

Abstract
The cysteine-rich protein CCN6 [or Wnt-1-induced signaling protein 3 (WISP3)] exerts tumor-suppressive effects in aggressive inflammatory breast cancer. Loss of CCN6 is associated with poorly differentiated phenotypes and increased invasion. Here, we show that reduction of CCN6 expression occurs in 60% of invasive breast carcinomas and is associated with axillary lymph node metastases. Furthermore, low CCN6 expression in invasive carcinoma tissue samples correlates with reduced expression of E-cadherin. In vitro, RNA interference knockdown of CCN6 in two benign human mammary epithelial cell lines (HME and MCF10A) decreased expression of E-cadherin protein and mRNA and reduced activity of the E-cadherin promoter; this reduction was dependent on intact E-box elements. CCN6 knockdown in HME cells resulted in up-regulation of the E-cadherin transcriptional repressors Snail and ZEB1 and enhanced their recruitment and binding to the E-cadherin promoter as analyzed by chromatin immunoprecipitation assays. Small interfering RNA-mediated knockdown of ZEB1 or Snail blocked the down-regulation of E-cadherin caused by CCN6 inhibition. These data show, for the first time, that CCN6 expression is reduced or lost in a substantial number of invasive breast carcinomas and that CCN6 modulates transcriptional repressors of E-cadherin. Together, our results lead to a new hypothesis that Snail and ZEB1 are downstream of CCN6 and play a critical role in CCN6-mediated regulation of E-cadherin in breast cancer.
Afflicting one of eight women, breast cancer is the second leading cause of cancer-related deaths in women in the United States.1 Despite advances in the early detection and treatment of breast cancer, mortality for those 20% of patients with recurrences and/or metastases is nearly 100%.2 Discovering and characterizing key genes and pathways that define breast cancers with metastatic ability will help identify molecular markers that predict prognosis before metastasis develops and that may represent appropriate therapeutic and/or preventative targets.
Once cancer develops, the acquisition of invasive capabilities is important for the progression of disease. Tumor cell migration and metastasis is a highly coordinated process. It requires not only alteration of cell adhesion to extracellular matrix proteins, but also the disruption of cell-cell junctions and changes in cell morphology, which is termed epithelial-mesenchymal transition (EMT).3,4,5 During this complex and dynamic process, epithelial cells acquire fibroblast-like properties and show reduced intercellular adhesion and increased motility. The initiation of EMT in epithelial-derived cancer types is marked by E-cadherin repression.6,7 The key events that lead to the down-regulation of E-cadherin and initiation of EMT in breast cancer remain elusive.
CCN6 [Wnt-1 induced signaling protein (WISP3)] is a cysteine-rich protein down-regulated in the most lethal form of locally advanced breast cancer, inflammatory breast cancer, and in a group of advanced-stage noninflammatory breast cancer tumors.8 CCN6 re-expression restores differentiated epithelial phenotypes in the breast. Consistently, accumulated evidence shows that CCN6 inhibits tumor cell motility and invasion in vitro and inhibits tumor growth in vivo.9,10,11,12 CCN6 is also shown to inhibit tumor-induced angiogenesis.10,11 Recently, we demonstrated that CCN6 inhibition causes EMT of breast epithelial cells.12 This function of CCN6 correlates with its effect on cell motility and invasion. The current study describes the first mechanistic evidence that CCN6 regulates E-cadherin levels and that the transcriptional repressors Snail and ZEB1 are required for this function, and demonstrates that CCN6 expression in human breast tissues correlates with lymph node metastasis and E-cadherin levels.
Materials and Methods
Human Breast Tissue Samples and Immunohistochemistry
For tissue microarray (TMA) construction, 116 invasive breast carcinoma tissues obtained with Institutional Review Board approval were used. The TMA was constructed using triplicate tissue samples as previously described.13 Clinical and pathological variables were determined following well established criteria. All invasive carcinomas were graded according to the method described by Elston and Ellis.14 Standard biotin-avidin complex immunohistochemistry was performed on 4-μm-thick paraffin-embedded tissue sections of the TMA, using a primary anti-CCN6 antibody (dilution 1:150; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and a monoclonal anti-E-cadherin antibody (1:400; Zymed, Carlsbad, CA).9,13,15 CCN6 protein expression was scored using a standard, pathologist-based four-tiered scoring system as 1, negative; 2, weak; 3, moderate; and 4, strong.13,16 Negative and weak staining were considered low CCN6, and moderate and strong were considered high CCN6 based on our studies on the biology of CCN6.9,10,11 To analyze the expression of E-cadherin on tissue samples, we used a quantitative image analysis system (ACIS; ChromaVision Medical Systems, Inc., San Juan Capistrano, CA), which allows for a continuous rather than categorical scoring, and it is well suited to analyzed small changes in protein expression.17,18 Using this system, E-cadherin staining intensity was evaluated on a scale of 73 to 139 (mean, 100.5). A Wilcoxon exact test was used to compare the distributions of E-cadherin intensity according to CCN6 protein levels, and a P value <0.05 was considered statistically significant.
Xenografts
Paraffin embedded tissue sections of six xenografts derived from SUM149 breast cancer (CCN6-deficient) cells overexpressing CCN6 (four tumors) or vector controls (two tumors) were studied. We have previously found that xenografts derived from SUM149/CCN6 cells developed more slowly, were significantly smaller, and were better differentiated than SUM149/vector (CCN6-deficient) tumors.11 We analyzed CCN6 and E-cadherin expression on these xenografts using anti-CCN6 and anti-E-cadherin antibodies as described above.
Cell Culture
HME cells were cultured in Ham’s F-12 medium (Invitrogen, Carlsbad, CA) supplemented with 5% fetal bovine serum (Cambrex, Walkersville, MD), 1 μg/ml hydrocortisone, 5 μg/ml insulin, 10 ng/ml epidermal growth factor, and 100 ng/ml cholera toxin at 37°C under 10% CO2. MDA-MB-231 and HEK293 cells (American Type Culture Collection, Manassas, VA) were maintained in Dulbecco’s modified Eagle’s medium (Invitrogen) supplemented with 10% fetal bovine serum. MCF10A (American Type Culture Collection) cells were cultured in Dulbecco’s modified Eagle’s medium/Ham’s F-12 (Invitrogen) supplemented with 10% fetal bovine serum, 1 μg/ml hydrocortisone, 5 μg/ml insulin, and 10 μg/ml epidermal growth factor.12
Generation of Stable HME Cell Lines with CCN6 Knockdown
HME CCN6 knockdown stable cell lines were generated by small interfering RNA (siRNA-CCN6) and short hairpin RNA (shRNA-CCN6), respectively. The empty vectors were used as controls. The construction of HME siRNA-CCN6 has been described previously.12 shRNA-CCN6 plasmid was purchased from Sigma (St. Louis, MO), based on a lentiviral vector (pLKO.1-puro). The shRNA-CCN6 was packaged at University of Michigan Vector Core, and the virus-containing supernatant was diluted 1:1 with fresh medium and used to infect HME cells. Selection was initiated in 10 μg/ml puromycin (Sigma) 48 hours after infection of HME cells. Stable transfectants were established after 3 weeks. The shRNA-CCN6 target sequence was as follows: 5′-CCGGCCATTAGATACAACACCTGAACTCGAGTTCAGGTGTTGTA-3′.
Small Interfering RNA-Mediated RNA Interference
Snail and ZEB1 mRNA were blocked by using siRNA SMARTpool (Dharmacon, Lafayette, CO) following the manufacturer’s instructions. Briefly, HME shRNA-CCN6 or siRNA-CCN6 stable cells were grown to 70 to 80% confluence for 24 hours and were then treated with 100 nmol/L siRNA-Snail and 100 nmol/L siRNA-Zeb1, respectively. siCONTROL nontargeting siRNA pool (100 nmol/L; Dharmacon) was used as negative control. DharmaFECT Transfection reagent (Dharmacon) was used following the protocol described by the manufacturer. The medium was changed 24 hours after transfection, and the cells were incubated in fresh medium for an additional 48 to 72 hours. The following pairs (sense/antisense) of SMARTpool siRNA were used: siRNA-Snail, 5′-ACUCAGAUGUCAAGAAUAUU-3′/5′-PUACUUCUUGACAUCUGAGUUU-3′, 5′-GCAAAUACUGCAACAAGGAUU-3′/5′-PUCCUUGUUGCAGUAUUUGCUU-3′, 5′-GCUCGGACCUUCUCCCGAAUU-3′/5′-PUUCGGGAGAAGGUCCGAGCCUU-3′, and 5′-GCUUGGGCCAAGUGCCCAAUU-3′/5′-PUUGGGCACUUGGCCCAAGCUU-3′; and siRNA-Zeb1, 5′-GAACCACCCUUGAAAGUGAUU-3′/5′-PUCACUUUCAAGGGUGGUUCUU-3′, 5′-GAAGCAGGAUGUACAGUAAUU-3′/5′-PUUACUGUACAUCCUGCUUCUU-3′, 5′- AAACUGAACCUGUGGAUUAUU-3′/5′-PUAAUCCACAGGUUCAGUUUUU-3′, and 5′-GAUAGCACUUGUCUUCUGUUU-3′/5′-PACAGAAGACAAGUGCUAUCUU-3′.
Western Blot Analysis
Samples for analysis by immunoblot were prepared as described previously.9,10,11 In brief, cell lysates were prepared in lysis buffer containing 50 mmol/L Tris-HCl (pH 7.4), 1% Nonidet P-40, and a mixture of protease inhibitors (Roche, Indianapolis, IN). The proteins were resolved by SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose. After blocking with 5% nonfat milk in Tris-buffered saline/Tween 20 at room temperature for 1 hour, the membranes were incubated with the following antibodies: anti-CCN6 (Santa Cruz Biotechnology) at 1:1000 dilution, anti-Snail (rabbit polyclonal; Santa Cruz Biotechnology) at 1:1000 dilution, anti-Zeb1 (Santa Cruz Biotechnology) at 1:500 dilution, anti-E-cadherin (BD Transduction Laboratories, San Jose, CA) at 1:2500, and anti-β-actin (Sigma) at1:10.000. After washing in Tris-buffered saline/Tween 20, the blot was incubated with horseradish peroxidase-conjugated secondary antibodies at 1:2000 (Amersham Bioscience, Piscataway, NJ), and the antigen-antibody complexes were visualized by ECL system (Amersham Bioscience).
Total RNA Preparation, cDNA Synthesis, and Real-Time PCR Analysis
Total RNA was isolated from cells with a TriZol kit (Life Technologies, Inc., Gaithersburg, MD). cDNA was synthesized using a reverse transcription system (Promega, Madison, WI) and 1 μg of total RNA as template. For real-time PCR, relative gene expression was determined using SYB Green PCR Master Mix kit (Applied Biosystems, Foster City, CA) in an Applied Biosystems 7300 Real Time PCR system following the manufacturer’s protocol. Amplification was performed for 40 cycles of 3 minutes at 95°C, 60 seconds at 60°C, and 3 minutes at 72°C. The following pairs of primers (forward/reverse, 250 nmol/L of each) were used: SNAIL, 5′-GCGAGCTGCAGGACTCTAAT-3′/5′-CCRCTGTCCTCATCTGACA-3′; SLUG, 5′-TTCGGACCCACACATTACCT-3/5′-TTGGAGCAGTTTTTGCACTG- 3′; TWIST1, 5′-GGAGTCCGCAGTCTTACGAG-3′/5′-TGGAGGACCTGGTAGAGGAA-3′; TWIST2, 5′-AGCAAGAAGTCGAGCTAAGA-3′/5′-CAGCTTGAGCGTCTGGATCT-3′; smad-interacting protein-1, 5′-AATGGCAACAGCAACAAGTG-3′/5′-CCCCGTCAGCACATAACTTT-3′; ZEB1, 5′-GCACAACCAAGTGCAGAAGA-3′/5′-CATTTGCAGATTGAGGCTGA-3′; E-CADHERIN, 5′-CGACCAACCCAAGAATCTA-3′/5′-AGGCTGTGCCTTCCTACAGA-3′; and β-ACTIN, 5′-TCCCTGGAGAAGAGCTACGA-3′/5′-AGCACTGTGTTGGCGTACAG-3′. The quantity of DNA in each sample was calculated by interpolating its threshold cycle value versus a standard curve of threshold cycle values obtained from serially diluted cDNA from a mixture of all of the samples. The calculated quantity of the target gene for each sample was then divided by the average calculated quantity of β-actin corresponding to each sample to give a relative expression of the target gene for each sample.
Luciferase Reporter Assays
HME, MCF10A, and HEK293 cell transfections were performed in six-well plates using FuGENE6 Transfection Reagent (Roche) following the protocol described by the manufacturer. HEK293 and MCF10A cells were cotransfected with 1 g of reporter gene Ecad(−108)-Luc (kind gift of Dr. Eric Fearon) and 0.5 g of pSilencer2.1-U6-CCN6-siRNA5 or pSilencer2.1-U6 vector. HME siRNA-vector and HME siRNA-CCN6 stable clones were transfected 1.0 g of reporter gene Ecad(−108)-Luc only. Luciferase assays were performed 24 hours after transfection using a Dual Luciferase Assay System (Promega) and normalized by measuring Renilla activities (cotransfected with 4 ng of pSV-40). E-cadherin promoter activities were presented as relative light units to that obtained from pGL3-transfected cells. Triplicate samples were run in all of the experiments, which were repeated at least three times.
To examine the transcriptional regulation of E-cadherin in CCN6 knockdown clones, we used the wild-type [Ecad(−108)-Luc] and E-box-mutated E-cadherin reporter gene constructs [Ecad(−108)-AMut, Ecad(−108)-CMut, and Ecad(−108)-ABCMut; kind gift of Dr. Eric Fearon].6,19 HME siRNA-vector and HME siRNA-CCN6 stable clones were transiently transfected with either Ecad(−108)-Luc or Ecad(−108)-Ebox mutants as above along with the pRL-null vector. Cells were lysed 48 hours later, and luciferase assays were performed using the dual luciferase assay system (Promega). Each experiment was performed in triplicate.
Immunofluorescence and Confocal Microsocopy
Cells were grown on chamber slides (Nalgen Nunc International, Naperville, IL) and fixed with 3.7% formaldehyde in PBS (pH 7.4) for 10 minutes at room temperature and then permeabilized with PBS containing 0.5% Triton X-100 for 10 minutes at room temperature. The cells were blocked with 5% bovine serum albumin in PBS for 1 hour at room temperature. After washing the slides with PBS, a mixture of mouse monoclonal anti-E-cadherin antibody at 1:500 dilution (BD Transduction Laboratories) and rabbit polyclonal anti-Snail antibody at 1:200 dilution (Abcam, Cambridge, UK) was added and incubated overnight at 4°C. After washing the slides, a mixture of secondary donkey anti-rabbit Alexa 488 and donkey anti-mouse Alexa 555 (Molecular Probes, Eugene, OR) was applied at 1:1500 dilution and incubated in the dark for 1 hour. Washing the slides with PBS, anti-fade with 4′6-diamidino-2-phenylindole was applied to stain nuclei and covered by a glass coverslip. Confocal images were taken with a Zeiss LSM510 META imaging system using UV Argon and Helium Neon 1 light source.
Chromatin Immunoprecipitation Assay
Chromatin immunoprecipitation analysis was performed using the ChIP-IT Enzymatic kit (Active Motif, Carlsbad, CA) following the manufacturer’s protocol. Briefly, HME shRNA-CCN6 stable clones and control HME shRNA-vector were grown to 70 to 80% confluence on 150-mm plates. The cells were fixed with 1.0% formaldehyde, and the nuclei were released using lysis buffer. The chromatin was enzymatically sheared into small uniform fragments by incubation at 37°C for 10 minutes, and the protein/DNA complexes were then immunoprecipitated overnight at 4°C using anti-Snail (rabbit polyclonal; Abcam), anti-Zeb1 (Santa Cruz Biotechnology), and control IgG (normal Rabbit IgG) antibodies, respectively, with protein G magnetic beads. The beads were then collected by magnetic pull-down. The cross-linked protein/DNA complexes were eluted from G beads at 94°C for 15 minutes and then treated with proteinase K at 37°C for 1 hour to reverse protein/DNA complex. Stop buffer was subsequently added to inhibit proteinase K function. The resulting DNA was subjected directly to PCR analysis for 36 cycles using the following conditions: 5 minutes at 94°C, 30 seconds at 94°C, 30 seconds at 55°C, 20 seconds at 72°C, and 7 minutes at 72°C. The primers (400 nmol/L) for E-cadherin promoter were synthesized as follows: forward, 5′-TAGAGGGTCACCGCGTCTAT-3′ (−170∼−151); reverse, 5′-TCACAGGTGCTTTGCAGTTC-3′ (+10∼+39). The PCR product covers the A, B, and C E-box elements in the proximal E-cadherin: E-box A, 5′-CAGGTG-3′ (−79∼−74); E-box B, 5′-CACCTG-3′ (−59∼−54); and E-box C, 5′-CACCTG-3′ (+21∼+26).
Results
CCN6 Protein Expression Is Associated with Axillary Lymph Node Metastasis and E-Cadherin Expression in Human Invasive Breast Carcinoma Tissue Sections
Using high-density TMAs, we evaluated the expression of CCN6 and E-cadherin proteins on 116 unselected invasive carcinoma tissue samples to characterize its expression in situ by immunohistochemistry. Clinical and pathological characteristics of the patients can be found in Table 1. CCN6 protein expression was observed primarily in the cytoplasm of breast cancer cells and less frequently in the nucleus (Figure 1). Occasional stromal cells also expressed CCN6 protein. Invasive carcinomas expressing high levels of CCN6 (scores 3 to 4, CCN6+) and those that expressed low levels (scores 1 to 2, CCN6−) were readily apparent (Figure 1). We noted that of the 102 invasive carcinomas with available tumor for immunohistochemistry, CCN6 expression was lost or reduced (scores 1 to 2) in 62 (60.7%) tumors. Furthermore, invasive carcinomas with low CCN6 had increased incidence of lymph node metastasis when compared with tumors with high CCN6 (χ2 test, P = 0.04). CCN6 expression was not associated with other tumor characteristics, including histological type, grade, size, hormonal receptor status, or HER-2/neu overexpression (Table 2).
Table 1.
Clinical and Pathological Characteristics of the Patients Included in This Study
| Parameter | Value |
|---|---|
| No. of patients | 116 |
| Median age [years (range)] | 52 (30–80) |
| Histopathological grade [n (%)] | |
| 1 | 24 (24.49) |
| 2 | 58 (59.18) |
| 3 | 16 (16.33) |
| Histopathological type [n (%)] | |
| Ductal | 85 (73.28) |
| Lobular | 19 (16.38) |
| Mixed ductal and lobular | 11 (9.48) |
| Tubular | 1 (0.86) |
| Median tumor size [cm (range)] | 1.7 (0.3–8.0) |
| Lymph nodes [n (%)] | |
| Negative | 34 (30.91) |
| Positive | 76 (69.09) |
| ER status [n (%)] | |
| Negative | 14 (14.00) |
| Positive | 86 (86.00) |
| PR status [n (%)] | |
| Negative | 22 (22.00) |
| Positive | 78 (78.00) |
| Her-2/neu status [n (%)] | |
| Not overexpressed | 57 (66.28) |
| Overexpressed | 29 (33.72) |
| CCN6 [n (%)] | |
| Normal | 40 (39.2) |
| Reduced | 62 (60.8) |
Figure 1.

CCN6 expression is associated with E-cadherin protein in human invasive carcinomas of the breast. TMA sections stained with CCN6 and E-cadherin antibodies showing a representative invasive ductal carcinoma with high cytoplasmic CCN6 and positive E-cadherin at the cytoplasmic membranes (top) and another invasive ductal carcinoma with low CCN6 and reduced/neg E-cadherin (bottom). Immunohistochemistry was analyzed using pathology-based interpretation and an automated image analysis system (ChromaVision Medical Systems), consisting of an automated robotic bright-field microscope linked to a computer through a Microsoft Windows NT-based software interface. This system allows more reproducible and objective scoring than manual interpretation and provides a continuous rather than categorical score. Magnification, ×400.
Table 2.
Analysis of CCN6 Expression According to Clinical and Pathological Characteristics of the Patient Cohort
| Parameter | CCN6
|
P value* | |||
|---|---|---|---|---|---|
| Low
|
High
|
||||
| n | % | n | % | ||
| Histopathological grade | 0.18 | ||||
| 1 | 15 | 29.4 | 6 | 16.7 | |
| 2 | 29 | 56.9 | 23 | 63.9 | |
| 3 | 7 | 13.7 | 7 | 19.4 | |
| Tumor size | 0.37 | ||||
| <2 cm | 35 | 59.3 | 24 | 68.6 | |
| ≥2 cm | 24 | 40.7 | 11 | 21.4 | |
| Lymph nodes | 0.04 | ||||
| Negative | 27 | 46.6 | 8 | 24.2 | |
| Positive | 31 | 53.4 | 25 | 75.8 | |
| ER status | 0.21 | ||||
| Negative | 5 | 9.4 | 7 | 19.4 | |
| Positive | 48 | 90.6 | 29 | 80.6 | |
| PR status | 0.08 | ||||
| Negative | 8 | 15.1 | 11 | 30.6 | |
| Positive | 45 | 84.9 | 25 | 69.4 | |
| HER-2/neu status | 0.37 | ||||
| Not overexpressed | 30 | 68.2 | 18 | 58.1 | |
| Overexpressed | 14 | 31.8 | 13 | 41.9 | |
P value computed using χ2 test.
As expected, E-cadherin expression had a crisp membranous staining pattern. The intensity of the staining was measured using an image analysis system in a continuous scale, allowing for high reproducibility and precise scoring of protein expression.17 There was a strong association between CCN6 and E-cadherin proteins. Invasive carcinomas with low CCN6 expression also had reduced or absent E-cadherin protein at the cell membrane. Invasive carcinomas with low CCN6 had a mean E-cadherin expression of 98, whereas the invasive carcinomas with high CCN6 had a mean E-cadherin expression of 105.3 (t-test, P = 0.02; Figure 1). Collectively, these data show that CCN6 down-regulation is a frequent event in invasive breast carcinomas and that it is associated with lymph node metastasis, and they highlight the strength of the association between CCN6 and E-cadherin proteins in human breast cancer.
CCN6 Regulates E-Cadherin mRNA and Protein Expression in Benign and Malignant Breast Cells in Vivo and in Vitro
Immunoblot analysis of a panel of benign breast cells and breast cancer cells showed that CCN6 expression is highest in benign mammary epithelial cells (HME and MCF10A). Supporting our findings in breast tissue samples, CCN6 protein is decreased in breast cancer cell lines. When compared with benign breast cells, the well differentiated, noninvasive MCF-7 breast cancer cells have slightly decreased levels of CCN6 protein, whereas CCN6 is greatly decreased in the invasive and metastasizing MDA-MB-231 cell line and is almost absent in the poorly differentiated SUM149 inflammatory breast cancer cell line (Figure 2).
Figure 2.

siRNA- and shRNA-mediated disruption of CCN6 down-regulates E-cadherin in benign breast and nonbreast epithelial cells. A: CCN6 protein levels of HME cells, MCF10A benign breast cells, and a panel of breast cancer cell lines. B: Stable down-regulation of CCN6 in HME cells by siRNA and shRNA. Using these two strategies, CCN6 protein levels were reduced in HME cells. C: CCN6 inhibition induces EMT of HME cells. Phase contrast microscopy showing that whereas HME controls are oval, CCN6-deficient HME cells (either by si- or shRNA) have cytoplasmic extensions and mesenchymal cell shape. D: CCN6-deficient clones have significantly reduced levels of E-cadherin protein (top) and E-cadherin mRNA (bottom) when compared with the empty vector-transfected HME cells. E: Inhibition of CCN6 markedly reduces the activity of the E-cadherin gene promoter. The effect of CCN6 inhibition on the activity of the E-cadherin promoter was assessed on HME, MCF10A, and HEK293 cells with the reporter construct, Ecad(−108)-Luc, which contains the wild-type promoter sequence from nucleotides −108 to +125 of the endogenous E-cadherin promoter.
We have stably knocked down CCN6 in HME cells using siRNA and shRNA (Figure 2B). Stable CCN6 knockdown in HME cells resulted in phenotypic features of EMT and in marked down-regulation of E-cadherin protein and mRNA (Figure 2, B–D). Furthermore, CCN6 inhibition in HME and MCF10A benign breast cells caused greater than twofold reduction in E-cadherin promoter activity (Figure 2E). The effect of CCN6 inhibition on E-cadherin promoter activity is not exclusive to mammary cells and can be extended to other epithelial cells, as demonstrated by the significantly reduced activity of the E-cadherin promoter in HEK293 cells with transient inhibition of CCN6 (Figure 2E, right panel).
We postulated that the observed down-regulation of E-cadherin is a specific event triggered by CCN6 inhibition. To test this hypothesis, we investigated whether restoration of CCN6 in SUM149 cells was able to up-regulate E-cadherin expression in vivo. SUM149 cells derive from a primary inflammatory breast cancer and have very low CCN6 protein expression in the wild type (Figure 2A). We have previously shown that restoration of CCN6 expression in SUM149 cells decreases their invasiveness and tumorigenic ability in vivo and in vitro.11 When injected in the mammary fat pads of athymic nude mice, SUM149/vector cells formed large tumors when compared with significantly smaller tumors formed by the SUM149/CCN6+ cells.11 In addition to their smaller size, SUM149/CCN6+ mammary xenografts were better differentiated than the SUM149/vector xenografts. In the former, the cancer cells were arranged in glandular structures and had less atypia than in the latter, which was characterized mainly by sheets of highly pleomorphic cancer cells (Figure 3). Immunohistochemical staining on six mammary xenografts of SUM149/CCN6+ or SUM149/vector control cells showed that restoration of CCN6 expression in SUM149 cells induced the up-regulation of E-cadherin protein in the cell membranes (Figure 3).
Figure 3.

Restoration of CCN6 protein in inflammatory breast cancer cells SUM149 results in up-regulation of E-cadherin protein in vivo. Mammary gland xenografts derived from SUM149/CCN6+ cells or SUM149/empty vector controls (CCN6 negative) were analyzed histologically and assayed by immunohistochemistry to investigate the expression levels of CCN6 and E-cadherin proteins. Top panels show a SUM149/vector tumor with negative CCN6 and E-cadherin proteins. Middle and bottom panels show two representative xenografts derived from SUM149/CCN6+ cells showing expression of CCN6 and a concomitant up-regulation of E-cadherin protein localized to the cell membrane. Note the gland formation in the CCN6+ xenografts best seen on the bottom panel. Magnification, ×400 and ×600.
CCN6-Mediated Regulation of E-Cadherin Requires Intact E-Box Elements in the E-Cadherin Promoter
The E-cadherin promoter contains multiple characterized elements, including three E-boxes, a CCAAT box, and a GC-rich element. The specific sequences contained in these E-boxes have been shown to be critical in transcriptional repression of the E-cadherin gene.5,19 The observed reduction of E-cadherin promoter activity on CCN6 knockdown led us to hypothesize that CCN6 may function directly or indirectly through the E-boxes in the E-cadherin promoter. To test this hypothesis, we investigated the E-cadherin promoter activity using an intact promoter [Ecad(−108)-Luc] or a promoter with specific mutations in the E-box elements Ecad(−108)/EboxA, Ecad(−108)/EboxC, and Ecad(−108)/Ebox 3× Mut.6,19 As shown in Figure 4A, mutation of the E-boxes abrogated the effect of CCN6 on the activity of the E-cadherin promoter. Taken together, these data demonstrate that CCN6 inhibition decreases E-cadherin gene transcription and that this effect requires intact proximal E-box elements in the promoter of the E-cadherin gene.
Figure 4.

CCN6-mediated E-cadherin repression is dependent on Snail and ZEB1. A: E-cadherin promoter activity of wild-type (left) and E-box mutant (right) constructs was determined in HME cells transfected with the empty vector and HME cells with stable CCN6 knockdown. CCN6-mediated reduction in the activity of the E-cadherin promoter was abrogated by the introduction of mutations in the E-boxes. B: Quantitative SYBR green real-time RT-PCR of the E-cadherin transcriptional repressors SNAIL, SLUG, TWIST1, TWIST2, smad-interacting protein-1, and ZEB1 was performed on HME cells transfected with the empty vector and HME cells with stable CCN6 knockdown. Each sample was tested in duplicate, and a ratio was calculated relative to the housekeeping gene GAPDH. C: Western immunoblot showing that CCN6 knockdown results in a marked up-regulation of Snail and ZEB1 proteins. Snail–E-cadherin localization was determined by confocal laser microscopy. CCN6 siRNA inhibition caused marked nuclear accumulation of Snail protein (red) and loss of E-cadherin (green) protein at the cell membranes compared with the HME vector control cells. D: Snail and ZEB1 are required for CCN6-mediated down-regulation of E-cadherin. siRNA-mediated knockdown of Snail expression decreased Snail protein and increased E-cadherin. In control siRNA-treated cells (si-Co), Snail is increased and E-cadherin is reduced. siRNA-mediated knockdown of Snail expression interrupted the effect of CCN6 down-regulation of E-cadherin expression. Similarly, knockdown with ZEB1 siRNA decreased ZEB1protein and increased E-cadherin levels. siRNA-mediated knockdown of ZEB1 expression interrupted the effect of CCN6 down-regulation of E-cadherin expression.
CCN6 Knockdown Leads to Up-Regulation of E-Cadherin Transcriptional Repressors ZEB1 and Snail
Based on the above findings, we next wished to investigate whether CCN6 knockdown had an effect on the levels of the E-cadherin transcriptional repressors, which have been demonstrated to bind to the E-cadherin proximal promoter E-boxes.5,19 As determined by quantitative real-time PCR, CCN6 knockdown up-regulated SNAIL and ZEB1, whereas it had no effect on the mRNA levels of SLUG, TWIST1, TWIST2, and smad-interacting protein-1 (Figure 4B). Western immunoblot analysis showed that CCN6 knockdown in HME cells, by either si- or shRNA, caused a marked increase in Snail and ZEB1 protein levels (Figure 4C).
In situ analysis of the expression of E-cadherin and Snail proteins was performed using immunofluorescence. HME cells transfected with the empty vector had low levels of Snail protein in the nucleus and prominent E-cadherin expression in the cell membranes. In stark contrast, HME cells with CCN6 knockdown exhibited increased Snail protein in the nucleus coupled with almost complete down-regulation of membranous E-cadherin. Furthermore, although Snail was faintly and evenly distributed in the nucleus of the parental HME/vector cells, it accumulated forming large nuclear clumps in HME cells that had undergone CCN6 knockdown (Figure 4C).
To provide evidence that Snail and ZEB1 are required for CCN6-mediated E-cadherin down-regulation, we used siRNA to knockdown Snail and ZEB1 expression in HME cells (Figure 4D). Snail siRNA resulted in a marked reduction of Snail protein compared with cells transfected with the empty vector. Concomitantly, siRNA-mediated knockdown of Snail led to a marked increase in E-cadherin protein. Transfection with ZEB1 siRNA resulted in a significant decrease in ZEB1 protein compared with vector-transfected cells. Concomitantly, siRNA-mediated knockdown of ZEB1 led to increased E-cadherin levels. These results implicate CCN6 as a modulator of ZEB1 and Snail and define a pathway by which CCN6 knockdown decreases E-cadherin expression in breast cancer.
CCN6 Knockdown Enhances Snail and ZEB1 Binding to the E-Cadherin Promoter
To investigate whether the up-regulation of Snail and ZEB1 by CCN6 knockdown is associated with an increase in binding to the E-cadherin promoter, we examined the binding of these transcriptional repressors to E-boxes by chromatin immunoprecipitation assays. As expected, the Snail antibody pulled down Snail protein complexes. Similarly, anti-ZEB1 antibody (directed against the N-terminal domain), efficiently pulled down ZEB1 protein complexes. Of note, an enhanced expression of Snail and ZEB1 in CCN6 knockdown cells led to an increase in chromatin binding (Figure 5).
Figure 5.
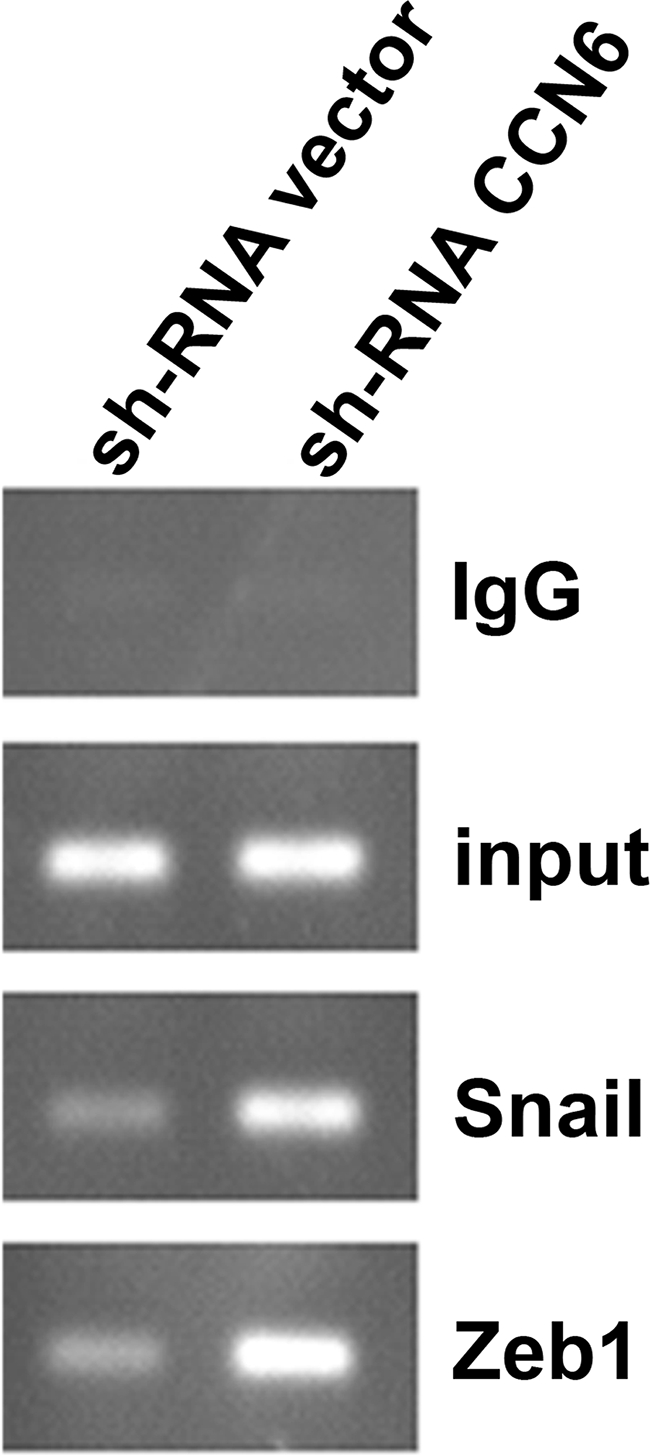
CCN6 knockdown enhances Snail and ZEB1 recruitment and binding to the E-cadherin promoter. Chromatin immunoprecipitation assay shows that ZEB1 and Snail associated with the E-cadherin promoter at the chromatin level. HME cells were subjected to chromatin immunoprecipitation analyses using antibodies to Snail, ZEB1, and control IgG.
Discussion
CCN6, also termed WISP3, is one of six members of the highly conserved CCN family of growth factors, which also include Cyr61 (CCN1), connective tissue growth factor (CCN2), Nov (CCN3), WISP1 (CCN4), and WISP2 (CCN5). CCN proteins have intracellular and extracellular functions20,21 and have been shown to mediate epithelial and stromal cross talk.22,23,24,25,26,27 Our laboratory and other investigators have demonstrated that deregulation of this protein family can lead to cancer.11,28,29
Our laboratory reported that CCN6 has tumor inhibitory functions in an aggressive form of breast cancer, inflammatory breast cancer, in vivo and in vitro.11 CCN6 inhibition by stable siRNA in benign mammary epithelial cells causes an EMT and triggers anchorage-independent growth, invasion, and motility of HME cells.12 Collectively, these results provide evidence that CCN6 has tumor suppressor functions in the breast and regulates the epithelial phenotype in mammary epithelial cells and that further studies on the role of CCN6 in breast cancer are warranted.
In this study, using high-density TMAs and immunohistochemistry on unselected human breast cancer tissue samples, we show CCN6 protein is predominantly detected in the cytoplasm of the cancer cells and in some cancer cell nuclei. Reduction or loss of CCN6 protein levels are frequent events in invasive carcinomas of the breast irrespective of the histological type. Sixty percent of all invasive carcinomas in our cohort showed low or absent CCN6 protein expression. CCN6 was not associated with histological type or grade or hormonal receptor status. Importantly, we found a significant association between CCN6 expression and the presence of axillary lymph metastasis. Invasive carcinomas with low CCN6 had increased incidence of positive lymph nodes when compared with tumors with high CCN6 expression. We also noted a strong association between CCN6 and E-cadherin proteins. Invasive carcinomas with low CCN6 had reduced E-cadherin protein in the cellular membrane. These data led us to hypothesize that CCN6 loss induces an invasive breast cancer phenotype by regulating E-cadherin expression, and we set out to elucidate the underlying mechanism.
In our initial experiment, analysis of CCN6 expression in breast cell lines revealed that benign cells, HME and MCF10A, have strong CCN6 expression, whereas invasive carcinoma cells have decreased CCN6 levels according to their degree of differentiation and invasive potential. In addition, CCN6-deficient HME cells have features of a typical EMT.12 We found that this morphological and immunophenotypical change is associated with a strong suppression of E-cadherin mRNA and protein. The effect of CCN6 on E-cadherin expression was further investigated in vivo using xenografts derived from the SUM149 cell line. These cells derive from inflammatory breast cancer and have minimal to absent expression of CCN6.8,9 Mammary xenografts derived from SUM149 cells overexpressing CCN6 showed an up-regulation of membranous E-cadherin by immunohistochemistry, whereas the control xenografts lacking CCN6 showed absent E-cadherin protein expression. Taken together, these data strongly support the hypothesis that CCN6 regulates E-cadherin expression in normal breast and in breast cancer.
The suppression of E-cadherin mRNA induced by CCN6 knockdown suggested to us that CCN6 may influence the transcription of the E-cadherin gene. Although inactivating mutations or promoter hypermethylation have been observed to account for loss of E-cadherin function in invasive lobular and invasive ductal carcinomas of the breast, transcriptional repression has emerged as one of the important mechanisms for the down-regulation of E-cadherin during breast cancer development and progression.5 To investigate whether CCN6 knockdown modulates the activity of the E-cadherin promoter, we performed luciferase reporter assays on two benign breast cell lines, HME and MCF10A, and a benign nonmammary kidney-liver epithelial cell, HEK293 cells. These experiments showed that CCN6 knockdown was followed by a significant decrease in the activity of the E-cadherin promoter.
The complexities of the intracellular pathways that regulate E-cadherin expression are not yet fully understood. The proximal E-cadherin promoter-containing sequences extending to −108 of the E-cadherin gene are critical for E-cadherin transcription.6,19 Recently, several E-cadherin transcriptional repressors have been characterized and shown to interact with proximal E-boxes of the E-cadherin promoter, including Snail, Slug, ZEB1(∂EF1), smad-interacting protein-1, Twist1, and Twist2.3,5,30,31 To specifically address whether CCN6-mediated E-cadherin repression is exerted through the E-box elements in the proximal E-cadherin gene promoter, we used several well characterized E-box mutants and luciferase reporter assays. Our results show that CCN6-mediated effect on the E-cadherin promoter activity requires intact E-box elements.
It has been postulated that the relevance of the E-cadherin transcriptional regulators is dependent on the cell and tissue context. In breast cancer, the Snail family of transcription factors plays a crucial role in the regulation of E-cadherin. Snail up-regulation is associated with the EMT and correlates with disappearance of adherens junctions, profound morphological changes, and enhanced migratory and invasive capabilities of breast cells.30 Furthermore, Snail is an important predictor of breast cancer metastatic potential and recurrence.32 Although the role of ZEB1 during cancer progression is less clear, and little is known about the importance of ZEB1 in breast cancer, recent studies support the notion that ZEB1 is an important regulator of cell polarity and differentiation in invasive ductal and lobular carcinomas of the breast.33 Our data show that knockdown of CCN6 in HME cells results in increased Snail and ZEB1 mRNA and protein levels. To determine the importance of Snail and ZEB1 in the CCN6-mediated regulation of E-cadherin, we used siRNA to knockdown Snail and ZEB1 in HME cells with stable CCN6 knockdown and controls. Snail knockdown prevented the down-regulation of E-cadherin caused by CCN6 knockdown. Similarly, ZEB1 knockdown prevented the effect of CCN6 down-regulation on E-cadherin. Collectively, these results reveal that Snail and ZEB1 are downstream of CCN6 and are necessary for CCN6-mediated inhibition of E-cadherin.
Snail and ZEB1 bind to the E-boxes in the E-cadherin promoter and thus repress E-cadherin transcription.34 We directly investigated whether CCN6 knockdown results in increased recruitment of Snail and ZEB1 to the E-cadherin promoter using chromatin immunoprecipitation assays. Our results show that the increased levels of Snail and ZEB1 that result from CCN6 knockdown are associated with enhanced chromatin binding of these proteins to the E-cadherin promoter. This is the first report implicating CCN6 in the regulation of E-cadherin through transcriptional mechanisms in cancer.
The molecular details of how Snail and ZEB1 are regulated in benign and cancer cells are being actively investigated but still remain elusive.7,35 Several transcriptional mechanisms have been implicated in the regulation and function of Snail and ZEB1.34 Recent studies have also shown that posttranscriptional mechanisms may play an important role in the regulation of both factors.34 The subcellular localization of Snail can be modulated by phosphorylation involving the p21-activated kinase 1.36 Glycogen synthase kinase 3β phosphorylation of the Snail nuclear export signal and destruction box provokes its cytoplasmic export and ubiquitin-mediated proteasome degradation.7,37 In addition to phosphorylation, other interactions also appear to influence the stability of Snail, such as cooperation with lysyl-oxidase like-2 and -3 to repress E-cadherin and to induce EMT.34 Our immunofluorescence studies in situ show that CCN6 knockdown in HME cells resulted in a marked accumulation of Snail in the nuclei and almost no Snail protein in the cytoplasm. The marked accumulation of this protein in the nuclei was accompanied by almost complete loss of E-cadherin at the cell membranes. These data lead us to propose the novel hypothesis that CCN6 may affect the nuclear-cytoplasmic export of Snail and facilitate their degradation in the cytoplasm, which warrants further investigation.
CCN6 is a secreted protein with high sequence homology to CCN4 (WISP1), which was initially found to be up-regulated in Wnt-1-expressing C57MG mouse mammary epithelial cells when compared with parental C57MG cells.38 Recently, Yook et al6,37 provided the first link between Wnt-1 signaling with Snail regulation. The investigators showed that Wnt-1-conditioned cell extracts inhibit Snail phosphorylation and consequently increase Snail protein levels and activity and drive an EMT on HEK293 cells and breast cancer cells MCF7.6 Based on these novel data, further experiments will aim to determine whether CCN6 affects the Wnt signaling pathway.
Our finding that CCN6 loss is associated with a reduction of E-cadherin protein in tumors with ductal and lobular histology is intriguing. The majority of invasive lobular carcinomas have genetic and/or epigenetic alterations of the E-cadherin gene. More than 50% of invasive lobular carcinomas are reported to possess an E-cadherin mutation, and in accordance with the two-hit hypothesis, the majority of mutations are found in combination with loss of heterozygosity of the wild-type E-cad locus. Interestingly, invasive lobular carcinomas also have a high rate of epigenetic inactivation of E-cadherin, which occurs in the absence of mutations. It has been proposed that the frequency with which methylation targets the E-cadherin promoter increases when the promoter activity is down-regulated by other mechanisms, such as transcriptional repression via Snail.39 Furthermore, Aigner et al33 reported that the E-cadherin transcriptional repressor ZEB1 is markedly up-regulated in invasive lobular carcinomas of the breast. These data strongly suggest that transcriptional repression is involved in E-cadherin regulation in invasive lobular carcinomas, in addition to its well established role in the pathogenesis of invasive ductal carcinomas. Collectively, these data strongly support our hypothesis that CCN6 loss plays a role in the repression of E-cadherin in invasive ductal and lobular carcinomas of the breast.
In summary, we have found that CCN6 is lost in 60% of invasive carcinomas of the breast and that low CCN6 is associated with the presence of axillary lymph node metastasis and decreased expression of E-cadherin. We provide mechanistic evidence that CCN6 loss results in up-regulation of two important E-cadherin transcriptional repressors, Snail and ZEB1, and recruits them to the E-cadherin promoter thereby regulating E-cadherin expression in benign breast cells and in breast cancer. The discovery of CCN6 as a novel regulator of E-cadherin, Snail, and ZEB1 may have profound implications in breast tumorigenesis. Specifically, modulation of CCN6 levels may in the future lead to prevention of breast cancer invasion and metastases.
Acknowledgments
We thank Dr. Eric Fearon for the luciferase reporter constructs Ecad(−108)-Luc), Ecad(−108)-AMut, Ecad(−108)-CMut, and Ecad(−108)-ABCMut, and for advice. We thank Maria E. Gonzalez and Quintin Pan for helpful suggestions and critical discussion during the execution of this project. We thank Yipin Wu for assistance with the immunofluorescence experiments.
Footnotes
Address reprint requests to Dr. Celina G. Kleer, University of Michigan Medical School, Department of Pathology, 3510C MSRB1, 1150 W. Medical Center Dr., Ann Arbor, MI 48109-0605. E-mail: kleer@umich.edu.
Supported by National Institutes of Health grants K08 CA090876 and R01 CA107469 (to C.G.K.), and R01 CA66712 (to S.D.M.), by Department of Defense grant DAMD17-01-1-490 (to C.G.K.), and by a grant from the Burroughs Wellcome Fund (to S.D.M.).
References
- Jemal A, Murray T, Samuels A, Ghafoor A, Ward E, Thun MJ. Cancer statistics, 2003. CA Cancer J Clin. 2003;53:5–26. doi: 10.3322/canjclin.53.1.5. [DOI] [PubMed] [Google Scholar]
- Battaglia C, Salani G, Consolandi C, Bernardi LR, De Bellis G. Analysis of DNA microarrays by non-destructive fluorescent staining using SYBR green II [In Process Citation]. Biotechniques. 2000;29:78–81. doi: 10.2144/00291st01. [DOI] [PubMed] [Google Scholar]
- Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De Herreros A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- Hajra KM, Chen DY, Fearon ER. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002;62:1613–1618. [PubMed] [Google Scholar]
- Yook JI, Li XY, Ota I, Fearon ER, Weiss SJ. Wnt-dependent regulation of the E-cadherin repressor snail. J Biol Chem. 2005;280:11740–11748. doi: 10.1074/jbc.M413878200. [DOI] [PubMed] [Google Scholar]
- Zhou BP, Deng J, Xia W, Xu J, Li YM, Gunduz M, Hung MC. Dual regulation of Snail by GSK-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat Cell Biol. 2004;6:931–940. doi: 10.1038/ncb1173. [DOI] [PubMed] [Google Scholar]
- van Golen KL, Davies S, Wu ZF, Wang Y, Bucana CD, Root H, Chandrasekharappa S, Strawderman M, Ethier SP, Merajver SD. A novel putative low-affinity insulin-like growth factor-binding protein, LIBC (lost in inflammatory breast cancer), and RhoC GTPase correlate with the inflammatory breast cancer phenotype. Clin Cancer Res. 1999;5:2511–2519. [PubMed] [Google Scholar]
- Kleer CG, Zhang Y, Pan Q, Merajver SD. WISP3 (CCN6) is a secreted tumor-suppressor protein that modulates IGF signaling in inflammatory breast cancer. Neoplasia. 2004;6:179–185. doi: 10.1593/neo.03316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleer CG, Zhang Y, Pan Q, Gallagher G, Wu M, Wu ZF, Merajver SD. WISP3 and RhoC guanosine triphosphatase cooperate in the development of inflammatory breast cancer. Breast Cancer Res. 2004;6:R110–R115. doi: 10.1186/bcr755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleer CG, Zhang Y, Pan Q, van Golen KL, Wu ZF, Livant D, Merajver SD. WISP3 is a novel tumor suppressor gene of inflammatory breast cancer. Oncogene. 2002;21:3172–3180. doi: 10.1038/sj.onc.1205462. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Pan Q, Zhong H, Merajver SD, Kleer CG. Inhibition of CCN6 (WISP3) expression promotes neoplastic progression and enhances the effects of insulin-like growth factor-1 on breast epithelial cells. Breast Cancer Res. 2005;7:R1080–R1089. doi: 10.1186/bcr1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA, Chinnaiyan AM. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606–11611. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elston EW, Ellis IO. Method for grading breast cancer. J Clin Pathol. 1993;46:189–190. doi: 10.1136/jcp.46.2.189-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski PJ, Rubin MA, Kleer CG. E-cadherin expression in primary carcinomas of the breast and its distant metastases. Breast Cancer Res. 2003;5:R217–R222. doi: 10.1186/bcr651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleer CG, Griffith KA, Sabel MS, Gallagher G, van Golen KL, Wu ZF, Merajver SD. RhoC-GTPase is a novel tissue biomarker associated with biologically aggressive carcinomas of the breast. Breast Cancer Res Treat. 2005;93:101–110. doi: 10.1007/s10549-005-4170-6. [DOI] [PubMed] [Google Scholar]
- Witkiewicz AK, Varambally S, Shen R, Mehra R, Sabel MS, Ghosh D, Chinnaiyan AM, Rubin MA, Kleer CG. Alpha-methylacyl-CoA racemase protein expression is associated with the degree of differentiation in breast cancer using quantitative image analysis. Cancer Epidemiol Biomarkers Prev. 2005;14:1418–1423. doi: 10.1158/1055-9965.EPI-04-0607. [DOI] [PubMed] [Google Scholar]
- Camp RL, Dolled-Filhart M, King BL, Rimm DL. Quantitative analysis of breast cancer tissue microarrays shows that both high and normal levels of HER2 expression are associated with poor outcome. Cancer Res. 2003;63:1445–1448. [PubMed] [Google Scholar]
- Hajra KM, Ji X, Fearon ER. Extinction of E-cadherin expression in breast cancer via a dominant repression pathway acting on proximal promoter elements. Oncogene. 1999;18:7274–7279. doi: 10.1038/sj.onc.1203336. [DOI] [PubMed] [Google Scholar]
- Perbal B. NOV (nephroblastoma overexpressed) and the CCN family of genes: structural and functional issues. Mol Pathol. 2001;54:57–79. doi: 10.1136/mp.54.2.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigstock DR, Goldschmeding R, Katsube KI, Lam SC, Lau LF, Lyons K, Naus C, Perbal B, Riser B, Takigawa M, Yeger H. Proposal for a unified CCN nomenclature. Mol Pathol. 2003;56:127–128. doi: 10.1136/mp.56.2.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CG, Chen CC, Leu SJ, Grzeszkiewicz TM, Lau LF. Integrin-dependent Functions of the angiogenic inducer NOV (CCN3): implication in wound healing. J Biol Chem. 2005;280:8229–8237. doi: 10.1074/jbc.M404903200. [DOI] [PubMed] [Google Scholar]
- Lin CG, Leu SJ, Chen N, Tebeau CM, Lin SX, Yeung CY, Lau LF. CCN3 (NOV) is a novel angiogenic regulator of the CCN protein family. J Biol Chem. 2003;278:24200–24208. doi: 10.1074/jbc.M302028200. [DOI] [PubMed] [Google Scholar]
- Lau LF, Lam SC. The CCN family of angiogenic regulators: the integrin connection. Exp Cell Res. 1999;248:44–57. doi: 10.1006/excr.1999.4456. [DOI] [PubMed] [Google Scholar]
- Sakamoto K, Yamaguchi S, Ando R, Miyawaki A, Kabasawa Y, Takagi M, Li CL, Perbal B, Katsube K. The nephroblastoma overexpressed gene (NOV/ccn3) protein associates with Notch1 extracellular domain and inhibits myoblast differentiation via Notch signaling pathway. J Biol Chem. 2002;277:29399–29405. doi: 10.1074/jbc.M203727200. [DOI] [PubMed] [Google Scholar]
- Li CL, Martinez V, He B, Lombet A, Perbal B. A role for CCN3 (NOV) in calcium signalling. Mol Pathol. 2002;55:250–261. doi: 10.1136/mp.55.4.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perbal B, Martinerie C, Sainson R, Werner M, He B, Roizman B. The C-terminal domain of the regulatory protein NOVH is sufficient to promote interaction with fibulin 1C: a clue for a role of NOVH in cell-adhesion signaling. Proc Natl Acad Sci USA. 1999;96:869–874. doi: 10.1073/pnas.96.3.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benini S, Perbal B, Zambelli D, Colombo MP, Manara MC, Serra M, Parenza M, Martinez V, Picci P, Scotlandi K. In Ewing’s sarcoma CCN3(NOV) inhibits proliferation while promoting migration and invasion of the same cell type. Oncogene. 2005;24:4349–4361. doi: 10.1038/sj.onc.1208620. [DOI] [PubMed] [Google Scholar]
- Manara MC, Perbal B, Benini S, Strammiello R, Cerisano V, Perdichizzi S, Serra M, Astolfi A, Bertoni F, Alami J, Yeger H, Picci P, Scotlandi K. The expression of ccn3(nov) gene in musculoskeletal tumors. Am J Pathol. 2002;160:849–859. doi: 10.1016/S0002-9440(10)64908-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowin P, Rowlands TM, Hatsell SJ. Cadherins and catenins in breast cancer. Curr Opin Cell Biol. 2005;17:499–508. doi: 10.1016/j.ceb.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 2005;24:2375–2385. doi: 10.1038/sj.onc.1208429. [DOI] [PubMed] [Google Scholar]
- Moody SE, Perez D, Pan TC, Sarkisian CJ, Portocarrero CP, Sterner CJ, Notorfrancesco KL, Cardiff RD, Chodosh LA. The transcriptional repressor Snail promotes mammary tumor recurrence. Cancer Cell. 2005;8:197–209. doi: 10.1016/j.ccr.2005.07.009. [DOI] [PubMed] [Google Scholar]
- Aigner K, Descovich L, Mikula M, Sultan A, Dampier B, Bonne S, van Roy F, Mikulits W, Schreiber M, Brabletz T, Sommergruber W, Schweifer N, Wernitznig A, Beug H, Foisner R, Eger A. The transcription factor ZEB1 (deltaEF1) represses Plakophilin 3 during human cancer progression. FEBS Lett. 2007;581:1617–1624. doi: 10.1016/j.febslet.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–428. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- Bachelder RE, Yoon SO, Franci C, de Herreros AG, Mercurio AM. Glycogen synthase kinase-3 is an endogenous inhibitor of Snail transcription: implications for the epithelial-mesenchymal transition. J Cell Biol. 2005;168:29–33. doi: 10.1083/jcb.200409067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Rayala S, Nguyen D, Vadlamudi RK, Chen S, Kumar R. Pak1 phosphorylation of snail, a master regulator of epithelial-to-mesenchyme transition, modulates snail’s subcellular localization and functions. Cancer Res. 2005;65:3179–3184. doi: 10.1158/0008-5472.CAN-04-3480. [DOI] [PubMed] [Google Scholar]
- Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK, Choi YJ, Kim J, Fearon ER, Weiss SJ. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8:1398–1406. doi: 10.1038/ncb1508. [DOI] [PubMed] [Google Scholar]
- Pennica D, Swanson TA, Welsh JW, Roy MA, Lawrence DA, Lee J, Brush J, Taneyhill LA, Deuel B, Lew M, Watanabe C, Cohen RL, Melhem MF, Finley GG, Quirke P, Goddard AD, Hillan KJ, Gurney AL, Botstein D, Levine AJ. WISP genes are members of the connective tissue growth factor family that are up-regulated in wnt-1-transformed cells and aberrantly expressed in human colon tumors. Proc Natl Acad Sci USA. 1998;95:14717–14722. doi: 10.1073/pnas.95.25.14717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathdee G. Epigenetic versus genetic alterations in the inactivation of E-cadherin. Semin Cancer Biol. 2002;12:373–379. doi: 10.1016/s1044-579x(02)00057-3. [DOI] [PubMed] [Google Scholar]