

Abstract
High levels of circulating soluble CD40 ligand (sCD40L) are frequently found in patients with hypercholesterolemia, diabetes, ischemic stroke, or acute coronary syndromes, predicting an increased rate of atherosclerotic plaque rupture and restenosis after coronary/carotid interventions. Clinical restenosis is characterized in part by exaggerated neointima formation, but the underlying mechanism remains incompletely understood. This study investigated the role of elevated sCD40L in neointima formation in response to vascular injury in an atherogenic animal model and explored the molecular mechanisms involved. apoE−/− mice fed a Western diet developed severe hypercholesterolemia, significant hyperglycemia, and high levels of plasma sCD40L. Neointima formation after carotid denudation injury was exaggerated in the apoE−/− mice. In vivo, blocking CD40L with anti-CD40L monoclonal antibody attenuated the early accumulation of Ly-6G+ neutrophils and Gr-1+ monocytes (at 3 days) and the late accumulation of Mac-2+ macrophages (at 28 days) in the denudated arteries; it also reduced the exaggerated neointima formation at 28 days. In vitro, recombinant CD40L stimulated platelet P-selectin and neutrophil Mac-1 expression and platelet-neutrophil co-aggregation and adhesive interaction. These effects were abrogated by anti-CD40L or anti-Mac-1 monoclonal antibody. Moreover, recombinant CD40L stimulated neutrophil oxidative burst and release of matrix metalloproteinase-9 in vitro. We conclude that elevated sCD40L promotes platelet-leukocyte activation and recruitment and neointima formation after arterial injury, potentially through enhancement of platelet P-selectin and leukocyte Mac-1 expression and oxidative activity.
Overexpression of CD40 ligand (CD40L; CD154), a key immune and inflammatory mediator, has emerged as an important contributor to inflammation and atherosclerosis.1,2,3,4,5,6,7,8 Clinically, elevated levels of circulating soluble CD40L (sCD40L) have been associated with hypercholesterolemia, diabetes, ischemic stroke and acute coronary syndromes, and predict increased restenosis after percutaneous coronary (and carotid) intervention (PCI).8,9,10,11,12 Whereas the role of CD40L in experimental atherosclerosis has been examined extensively, relatively little is known about the contribution of elevated CD40L to neointima formation and restenosis, particularly in an atherogenic environment.
A growing body of clinical evidence supports the potential importance of elevated plasma sCD40L in the mechanism of restenosis after PCI. Cipollone et al13 reported that preprocedural levels of plasma sCD40L were significantly higher in patients that developed restenosis than in patients with favorable outcomes, and raised sCD40L at baseline was significantly correlated with circulating adhesion molecules (sICAM-1, sVCAM-1, sE-selectin), plasma monocyte chemoattractant protein-1 after PCI, and with lumen loss at 6-month follow-up. Turker et al14 reported that preprocedural levels of plasma sCD40L were significantly higher and more prolonged in patients with in-stent restenosis compared with patients without in-stent restenosis at 6-month follow-up. In the same study, plasma sCD40L levels were further increased after PCI. Aggarwal et al15 showed that plasma sCD40L was raised in patients shortly (10 minutes) after coronary angioplasty and stent placement. L’Allier et al16 reported that elevation of plasma sCD40L persisted for at least 1 month after angioplasty and correlated with angiographic late lumen loss at 6-month follow-up. Although these clinical observations are suggestive of a causal relationship, whether and how elevated plasma sCD40L contributes to neointima formation after arterial injury have not been investigated.
Because most patients who undergo PCI have underlying atherosclerosis and also frequently have hypercholesterolemia or diabetes, studies of the mechanisms of neointima formation and restenosis in preclinical models should attempt to mimic clinically relevant conditions. We and others have previously reported that atherosclerosis-prone apoE-deficient (apoE−/−) mice develop severe hypercholesterolemia, accelerated atherosclerosis, significant insulin resistance, and hyperglycemia when fed an atherogenic Western diet (WD).17,18,19 The present study further showed that WD-fed apoE−/− mice developed high levels of plasma sCD40L as well as exaggerated neointima formation after carotid wire denudation injury compared with chow-fed apoE−/− mice. Therefore, this model provides a unique opportunity for understanding the role of elevated circulating CD40L in neointima formation after vascular injury under clinically relevant conditions.
Leukocyte recruitment plays an essential role in the mechanism of neointima formation and restenosis.20,21 Mac-1 (CD11b/CD18, αMβ2) is the predominant leukocyte-specific β2 integrin. It is up-regulated on activated leukocytes and binds to multiple ligands or counter-receptors such as fibrinogen, platelet glycoprotein Ibα (GPIbα), and intercellular adhesion molecule-1 (ICAM-1) and ICAM-2.22,23,24 Mac-1 has been shown to be a crucial regulator of leukocyte recruitment by promoting leukocyte firm adhesion to adherent platelets and fibrinogen at the sites of injured arteries, and has been associated with neointimal hyperplasia after vascular injury.24 Clinical studies demonstrate that activation of circulating neutrophils (as determined by enhanced Mac-1 expression) is associated with late lumen loss and restenosis.25,26,27,28,29 However, the mechanism underlying neutrophil activation and recruitment in the response to vascular injury is largely unknown. The present study investigated the role of elevated sCD40L in leukocyte recruitment and neointimal formation in response to vascular injury in an atherogenic animal model, and explored the molecular mechanisms involved.
Materials and Methods
Reagents and Antibodies
Recombinant mouse and human soluble CD40 ligand (rm-CD40L, rh-CD40L) was purchased from R&D Systems Inc. (Minneapolis, MN). Other reagents were from Sigma Chemical Co. (St. Louis, MO), unless otherwise specified. Function-blocking anti-human CD40L (TRAP1) and anti-human CD11b/Mac-1 (ICRF44) monoclonal antibodies (mAbs) were purchased from BD Biosciences (San Jose, CA). Function-blocking anti-mouse CD40 (HM40–3) and anti-mouse CD40L (MR1) mAbs were purchased from eBiosciences Inc. (San Diego, CA) and Taconic Biotechnology (Germantown, MD), respectively. Function-blocking anti-mouse integrin CD11b mAb (M1/70, αM chain of Mac-1) was from eBioscience Inc. Normal Armenian hamster IgG was from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Fluorescence-conjugated antibodies were from BD Biosciences PharMingen (San Diego, CA), unless otherwise specified.
Mouse Injury Model and Antibody Treatment
Male C57/BL6 apoE−/− mice 8 to 10 weeks of age (18–20 g; stock no. 002052, The Jackson Laboratory, Bar Harbor, ME) were used for these experiments. Mice were fed with a Western-type diet (TD 88137; Harlan-Teklad, Madison, WI) containing 21% fat by weight (0.15% cholesterol and 19.5% casein without sodium cholate) for 1 week before and 4 weeks after carotid wire injury, as previously described.17,30
The mouse carotid artery wire injury model of Lindner et al31 was used with minor modification, as previously published.17,30 Animals were handled in compliance with the Guiding Principles in the Care and Use of Animals. Protocol approval was obtained from the Animal Research Committee of the University of Virginia Health System.
In the in vivo study, mice were randomly divided into three experimental groups: 1) MR1 mAb-treated group, 2) (hamster IgG) isotype control-treated group, and 3) no antibody treatment. Starting 3 hours before carotid injury, each mouse was given 250 μg of MR1 mAb or isotype control Ab every 3 days until sacrifice via intraperitoneal injection with the operator blinded to treatment.
Morphometric Analysis and Immunohistochemistry
The arterial segments were dehydrated in ethanol and xylene and embedded in paraffin. Sections (5 μm thick) were stained by the Movat method.32 Histomorphometric analysis was performed by individuals blinded to treatment group. For quantitative histopathological comparisons, the mean data of 10 sections was taken. The area of the lumen, internal elastic lamina, and external elastic lamina were determined by planimetry using Image Pro Plus 3.0 (Media Cybernetics), and the lumen area, plaque area, medial area, intima to media ratio, and overall vessel area were calculated. Neointima area was calculated by subtracting lumen area from the internal elastic lamina area, and medial area was determined by subtracting the internal elastic lamina area from the external elastic lamina area. Arterial size was measured by tracing the circumference of the external elastic lamina.
Paraffin-embedded arterial sections were processed for immunohistochemistry with the avidin-biotin-peroxidase method (Vector Laboratories) using smooth muscle α-actin mAb (α-SMA; Sigma Chemical Co.) to detect vascular smooth muscle cells (SMCs); Mac-2 (M3/38, Accurate) was used to detect macrophages. For quantitative comparisons of macrophage or smooth muscle cell content, sections were digitized and the number of positively stained pixels were counted and normalized to total neointima and medial area using Image Pro Plus 3.0 (Media Cybernetics).
Flow Cytometry Analysis of Leukocytes Accumulated in Mouse Carotid Arteries
Flow cytometry was performed to assess the total leukocyte content and the subpopulations of leukocytes accumulated within the arterial wall. Arterial cells were isolated from carotid arteries of apoE−/− mice at 3 days after wire injury using an enzymatic disaggregation method as previously reported.33,34 The number of cells obtained from a pool of five common carotid arteries ranged from 3 × 105 to 5 × 105. Cell suspensions were washed twice with phosphate-buffered saline containing 3% bovine serum albumin (BSA) and divided into aliquots (approximately 1 × 105 cells per tube). All fluorescence conjugated antibodies were obtained from BD PharMingen unless specified otherwise. For assessing total leukocyte content, cells were stained for 15 minutes at 4°C with the allophycocyanin-conjugated anti-mouse CD45 mAb (clone 30-F11, 1/450) or an isotype control rat IgG2b,κ. For assessing leukocyte subpopulations, cells were simultaneously stained with the allophycocyanin-conjugated anti-mouse CD45 mAb and either fluorescein isothiocyanate (FITC)-conjugated anti-mouse LY-6G mAb (for granulocytes/neutrophils, clone 1A8, 1/100), or FITC-conjugated anti-mouse Mac-3 (for macrophages, clone M3/84, 1/50, Santa Cruz). To exclude dead cells, 20 μl of propidium iodide (40 μg/ml, Calbiochem) was added to each aliquot immediately before flow cytometry. Propidium iodide-stained cells were excluded from further analysis. Samples were analyzed with a Becton Dickinson FACSCalibur flow cytometer. Data were expressed as percentage of total leukocytes and individual subpopulations in total live cells counted.
Blood Collection and Biochemical Analysis
Blood samples were harvested by cardiac puncture into EDTA-containing Microtainer tubes (Becton-Dickinson) at the time of euthanasia. Complete blood counts and automated differential leukocyte counts were performed by the University of Virginia Clinical Pathology Laboratory. For lipoprotein levels, blood samples at the time of death were drawn by cardiac puncture and placed in serum separator tubes. Lipid levels were determined by the University of Virginia Clinical Pathology Laboratory. Fasting blood glucose was measured by glucometer (Accucheck Advantage; Roche Diagnostics, Indianapolis, IN). Plasma sCD40L was measured by a mouse soluble CD40L enzyme-linked immunosorbent assay kit (AXXORA). The lower limit of detection for the sCD40L assay was 0.14 ng/ml, and the overall intra-assay coefficient of variation was 6.5%. The specificity of the sCD40L assay was confirmed by using plasma obtained from CD40L−/− mice in which sCD40L was undetectable by the enzyme-linked immunosorbent assay.
In some experiments, the presence of plasma sCD40L was detected by immunoprecipitation and Western blot analysis. Briefly, whole blood (0.8–1.0 ml per mouse) was collected from anesthetized mice by cardiac puncture and anticoagulated with 3.8% trisodium citrate (1:10 vol). Anticoagulated blood was centrifuged immediately at 14,000 rpm for 5 minutes in an Eppendorf microfuge. Supernatant plasma was collected, pooled (>1.0 ml, from three mice), and stored at −80°C until analysis. The pooled plasma was subjected to immunoprecipitation for CD40L with a polyclonal antibody against CD40L (K-19, Santa Cruz) and subjected to 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose, probed with purified MR1 mAb, and visualized with the enhanced chemiluminescence system (Amersham).
Preparation of Mouse Neutrophils and Platelets
Peritoneal neutrophils were prepared from mice as previously described.23 Mice were injected with 2.5 ml of sterile 3% thioglycolate broth solution (Sigma Chemical Co.) by intraperitoneal administration, and 5 hours after injection neutrophils were harvested and purified using a two-layer Percoll gradient (80% over 64%). Cytospin preparations stained with Giemsa revealed that >90% of the cells were neutrophils. Cell viability (>95%) was assessed using trypan blue exclusion. Isolated neutrophils were resuspended in RPMI 1640 containing 0.5% (w/v) low-endotoxin BSA.
Platelet-rich plasma and washed platelets were prepared from mice as previous described.35 Washed platelets were resuspended in Tyrode’s buffer (10 mmol/L 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 12 mmol/L NaHCO3, 137 mmol/L NaCl, 2.7 mmol/L KCl, 0.42 mmol/L NaH2PO4, and 5.5 mmol/L glucose, pH 7.4) containing 0.1% BSA. Platelet count was adjusted to 2 × 108/ml.
Detection of CD40 Receptor Expression in Mouse Neutrophils
For detection of CD40 mRNA expression, total cellular RNA was extracted from fresh isolated mouse peritoneal neutrophils with TRIzol (Invitrogen, Carlsbad, CA) treated with DNase I to remove genomic DNA, then purified with the use of a RNA purification kit (Invitrogen), and reverse-transcribed using the Superscript II (Life Technologies Inc., Rockville, MD) and oligo-dT primers. The following mouse polymerase chain reaction primers36 were used: CD40 forward, 5′-TCCCTGCCCAGTCGGCTTCT-3′, and reverse, 5′-CTGTCTTGGCTCATCTCAAA-3′ (505 bp); β-actin forward, 5′-GTGGGCCGCTCTAGGCACCAA-3′, and reverse, 5′-CTCTTTGATGTCACGCACGATTTC-3′ (540 bp). Polymerase chain reaction was performed by an initial cycle at 95°C for 4 minutes followed by 30 cycles for CD40 or 18 cycles for β-actin of 94°C for 45 seconds, 58°C for 45 seconds, 72°C for 1 minute, and ending with a 5-minute extension at 72°C. The amplified products were separated on 1.5% agarose gels.
For detection of CD40 protein expression, cellular lysates were prepared from fresh isolated mouse peritoneal neutrophils with RIPA buffer (phosphate-buffered saline containing 1% Nonidet P-40, 0.1% sodium dodecyl sulfate, and 0.5% sodium deoxycholate). Protein concentration was measured using a BCA protein assay kit (Pierce, Rockford, IL). Equal amounts (20 μg/lane) of protein lysates were run out on a 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (Amersham). Membranes were blocked with blocking solution (Tris-buffered saline/Tween 20 containing 5% nonfat dry milk), blotted with a goat polyclonal CD40 antibody (T-20, Santa Cruz) and visualized with the enhanced chemiluminescence system (Amersham).
Flow Cytometry Analysis of Neutrophil Mac-1 Expression and Oxidative Burst
Isolated neutrophils (1 × 106/ml, in RPMI 1640/0.1% BSA) were incubated with 3 μl of FITC-conjugated rat anti-mouse CD11b mAb (M1/70 against integrin αM chain of Mac-1), or control FITC-rat IgG2b,κ, for 30 minutes at room temperature in the dark. Neutrophils were then incubated with rm-CD40L in the presence or absence of anti-CD40 mAb (HM40–3), anti-CD40L mAb (MR1), or hamster IgG (Iso) for 15 minutes at 37°C. Neutrophils incubated with phorbol 12-myristate 13-acetate (50 ng/ml) for 5 minutes served as a positive control. Samples were then fixed in 1% buffered paraformaldehyde and analyzed for neutrophil αM integrin expression by a Becton Dickinson FACScalibur with CellQuest software.
Neutrophil oxidative burst was measured by a flow cytometric approach using dihydrorhodamine 123 (DHR123) as a fluorescent probe as previously described.37 Briefly, neutrophils were isolated from blood of adult human healthy donors and suspended in Hanks’ balanced salt solution containing 0.25% BSA. Aliquots of neutrophils (2 × 105 cells in 200 μl) were loaded with 1 μmol/L DHR123 (Molecular Probes) for 5 minutes at 37°C and then stimulated with rh-CD40L under defined conditions. Samples were analyzed immediately by flow cytometry. Data are expressed as mean fluorescence intensity in FL-1 channel for DHR.
Neutrophil Firm Adhesion to Surface-Adherent Platelets
Isolated neutrophil adhesion to activated surface-adherent platelets was performed essentially as previously described.23 Washed mouse platelets (1.5 × 107 in 100 μl of phosphate-buffered saline) were loaded into 96-well microtiter plates precoated with 4% solution of 3-aminopropyltriethoxysilane (Sigma Chemical Co.). Platelets were allowed to settle, adhere, and spread onto the surface of the 3-aminopropyltriethoxysilane-coated wells for 45 minutes and then stimulated with thrombin (0.2 U/ml) for 5 minutes to activate platelets. After that, unbound platelets were removed by washing with RPMI 1640/0.1% BSA. Neutrophils (1.5 × 105 in 100 μl of RPMI 1640/0.1% BSA) were loaded with 1.0 μmol/L calcein AM (Molecular Probes) for 20 minutes, washed twice, and then added to each platelet-coated microtiter well for 60 minutes. To determine the effects of CD40L or mAbs on neutrophil adhesion, the calcein AM-loaded neutrophils were incubated without or with rm-CD40L, anti-CD40 mAb (HM40–3) or anti-CD11b mAb (M1/70), or rm-CD40L plus mAbs. Neutrophil adhesion was assessed by a fluorescence microplate reader at 485-nm excitation and 530-nm emission. Data were expressed as the percentage of adherent cells in total cells by calculating the ratio of the fluorescence of calcein AM-loaded cells in each well before and after washing, respectively.
Statistical Analysis
Statistical analysis was performed with one-way analysis of variance or Student’s t-test as appropriate. The sample size was determined by using a statistical power analysis based on the number of carotid arteries in each group needed to detect statistically significant difference with a power of 0.85 and α = 0.05, and data including plaque area and percentage area of macrophages and SMCs are expressed as the mean ± SEM. A value of P < 0.05 was considered significant.
Results
High Levels of Plasma sCD40L in Western Diet-Fed apoE−/− Mice
Because the presence of sCD40L in mouse plasma has not been reported, we initially measured mouse plasma sCD40L by Immunoprecipitation-Western blot analysis. sCD40L was readily detectable in WD-fed apoE−/− mice without injury (Figure 1A, lane 2) and was further increased at 1 hour after the carotid wire injury (Figure 1A, lane 3). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis showed a single band at approximately 18 kd, indicating that the protein detected is monomeric soluble CD40L.
Figure 1.

A: Immunoprecipitation/Western blot analysis of soluble CD40L protein in pooled plasma from uninjured (lane 2) and wire-injured (lane 3) apoE−/− mice. Plasma from wire-injured apoE−/−CD40L−/−mice was used as a negative control (lane 1). Similar results were obtained in two independent experiments. B: Comparison of levels of total cholesterol, glucose, and sCD40L in blood samples from chow-fed C57Bl/6, chow-fed apoE−/− and Western diet-fed apoE−/− mice. *P < 0.01 and **P > 0.05 versus bar 1, #P < 0.01 versus bar 2.
To ensure the effects of the respective diets on blood lipids, glucose, and sCD40L levels, we measured the levels of total cholesterol, glucose, and sCD40L in normal wild-type C57BL6 mice (n = 10) and apoE−/− mice fed on a chow (n = 10) or an atherogenic WD (n = 10) (Figure 1B). Blood measurements were performed after 5 weeks of feeding WD. Results demonstrated that WD-fed apoE−/− mice developed not only severe hypercholesterolemia and significant hyperglycemia but also high levels of plasma sCD40L, as has previously been demonstrated in patients.9,10,11,12
Platelet Deposition and Activation Supports the Early Recruitment of Leukocytes to the Sites of Vascular Injury
After arterial denudation injury, platelet deposition precedes leukocyte accumulation at sites of vascular injury.20 Early recruitment of leukocytes is mediated through platelet-leukocyte interactions followed by leukocyte transmigration through the surface-adherent platelet monolayer, resulting in leukocyte infiltration into vessel wall.38,39 Consistent with this notion, scanning electron microscopic examination showed that large numbers of platelets were deposited on the luminal surface of denudated carotid arteries at 4 hours after the injury (see Supplemental Figure S1A at http://ajp.amjpathol.org), and that most of the deposited platelets appeared to be in an activated state, as judged by platelet shape seen under a high-power magnification (×10,000, panel d). These changes are typical of activated platelets.40 Immunohistochemical staining showed significant platelet deposition in the medial layer and on the luminal surface of the 1-day denudated carotid artery (see Supplemental Figure S1B, panel a, at http://ajp.amjpathol.org). P-selectin staining was restricted to the luminal surface (see Supplemental Figure S1B, panel b, at http://ajp.amjpathol.org), which is consistent with the known propensity of activated platelets to shed P-selectin41 and supported by immunostaining data in a recent report.42 These results suggest that activated platelets may provide an adhesive surface, mediating leukocyte rolling adhesion potentially through the interaction of P-selectin expressed on activated platelets with its ligand PSGL-1 expressed by leukocytes.20
Blocking CD40L Reduces Early Leukocyte Recruitment after Arterial Injury
The process of leukocyte recruitment to the site of vascular injury is characterized by the early neutrophil/monocyte accumulation and the late macrophage accumulation. Therefore, we further determine whether blocking CD40L by anti-CD40L mAb reduces the early recruitment of leukocytes by flow cytometry analysis of cells isolated from carotid arteries 3 days after wire injury. The results show an increase in accumulation of total CD45+ leukocytes (fourfold), Ly-6G+ neutrophils (19-fold), and Mac-3+ macrophages (2.4-fold) in the injured carotid arteries compared with non-injured controls (Figure 2). Antibody blockade of CD40L reduced Ly-6G+ neutrophils and Mac-3+ cell accumulation by 44% (P = 0.011) and 18% (P = 0.038), respectively, compared with the antibody-untreated injured vessels. Because so far there is a lack of specific antigen markers to distinguish mouse monocytes from other inflammatory cells, Gr-1+ cells (monocytes and some granulocytes) were analyzed in the same samples. The results show an 18.7-fold increase in accumulation of the phycoerythrin-Gr-1+ cells that was reduced by 49% with antibody blockade of CD40L (data not shown). These results support the notion that CD40L is a crucial regulator of early leukocyte recruitment to the site of vascular injury.
Figure 2.

A: Flow cytometry of leukocyte subpopulations isolated from injured left and uninjured right carotid arteries of apoE−/− mice 3 days after wire injury. Uninjured (a, d) and injured carotid arteries without (b, e) or with (c, f) MR1 antibody treatment. FITC-LY-6G-positive neutrophils (a–c) and the FITC-Mac-3-positive macrophages (d–f) appear in the upper right quadrant (UR) as indicated. The number shown in each UR indicates the percentage of total live cells counted. Note the increase in both neutrophils and macrophages after arterial injury and the reduction by MR-1 treatment. B: Statistical data analysis from three separate experiments. Data were expressed as percentage ± SD of the indicated leukocyte subpopulations in total live cells counted. Note the increase in both Ly-6G+ neutrophils and Mac-3+ macrophages after arterial injury and the reduction by MR-1 treatment, *P < 0.001 and **P < 0.01 versus specific uninjured controls. #P < 0.02 and ##P < 0.05 versus specific injured groups.
Blocking CD40L Reduces Exaggerated Neointimal Hyperplasia after Arterial Injury
To determine the role of elevated CD40L in the development of neointimal hyperplasia after arterial injury, WD-fed apoE−/− mice were treated with anti-CD40L mAb or isotype control antibody. Wire denudation injury elicited mild neointimal formation in chow-fed apoE−/− mice (data not shown) but exaggerated neointimal formation in the Western diet-fed apoE−/− mice (Figure 3A). The size of the neointimal lesions was reduced by >50% (P < 0.01) in the MR1-treated group compared with the isotype IgG-treated or untreated groups (Figure 3A), supporting the contribution of elevated sCD40L to the exaggerated neointima formation in the WD-fed apoE−/− mice.
Figure 3.

A: Movat-stained representative cross-sections of carotid arteries of apoE−/− mice at 28 days after wire injury. a: An injured, untreated vessel. b: An injured, isotype IgG-treated vessel. c: An injured, MR1-treated vessel. Magnification, ×200. Panel d summarizes the quantitative histomorphometry of plaque area in the three groups. Note the significant reduction in neointimal plaque area in the MR1-treated group; *P < 0.05. B: Quantitative immunocytochemistry of macrophage (Mac-2) and SMC (SM α-actin) content in the neointimal plaques of carotid arteries of apoE−/− mice 4 weeks after denudation injury and 5 weeks on the atherogenic Western diet. Note the significant reduction in macrophage content without significant change in SMC content in the MR-1 treated group, *P < 0.05. C: Lipid profiles in apoE−/− mice fed a Western-type diet 1 week before and 4 weeks after carotid wire injury. Whole blood samples were collected by direct cardiac puncture at 28 days after injury. No significant differences were seen between the MR1-treated, isotype IgG-treated, and non-treated groups with respect to the levels of total cholesterol, low density lipoprotein cholesterol (LDL-C), high density lipoprotein cholesterol (HDL-C), and triglycerides (TG).
Similar to our previous reports,17,30 the percentage area of Mac-2+ macrophages and α-SMA+ vascular SMCs within the neointimal lesions was approximately 25% and 20%, respectively, in the control groups, which indicates a substantial contribution of macrophage accumulation to the development of neointimal hyperplasia in this injury model. Antibody blockade of CD40L resulted in a 56% reduction (P < 0.01) in the neointimal macrophage content but had no significant effect on the neointimal vascular SMC content (Figure 3B). This latter observation was consistent with in vitro data from our study and others. The present study examined the effect of sCD40L on mouse aortic SMC migration using two approaches: the scratch wound random migration (see Supplemental Figure S2A at http://ajp.amjpathol.org) and the modified Boyden chamber chemotaxis migration (see Supplemental Figure S2B at http://ajp.amjpathol.org). We observed that sCD40L (0.1, 1.0, 10 μg/ml) did not induce mouse SMC migration in both cell migration models. Hermann et al43 examined the effects of CD40L on proliferation and migration of cultured bovine coronary SMCs. Their results showed that sCD40L stimulation caused a small but significant increase in DNA synthesis. However, sCD40L-induced DNA synthesis was not followed by proliferation (increase in cell number). Furthermore, sCD40L did not induce SMC migration. In addition, antibody blockade of CD40L had no significant effects on blood lipid profiles (Figure 3C) or complete blood counts (Table 1). These data suggest that CD40L contributes to the exaggerated neointima formation possibly through modulating leukocyte recruitment to the site of vascular injury.
Table 1.
Complete Blood Cell Counts from Anti-CD40L Antibody (MR1)-Treated and Antibody-Untreated Mice
| Mice | White blood cells | Neutrophils | Lymphocytes | Monocytes | Platelets |
|---|---|---|---|---|---|
| Untreated | 4.81 ± 0.56 | 2.57 ± 0.32 | 1.79 ± 0.21 | 0.28 ± 0.13 | 735 ± 92 |
| MR1-treated | 4.45 ± 0.48 | 2.32 ± 0.45 | 1.82 ± 0.23 | 0.26 ± 0.14 | 818 ± 108 |
Values are 1000/μl.
Expression of CD40 Receptor in Mouse Neutrophils in Vitro
CD40 is a 45- to 50-kd type I transmembrane glycoprotein. To our knowledge, CD40 is the only known receptor for CD40L that has been identified in neutrophils to date, although CD40L has been shown to bind to other receptors such as αIIbβ3 in platelets44 and Mac-1 in human monocytes.45 Because the presence of CD40 in mouse neutrophils has not been reported, we examined the expression of CD40 in isolated mouse peritoneal neutrophils. As expected, CD40 mRNA was not found in neutrophils isolated from CD40−/− mice, but its expression was readily detectable in neutrophils isolated from C57BL6 mice and in the murine monocytic J774A.1 cells (TIB-67; American Type Culture Collection, Manassas, VA), as analyzed by reverse transcriptase-polymerase chain reaction analysis (Figure 4A). Consistent with mRNA expression, CD40 protein was not measurable in neutrophils isolated from CD40−/− mice (data not shown) but was readily detectable in neutrophils isolated from C57BL6 mice and in J774A.1 cells, as measured by Western blotting (Figure 4B).
Figure 4.
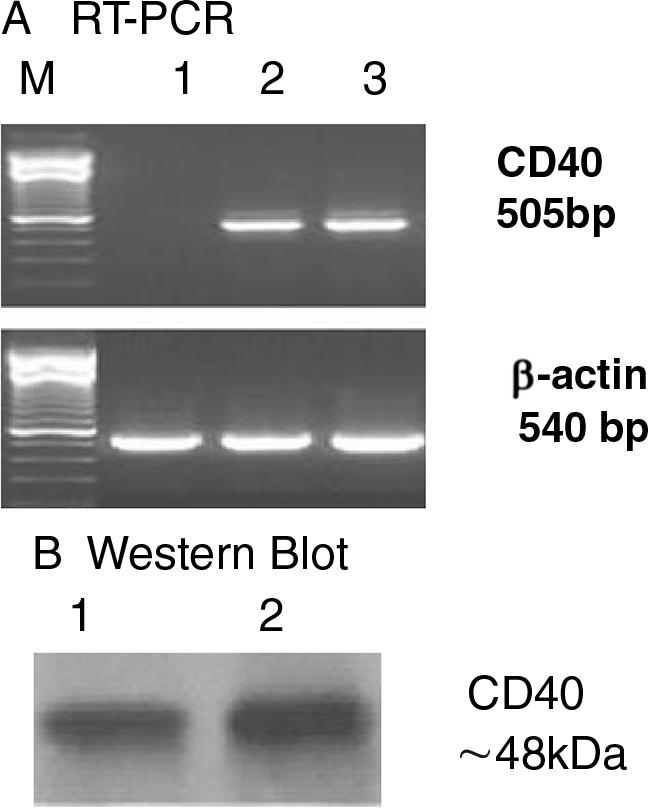
A: Reverse transcriptase-polymerase chain reaction analysis of CD40 mRNA expression. Total cellular RNA was extracted from thioglycolate-elicited peritoneal neutrophils in CD40-deficient mice (lane 1), wild-type C57BL6J mice (lane 2), and from murine monocytic J774A.1 cells (lane 3, positive control), respectively. Note the presence of CD40 mRNA in peritoneal neutrophils from C57Bl/6J mice. M, 100-bp polymerase chain reaction DNA marker. β-Actin served as a loading control. B: Western blot analysis of CD40 protein expression. Total cellular protein was extracted from thioglycolate-elicited peritoneal neutrophils in C57BL/6J mice (lane 1) and from the murine monocytic J774A.1 cells (lane 2, positive control), respectively. Proteins from whole cell lysates (20 μg protein/lane) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and were immunoblotted with a goat polyclonal CD40 antibody (T-20). The CD40 glycoprotein band runs at 48 kd. Note the presence of CD40 protein in peritoneal neutrophils from C57Bl/6J mice.
CD40L Promotes Leukocyte Mac-1 Expression and Leukocyte Firm Adhesion to Activated Surface-Adherent Platelets in Vitro
To explore the molecular mechanisms by which CD40L promotes the early leukocyte recruitment to the sites of vascular injury, we tested the hypothesis that CD40L up-regulates Mac-1 integrin expression and thus promotes neutrophil firm adhesion to adherent platelets in vitro. Flow cytometry analysis shows that rm-CD40L (100 ng/ml) significantly increased the expression of CD11b (αM chain of Mac-1) on mouse peritoneal neutrophils. This effect was abrogated by either 10 μg/ml of anti-CD40L mAb (MR1) or anti-CD40 mAb (HM40–3) (Figure 5A). Further, we assessed neutrophil firm adhesion to activated surface-adherent platelets in vitro. As shown in Figure 5B, rm-CD40L (100 ng/ml) significantly stimulated neutrophil adhesion to the adherent platelets, and this effect was abrogated by 10 μg/ml anti-CD11b mAb (M1/70). These results suggest a Mac-1 activation-dependent mechanism by which CD40-CD40L interaction promotes neutrophil firm adhesion to the surface-adherent platelets at the site of vascular injury.
Figure 5.

A: CD40L stimulates neutrophil Mac-1 integrin expression. Peritoneal neutrophils isolated from C57BL6 mice were stimulated with rm-CD40L (100 ng/ml), without or with pretreatment (30 minutes) by 10 μg/ml of anti-mouse CD40 mAb (HM40–3), anti-mouse CD40L mAb (MR1), or hamster IgG (Iso). Stimulation of neutrophils with phorbol 12-myristate 13-acetate (50 ng/ml) was used a positive control. Mac-1 expression was determined by fluorescence-activated cell sorting analysis of CD11b (αM chain of Mac-1). Data shown are mean fluorescence intensity from three separate experiments and expressed as mean ± SD *P < 0.01 versus bar 1; **P < 0.05 versus bar 3. B: CD40L promotes neutrophil firm adhesion to activated surface-adherent platelets. Peritoneal neutrophils isolated from C57BL6 mice were added to surface-adherent platelets. Neutrophil adhesion was stimulated with rm-CD40L (100 ng/ml) without or with pretreatment (30 minutes) by 10 μg/ml of anti-mouse CD11b mAb (M1/70, αM chain of Mac-1), or rat IgG2b (Iso). Stimulation of neutrophils with phorbol 12-myristate 13-acetate (50 ng/ml) was used a positive control. Neutrophil adhesion was measured as described in Materials and Methods. Data shown are mean ± SD from three separate experiments. *P < 0.01 versus bar 1 indicated condition; **P < 0.05 versus bar 3 indicated condition.
CD40L Promotes Human Neutrophil Oxidative Burst and Release of Matrix Metalloproteinase-9 in Vitro
Neutrophil oxidative burst was measured by flow cytometry using DHR123 as a fluorescent probe. Results show that recombinant human CD40L (100 ng/ml) significantly enhanced neutrophil DHR123 fluorescence, and this effect was blocked by both anti-human CD40L and anti-human CD11b/Mac-1 monoclonal antibodies (Figure 6). Furthermore, this effect was abrogated by either the phosphatidylinositol 3-kinase inhibitor (Ly294002, 1 μmol/L) or the Src kinase inhibitor (PP2, 1 μmol/L), suggesting through the phosphatidylinositol 3-kinase and Src kinase-dependent signaling pathways (Figure 6). The phosphatidylinositol 3-kinase and Src kinase signaling pathways are the known major signaling pathways responsible for leukocyte activation.42,46 matrix metalloproteinase-9 was measured by enzyme-linked immunosorbent assay kits (R&D Systems) by following the manufacturer’s directions. Briefly, isolated human neutrophils were incubated with rh-CD40L (100 ng/ml) for 30 minutes and after centrifugation cell supernatants were collected for enzyme-linked immunosorbent assay. Results show the rh-CD40L stimulation increased the release of matrix metalloproteinase-9 by 146 ± 24% (n = 3), compared with a vehicle-treated control (P < 0.01).
Figure 6.

CD40L promotes neutrophil oxidative burst via phosphatidylinositol 3-kinase and Src kinase-dependent signaling pathways. Aliquots of neutrophils (2 ′ 105 cells in 200 ml) were loaded with 1 mmol/L DHR123 for 5 minutes at 37°C, followed by stimulation with 100 ng/ml rh-CD40L in combination with anti-human CD40L mAb (TRAP1, 1 mg/ml) or anti-human CD11b/Mac-1 (ICRF44, 1 mg/ml) mAb, or in the presence of the phosphatidylinositol 3-kinase inhibitor (LY294002, 1.0 mmol/L) or the Src kinase inhibitor (PP2, 1.0 mmol/L) for 30 minutes at 37°C. Numbers indicate the mean fluorescence intensity of DHR in green fluorescence (FL1) channel by flow cytometry. Similar results were observed from three separate experiments.
Discussion
Clinical studies have clearly shown that elevated plasma sCD40L is associated with an increased rate of restenosis in patients after PCI, as determined by coronary angiography for late lumen loss at 6-month follow-up.13,14,15,16 However, it remains unclear whether and how elevated sCD40L contributes to neointima formation, a prominent feature of restenosis. There are several important findings in the present study. The first significant finding is that elevated circulating sCD40L made a substantial contribution to the exaggerated neointima formation in the apoE−/− mouse model of vascular injury. To our knowledge, levels of plasma sCD40L in mouse models have not been reported to date. In the present study, we detected plasma levels of sCD40L in atherosclerosis-prone apoE−/− mice and found that WD-fed apoE−/− mice developed not only severe hypercholesterolemia and significant hyperglycemia, but also high levels of plasma sCD40L, and that plasma sCD40L level was further increased (1 hour) after arterial injury. These findings from the WD-fed apoE−/− mouse model are close to those found in patients who had hypercholesterolemia and diabetes and underwent PCI.9,10,11,12 Further, the present study showed that neointima formation was exaggerated in the WD-fed apoE−/− mice compared with normal chow-fed apoE−/− mice. This finding is agreement with clinical restenosis that is characterized by exaggerated neointima formation in patients with diabetes.47,48,49 In addition, the extent of macrophage content in the neointimal lesions from the WD-fed apoE−/− mouse model was close to that found in human restenotic lesions. Moreno et al50 reported that macrophage-rich areas were ∼20% in restenotic lesions from patients with restenosis. Therefore, this model provides a unique opportunity for understanding the mechanistic role of elevated sCD40L in the development of neointimal plaque formation after arterial injury in the context of atherosclerosis associated with hypercholesterolemia and diabetes. To investigate the contribution of CD40L to neointima formation, WD-fed apoE−/− mice were treated with the neutralizing monoclonal antibody (MR1) against murine CD40L. This MR1 antibody was chosen because it has been used in numerous published studies including mouse models of atherosclerosis and transplant-associated vasculopathy.2,3,4,5,51 For example, administration of the anti-CD40L antibody (MR1) to either apoE−/− mice or LDL receptor-deficient mice reduced the size of atherosclerotic lesions and macrophage content on high-cholesterol diets.2,3,4,5 The present study showed that blocking CD40L by MR1 significantly reduces the exaggerated neointima formation, with a >50% reduction in neointimal size and 56% reduction in neointimal macrophage content (Figure 3).
The second significant finding is that CD40L is an important regulator of leukocyte recruitment to sites of vascular injury, and the CD40L effects may be mediated through multiple pathways including P-selectin- and Mac-1-dependent mechanisms. Abundant experimental and clinical evidence supports a critical role for platelet activation and leukocyte recruitment in the initiation and progression of neointima formation after arterial injury. Following arterial denudation injury, platelets are quickly deposited on the luminal surface of denudated arteries, activated and release proinflammatory mediators, which promote leukocyte adhesion.20,21 Inhibition of platelet-derived inflammatory molecules such as P-selectin and CCL5/RANTES has been shown to reduce leukocyte recruitment and neointima formation after arterial injury.52,53 Inhibition of leukocyte adhesion and accumulation results in a significant decrease in neointimal thickening and a lower rate of restenosis in experimental and clinical studies.24,54,55,56,57 The present study showed large numbers of activated platelets deposited on the luminal surface of the denudated carotid artery during the early hours after injury by scanning electron microscopy. Activated surface-adherent platelets not only release inflammatory mediators but also provide an adhesive surface, mediating leukocyte rolling along the denudated vessel wall mainly through the interaction of P-selectin expressed on activated platelets with its ligand PSGL-1 expressed on leukocytes.20 As evidence supporting this notion and supported by data from a recent report,42 immunohistochemistry showed robust P-selectin staining on the luminal surface (see Supplemental Figure S1B; panel b, at http://ajp.amjpathol.org), which is consistent with the known propensity of activated platelets to shed P-selectin.41 A recent study showed that recombinant soluble CD40L enhanced human platelet P-selectin expression, aggregation, and platelet-leukocyte conjugation in vitro.58 Similarly, our data demonstrated that recombinant sCD40L robustly stimulated mouse platelet P-selectin expression and this effect was blocked by MR1 antibody pretreatment (see Supplemental Figure S3A at http://ajp.amjpathol.org). Furthermore, sCD40L induced platelet-neutrophil co-aggregation in mouse whole blood (see Supplemental Figure S3B at http://ajp.amjpathol.org), and this effect was abrogated by anti-P-selectin antibody (RB40.34). Taken together, these observations suggest a P-selectin-dependent mechanism responsible for the CD40L-mediated effects.
Distinct adhesion molecules regulate different stages of leukocyte trafficking in a multistep process at sites of inflammation and vascular injury.20 Leukocyte Mac-1 activation and up-regulation is critical for mediating leukocyte firm adhesion and recruitment in vitro and in vivo. In clinical studies, Mac-1 expression on the surface of neutrophils is up-regulated locally and systemically in the early phase after angioplasty/stenting and is associated with neointimal thickening and restenosis.26,27 However, the mechanism of Mac-1 up-regulation on neutrophils is poorly understood. Activated platelets are the primary source of both P-selectin and CD40L in the circulation. Ma et al59 showed that P-selectin enhanced the Mac-1 integrin activation and human neutrophil adhesion to immobilized fibrinogen in vitro. Zirlik et al45 recently reported that CD40L interacts with the integrin Mac-1, which results in Mac-1-dependent adhesion and migration of human monocytes as well as myeloperoxidase release in vitro, but they did not examine effects of CD40L on Mac-1 expression. In our study, we have observed that incubation of mouse neutrophils with activated platelets enhanced neutrophil Mac-1 expression in vitro, and this effect was inhibited by anti-CD40L and anti-P-selectin mAbs (data not shown). We demonstrated that sCD40L stimulates Mac-1 integrin expression on mouse neutrophils as well as promoting neutrophil firm adhesion to activated surface-adherent platelets. These findings suggest a novel molecular mechanism for CD40L-mediated platelet-leukocyte adhesive interaction and leukocyte recruitment via up-regulating Mac-1 expression.
Oxidative stress is widely considered as a common signaling mechanism of the vascular response to injury. Oxidative stress is enhanced in patients with hypercholesterolemia and hyperglycemia, which have been associated with elevated levels of plasma-soluble CD40L.60 Vascular oxidative stress is regulated by many enzyme systems including NAD(P)H oxidase (Nox) and nitric oxide synthase (NOS). For example, in Nox2−/− (gp91phox) mice, absence of Nox2 led to a significant reduction in vascular superoxide (O2−) production and attenuates neointima formation after artery injury.61 In patients with inherited Nox2 deficiency, collagen- and thrombin-stimulated platelets showed an almost complete absence of O2− and CD40L expression.46 The importance of CD40L in vascular oxidative stress was suggested by previous studies. Platelets isolated from CD40L-deficient mice had decreased agonist-induced O2− generation compared with wild-type mice.58 In vitro, CD40L has been shown to enhance reactive oxygen species generation in platelets and leukocytes, and to reduce endothelial eNOS expression and nitric oxide bioavailability in vascular endothelial cells, leading to inhibition of endothelial cell migration.62 These effects may critically affect endothelial regeneration after plaque erosion or denudation and thereby may contribute to the increased risk for development of acute coronary events in patients with high circulating levels of CD40L.62
Neutrophil activation associated with an oxidative burst has been associated with neointimal thickening in patients undergoing coronary stenting.26,27 Early neutrophil recruitment occurs at the sites of arterial injury and endothelial denudation where platelets and fibrin(ogen) have been deposited.38,55 Neutrophils participate in the vascular injury response possibly through their ability to generate an oxidative burst and release metalloproteinases.63 The present study showed that carotid denudation injury induced a significant neutrophil recruitment at 3 days in injured carotid arteries, which was reduced by CD40L blockade. Our in vitro data showed that recombinant sCD40L enhanced oxidative burst and matrix metalloproteinase-9 release in human neutrophils via the phosphatidylinositol 3-kinase and Src kinase signaling pathways, known major signaling mechanisms responsible for leukocyte activation.42,58 These observations suggest that neutrophils are a major source of oxidative stress in the response to vascular injury and inflammation. In conclusion, the present study provides the first demonstration that elevated sCD40L promotes platelet-leukocyte activation and recruitment and neointima formation after arterial injury in an atherogenic animal model, potentially through enhancing platelet P-selectin and leukocyte Mac-1 expression and oxidative activity.
Footnotes
Address reprint requests to Ian J. Sarembock or Guohong Li, The Ohio Heart and Vascular Center, 2123 Auburn Ave., Suite 136, Cincinnati, OH 45219. E-mail: sarembock@ohioheart.org and gli@lsuhsc.edu.
Supported by National Institutes of Health grants HL-66264 (to I.J.S.) and HL-58108 (to K.L.) and by American Heart Association grant 0530166N (to G.L.).
Supplemental material for this article can be found on http://ajp. amjpathol.org.
References
- Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller-Berghaus G, Kroczek RA. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature. 1998;391:591–594. doi: 10.1038/35393. [DOI] [PubMed] [Google Scholar]
- Mach F, Schonbeck U, Sukhova GK, Atkinson E, Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature. 1998;394:200–203. doi: 10.1038/28204. [DOI] [PubMed] [Google Scholar]
- Lutgens E, Gorelik L, Daemen MJ, de Muinck ED, Grewal IS, Koteliansky VE, Flavell RA. Requirement for CD154 in the progression of atherosclerosis. Nat Med. 1999;5:1313–1316. doi: 10.1038/15271. [DOI] [PubMed] [Google Scholar]
- Lutgens E, Cleutjens KB, Heeneman S, Koteliansky VE, Burkly LC, Daemen MJ. Both early and delayed anti-CD40L antibody treatment induces a stable plaque phenotype. Proc Natl Acad Sci USA. 2000;97:7464–7469. doi: 10.1073/pnas.97.13.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonbeck U, Sukhova GK, Shimizu K, Mach F, Libby P. Inhibition of CD40 signaling limits evolution of established atherosclerosis in mice. Proc Natl Acad Sci USA. 2000;97:7458–7463. doi: 10.1073/pnas.97.13.7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipps RP, Koumas L, Leung E, Reddy SY, Blieden T, Kaufman J. The CD40-CD40 ligand system: a potential therapeutic target in atherosclerosis. Curr Opin Invest Drugs. 2001;2:773–777. [PubMed] [Google Scholar]
- Garlichs CD, Kozina S, Fateh-Moghadam S, Handschu R, Tomandl B, Stumpf C, Eskafi S, Raaz D, Schmeisser A, Yilmaz A, Ludwig J, Neundörfer B, Daniel WG. Upregulation of CD40-CD40 ligand (CD154) in patients with acute cerebral ischemia. Stroke. 2003;34:1412–1418. doi: 10.1161/01.STR.0000074032.64049.47. [DOI] [PubMed] [Google Scholar]
- Kopp CW, Steiner S, Nasel C, Seidinger D, Mlekusch I, Lang W, Bartok A, Ahmadi R, Minar E. Abciximab reduces monocyte tissue factor in carotid angioplasty and stenting. Stroke. 2003;34:2560–2567. doi: 10.1161/01.STR.0000094425.06242.64. [DOI] [PubMed] [Google Scholar]
- Garlichs CD, Johm S, Schmeisser Eskafi S, Goppelt-Struebe M, Schmieder R, Daniel WG. Upregulation of CD40 and CD40 ligand (CD154) in patients with moderate hypercholesterolemia. Circulation. 2001;104:2395–2400. doi: 10.1161/hc4501.099312. [DOI] [PubMed] [Google Scholar]
- Sanguigni V, Pignatelli P, Lenti L, Ferro D, Bellia A, Carnevale R, Tesauro M, Sorge R, Lauro R, Violi F. Short-term treatment with atorvastatin reduces platelet CD40 ligand and thrombin generation in hypercholesterolemic patients. Circulation. 2005;111:412–419. doi: 10.1161/01.CIR.0000153810.81187.7D. [DOI] [PubMed] [Google Scholar]
- Cipollone F, Chiarelli F, Davi G, Ferri C, Desideri G, Fazia M, Iezzi A, Santilli F, Pini B, Cuccurullo C, Tumini S, Del Ponte A, Santucci A, Cuccurullo F, Mezzetti A. Enhanced soluble CD40 ligand contributes to endothelial cell dysfunction in vitro and monocyte activation in patients with diabetes mellitus: effect of improved metabolic control. Diabetologia. 2005;48:1216–1224. doi: 10.1007/s00125-005-1750-2. [DOI] [PubMed] [Google Scholar]
- Heeschen C, Dimmeler S, Hamm CW, van den Brand MJ, Boersma E, Zeiher AM, Simoons ML, (CAPTURE Study Investigators) Soluble CD40 ligand in acute coronary syndromes. N Engl J Med. 2003;348:1104–1111. doi: 10.1056/NEJMoa022600. [DOI] [PubMed] [Google Scholar]
- Cipollone F, Ferri C, Desideri G, Paloscia L, Materazzo G, Cuccurullo C, Pini B, Bucci M, Santucci A, Cuccurullo F, Mezzetti A. Preprocedural level of soluble CD40L is predictive of enhanced inflammatory response and restenosis after coronary angioplasty. Circulation. 2003;108:2776–2782. doi: 10.1161/01.CIR.0000103700.05109.0D. [DOI] [PubMed] [Google Scholar]
- Turker S, Guneri S, Akdeniz B, Ozcan MA, Baris N, Badak O, Kirimli O, Yuksel F. Usefulness of preprocedural soluble CD40 ligand for predicting restenosis after percutaneous coronary intervention in patients with stable coronary artery disease. Am J Cardiol. 2006;97:198–202. doi: 10.1016/j.amjcard.2005.08.024. [DOI] [PubMed] [Google Scholar]
- Aggarwal A, Blum A, Schneider DJ, Sobel BE, Dauerman HL. Soluble CD40 ligand is an early initiator of inflammation after coronary intervention. Coron Artery Dis. 2004;15:471–475. doi: 10.1097/00019501-200412000-00003. [DOI] [PubMed] [Google Scholar]
- L’Allier PL, Tardif JC, Gregoire J, Joyal M, Lesperance J, Fortier A, Guertin MC. Sustained elevation of serum CD40 ligand levels one month after coronary angioplasty predicts angiographic restenosis. Can J Cardiol. 2005;21:495–500. [PubMed] [Google Scholar]
- Phillips JW, Barringhaus KG, Sanders JM, Yang Z, Chen M, Hesselbacher S, Czarnik AC, Ley K, Nadler J, Sarembock IJ. Rosiglitazone reduces the accelerated neointima formation after arterial injury in a mouse injury model of type 2 diabetes. Circulation. 2003;108:1994–1999. doi: 10.1161/01.CIR.0000092886.52404.50. [DOI] [PubMed] [Google Scholar]
- Su Z, Li Y, James JC, Matsumoto AH, Helm GA, Lusis AJ, Shi W. Genetic linkage of hyperglycemia, body weight and serum amyloid-P in an intercross between C57BL/6 and C3H apolipoprotein E-deficient mice. Hum Mol Genet. 2006;15:1650–1658. doi: 10.1093/hmg/ddl088. [DOI] [PubMed] [Google Scholar]
- Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ, Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation. 2007;115:1275–1284. doi: 10.1161/CIRCULATIONAHA.106.663120. [DOI] [PubMed] [Google Scholar]
- Shah PK. Inflammation, neointimal hyperplasia, and restenosis: as the leukocytes roll, the arteries thicken. Circulation. 2003;107:2175–2177. doi: 10.1161/01.CIR.0000069943.41206.BD. [DOI] [PubMed] [Google Scholar]
- Welt FG, Rogers C. Inflammation and restenosis in the stent era. Arterioscler Thromb Vasc Biol. 2002;22:1769–76. doi: 10.1161/01.atv.0000037100.44766.5b. [DOI] [PubMed] [Google Scholar]
- Weber C, Springer TA. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J Clin Invest. 1997;100:2085–2093. doi: 10.1172/JCI119742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, Ballantyne CM, Zhang L, Furman MI, Berndt MC, Lopez JA. Platelet glycoprotein ibalpha is a counterreceptor for the leukocyte integrin Mac-1 (CD11b/CD18). J Exp Med. 2000;192:193–204. doi: 10.1084/jem.192.2.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sakuma M, Chen Z, Ustinov V, Shi C, Croce K, Zago AC, Lopez J, Andre P, Plow E, Simon DI. Leukocyte engagement of platelet glycoprotein Ibalpha via the integrin Mac-1 is critical for the biological response to vascular injury. Circulation. 2005;112:2993–3000. doi: 10.1161/CIRCULATIONAHA.105.571315. [DOI] [PubMed] [Google Scholar]
- Neumann FJ, Ott I, Gawaz M, Puchner G, Schomig A. Neutrophil and platelet activation at balloon-injured coronary artery plaque in patients undergoing angioplasty. J Am Coll Cardiol. 1996;27:819–824. doi: 10.1016/0735-1097(95)00563-3. [DOI] [PubMed] [Google Scholar]
- Inoue T, Uchida T, Yaguchi I, Sakai Y, Takayanagi K, Morooka S. Stent-induced expression and activation of the leukocyte integrin Mac-1 is associated with neointimal thickening and restenosis. Circulation. 2003;107:1757–1763. doi: 10.1161/01.CIR.0000060487.15126.56. [DOI] [PubMed] [Google Scholar]
- Inoue T, Kato T, Hikichi Y, Hashimoto S, Hirase T, Morooka T, Imoto Y, Takeda Y, Sendo F, Node K. Stent-induced neutrophil activation is associated with an oxidative burst in the inflammatory process, leading to neointimal thickening. Thromb Haemost. 2006;95(1):43–48. [PubMed] [Google Scholar]
- Inoue T, Sakai Y, Hoshi K, Yaguchi I, Fujito T, Morooka S. Lower expression of neutrophil adhesion molecule indicates less vessel wall injury and might explain lower restenosis rate after cutting balloon angioplasty. Circulation. 1998;97:2511–2518. doi: 10.1161/01.cir.97.25.2511. [DOI] [PubMed] [Google Scholar]
- Inoue T, Sakai Y, Morooka S, Hayashi T, Takayanagi K, Takabatake Y. Expression of polymorphonuclear leukocyte adhesion molecules and its clinical significance in patients treated with percutaneous transluminal coronary angioplasty. J Am Coll Cardiol. 1996;28:1127–1233. doi: 10.1016/S0735-1097(96)00308-7. [DOI] [PubMed] [Google Scholar]
- Li G, Sanders JM, Phan ET, Ley K, Sarembock IJ. Arterial macrophages and regenerating endothelial cells express P-selectin in atherosclerosis-prone apolipoprotein E-deficient mice. Am J Pathol. 2005;167:1511–1518. doi: 10.1016/S0002-9440(10)61237-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner V, Fingerle J, Reidy MA. Mouse model of arterial injury. Circ Res. 1993;73:792–796. doi: 10.1161/01.res.73.5.792. [DOI] [PubMed] [Google Scholar]
- Movat H. Demonstration of all connective tissue elements in a single section. Arch Pathol Med. 1955;60:289–295. [PubMed] [Google Scholar]
- Hay C, Micko C, Prescott MF, Liau G, Robinson K, De Leon H. Differential cell cycle progression patterns of infiltrating leukocytes and resident cells after balloon injury of the rat carotid artery. Arterioscler Thromb Vasc Biol. 2001;21:1948–1954. doi: 10.1161/hq1201.100256. [DOI] [PubMed] [Google Scholar]
- Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J Exp Med. 2006;203:1273–1282. doi: 10.1084/jem.20052205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elzey BD, Tian J, Jensen RJ, Swanson AK, Lees JR, Lentz SR, Stein CS, Nieswandt B, Wang Y, Davidson BL, Ratliff TL. Platelet-mediated modulation of adaptive immunity: a communication link between innate and adaptive immune compartments. Immunity. 2003;19:9–19. doi: 10.1016/s1074-7613(03)00177-8. [DOI] [PubMed] [Google Scholar]
- Heinzel FP, Rerko RM, Hujer AM. Underproduction of interleukin-12 in susceptible mice during progressive leishmaniasis is due to decreased CD40 activity. Cell Immunol. 1998;184:129–142. doi: 10.1006/cimm.1998.1267. [DOI] [PubMed] [Google Scholar]
- Li G, Keenan AC, Young J, Fierschnaller V, Berlin H, Hall M, Steinhubl SR, Smyth SS. Effects of unfractionated heparin and glycoprotein IIb/IIIa antagonists versus bivalirdin on myeloperoxidase release from neutrophils. Arterioscler Thromb Vasc Biol. 2007;27:1850–1856. doi: 10.1161/ATVBAHA.107.144576. [DOI] [PubMed] [Google Scholar]
- Roque M, Fallon JT, Badimon JJ, Zhang WX, Taubman MB, Reis ED. Mouse model of femoral artery denudation injury associated with the rapid accumulation of adhesion molecules on the luminal surface and recruitment of neutrophils. Arterioscler Thromb Vasc Biol. 2000;20:335–342. doi: 10.1161/01.atv.20.2.335. [DOI] [PubMed] [Google Scholar]
- Osaka M, Hagita S, Haraguchi M, Kajimura M, Suematsu M, Yoshida M. Real time imaging of mechanically injured-femoral artery in mouse revealed a biphasic pattern of leukocyte accumulation. Am J Physiol Heart Circ Physiol. 2007;292:H1876–H1882. doi: 10.1152/ajpheart.00708.2006. [DOI] [PubMed] [Google Scholar]
- Inwald DP, McDowall A, Peters MJ, Callard RE, Klein NJ. CD40 is constitutively expressed on platelets and provides a novel mechanism for platelet activation. Circ Res. 2003;92:1041–1048. doi: 10.1161/01.RES.0000070111.98158.6C. [DOI] [PubMed] [Google Scholar]
- Dole VS, Bergmeier W, Mitchell HA, Eichenberger SC, Wagner DD. Activated platelets induce Weibel-Palade-body secretion and leukocyte rolling in vivo: role of P-selectin. Blood. 2005;106:2334–2339. doi: 10.1182/blood-2005-04-1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evangelista V, Pamuklar Z, Piccoli A, Manarini S, Dell’elba G, Pecce R, Martelli N, Federico L, Rojas M, Berton G, Lowell CA, Totani L, Smyth SS. Src family kinases mediate neutrophil adhesion to adherent platelets. Blood. 2007;109:2461–2469. doi: 10.1182/blood-2006-06-029082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann A, Schror K, Weber AA. CD40 ligand (CD40L) does not stimulate proliferation of vascular smooth muscle cells. Eur J Cell Biol. 2002;81:213–221. doi: 10.1078/0171-9335-00240. [DOI] [PubMed] [Google Scholar]
- Andre P, Prasad KS, Denis CV, He M, Papalia JM, Hynes RO, Phillips DR, Wagner DD. CD40L stabilizes arterial thrombi by a beta3 integrin–dependent mechanism. Nat Med. 2002;8:247–252. doi: 10.1038/nm0302-247. [DOI] [PubMed] [Google Scholar]
- Zirlik A, Maier C, Gerdes N, MacFarlane L, Soosairajah J, Bavendiek U, Ahrens I, Ernst S, Bassler N, Missiou A, Patko Z, Aikawa M, Schonbeck U, Bode C, Libby P, Peter K. CD40 ligand mediates inflammation independently of CD40 by interaction with Mac-1. Circulation. 2007;27:1571–1580. doi: 10.1161/CIRCULATIONAHA.106.683201. [DOI] [PubMed] [Google Scholar]
- Pignatelli P, Sanguigni V, Lenti L, Ferro D, Finocchi A, Rossi P, Violi F. gp91phox-dependent expression of platelet CD40 ligand. Circulation. 2004;110:1326–1329. doi: 10.1161/01.CIR.0000134963.77201.55. [DOI] [PubMed] [Google Scholar]
- Kornowski R, Mintz GS, Kent KM, Pichard AD, Satler LF, Bucher TA, Hong MK, Popma JJ, Leon MB. Increased restenosis in diabetes mellitus after coronary interventions is due to exaggerated intimal hyperplasia: a serial intravascular ultrasound study. Circulation. 1997;95:1366–1369. doi: 10.1161/01.cir.95.6.1366. [DOI] [PubMed] [Google Scholar]
- Kornowski R, Mintz GS, Abizaid A, Leon MB. Intravascular ultrasound observations of atherosclerotic lesion formation and restenosis in patients with diabetes mellitus. Int J Cardiovasc Intervent. 1999;2:13–20. doi: 10.1080/acc.2.1.13.20. [DOI] [PubMed] [Google Scholar]
- Radke PW, Friese K, Buhr A, Nagel B, Harland LC, Kaiser A, Remmel M, Hanrath P, Schunkert H, Hoffmann R. Comparison of coronary restenosis rates in matched patients with versus without diabetes mellitus. Am J Cardiol. 2006;98:1218–1222. doi: 10.1016/j.amjcard.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Moreno PR, Bernardi VH, Lopez-Cuellar J, Newell JB, McMellon C, Gold HK, Palacios IF, Fuster V, Fallon JT. Macrophage infiltration predicts restenosis after coronary intervention in patients with unstable angina. Circulation. 1996;94:3098–3102. doi: 10.1161/01.cir.94.12.3098. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mazer SP, Minamoto K, Takuma S, Homma S, Yellin M, Chess L, Fard A, Kalled SL, Oz MC, Pinsky DJ. Suppression of murine cardiac allograft arteriopathy by long-term blockade of CD40-CD154 interactions. Circulation. 2002;105:1609–1614. doi: 10.1161/01.cir.0000013022.11250.30. [DOI] [PubMed] [Google Scholar]
- Manka D, Forlow SB, Sanders JM, Hurwitz D, Bennett DK, Green SA, Ley K, Sarembock IJ. Critical role of platelet P-selectin in the response to arterial injury in apolipoprotein-E-deficient mice. Arterioscler Thromb Vasc Biol. 2004;24:1124–1129. doi: 10.1161/01.ATV.0000127619.04687.f4. [DOI] [PubMed] [Google Scholar]
- Schober A, Manka D, von Hundelshausen P, Huo Y, Hanrath P, Sarembock IJ, Ley K, Weber C. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation. 2002;106:1523–1529. doi: 10.1161/01.cir.0000028590.02477.6f. [DOI] [PubMed] [Google Scholar]
- Simon DI, Dhen Z, Seifert P, Edelman ER, Ballantyne CM, Rogers C. Decreased neointimal formation in Mac-1(−/−) mice reveals a role for inflammation in vascular repair after angioplasty. J Clin Invest. 2000;105:293–300. doi: 10.1172/JCI7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welt FG, Edelman ER, Simon DI, Rogers C. Neutrophil, not macrophage, infiltration precedes neointimal thickening in balloon-injured arteries. Arterioscler Thromb Vasc Biol. 2000;20:2553–2558. doi: 10.1161/01.atv.20.12.2553. [DOI] [PubMed] [Google Scholar]
- Rogers C, Welt FG, Karnovsky MJ, Edelman ER. Monocyte recruitment and neointimal hyperplasia in rabbits. Arterioscler Thromb Vasc Biol. 1996;16:1312–1318. doi: 10.1161/01.atv.16.10.1312. [DOI] [PubMed] [Google Scholar]
- Inoue T, Uchida T, Sakuma M, Imoto Y, Ozeki Y, Ozaki Y, Hikichi Y, Node K. Cilostazol inhibits leukocyte integrin Mac-1, leading to a potential reduction in restenosis after coronary stent implantation. J Am Coll Cardiol. 2004;44:1408–1414. doi: 10.1016/j.jacc.2004.06.066. [DOI] [PubMed] [Google Scholar]
- Chakrabarti S, Varghese S, Vitseva O, Tanriverdi K, Freedman JE. CD40 ligand influences platelet release of reactive oxygen intermediates. Arterioscler Thromb Vasc Biol. 2005;25:2428–2434. doi: 10.1161/01.ATV.0000184765.59207.f3. [DOI] [PubMed] [Google Scholar]
- Ma YQ, Plow EF, Geng JG. P-selectin binding to P-selectin glycoprotein ligand-1 induces an intermediate state of alphaMbeta2 activation and acts cooperatively with extracellular stimuli to support maximal adhesion of human neutrophils. Blood. 2004;104:2549–2556. doi: 10.1182/blood-2004-03-1108. [DOI] [PubMed] [Google Scholar]
- Sanguigni V, Ferro D, Pignatelli P, Del Ben M, Nadia T, Saliola M, Sorge R, Violi F. CD40 ligand enhances monocyte tissue factor expression and thrombin generation via oxidative stress in patients with hypercholesterolemia. J Am Coll Cardiol. 2005;45:35–42. doi: 10.1016/j.jacc.2004.09.047. [DOI] [PubMed] [Google Scholar]
- Chen Z, Keaney JF, Jr, Schulz E, Levison B, Shan L, Sakuma M, Zhang X, Shi C, Hazen SL, Simon DI. Decreased neointimal formation in Nox2-deficient mice reveals a direct role for NADPH oxidase in the response to arterial injury. Proc Natl Acad Sci USA. 2004;101:13014–13019. doi: 10.1073/pnas.0405389101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbich C, Dernbach E, Aicher A, Zeiher AM, Dimmeler S. CD40 ligand inhibits endothelial cell migration by increasing production of endothelial reactive oxygen species. Circulation. 2002;106:981–986. doi: 10.1161/01.cir.0000027107.54614.1a. [DOI] [PubMed] [Google Scholar]
- Lee MS, David EM, Makkar RR, Wilentz JR. Molecular and cellular basis of restenosis after percutaneous coronary intervention: the intertwining roles of platelets, leukocytes, and the coagulation-fibrinolysis system. J Pathol. 2004;203:861–870. doi: 10.1002/path.1598. [DOI] [PubMed] [Google Scholar]