible ends for NheI (in the 5′ end) and BglII (in the 3′ end) endonucleases, was obtained by annealing the complementary single stranded oligonucleotides: 5′-CTA GCA CAG AAA TAC TAG ACA CAG TAA TCC ACA CAA GGT TAA CAA AAC AGT GGG AAA TAT GGA AAC AAA TAC CTA ATG CCC CGC CTG GAT TCG TTC TTT T-3′ and 5′-GAT CAA AAG AAC GAA TCC AGG CGG GGC ATT AGG TAT TTG TTT CCA TAT TTC CCA CTG TTT TGT TAA CCT TGT GTG GAT TAC TGT GTC TAG TAT TTC TGT G-3′. The purified 100-bp double-stranded DNA fragment was ligated in the pGL3-promoter vector (Promega), previously digested with NheI and BglII restriction endonucleases, upstream of the SV40 promoter, yielding pGL3-SV40-Prx III-FDBE (–300 to –200).
Plasmid Transfection—For electromobility shift assay (EMSA) and chromatin immunoprecipitation (ChIP) assay, HEK-293 cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Approximately 2 × 106 cells were transfected with 3 μg of pcDNA3-FLAG-FOXO3A (15) or with pcDNA3 empty vector (as control) in a 100-mm cell culture dish at 60% confluency by using Efectene reagent (Qiagen, Hilden, Germany). 36 h post-transfection, cells were serum-deprived for another 8 h, harvested, and used for the preparation of nuclear extracts (described below).
For luciferase reporter promoter assays, human cardiac fibroblasts were plated at density of 200,000 cells/35-mm dish (30% confluency) and 24 h later co-transfected with 2 μg of either full-length Prx III promoter luciferase construct (pGL3-Basic-Prx III) or Prx III-FDBE luciferase plasmid (pGL3-SV40-Prx III-FDBE (–300 –200) together with pcDNA3-FLAG-FOXO3A or with pcDNA3 empty vector (as control) by using Effectene reagent (Qiagen). To control the efficiency of transfection, cells were co-transfected simultaneously with Renilla luciferase. Twenty-four hours later, cells were changed to serum-free medium and after another 24 h were harvested and processed for luciferase assay (as described below).
Transfection with Small Interference RNA—HEK-293 cells were trypsinized and plated on 60-mm dishes (for RNA extraction) or 35-mm dishes (for Amplex Red assay) at 30% confluence for transfection. To down-regulate FOXO3A or/and Prx III, siFOXO3A (sense sequence: 5′-UCA CCU UCA GUA AGC AAG CCG UGC A-3′; antisense sequence: 5′-UGC ACG GCU UGC UUA CUG AAG GUG A-3′) were purchased from Invitrogen. siPrx III (sense sequence, 5′-GGG UAC AGC CGU UGU CAA Utt-3′ antisense sequence, 5′-AUU GAC AAC GGC UGU ACC Ctt-3′) were purchased from Qiagen. Individual small interference RNA (siRNA), oligofectamine, and Opti-MEM (Invitrogen) were mixed and incubated at room temperature for 20 min. siRNA-oligofectamine complexes were incubated with the cells for 5 h, in Opti-MEM with 10% fetal bovine serum (siRNA at 50 nm final concentration). After 72 h, siRNA-oligofectamine complexes were removed and cells placed in serum-free Dulbecco's modified Eagle's medium (ATCC) for 40 h. Cells were processed for Amplex Red assay or RNA extraction, or fluorescence-activated cell sorter analysis as described.
Human cardiac fibroblasts were trypsinized and plated on 100-mm dishes at 30–50% confluence 24 h prior to transfection. Individual siRNAs (at 50 nm final concentration), oligofectamine, and Opti-MEM (Invitrogen) were mixed and incubated at room temperature for 20 min. siRNA-oligofectamine complexes were incubated with the cells for 96 h, after which siRNA-oligofectamine complexes were removed and cells placed in serum-free fibroblast growth medium medium (Cambrex) for 24 h. To control for possible nonspecific effects of siRNAs, we used the universal negative control siRNA commercially available from Ambion.
Preparation of Nuclear Extracts—Isolation of nuclear fractions was performed as described previously by Neufeld and White (16). Briefly, human cardiac fibroblasts or HEK-293 cells were scraped into ice-cold PBS, rinsed, and re-suspended in L-buffer (PBS containing 0.1% Triton X-100, 0.1% Nonidet P-40, pH 7.4) with protease inhibitors. Nuclei and non-lysed cells were pelleted at 1000 × g for 10 min at 4 °C. The supernatant fraction was collected and classified as the non-nuclear fraction. The nuclear pellet was purified by passage through a 0.21-gauge needle 5 times and passage through 0.85 m sucrose cushion in PBS (20,800 × g, microcentrifuge; 15 min at 4 °C). Nuclei in the pellet were lysed by sonication, treated with DNase I (500 units total), and re-sonicated to prepare a nuclear lysate. Non-nuclear fractions were characterized by immunoblotting for the cytoskeletal marker β-tubulin (Sigma) and nuclear extracts by immunoblotting for histone H3 (Cell Signaling).
Western Blotting—Human cardiac fibroblasts were harvested in lysis buffer and processed for Western blotting as published previously (17). After separation by SDS-PAGE and transfer to nitrocellulose membranes, signals were detected with specific corresponding antibodies, visualized with enhanced chemiluminescence, and quantified by laser densitometry (17).
Quantitative Real-time PCR—Quantification of human Prx III and 18 S rRNA was performed by amplification of cDNA using the LightCycler thermocycler (Roche, Nutley, NJ) as described previously (17, 18). The primers used for detection of Prx III mRNA message were forward: 5′-GAT GTG AAC TGT GAA GTT GTC GCA GTC TC-3′ and reverse: 5′-GCT GGA CTT GGC TTG ATC GTA GGA GAA T-3′.
EMSA—Complementary single-stranded oligonucleotides were annealed, and the resulted double-stranded DNA fragments were 5′-end labeled with [γ-32P]ATP by using the Ready-to-Go polynucleotide kinase kit according to the instructions of the supplier (GE Healthcare). 32P-labeled probes were incubated (binding buffer: KCl 100 mm, dithiothreitol 0.1 mm, EDTA 0.1 mm in Tris-HCl 20 mm, pH 8.3, supplemented with sonicated salmon sperm DNA (1 μg/30 μl binding reaction)) with total nuclear extracts (prepared as described above) for 30 min on ice. Binding reactions were stopped by the addition of sample buffer and subjected to electrophoresis.
ChIP Assay—For ChIP assays, we used the chromatin immunoprecipitation assay kit, purchased from Upstate and followed the instructions of the supplier. Briefly, cells were fixed with 1% formaldehyde for 10 min, washed, and harvested in SDS lysis buffer. After sonication, lysates containing soluble chromatin were incubated overnight with 4 μg of anti-FOXO3A antibody (H-144, Santa Cruz Biotechnology) or with 4 μg of anti-FLAG antibody (Sigma) for the overexpressed FOXO3A. DNA-protein immunocomplexes were precipitated with protein A-agarose beads, washed, and eluted. An overnight incubation with sodium chloride (final concentration of 0.2 m) at 65 °C was performed to reverse cross-link DNA-protein complex. The samples were proteinase K-digested as recommended, purified with phenol/chloroform, and precipitated with ethanol. The eluates were used as templates in PCR reactions using the primers 5′-CGG ACT AAA ACT GCA TTT GTA ATT A-3′ and 5′-CAC GTG GTT TTC CTA CTG TC-3′. The expected DNA fragment was 214 bp in length and amplified region of the Prx III promoter, which encompasses the FOXO3A binding sites.
Flow Cytometry—Early apoptosis was measured by staining cells with Annexin V-phycoerythrin (for apoptosis), 7-amino-actinomycin D for necrosis) using Annexin V-PE apoptosis detection kit I (559763; BD Biosciences) following the manufacturer's instructions. In summary, after transfection, HEK-293 cells were trypsinized, washed with cold PBS twice, and resuspended in 1× binding buffer. 100 μl (0.1 million cells) of cell suspension were incubated with 5 μl of Annexin V-PE and 5 μl of 7-amino-actinomycin D. After staining, 10,000 cells were analyzed by flow cytometry within one hour, and results were reported as percent cells positive for Annexin V and negative for 7-amino-actinomycin D (indicative of early apoptosis).
Measurement of Hydrogen Peroxide (H2O2) using Amplex Red Assay—H2O2 production from intact cells was measured using the oxidation of the fluorogenic indicator Amplex Red (Amplex Red hydrogen peroxide/peroxidase assay kit (A22188; Molecular Probes Inc., Eugene, OR) in the presence of horseradish peroxidase (HRP) as per the manufacturer's instructions. The concentrations of horseradish peroxidase and Amplex Red in the incubation were 0.1 unit/ml and 50 μm, respectively. Fluorescence was recorded in a microplate reader (CytoFluor II; Applied Biosystems, Foster City, CA) with 530 nm excitation and 590 nm emission wavelengths. Standard curves obtained by adding known amounts of H2O2 to assay medium in the presence of the reactants (Amplex Red and horseradish peroxidase) were linear up to 100 μm. Background fluorescence was measured in the presence or absence of cells and presented as fluorescence minus background and reported (μm/106 cells/30 min). After transfection with various siRNAs and serum deprivation as above, medium was removed, and 500 μl of 1× reaction buffer was added to 35-mm dish and incubated with cells for 1 h at 37 °C. Next, 50 μl of reaction mixture from each dish was aliquoted in triplicates and placed into individual wells of a microplate (96-wells plate), and then 50 μl of the Amplex Red reagent/HRP working solution were added to each well. The reaction was incubated at room temperature for 30 min, protected from light, and then fluorescence was measured. Separately, cells treated identically for each sample from experimental setup were incubated with polyethylene glycol-catalase (100 units/ml) overnight after which media was removed and cells processed for Amplex Red assay as described. For Fig. 4A, the final signal reported in graph is the catalase-inhibitable cellular H2O2 production for each cellular sample normalized to cell number per dish.
FIGURE 4.

Peroxiredoxin III protects against apoptosis by reducing oxidative stress triggered by serum deprivation. HEK-293 cells were transfected at 40% confluence with siCon, siPrx III, siFOXO3A, or combined as described under “Experimental Procedures.” Next, cells were serum-deprived for 40 h, harvested, stained, and 10,000 cells per sample were assayed for Annexin V-PE fluorescence staining as described under “Experimental Procedures” (Fig. 4A). Data are mean values ± S.E. from four experiments. *, p < 0.05 versus control. Alternatively, in separate experiments, cells were harvested after 24 h of serum deprivation for H2O2 measurement using Amplex Red assay as described under “Experimental Procedures” (B). Data are mean ± S.E. from six experiments. *, p < 0.05 versus control.
Luciferase Measurements—Detection of luciferase activity in human cardiac fibroblasts was performed by using the dualluciferase reporter assay system from Promega and following strictly the instructions of the supplier. Briefly, cells were lysed in lysis buffer, and 20 μl of the cell lysate were used for the detection of the chemiluminescence signal by incubation at room temperature with 100 μl of luciferase substrate. The signals were read in a TD-20/20 Luminometer (Turner Designs, Sunnyvale, CA), and values obtained from firefly luciferase activity were normalized to Renilla luciferase and protein concentration.
Statistical Analysis—Results are expressed as mean ± S.E. Statistical significance was assessed by using paired Student's t test or one-way analysis of variance using GraphPad Prism software version 4.0 for Mac (San Diego, CA). A value of p < 0.05 was considered to be statistically significant.
RESULTS
FOXO3A Is Required for Expression of Prx III in Human Cardiac Fibroblasts—It is well established that FOXO3A is involved in cellular resistance to oxidative stress in response to serum deprivation by increasing the expression of antioxidant enzymes catalase and MnSOD (2, 3). We hypothesized that FOXO3A also regulates Prx III (which scavenges H2O2 and is localized exclusively in mitochondria). To test whether FOXO3A is necessary for the expression of Prx III, we depleted FOXO3A by transfecting human cardiac fibroblasts (HCFb) with specific siRNA. We first confirmed that FOXO3A protein was reduced by using Western blotting. Depletion of FOXO3A caused 70% reduction in expression of Prx III protein (Fig. 1A). In contrast, expression of Prx I, a cytosolic homologue of Prx III (4, 5), was not changed by depletion of FOXO3A (data not shown). This strongly supports the notion that FOXO3A regulates expression of Prx III in HCFb. Because FOXO3A is a transcription factor, we tested whether it is required for transcription of Prx III. We therefore performed quantitative PCR using RNA extracted from HCFb transfected with either control siRNA or siFOXO3A. Depletion of FOXO3A was associated with a 67% reduction in Prx III mRNA (Fig. 1B), consistent with protein down-regulation. Thus, FOXO3A is important for regulation of Prx III mRNA and protein in serum-deprived HCFb.
FIGURE 1.

FOXO3A is required for expression of Prx III in human cardiac fibroblasts. A, human cardiac fibroblasts were transfected with non-silencing siRNA (siCon) or siRNA against FOXO3A (siFOXO3A). Western blotting was performed for FOXO3A and Prx III. CDK4 served as a loading control. The blot shown is representative of three similar experiments. B, human cardiac fibroblasts were transfected as above and harvested for RNA after 48 h. Prx III mRNA was quantified using quantitative real-time PCR. Data are mean ± S.E. from three experiments normalized to 18 S. *, p = 0.001 versus control. C, a scan of Prx III gene promoter, performed with the GENOMATIX software revealed the presence of two putative binding sites for the transcriptional factor FOXO3A. The binding sites (bold in the sequence) are located –267 to –259 and –244 to –236. Mismatches from the published consensus sequence are underlined.
FOXO3A Binds Its Cognate Sequences within the Prx III Promoter—Because FOXO3A may modulate Prx III transcription by binding to its promoter, we scanned the Prx III promoter for putative FOXO3A binding sites using the Genomatix software. We found two putative FOXO regulatory sequences, the first located from –267 to –259 (matrix similarity of 0.886) and the second located from –244 to –236 (matrix similarity of 0.891) relative to the start codon (Fig. 1C).
To determine whether FOXO3A binds to Prx III promoter at the sites predicted by the Genomatix software, we first performed EMSA. We found that a 50 bp double-stranded oligonucleotide probe encompassing both putative FOXO3 binding sites in the Prx promoter (underlined in Fig. 2A, WT) exhibited an electromobility shift (Fig. 2B) when incubated with a nuclear extract prepared from serum-deprived human cardiac fibroblasts. We then sought to determine the specificity of the FOXO binding sites within the Prx III promoter. The two putative binding sites were replaced with random-generated sequences and tested using EMSA. We found maximal binding to the wild-type probe (Fig. 2C) and decreased binding when the probe was mutated, either at the distal binding site (M01; Fig. 2A) or at the proximal binding site (M02; Fig. 2A). When both binding sites were mutated, the electromobility shift was minimal (Fig. 2C). These data demonstrate that binding of FOXO requires both –267 to –259 and –244 to –236 promoter sequences. We next depleted FOXO3A with siFOXO3A in HEK-293 cells and performed EMSA using the corresponding nuclear extracts. As expected, the band was drastically reduced by depletion of FOXO3A when compared with control siRNA transfected cells (Fig. 2D). We also overexpressed FOXO3A by transfecting with plasmid encoding wild-type FOXO3A and found a dramatic increase in the intensity of electromobility shift (data not shown). These results suggest that endogenous FOXO3A binds to the Prx III promoter.
FIGURE 2.

FOXO3A binds its cognate sequences within the Prx III promoter. A and B, binding of FOXO3A to Prx III promoter in vitro. Representative EMSA demonstrating that a 50 bp double-stranded oligonucleotide encompassing both putative FOXO3A binding sites (sequence WT-FDBE in panel A) exhibits an electromobility shift when incubated with nuclear extracts from human cardiac fibroblasts (see under “Experimental Procedures” for EMSA). C, binding of FOXO3A to Prx III is specific to –267 and –259 sites. To show specificity for binding, the FOXO3A binding sites within Prx III promoter were replaced with random sequences (sequences M01, M02, and M03 as indicated in A) and used in EMSA performed on HEK-293 cells nuclear extracts. D, endogenous FOXO3A binds to Prx III promoter. To test whether endogenous FOXO3A is required, HEK-293 cells were transfected (as described under “Experimental Procedures”) with siCon or siFOXO3A, and 96 h later cells were harvested and processed, and the corresponding nuclear extracts (10 and 20 μg, respectively) were employed in mobility shift assay (D). Each blot is representative of three similar experiments. E, binding of FOXO3A to the Prx III promoter is impaired by IGF-1 treatment. HEK-293 cells were treated with IGF-1 (40 ng/ml medium) for 8 h and the corresponding nuclear extracts employed in EMSA as described under “Experimental Procedures.” Shown are increasing amounts of nuclear lysates. F, FOXO3A binds in vivo to Prx III promoter. Chromatin immunoprecipitation assays were performed in HEK-293 cells as described under “Experimental Procedures.” HEK-293 cells were transfected with control or FOXO3A plasmids and harvested for ChIP assay after 48 h. FOXO3A was immunoprecipitated either with an antibody against FLAG from cells transfected with pcDNA3-FLAG-FOXO3 (upper panel) or with FOXO3A antibody (middle and lower panels). This is representative of three similar experiments. The lower panel is identical with middle panel; however the image was inverted to enhance the intensity of FOXO3A PCR band.
It is well established that IGF-1 or serum activate the phosphatidylinositol 3-kinase/Akt pathway, leading to inactivation of FOXO3A by serine phosphorylation and subsequent nuclear exclusion (19). Therefore, we hypothesized that treatment with IGF-1 will inhibit the electromobility shift of DNA probes containing FOXO binding sites. Indeed, IGF-1 treatment (40 ng/ml) for 8 h caused a substantial decrease in the amount of shifted DNA in treated cells versus the serum-free cells (Fig. 2E).
These results demonstrate that FOXO3A binds its cognate sequences within the Prx III promoter in vitro. To confirm that FOXO3A also binds to the Prx III promoter in vivo, we performed ChIP. First, we transfected HEK-293 cells with a plasmid encoding the FLAG-tagged wild-type FOXO3A and performed PCR amplification of the 214 bp DNA fragment encompassing the double FOXO binding site of Prx III promoter in cell lysates immunoprecipitated with antibody against the Flag epitope. As expected, in the presence of antibody, the DNA fragment could be amplified by PCR (Fig. 2F, top panel) whereas no amplification was detected in the absence of the Flag antibody. We also tested whether endogenous FOXO3A binds to Prx III promoter by performing ChIP after immunoprecipitating with antibody against FOXO3A and obtained similar results (Fig. 2F, middle and lower panels). Taken together, these results confirm that FOXO3A binds to the Prx III promoter.
FOXO3A Is an Activator of the Prx III Promoter in Human Cardiac Fibroblasts in Response to Serum Deprivation—After establishing that FOXO3A binds to Prx III promoter, we wanted to determine the functional consequence of this binding in human cardiac fibroblasts. We first cloned the promoter for human Prx III (see under “Experimental Procedures”) upstream of luciferase in the pGL3 Basic plasmid (pGL3-Prx III promoter). Then, we co-transfected HCFb with this construct and either a control or a FOXO3A-expressing plasmid and measured luciferase activity 24 h later. We observed a 4.5-fold increase in Prx III promoter activity when cells were co-transfected with FOXO3A versus cells co-transfected with the control plasmid (Fig. 3). In a separate set of experiments we replaced the whole Prx III promoter with a short sequence containing FOXO3A binding sites (pGL3-Prx III FDBE; see “Experimental Procedures” and Fig. 3) upstream of luciferase, co-expressed it with FOXO3A plasmid and obtained similar results (Fig. 3). Altogether, these experiments demonstrate that FOXO3A is a potent activator of Prx III in serum-free conditions by directly binding to Prx III promoter in HCFb.
FIGURE 3.

FOXO3A increases peroxiredoxin III promoter activity in human cardiac fibroblasts. Human cardiac fibroblasts were co-transfected at 40% confluence with a reporter plasmid containing the DNA-binding elements of FOXO3A in the Prx III promoter (pGL3-Prx III FDBE) and a plasmid containing human FOXO3A (pcDNA3-FLAG-FOXO3A). Alternatively, cells were transfected with a reporter plasmid containing the full-length Prx III promoter (pGL3-Prx III promoter) with or without pcDNA3-FLAG-FOXO3A for 48 h. To control for transfection efficiency, all cells were also co-transfected in 1:10 ratio with Renilla luciferase plasmid. Cells were serum-deprived for 24 h and harvested for luciferase assays as described under “Experimental Procedures.” The data represent mean ± S.E. of luciferase relative luminescence units (RLU) from four different experiments normalized to corresponding Renilla luciferase activity. *, **, p < 0.005 versus control.
Prx III Protects against Apoptosis by Scavenging H2O2 in Response to Serum Deprivation—In HEK-293 cells FOXO3A protects from serum-induced oxidative stress and apoptosis by up-regulating mitochondrial superoxide dismutase. To test whether the up-regulation of Prx III by FOXO3A is required for protection against serum deprivation-induced apoptosis, we depleted serum-starved HEK-293 cells from Prx III, FOXO3A or both by performing transfection with siRNA against Prx III and FOXO3A and measured apoptosis by performing fluorescence-activated cell sorter analysis for Annexin V, a marker of early apoptosis. We found that deletion of Prx III, increases apoptosis by 54%, deletion of FOXO3A enhances apoptosis by 275% and combination of both further augments apoptosis by 431% (n = 4, p < 0. 05, Fig. 4A). The pro-apoptotic effect caused by lack of Prx III and FOXO3A was associated with an increased in total cellular level of H2O2 as assayed by Amplex Red (Fig. 4B). Similar results were obtained in human cardiac fibroblasts (data not shown). Deletion of FOXO3A had a more potent effect on both apoptosis and oxidative stress. Taken together, these data demonstrate that Prx III is required to protect against oxidative stress-induced apoptosis in response to serum deprivation and quiescence.
DISCUSSION
In the present study, we provide evidence that FOXO3A is essential for expression of Prx III in response to serum deprivation in human cardiac fibroblasts. We further demonstrate that FOXO3A binds to the Prx III promoter at two obligatory sites: –267 to –259 and –244 to –236 upstream of the ATG of the first exon of Prx III (Fig. 1C). The binding of FOXO3A to Prx III promoter was impaired upon stimulation with IGF-1 (which inactivates and excludes FOXO3A from the nucleus) and was enhanced by serum deprivation, a known stimulus for activation and nuclear translocation of FOXO3A (20).
These effects were mediated by modulation of FOXO3A activity because overexpression of FOXO3A enhanced the activity of Prx III promoter (Fig. 3) whereas depletion of endogenous FOXO3A inhibited expression of Prx III mRNA and protein in serum-deprived fibroblasts (Fig. 1, A and B). These new findings support the notion that FOXO3A is a critical regulator of Prx III expression in human cardiac fibroblasts.
Mitochondria are key sources of oxidative stress in physiological response to serum deprivation, growth factor stimulation, or apoptosis. Recent evidence has accumulated to demonstrate that Prx III functions in parallel with mitochondrial glutathione peroxidases to protect mitochondria against oxidative stress by scavenging H2O2 (21, 22). Mitochondria contain exclusively not only Prx III (30 times more abundant than glutathione peroxidase 1) but also thioredoxin 2 (Trx-2) and thioredoxin reductase 2, required to regenerate oxidized Prx III by transferring electrons from GSH and NADPH (10). Both Prx III and Trx-2 are necessary to prevent apoptosis induced by oxidative stress in mitochondria of HeLa cells exposed to tumor necrosis factor-α (21), glutathione depletion (22), or staurosporine (10). Furthermore, Prx III can reduce mitochondrial oxidative stress and apoptosis by inactivating H2O2 as observed by transfecting HeLa cells with Prx III or mitochondrial-targeted catalase (10).
Several mechanisms have been identified that regulate the function of peroxiredoxins. These enzymes are reversibly oxidized and inactivated by H2O2, and their reactivation is mediated by either thioredoxins or sulfiredoxin, depending on the level of oxidation (4). Phosphorylation of peroxiredoxins 1, 2, and 3 at Thr-90 by cyclin-dependent kinases also inactivates them during progression through the M phase of the cell cycle (23). Wonsey et al. (12) have recently shown that Prx III expression is also regulated by the pro-oncogenic transcription factor c-myc. Overexpression of c-myc in rat fibroblasts causes oncogenic transformation and is mediated by enhanced expression of Prx III by direct binding of c-myc to the Prx III promoter. Deletion of Prx III in c-myc-transformed fibroblasts stimulated with serum leads to severe mitochondrial dysfunction by reducing the mitochondrial redox potential, causing impaired mitochondrial biogenesis, reduced proliferation rate, and ability to promote tumor formation (12). The up-regulation of Prx III occurred in response to serum stimulation (12). In contrast, we found that serum withdrawal stimulates binding of FOXO3A to Prx III promoter, and the DNA consensus sequence for FOXO3A is different from the one for c-myc on Prx III promoter. Together these data suggest that c-myc activates Prx III expression during proliferation, whereas FOXO3A regulates Prx III expression in response to serum deprivation.
FOXO3A has been recently shown to be a key transcription factor involved in
resistance to oxidative stress in quiescent 3T3 fibroblasts exposed to serum
withdrawal (2). Both serum
withdrawal and serum stimulation cause oxidative stress in cells. However,
resistance to oxidative stress is mediated by different mechanisms in each
condition. In serum or growth factors stimulated cells, protection is
conferred by activation of protein kinase B/Akt, which prevents generation of
oxidative stress in mitochondria by coupling the glycolytic pathway with
inhibition of mitochondrial release of cytochrome c
(24,
25). In contrast, in quiescent
or serum-deprived cells, the forkhead transcription factor FOXO3A supports
resistance to oxidative stress by increasing expression of MnSOD
(2) and catalase
(3) by directly interacting
with their promoters. Our observation that FOXO3A also regulates expression of
Prx III, another H2O2 scavenging antioxidant enzyme is
consistent with these studies. Furthermore, Prx III up-regulation by FOXO3A is
important for the survival response and protects against oxidative stress
caused by serum deprivation (Fig. 4,
A and B). MnSOD is ubiquitously expressed and
confers protection by converting
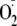 to H2O2 released by mitochondria during serum
deprivation, whereas catalase protects by converting
H2O2 to H2O. However, catalase expression is
limited to peroxisomes, and therefore its antioxidant capacity depends on
diffusion of H2O2 generated in mitochondria or other
cellular compartments. It is tempting to speculate that simultaneous
up-regulation of MnSOD and Prx III (both have the same ubiquitous tissue
distribution) by FOXO3A provides the ability to scavenge excessive
H2O2 produced by enhanced MnSOD activation in
mitochondria during serum deprivation. In contrast, up-regulation of catalase
by FOXO3A may serve the purpose of inactivating the H2O2
generated locally in peroxisomes during fatty acid oxidation in cells that
switch from glucose to lipid-based metabolism as proposed by Burgering et
al. (26).
to H2O2 released by mitochondria during serum
deprivation, whereas catalase protects by converting
H2O2 to H2O. However, catalase expression is
limited to peroxisomes, and therefore its antioxidant capacity depends on
diffusion of H2O2 generated in mitochondria or other
cellular compartments. It is tempting to speculate that simultaneous
up-regulation of MnSOD and Prx III (both have the same ubiquitous tissue
distribution) by FOXO3A provides the ability to scavenge excessive
H2O2 produced by enhanced MnSOD activation in
mitochondria during serum deprivation. In contrast, up-regulation of catalase
by FOXO3A may serve the purpose of inactivating the H2O2
generated locally in peroxisomes during fatty acid oxidation in cells that
switch from glucose to lipid-based metabolism as proposed by Burgering et
al. (26).
Acknowledgments
We are indebted to Dr. Bernard Lassègue for his critical reading of the manuscript.
This work was supported by an American Heart Association (AHA) Scientist Development Grant (0335244N), AHA Grant-in-aid (0755321B), and National American Federation of Aging Research grant (to D. S.), AHA post-doctoral fellowship Grant (0625309B) (to C. B. C.), and by the National Institutes of Health cardiovascular training grant (to I. C.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: Prx, peroxiredoxins; FDBE, FOXO3A DNA binding elements; EMSA, electromobility shift assay; ChIP, chromatin immunoprecipitation; HEK, human embryonic kidney; si, small interference; PBS, phosphate-buffered saline; PE, phycoerythrin; HRP, horseradish peroxidase; MnSOD, mitochondrial superoxide dismutase; HCFb, human cardiac fibroblasts; siRNA, small interference RNA; IGF, insulin-like growth factor; Trx, thioredoxin.
References
- 1.Li, P. F., Dietz, R., and von Harsdorf, R. (1999) FEBS Lett. 448 206–210 [DOI] [PubMed] [Google Scholar]
- 2.Kops, G. J., Dansen, T. B., Polderman, P. E., Saarloos, I., Wirtz, K. W., Coffer, P. J., Huang, T. T., Bos, J. L., Medema, R. H., and Burgering, B. M. (2002) Nature 419 316–321 [DOI] [PubMed] [Google Scholar]
- 3.Nemoto, S., and Finkel, T. (2002) Science 295 2450–2452 [DOI] [PubMed] [Google Scholar]
- 4.Rhee, S. G., Chae, H. Z., and Kim, K. (2005) Free Radic. Biol. Med. 38 1543–1552 [DOI] [PubMed] [Google Scholar]
- 5.Wood, Z. A., Poole, L. B., and Karplus, P. A. (2003) Science 300 650–653 [DOI] [PubMed] [Google Scholar]
- 6.Chae, H. Z., Kim, H. J., Kang, S. W., and Rhee, S. G. (1999) Diabetes Res. Clin. Pract. 45 101–112 [DOI] [PubMed] [Google Scholar]
- 7.Kang, S. W., Chae, H. Z., Seo, M. S., Kim, K., Baines, I. C., and Rhee, S. G. (1998) J. Biol. Chem. 273 6297–6302 [DOI] [PubMed] [Google Scholar]
- 8.Chae, H. Z., Kang, S. W., and Rhee, S. G. (1999) Methods Enzymol. 300 219–226 [DOI] [PubMed] [Google Scholar]
- 9.Wood, Z. A., Schroder, E., Robin Harris, J., and Poole, L. B. (2003) Trends Biochem. Sci. 28 32–40 [DOI] [PubMed] [Google Scholar]
- 10.Chang, T. S., Cho, C. S., Park, S., Yu, S., Kang, S. W., and Rhee, S. G. (2004) J. Biol. Chem. 279 41975–41984 [DOI] [PubMed] [Google Scholar]
- 11.Araki, M., Nanri, H., Ejima, K., Murasato, Y., Fujiwara, T., Nakashima, Y., and Ikeda, M. (1999) J. Biol. Chem. 274 2271–2278 [DOI] [PubMed] [Google Scholar]
- 12.Wonsey, D. R., Zeller, K. I., and Dang, C. V. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 6649–6654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pierrou, S., Hellqvist, M., Samuelsson, L., Enerback, S., and Carlsson, P. (1994) EMBO J. 13 5002–5012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nemoto, S., Combs, C. A., French, S., Ahn, B. H., Fergusson, M. M., Balaban, R. S., and Finkel, T. (2006) J. Biol. Chem. 281 10555–10560 [DOI] [PubMed] [Google Scholar]
- 15.Brunet, A., Sweeney, L. B., Sturgill, J. F., Chua, K. F., Greer, P. L., Lin, Y., Tran, H., Ross, S. E., Mostoslavsky, R., Cohen, H. Y., Hu, L. S., Cheng, H. L., Jedrychowski, M. P., Gygi, S. P., Sinclair, D. A., Alt, F. W., and Greenberg, M. E. (2004) Science 303 2011–2015 [DOI] [PubMed] [Google Scholar]
- 16.Neufeld, K. L., and White, R. L. (1997) Proc. Natl. Acad. Sci. U. S. A. 94 3034–3039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cucoranu, I., Clempus, R., Dikalova, A., Phelan, P. J., Ariyan, S., Dikalov, S., and Sorescu, D. (2005) Circ. Res. 97 900–907 [DOI] [PubMed] [Google Scholar]
- 18.Sorescu, D., Weiss, D., Lassegue, B., Clempus, R. E., Szocs, K., Sorescu, G. P., Valppu, L., Quinn, M. T., Lambeth, J. D., Vega, J. D., Taylor, W. R., and Griendling, K. K. (2002) Circulation 105 1429–1435 [DOI] [PubMed] [Google Scholar]
- 19.Brunet, A., Bonni, A., Zigmond, M. J., Lin, M. Z., Juo, P., Hu, L. S., Anderson, M. J., Arden, K. C., Blenis, J., and Greenberg, M. E. (1999) Cell 96 857–868 [DOI] [PubMed] [Google Scholar]
- 20.Zheng, W. H., Kar, S., and Quirion, R. (2000) J. Biol. Chem. 275 39152–39158 [DOI] [PubMed] [Google Scholar]
- 21.Hansen, J. M., Zhang, H., and Jones, D. P. (2006) Toxicol. Sci. 91 643–650 [DOI] [PubMed] [Google Scholar]
- 22.Zhang, H., Go, Y. M., and Jones, D. P. (2007) Arch. Biochem. Biophys. 465 119–126 [DOI] [PubMed] [Google Scholar]
- 23.Chang, T. S., Jeong, W., Choi, S. Y., Yu, S., Kang, S. W., and Rhee, S. G. (2002) J. Biol. Chem. 277 25370–25376 [DOI] [PubMed] [Google Scholar]
- 24.Gottlob, K., Majewski, N., Kennedy, S., Kandel, E., Robey, R. B., and Hay, N. (2001) Genes Dev. 15 1406–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kennedy, S. G., Kandel, E. S., Cross, T. K., and Hay, N. (1999) Mol. Cell. Biol. 19 5800–5810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burgering, B. M., and Medema, R. H. (2003) J. Leukocyte Biol. 73 689–701 [DOI] [PubMed] [Google Scholar]