

Abstract
Mutations in the I-II loop of Cav3.2 channels were discovered in patients with childhood absence epilepsy. All of these mutations increased the surface expression of the channel, whereas some mutations, and in particular C456S, altered the biophysical properties of channels. Deletions around C456S were found to produce channels that opened at even more negative potentials than control, suggesting the presence of a gating brake that normally prevents channel opening. The goal of the present study was to identify the minimal sequence of this brake and to provide insights into its structure. A peptide fragment of the I-II loop was purified from bacteria, and its structure was analyzed by circular dichroism. These results indicated that the peptide had a high α-helical content, as predicted from secondary structure algorithms. Based on homology modeling, we hypothesized that the proximal region of the I-II loop may form a helix-loop-helix structure. This model was tested by mutagenesis followed by electrophysiological measurement of channel gating. Mutations that disrupted the helices, or the loop region, had profound effects on channel gating, shifting both steady state activation and inactivation curves, as well as accelerating channel kinetics. Mutations designed to preserve the helical structure had more modest effects. Taken together, these studies showed that any mutations in the brake, including C456S, disrupted the structural integrity of the brake and its function to maintain these low voltage-activated channels closed at resting membrane potentials.
Voltage-gated calcium (Cav) channels regulate calcium influx in response to membrane depolarization. Their main role is to couple the electrical activity of cells with intracellular processes such as contraction, secretion, neurotransmission, and gene expression in many different cell types (1). Cav channels have been divided in two subfamilies, comprising high voltage-activated and low voltage-activated or T-type channels (2–4). These proteins are composed of four or five distinct subunits that are encoded by multiple genes. The main subunit, α1, contains four homologous repeats (I–IV), each composed of six transmembrane segments (S1–S6). Each repeat shows significant homology to voltage-gated K+ channels, such as Shaker (5), and mutagenesis studies have established the conservation of function. For example, mutations in the pore loop between S5 and S6 affect channel permeation (6, 7). Similarly, mutations in the S4 voltage sensors of T-channels affect channel gating as in K+ channels (8). Therefore crystal structures of K+ channels (9), and models developed for Na+ voltage-gated channels (10) are likely to provide insights into T-channel gating. In these models a change in membrane potential triggers an outward movement of the S4 voltage sensors, a concomitant movement of the S4-S5 linker, and a widening of the pore walls formed by S6 segments (10–12). In addition, S6 segments also play a similar role in T-channel inactivation as observed in high voltage-activated channels. For example, mutations in IIIS6 affect inactivation from the open state in Cav3.1 much as they did in Cav2.2 channels (13). Mutations in Cav1.2 discovered in Timothy syndrome patients dramatically slow inactivation of Cav1.2 channels (14), and similarly located mutations in Cav3.1 produce a similar effect (15). Despite these advances, little is known about why T-channels activate at lower voltages than other voltage-gated channels.
Toward this end, a recent study (15) reported that the transfer of the I-II loop of a high to a low voltage-activated channel created a chimera that gated at even lower voltages than the low voltage-activated wild type (WT).4 A similar result was reported in a functional study of single nucleotide polymorphisms present in childhood absence epilepsy patients (16), where it was observed that the mutation C456S in Cav3.2 channels (located in the proximal I-II loop) shifted the voltage dependence of activation to more negative potentials (17). This observation was followed up with a set of deletions exploring the role of the I-II loop in Cav3.2 channels (18). This study revealed that the I-II loop has two separable roles: one to regulate surface expression and another to modulate the biophysical properties of Cav3.2 channels. The deletions performed within the amino-terminal region of this loop modified the voltage dependence of these channels, allowing them to open at even more negative potentials. Such results suggested that the proximal region of this loop normally functions as a “brake” that regulates channel opening. The goal of the present study was to establish the identity of the brake and to make inferences about its structure. For this purpose we used a combination of molecular biological and electrophysiological techniques to test the hypothesis that the region forms a helix-loop-helix motif. To localize the distal end of the brake, we designed a series of deletion mutants. To test the helical structures we made a series of mutations that either stabilized (polyalanine) or disrupted the putative helices (polyglycine and proline). To test the importance of the loop region, we attempted to straighten it by either replacement with an α-helix or by deletion, which would fuse helix 1 to helix 2. Functional analysis of these mutations and deletions revealed a similar contribution of each part of the motif to the activity of Cav3.2 channels. We conclude that the gating brake has a helix-loop-helix structure contained within the first 60 amino acids after IS6 and that the structural integrity of the brake is important in keeping T-channels closed at the resting membrane potential.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis—PCR-based mutagenesis was performed using Pfu Ultra DNA polymerase (Stratagene, La Jolla, CA) and oligonucleotide primers (Invitrogen). The template was a fragment of human Cav3.2a cDNA (GenBank™ accession number AF051946) cloned into pGEM-3 (Promega, Madison, WI). As reported previously, a silent mutation was introduced into this fragment to create a second BspEI site (18), which allows recloning of the full-length cDNA by moving the BspEI (880 residues)/BspEI (2638 residues) fragment into pEGFP-CI (Clontech, Mountain View, CA). All of the cloning steps were verified by restriction analysis, and the sequence of the BspEI fragment was verified using automated sequencing (University of Virginia Biomolecular Research Facility).
Transfections—Human embryonic kidney 293 cells (HEK-293, CRL-1573; American Type Culture Collection, Manassas, VA) were grown in Dulbecco's modified medium F-12 (Invitrogen) supplemented with 10% fetal calf serum, penicillin G (100 units/ml), and streptomycin (0.1 mg/ml). The cells were transiently transfected with plasmid DNAs encoding each Cav3.2 variant using JET-PEI (Polyplus, Illkirch, France). After ∼24 h, green fluorescent protein-positive cells were selected for electrophysiological recordings.
Electrophysiology—Whole cell patch clamp recordings were obtained at room temperature using an Axopatch 200A amplifier equipped with a CV201A head stage. The amplifier was connected to a computer through a Digidata 1200 A/D converter and controlled using pCLAMP 9.2 software (Molecular Devices, Palo Alto, CA). The data were filtered at 2 kHz and digitized at 5 kHz. Whole cell Ca2+ currents were recorded using the following external solution 5 mm CaCl2, 166 mm tetraethyl ammonium chloride, and 10 mm HEPES, pH adjusted to 7.4 with tetraethyl ammonium-OH. The internal pipette solution contained the following 125 mm CsCl, 10 mm EGTA, 2 mm CaCl2, 1 mm MgCl2, 4 mm Mg-ATP, 0.3 mm Na3GTP, and 10 mm HEPES, pH adjusted to 7.2 with CsOH. The pipettes were made from TW-150-3 capillary tubing (World Precision Instruments, Inc., Sarasota, FL). There was no correction of the –9.4 mV junction potential. Under these solution conditions the pipette resistance was typically 2–3 MΩ. Access resistance and cell capacitance were calculated using on-line exponential fits to the capacitance transient induced by a 20-mV depolarization (Membrane Test, pCLAMP software). Cell capacitance averaged 10 picofarads. Access resistance averaged 4.2 mΩ. The cells where the access resistance exceeded 5.5 mΩ were not investigated. Total series resistance was compensated 70%, resulting in a maximal residual voltage error of less than 2 mV. Activation and inactivation kinetics were calculated simultaneously using double exponential fits to current traces using Clampfit (pCLAMP software). Peak currents (I) at each voltage step (Vm) were used to calculate the voltage dependence of activation for each cell using the following equation,
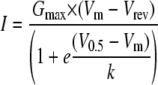 |
(Eq. 1) |
where Vrev is the extrapolated reversal potential, Gmax is the maximal conductance, V0.5 is the midpoint of activation, and k is the slope of the relationship. The voltage dependence of steady state inactivation was estimated using 15-s prepulses to varying potentials followed by a test pulse to –20 mV to measure channel availability. The current elicited during each test pulse was normalized to that observed when the holding potential was –110 mV (I/Imax), and the data from each cell was fit with the following Boltzmann equation using Prism® software (Graphpad, San Diego, CA).
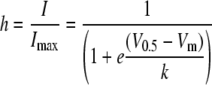 |
(Eq. 2) |
Conductance was also calculated at each test potential because some of the deletion mutants shifted the voltage dependence of activation to more negative potentials where the driving force for Ca2+ ions is larger. To allow comparisons of current density, conductance (G) was normalized to cell size (F). All of the results are presented as the means ± S.E. Significant differences in the average data were analyzed using one-way analysis of variance followed by Dunnett's multiple comparison test against WT channels or Bonferonni's multiple comparison test (GraphPad Prism).
Circular Dichroism Spectroscopy—The DNA fragment encoding amino acids 424–528 within the proximal region of the I-II loop of Cav3.2 α1 subunit was cloned into the maltose-binding protein (MBP) expression vector pMAL-c2x (New England BioLabs Inc., Beverly, MA). The subcloning was performed downstream of the MBP to express a MBP fusion protein. Afterward, this plasmid was transformed into Escherichia coli BL21 (Stratagene, La Jolla, CA). Positives clones were identified by restriction analysis, and single colonies were grown in Luria-Bertani broth in the presence of 100 μg/ml ampicillin. When the cell suspension reached an absorbance of 0.4 at 600 nm, protein expression was induced by the addition of 0.5 mm isopropyl thiogalactoside for 2 h at 37 °C. The cells were harvested, lysed with mild sonication, and centrifuged at 20,000 rpm. The lysate supernatant was loaded onto a amylase affinity column (New England Biolabs, Inc.), and the fusion protein was eluted with 10 mm maltose. After purification, the protein was cleaved from MBP with protease Xa and then purified by fast performance liquid chromatography on a Superdex S200 column. The CD measurements were collected using a freshly dissolved peptide solution (0.05 mg/ml) in the presence of 20 mm Tris-HCl, 200 mm NaCl buffer, pH 7.4, plus 0.17 mm n-dodecyl-β-D maltopyranoside. Similar results were obtained in the additional presence of 15% 2,2,2-trifluoroethanol. The CD spectra were obtained with an AVIV model 215 CD spectro-photometer (Aviv Biomedical, Lakewood, NJ). Three scans were recorded at 25 °C (resolution of 1 nm), averaged, and base line-corrected. The CD signals were corrected by subtracting the cuvette plus buffer blank signal.
RESULTS
To date, there is no structural data available on the I-II loop of T-type calcium channels. Therefore, we used secondary structure prediction, CD, homology modeling, and site-directed mutagenesis techniques to obtain insights into the possible structure of the Cav3.2 gating brake.
Secondary Structure of the Brake Region—Using SOPMA, a program that predicts secondary structure (19), it was found that the proximal I-II loop might form a helix-loop-helix structure. Predictions showed that the first α-helix (H1) runs from Thr420 to Leu441 and that the second one (H2) from Cys456 to Trp482 of Cav3.2 channels (Fig. 1). The program also predicted that 52% of the residues are contained in α-helical structures, 37% are in random coils, and only 11% are in β-turns. Homology modeling using the BioinfoBank MetaServer predicted a similar structure (20) and identified a crystallized protein, fumarase (Research Collaboratory for Structural Bioinformatics Protein Data Bank code 1YFM), whose structure contains a helix-loop-helix structure (Fig. 1B). Notably, the SOPMA program correctly predicts the structure of fumarase (Fig. 1A), lending support for a model where the helices are in an anti-parallel orientation, and helix 2 extends back toward the membrane (Fig. 1B).
FIGURE 1.

Secondary structure of the brake region within the proximal I-II loop of Cav3.2 calcium channels. A, alignment of the three human Cav3 channels. Asterisks highlight the amino acids that are conserved among all three T-channels. The predicted secondary structure of Cav3.2 is shown, where h stands for α-helix, c stands for coil, e stands for β-sheet, and t stands for turn. Also shown is the amino acid sequence of the region of fumarase that forms a helix-loop-helix structure, as well its predicted secondary structure. B, schematic of the initial region of I-II loop made using the crystal structure of fumarase and the potassium channel (MthK). The fumarase structure contains five helices packed around a hydrophobic core, and only two of the helices are shown in the schematic. C, gel of the purified I-II loop peptide (amino acids 424–528 of Cav3.2, GenBank™ accession number AF051946). The protein was purified as a fusion protein with MBP and then cleaved. D, CD spectra of the I-II loop peptide. The CD and SOPMA estimations of the dominant organized structures of this intracellular loop are shown in percentages of α-helix, β-turn, and random coil above the CD spectra.
The presence of α-helical content in this brake region was confirmed by circular dichroism. A peptide corresponding to amino acids 424–528 of the I-II loop of the Cav3.2 channel was purified in E. coli (Fig. 1C). CD spectra of the peptide displayed a large negative peak between 210 and 230 nm (Fig. 1D), indicating the presence of an α-helical structure. The secondary structure was deduced from the CD signal between 200 and 260 nm using the deconvolution program CDNN. The most prevalent structures in this intracellular segment were α-helix (52%) and coil (31%), whereas β-turn content was less abundant (16%). These results are consistent with the predictions made by the SOPMA algorithm and provided support for the model.
Mapping the Carboxyl Terminus of the Brake—In a previous study we showed that deletions of the proximal I-II loop (amino acids 429–491) affected gating, whereas deletions in the distal loop (amino acids 492–772) only affected surface expression (18). To map precisely the end of the brake region, we constructed a series of deletions that extend proximally from the amino acid 492 border (Fig. 2). The gating properties of these mutant channels were determined using current-voltage (I-V; Fig. 3) and steady state inactivation (h∞) protocols (Fig. 4). Representative traces obtained during the I-V protocol are shown in Fig. 3A. These data were analyzed to find the peak current at each voltage and fit with double exponentials to determine the kinetics of activation and inactivation. The raw peak current data were fit with a form of the Boltzmann equation that also calculates maximal conductance and the apparent reversal potential (see “Experimental Procedures”). In this manner the midpoint of activation (V0.5) was calculated from untransformed data for each cell and then averaged (Table 1). To illustrate the shifts in the IV curve, we also normalized the data for each cell to the maximum observed and present the average in Fig. 3B. These results show that deletions D2b (amino acids 466–491) and D2c (amino acids 475–491) had a profound effect on the voltage dependence of activation and kinetics, whereas D2e (amino acids 484–491) had no significant effect. A hallmark of T-channel kinetics is that they accelerate with stronger depolarizations, producing criss-crossing traces during the IV protocol (21). Therefore mutations that shift the voltage dependence of activation should produce a similar shift in the voltage dependence of kinetics, and this was observed with the D2b, D2c, and D2d channels (Fig. 3, D and E). These mutations also increased the maximum peak current observed (pA/picofarads; results not shown). Because part of this can be explained by the increased driving force where these channels open relative to WT, we calculated the conductance and normalized it to cell size as estimated by capacitance (Fig. 3C). D2b and D2c increased maximal conductance 2-fold (Table 1).
FIGURE 2.

Location of mutations made in the proximal region of the I-II loop of a human Cav3.2 channel. Amino acid sequences of each of the mutants beginning at residue 409 in the middle of the IS6 segment. The bold underlined letters in the mutants highlight the amino acids changed, whereas the periods represent the amino acids deleted. The residues involved in forming a putative salt bridge are also underlined and in bold type in the WT sequence.
FIGURE 3.

Mapping the distal terminus of the brake region. A, representative families of whole cell Ca2+ currents recorded from HEK-293 cells expressing WT (upper records) and D2b mutant (bottom records) Cav3.2 channels. The currents were activated by depolarizing steps from –80 to +20 mV from a holding potential of –100 mV. The traces obtained during steps to –50 mV are shown in bold type to emphasize the shift in the position of the I-V curve. The scale bar applies to both families of traces. B, normalized I-V curves for WT and deletion mutants. Smooth curves represent modified Boltzmann (see “Experimental Procedures”) fits to the average data. The data in Table 1 show the averages obtained from fits to the raw current data from each individual cell. C, plots of conductance normalized to current densities versus test potential. D and E, voltage dependence of the time constants of activation (D) and inactivation (E) kinetics. Time constants (obtained from two exponential fits of the raw traces) were plotted as a function of membrane potential. The symbols used to represent WT and mutants are given in D.
FIGURE 4.

Effects of helix 2 deletions on steady state inactivation. A, examples of Ca2+ currents recorded at –20 mV after 15-s prepulses to increasing values of voltages between –110 and –40 mV in HEK-293 cells expressing WT (upper records) and D2b mutant (bottom records) Cav3.2 channels. B, steady state inactivation curves. Maximal current (Imax) during the test pulse was obtained when the prepulse potential was –110 mV. Channel availability was calculated by dividing the remaining current (I) at –20 mV by Imax and expressed as a function of the prepulse potential. The smooth curves represent Boltzmann fits to the average data. The results from the averages of individual fits to each cell are reported in Table 1.
TABLE 1.
Electrophysiological properties of the I-II loop mutants
Statistical significance was determined using either one-way analysis of variance followed by Dunnett's multiple comparison test against WT or Student's t test. The Gmax, V50 of activation, and kinetic parameters were all determined from the I-V protocol and therefore have the same number of cells in each measurement.
|
Density (Gmax)
|
Activation
|
Inactivation
|
Kinetics at 0 mV
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| V50 | k | n | V50 | k | n | τ activation | τ inactivation | ||
| nS/picofarad | mV | mV | mV | mV | ms | ms | |||
| WT | 1.7 ± 0.2 | -42.7 ± 0.7 | 6.0 ± 0.1 | 16 | -77.7 ± 0.8 | -5.7 ± 0.2 | 12 | 2.1 ± 0.2 | 17.3 ± 0.7 |
| D2b | 3.8 ± 0.5a | -56.8 ± 1.1a | 5.3 ± 0.2a | 14 | -89.2 ± 0.8a | -4.0 ± 0.2a | 7 | 1.6 ± 0.1a | 18.5 ± 1.2 |
| D2c | 3.4 ± 0.6a | -51.8 ± 0.8a | 5.6 ± 0.2 | 13 | -80.6 ± 1.4 | -4.6 ± 0.3a | 9 | 1.3 ± 0.1a | 14.7 ± 1.0b |
| D2d | 2.3 ± 0.5 | -50.6 ± 0.4a | 5.3 ± 0.3 | 5 | -80.6 ± 0.7 | -4.7 ± 0.1b | 4 | 1.8 ± 0.1 | 17.3 ± 0.7 |
| D2e | 2.1 ± 0.3 | -43.4 ± 1.6 | 5.8 ± 0.2 | 14 | -73.6 ± 1.4 | -5.4 ± 0.2 | 8 | 2.1 ± 0.1 | 15.0 ± 0.6b |
| PA61 | 3.1 ± 0.3b | -52.9 ± 0.9a | 6.2 ± 0.2 | 12 | -82.5 ± 1.2b | -4.1 ± 0.1a | 8 | 1.7 ± 0.2b | 17.2 ± 0.9 |
| PPG1 | 1.9 ± 0.4 | -50.4 ± 0.7a | 5.9 ± 0.2 | 6 | -82.7 ± 1.3b | -4.9 ± 0.5b | 6 | 2.0 ± 0.2 | 19.8 ± 0.7b |
| PA64 | 2.2 ± 0.3 | -44.6 ± 0.7 | 6.4 ± 0.2a | 9 | -73.5 ± 1.0b | -5.8 ± 0.3 | 6 | 1.3 ± 0.1a | 15.2 ± 0.9 |
| PPG4 | 2.0 ± 0.2 | -50.7 ± 1.1a | 6.0 ± 0.3 | 6 | -81.3 ± 0.6b | -5.2 ± 0.2 | 4 | 0.9 ± 0.1a | 12.3 ± 1.6a |
| PA62 | 2.6 ± 0.2b | -50.5 ± 0.9a | 5.5 ± 0.1 | 12 | -80.4 ± 0.8 | -5.0 ± 0.1 | 8 | 1.8 ± 0.2 | 14.7 ± 0.6b |
| PPG2 | 2.4 ± 0.5 | -55.5 ± 0.4a | 5.9 ± 0.1 | 7 | -86.1 ± 0.9a | -4.1 ± 0.1a | 6 | 1.4 ± 0.1a | 18.5 ± 0.6 |
| H1A1 | 2.6 ± 0.3b | -51.1 ± 1.2a | 5.8 ± 0.2 | 18 | -82.3 ± 1.6a | -4.6 ± 0.2a | 17 | 1.5 ± 0.1a | 14.2 ± 0.8b |
| H1A2 | 2.1 ± 0.4 | -54.5 ± 1.2a | 5.7 ± 0.4 | 6 | -86.2 ± 0.7a | -3.9 ± 0.1a | 6 | 1.5 ± 0.2b | 19.2 ± 2.3a |
| H1A3 | 3.2 ± 0.3a | -53.9 ± 1.2a | 5.6 ± 0.2 | 16 | -84.0 ± 0.8a | -4.4 ± 0.2a | 16 | 1.4 ± 0.1a | 14.3 ± 0.8a |
| PA6T1 | 2.7 ± 0.4b | -52.9 ± 1.0a | 5.6 ± 0.2 | 12 | -84.4 ± 1.1a | -4.3 ± 0.1a | 9 | 1.2 ± 0.1a | 13.5 ± 0.5a |
| PA6T2 | 2.8 ± 0.5b | -50.9 ± 0.7a | 5.3 ± 0.2a | 14 | -81.0 ± 0.9 | -4.7 ± 0.2b | 9 | 1.4 ± 0.1a | 13.1 ± 0.5a |
| PA6T12 | 2.1 ± 0.5 | -55.0 ± 1.2a | 5.7 ± 0.2 | 10 | -88.8 ± 2.1a | -4.2 ± 0.1a | 7 | 1.2 ± 0.1a | 11.7 ± 0.5a |
| DC1 | 2.3 ± 0.3a | -57.3 ± 0.9a | 5.5 ± 0.2 | 10 | -85.6 ± 1.0a | -4.2 ± 0.1a | 6 | 1.3 ± 0.1a | 13.9 ± 0.3a |
| PA63 | 2.6 ± 0.5b | -52.7 ± 1.1a | 5.3 ± 0.4 | 7 | -82.8 ± 1.6b | -3.8 ± 0.1a | 4 | 1.1 ± 0.1a | 11.6 ± 0.3a |
Statistical significance when using analysis of variance.
Statistical significance when using t test.
Steady state inactivation was estimated using 15-s prepulses, followed by a test pulse to –20 mV to assess channel availability (Fig. 4A). The largest deletion of this series, D2b, again produced the largest effect, shifting the midpoint of the h∞ curve by –11.2 mV (Fig. 4B and Table 1). In contrast, the other deletions did not have a significant effect on this measure of closed state inactivation. These results indicate that the gating brake terminates around residue 484. As observed with D2d, any deletions proximal to this end have large effects on channel gating. These results also show that the brake plays a bigger role in channel activation than in inactivation from closed states.
Testing the Importance of the Putative α-Helices—The propensity of each amino acid to stabilize or disrupt secondary structure has been studied extensively, and a common finding is that alanine stabilizes α-helices, whereas prolines and glycines disrupt them (22, 23). In a previous study, we used circular dichroism to confirm the predicted changes induced in the structure of a peptide when six consecutive residues of an α-helix are replaced with alanine or proline (15). In this study we replaced the central region of each helix with either six alanines (helix 1, PA61 and PA64; helix 2, PA62) or proline-glycine repeats (helix 1, PPG1 and PPG4; helix 2, PPG2). Unexpectedly, both PA61 and PPG1 mutations shifted the voltage dependence of activation and inactivation to a similar extent (Table 1). As discussed below, three-dimensional modeling indicated that these mutations disrupted a salt bridge between Glu424 in helix 1 and either Lys470 or Arg474 in helix 2. Therefore we generated a second set of helix 1 mutants that preserved Glu424 (PA64 and PPG4; see Fig. 2 for location). As predicted, PA64 was very similar to WT, with no shift in the IV curve, a small depolarizing shift in the h∞ curve, and only modest effects on kinetics. In contrast, the PPG4 mutation shifted the IV curve –8 mV and the h∞ curve –4 mV and accelerated activation and inactivation kinetics (Fig. 5 and Table 1). Similarly, the polyalanine substitution in helix 2 (PA62) largely preserved normal gating (small shift in IV curve, no shift in the h∞ curve, and only affected inactivation kinetics), whereas the PPG2 mutation produced dramatic shifts in both the IV and h∞ curves and affected both activation and inactivation kinetics (Table 1).
FIGURE 5.

Biophysical properties of mutants to probe the helical structure of putative helices 1 and 2. A and B, normalized I-V curves for PA and PPG mutations made in helix 1 (A) and helix 2 (B). Labeling of symbols in these panels applies to the entire figure. C and D, effect of these mutations on steady state inactivation. The data points were obtained by plotting the normalized peak Ca2+ current at –20 mV against the prepulse potential for the indicated mutants. The smooth curves represent Boltzmann fits to the average data. The results from the average of individual fits to each cell are reported in Table 1. E and F, voltage dependence of the time constants of activation (E) and inactivation kinetics (F) of the helix 1 mutants PA64 and PPG4. Time constants were obtained from two exponential fits of the raw traces and plotted against membrane potential. WT channel properties are represented with a dotted line.
If the brake region extends from IS6 as a rigid α-helix, then insertions should alter the orientation of the brake with respect to the channel. To test this hypothesis we inserted one (H1A1), two (H1A2), or three (H1A3) consecutive alanines into the putative helix 1 (Fig. 2). The additions were inserted six amino acids distal to IS6, because a previous Cav3.2 mutant that deleted these residues was nonfunctional.5 All three insertions disrupted channel gating, shifting the IV curve approximately –10 mV; shifting the h∞ curve approximately 6 mV, and significantly accelerated channel kinetics (Fig. 6 and Table 1). Notably the voltage dependence of activation kinetics for all three insertions was shifted to a greater extent than the IV (∼–15 mV), whereas the inactivation kinetics of H1A2 showed reduced voltage dependence.
FIGURE 6.

Biophysical properties of alanine insertion mutants designed to probe the orientation of the brake region. A, normalized I-V curves for H1A1, H2A2, and H1A3 channels. B, effects of alanine insertions on steady state inactivation. The data points were obtained by plotting the normalized peak Ca2+ current at –20 mV against the prepulse potential for the indicated mutants. The smooth curves represent Boltzmann fits to the average data. The results from the average of individual fits to each cell are reported in Table 1. C and D, voltage dependence of the time constants of activation and inactivation. Time constants were obtained from two exponential fits of the raw traces and plotted against membrane potential. The smooth curves represent Boltzmann fits to the data. WT channel properties are represented with a dotted line.
Testing the Importance of the Loop Region—Secondary structure prediction programs suggest that the loop connecting helix 1 to helix 2 may be composed of two hairpin turns separated by β sheet and random coil (Fig. 1). We hypothesized that the loop acts to orient helix 2 back toward the inner mouth of the channel. To test this hypothesis, we performed two types of mutations: one to straighten the turn by replacing it with α-helix promoting alanines and another by deleting the entire loop region, effectively fusing helix 1 onto helix 2 (DC1). We initially focused on the second turn (replacing the sequence SEPGSC in PA6T1), because it was conserved across all three Cav3 channels. The first turn was also mutated (HLSNDS in PA6T2), and both turns were mutated in PA6T12. In all four turn mutants the midpoints of activation were significantly shifted to more negative potentials relative to WT, especially DC1 and PA6T12 (approximately –14 mV). Also, these mutations caused large negative shifts in the midpoints of steady state of inactivation (Fig. 7B), with the mutant PA6T12 displaying the largest shift (–10.8 mV). As observed with the alanine insertion mutants, activation and inactivation kinetics were accelerated at all test potentials (Fig. 7, C and D, and Table 1). Notably, kinetics were faster in the voltage independent range, indicating that these changes were due to genuine changes in gating and not simply due to voltage-dependent shifts in activation gating.
FIGURE 7.

Biophysical properties of the mutations made to disrupt the putative loop of the brake region. A, normalized IV plots for the DC1, PA6T1, PA6T2, and PA6T12 mutants. B, effects of these mutations on steady state inactivation. The data points were obtained by plotting the normalized peak Ca2+ current at –20 mV against the prepulse potential for the indicated mutants. The smooth curves represent Boltzmann fits to the average data. The results from the average of individual fits to each cell are reported in Table 1. C and D, voltage dependence of the time constants of activation (C) and inactivation (D) kinetics. Time constants were obtained from two exponential fits of the raw traces and plotted against membrane potential.
Three-dimensional Model of the Brake—Taken together, the results shown thus far demonstrate how critical the structure of the brake region is to channel gating and provide support for the hypothesis that this region may form a helix-loop-helix structure. To gain insight into the effects of the mutations, we generated several de novo three-dimensional models (the supplemental information includes a Protein Data Bank file of the entire 103-amino acid region modeled). We also modeled all 18 of the individual mutants. The model of the WT sequence shows a small kink immediately distal to IS6, followed by a helix-loop-helix structure. The model also suggests the presence of a salt bridge between Glu424 in helix 1 and either Lys470 or Arg474 in helix 2. Disruption of this salt bridge may explain why PA61 affected activity more than expected. To test this prediction we made two additional mutants in helix 1 (PA64, PPG4, discussed above) and PA63, which neutralizes the positively charged residues in helix 2 (RKVKRR; Fig. 2). The voltage dependence of activation and inactivation of PA63 channels were shifted from WT to the same degree as PA61, whereas their kinetics were the fastest of any mutant (Table 1). Clearly, charged residues in helix 1 and helix 2 play critical roles in the function of the gating brake.
DISCUSSION
The goal of this study was to explore the structure-function relationships of the T-type calcium channel Cav3.2, focusing on the intracellular loop connecting repeats I-II. This loop is particularly interesting because 7 of the 12 mutations found in childhood absence epilepsy patients are localized to it (16). In previous studies, we have shown that these I-II loop mutations are capable of affecting both the gating and surface expression of Cav3.2 channels, thereby providing a plausible explanation for their role in epilepsy (17, 18). These studies also showed that deletions in the proximal I-II loop controlled gating, whereas deletions of the distal loop had no effect on gating but led to enhanced surface expression of the channel. The present study was focused on the proximal I-II loop, which because of its ability to shift the voltage dependence of activation and accelerate kinetics we call a gating brake. The hypothesis being tested in this study is that the gating brake is formed by a helix-loop-helix structure.
The Proximal Region of the I-II Loop Has a High Content of α-Helix—The BioInfoBank Meta server goes beyond structure prediction and identifies proteins of known structure that are likely to be similar to the query sequence (20). This three-dimensional jury analysis of the proximal Cav3.2 I-II loop led to the prediction that the proximal I-II loop would form a helix-loop-helix structure similar to that found in fumarase (Fig. 1B). As a first test of this prediction, we purified a fragment of the I-II loop from E. coli and studied its structure using circular dichroism, a versatile tool for examining protein structure (24). The circular dichroism spectrum indicated that the proximal I-II loop contains a high α-helical content, just as predicted. Although insufficient amounts of purified protein were obtained for NMR analysis, these results establish that the proximal loop has a high helical content and provide experimental support for the structure predictions.
Mutations Made to Test the Helix-Loop-Helix Model—Four types of mutations were used to test the model. First, we used deletion analysis to find the distal end of the gating brake. Second, we tested for the existence of α-helical regions by replacing six consecutive amino acids with prolines and glycines to disrupt the helix, and as a control we replaced the same residues with alanine to conserve the helix. Third, we tested for the existence of the loop by either converting it to an α-helix or by deleting it entirely, effectively fusing helix 1 to helix 2. Finally, we also tested whether the addition of alanines to the middle of helix 1 would alter the pitch of the helix and thereby alter the orientation of the helix-loop-helix with respect to the channel.
In a previous study we found that deletion of helix 1 (D1) produced a similar effect on gating as deleting helix 2 (D2) and that deletions made beyond residue 491 did not alter gating (18). To map the distal border we constructed a series of smaller deletions, anchoring the distal end at residue 491. Deletion of 24 amino acids from helix 2 (D2b) produced a profound shift in the voltage dependence of both activation and inactivation and accelerated kinetics. Similar effects were noted in the previous study with D1, D2, and D1–3 (18), indicating that the D2b deletion maximally disrupted the function of the gating brake. Moving the proximal border an additional seven amino acids (D2c) had a more modest effect on the IV, and no significant effect on the h∞ curve. Interestingly, both activation and inactivation kinetics of D2c were accelerated relative to WT channels. In general, mutations that shifted activation kinetics also shifted the voltage dependence of activation, but this was not always the case for inactivation, because some mutations affected apparent open state kinetics with little or no effect on the voltage dependence of inactivation. Moving the proximal border eight more residues (D2e) resulted in channels that gated normally. Taken together these results indicate that the seven extra residues deleted in D2b (FRKVKRR) play an important role in the function of the brake. Therefore, we tested the effect of either deleting these residues (D2d) or converting them to alanines (PA63). The PA63 mutation caused a dramatic disruption of channel gating, shifting the voltage dependence of activation and inactivation, and accelerating kinetics. In contrast, the D2d mutation had a smaller effect on channel gating, shifting the IV curve, but having no effect on the midpoint of the h∞ curve or on channel kinetics. A possible explanation is that Arg477 and/or Arg481 functionally replaced Lys470 and/or Arg474, thereby restoring the putative salt bridge. We conclude that the gating brake ends just after this highly charged region and that this region plays a critical role in its function.
The Effect of Mutations in the α-Helical Regions—The guiding hypothesis for these studies was that replacement of six consecutive residues of an α-helix with residues such as proline and glycine would destabilize the helices and disrupt the function of the gating brake, whereas replacement with residues such as alanine would preserve structure and function (22, 23). We targeted the central portion of the putative helix and replaced six residues with either a PPG repeat or alanines (PA6). As predicted, the PPG substitutions caused large shifts in the voltage dependence of activation and inactivation and accelerated kinetics. The largest effects were observed with the helix 2 mutations. Surprisingly, the alanine substitutions also disrupted gating. The structural model of these mutants provides a possible explanation for these effects; replacement of charged residues with alanines in helix 1 eliminated an important salt bridge between helix 1 and 2. In addition to being conserved across all three mammalian Cav3 channels, the residues involved in this putative salt bridge are also conserved in the Drosophila homolog (GenBank™ accession number NP_572296). Moving the helix 1 mutations two amino acids down confirmed these predictions, because now the alanine mutation had little or no effect, whereas the poly-proline-glycine continued to disrupt function (PA64 and PPG4, respectively).
The Effect of Mutations in the Loop Regions—Our studies of the loop region were complicated by the presence of two putative hairpin turns, because only one turn would be required to orient helix 2 in an antiparallel manner with respect to helix 1. Indeed, replacement of either turn region with six alanines produced channels with only modest effects on gating. In contrast, mutation of both turn regions (PA6T12) or deletion of the entire loop region (DC1) produced dramatic shifts in gating. The modeling studies agree well with the experimental findings; replacement of either turn was insufficient to disrupt the helix-loop-helix structure because of compensation by the remaining turn. Modeling of the PA6T12 mutant suggested that helix 1 would be more extended and that loss of Asp444 and Glu452 creates a more hydrophobic region, thereby allowing closer contact between the helices. Modeling of DC1 indicates that the loop was effectively removed and that helix 1 was fused to helix 2. The gating phenotype of this mutant is similar to the D2 mutant, which lacks helix 2; therefore we infer that function of the gating brake was totally disrupted. The model also predicts that the epilepsy mutation, C456S, is at a critical location in the brake, forming the start of helix 2, and projecting toward helix 1. An alternative hypothesis is that the α-helices are not in an anti-parallel orientation (as in transcription factors) and that the charged residues form contacts with parts of the channel outside of the brake region. In either case, the results show that the loop region is critical for the function of the brake and that disruption of its structure leads to functional effects on channel gating.
Insertion of Alanines into Helix 1 Also Disrupts Function of the Brake—A second test of the α-helical structure of the helix 1 region was to insert one, two, or three alanines into the middle of the region. Our hypothesis was that each alanine would change the orientation of the remainder of the helix by 100 ° and extend the helix 1.5 Å. As predicted, these insertions produced dramatic shifts in the voltage dependence of activation and inactivation and accelerated kinetics. The gating properties of H1A1, H1A2, and H1A3 were all significantly different from WT channels but not different from each other with the exception of inactivation, where H1A2 channels inactivated more slowly. These results support the hypothesis that the brake region is precisely oriented with respect to the channel.
Disruption of the Brake Also Leads to Increases in Current Density—Almost all of the mutations displayed larger peak currents than wild type channels, even when corrected for cell size, as estimated by cell capacitance (pA/picofarads). However, most of this increase could be accounted for by the greater driving force for Ca2+ ions because peak currents in the mutants were recorded at more negative potentials and hence further away from the reversal potential. To account for this shift, we calculated the chord conductance, and again normalized to cell size, and report the maximal observed conductance (Gmax; nS/picofarads). Even after this correction, D2b, D2c, H1A3, and DC1 all showed a significant doubling of current density. Two likely explanations for this increment are that these mutations lead to increases in the probability of channel opening (Po) and/or increases in the surface expression of channels. In our previous study we found evidence for both; deletion of helix 1 led to increased current density with no change in surface expression, implicating changes in Po, whereas deletion of helix 2 led to both higher surface expression and current density (18). Additional studies are required to elucidate the mechanisms controlling trafficking of Cav3.2 channels, but clearly the I-II loop is involved in this process.
In summary, our results show that the brake is contained within the first 60 amino acids of the I-II loop of Cav3.2 channels, and that the structural integrity of this region is critical for the normal low voltage activation of this T-type channel. Any disruption of this structure leads to channels that open at more negative potentials, inactivate at more negative potentials, and show faster kinetics. As noted previously, inactivation of T-channels is coupled to activation (25), and this may explain the concomitant shifts in steady state activation and inactivation observed in this study. Our results indicate that the normal function of this region is as a brake that prevents channel opening after small depolarizations. Previous measurements of T-channel gating currents indicate that 80% of the channels open after only 20% of total charge movement (8, 26). This suggests that T-channels open after minimal movement of their voltage sensors, and modeling studies suggest that this might explain why T-channel kinetics are so voltage-dependent (8, 25, 27). We suggest that the brake region stabilizes the closed state, adopting the role of one of the S4-S5 linkers to keep the pore closed at resting membrane potentials. Disruption of the brake allows channels to open at more negative membrane potentials and open faster. Finally, helix 2 of the brake region is much less conserved among Cav3 channels than helix 1, suggesting interesting differences in the role of the brake in setting the low voltage gating activation of T-type channels.
Supplementary Material
This work was supported by National Institutes of Health Grant NS038691 (to E. P.-R.) and Consejo Nacional de Ciencia y Tecnología Grant J50250-Q (to J. C. G.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
Footnotes
The abbreviations used are: WT, wild type; PPG, poly-proline-glycine; PA, polyalanine; MBP, maltose-binding protein.
J. M. Arias, I. Vitko, and E. Perez-Reyes, unpublished observations.
References
- 1.Catterall, W. A., Perez-Reyes, E., Snutch, T. P., and Striessnig, J. (2005) Pharmacol. Rev. 57 411–425 [DOI] [PubMed] [Google Scholar]
- 2.Bean, B. P. (1989) Annu. Rev. Physiol. 51 367–384 [DOI] [PubMed] [Google Scholar]
- 3.Matteson, D. R., and Armstrong, C. M. (1986) J. Gen. Physiol. 87 161–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carbone, E., and Lux, H. D. (1984) Nature 310 501–502 [DOI] [PubMed] [Google Scholar]
- 5.Jan, L. Y., and Jan, Y. N. (1990) Nature 345 672. [DOI] [PubMed] [Google Scholar]
- 6.Cataldi, M., Perez-Reyes, E., and Tsien, R. W. (2002) J. Biol. Chem. 277 45969–45976 [DOI] [PubMed] [Google Scholar]
- 7.Talavera, K., Janssens, A., Klugbauer, N., Droogmans, G., and Nilius, B. (2003) J. Gen. Physiol. 121 529–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lam, A. D., Chikina, M. D., McNulty, M. M., Glaaser, I. W., and Hanck, D. A. (2005) Pfluegers Arch. Eur. J. Physiol. 451 349–361 [DOI] [PubMed] [Google Scholar]
- 9.Long, S. B., Campbell, E. B., and Mackinnon, R. (2005) Science 309 897–903 [DOI] [PubMed] [Google Scholar]
- 10.Yarov-Yarovoy, V., Baker, D., and Catterall, W. A. (2006) Proc. Natl. Acad. Sci. U. S. A. 103 7292–7297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Long, S. B., Campbell, E. B., and Mackinnon, R. (2005) Science 309 903–908 [DOI] [PubMed] [Google Scholar]
- 12.Tombola, F., Pathak, M. M., and Isacoff, E. Y. (2006) Annu. Rev. Cell Dev. Biol. 22 23–52 [DOI] [PubMed] [Google Scholar]
- 13.Marksteiner, R., Schurr, P., Berjukow, S., Margreiter, E., Perez-Reyes, E., and Hering, S. (2001) J. Physiol. (Lond.) 537 27–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Splawski, I., Timothy, K. W., Sharpe, L. M., Decher, N., Kumar, P., Bloise, R., Napolitano, C., Schwartz, P. J., Joseph, R. M., Condouris, K., Tager-Flusberg, H., Priori, S. G., Sanguinetti, M. C., and Keating, M. T. (2004) Cell 119 19–31 [DOI] [PubMed] [Google Scholar]
- 15.Arias, J. M., Murbartián, J., Vitko, I., Lee, J. H., and Perez-Reyes, E. (2005) FEBS Lett. 579 3907–3912 [DOI] [PubMed] [Google Scholar]
- 16.Chen, Y. C., Lu, J. J., Pan, H., Zhang, Y. H., Wu, H. S., Xu, K. M., Liu, X. Y., Jiang, Y. W., Bao, X. H., Yao, Z. J., Ding, K. Y., Lo, W. H. Y., Qiang, B. Q., Chan, P., Shen, Y., and Wu, X. R. (2003) Ann. Neurol. 54 239–243 [DOI] [PubMed] [Google Scholar]
- 17.Vitko, I., Chen, Y., Arias, J. M., Shen, Y., Wu, X. R., and Perez-Reyes, E. (2005) J. Neurosci. 25 4844–4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vitko, I., Bidaud, I., Arias, J. M., Mezghrani, A., Lory, P., and Perez-Reyes, E. (2007) J. Neurosci. 27 322–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Geourjon, C., and Deleage, G. (1995) Comput. Appl. Biosci. 11 681–684 [DOI] [PubMed] [Google Scholar]
- 20.Ginalski, K., Elofsson, A., Fischer, D., and Rychlewski, L. (2003) Bioinformatics 19 1015–1018 [DOI] [PubMed] [Google Scholar]
- 21.Randall, A. D., and Tsien, R. W. (1997) Neuropharmacol. 36 879–893 [DOI] [PubMed] [Google Scholar]
- 22.MacArthur, M. W., and Thornton, J. M. (1991) J. Mol. Biol. 218 397–412 [DOI] [PubMed] [Google Scholar]
- 23.O'Neil, K. T., and DeGrado, W. F. (1990) Science 250 646–651 [DOI] [PubMed] [Google Scholar]
- 24.Kelly, S. M., and Price, N. C. (2000) Curr. Prot. Pept. Sci. 1 349–384 [DOI] [PubMed] [Google Scholar]
- 25.Talavera, K., and Nilius, B. (2006) Pfluegers Arch. Eur. J. Physiol. 453 189–201 [DOI] [PubMed] [Google Scholar]
- 26.Lacinova, L., Klugbauer, N., and Hofmann, F. (2002) FEBS Lett. 531 235–240 [DOI] [PubMed] [Google Scholar]
- 27.Frazier, C. J., Serrano, J. R., George, E. G., Yu, X., Viswanathan, A., Perez-Reyes, E., and Jones, S. W. (2001) J. Gen. Physiol. 118 457–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.