

Abstract
The application of anti-CD3 F(ab′)2 monoclonal antibodies has recently been expanded to treat established autoimmune diseases, including type 1 diabetes. However, the mechanism underlying their effect remains largely unclear. We report that short-phase administration of anti-CD3 F(ab′)2 antibodies efficiently allowed 80% of new-onset, nonobese diabetic (NOD) mice to significantly regain both normoglycemia and pancreatic β cell-specific autoantigen (ie, glutamic acid decarboxylase and insulin) tolerance, with both effects lasting more than 40 weeks. The responsible mechanism appears to involve the induction and maintenance of a population of immunoregulatory CD1d-restricted natural killer T (NKT) cells, which were marked by an enhanced Th2 response and secretion of elevated levels of interleukin-10. In vivo neutralization of interleukin-4 and/or interleukin-10 bioactivity abrogated this anti-CD3-mediated effect. Importantly, when the cotransfer of NKT cells from the livers of anti-CD3-treated mice and splenocytes from untreated, acutely diabetic NOD mice was performed in NOD-severe combined immunodeficient mice, the NKT cells were sufficient to either delay or prevent the onset of diabetes compared with controls where only splenocytes were introduced. These data suggest that CD1d-restricted NKT cells may play a critical role in anti-CD3 antibody-induced diabetes remission and the restoration of immune tolerance.
Type 1 diabetes is an autoimmune disease characterized by immunological destruction of insulin-producing pancreatic β cells and subsequent hyperglycemia.1 The development of type 1 diabetes is influenced by multiple genetic and environmental factors, most of which remain unknown.2,3 The nonobese diabetic (NOD) mouse strain spontaneously develops a disease similar to human type 1 diabetes as a consequence of the loss of basic tolerogenic processes designed to control self/nonself discrimination.4
Many studies have demonstrated that defects of several regulatory lymphocytes contribute to the pathogenesis of type 1 diabetes, including CD4+ regulatory T cells (Tregs) and natural killer T (NKT) cells coexpressing T and NK cell receptors.5,6,7 The number and function of these cells are impaired in NOD mice and individuals at risk for type 1 diabetes. Increasing the number by adoptive transfer,8,9 or by augmenting function of these regulatory cells by T cell receptor engagement,10 has potential for the prevention and treatment of diabetes. Manipulation of CD4+ Tregs or NKT cells has been shown to be involved in antigen-induced tolerance to self islets.11
Anti-CD3 monoclonal antibody (mAb) (OKT3) is commonly considered as a category of immunomodulatory agents for silencing acute allograft rejection. Its therapeutic role in autoimmune diabetes has been recently reported that the treatment of new-established diabetes with anti-CD3 F(ab′)2 resulted in durable disease remission in animals or reduced usage of insulin in humans involving Th2 deviation.12,13 A population of CD4+CD25+ Tregs has been proposed to be implicated in this effect.14 However, whether other immunoregulatory pathways required for regulation of glycemia by anti-CD3 exist need to be determined. Here, we found that CD1d-restricted NKT cells were involved in anti-CD3-mediated diabetes remission. The studies on the role of NKT cells in the induction and maintenance of self tolerance after anti-CD3 F(ab′)2 treatment suggested that the number of CD1d-restricted NKT cells increased significantly in remitting NOD mice and their response deviated to Th2 phenotype. Notably, active inhibition of diabetes transfer by these cells indicated the generation of a population of protective NKT cells after anti-CD3 treatment.
Materials and Methods
Mice and Glycemia Screening
NOD.scid mice were obtained originally from the Jackson Laboratory (Bar Harbor, ME) and bred in our facilities under specific pathogen-free conditions. Care, use, and treatment of mice in this study were in strict agreement with the guidelines in the care and use of laboratory animals set forth by Institute of Basic Medical Sciences. The incidence of diabetes in these mice is 80 to 90% by 30 weeks of age. At 10 weeks of age, NOD mice were monitored for fasting blood glucose weekly. Diabetes was defined ≥11.3 mmol/L on two consecutive measurements.
Preparation of Anti-CD3 F(ab′)2 Antibodies
Hamster anti-murine CD3 mAb (145 2C11) was stored in our laboratory. Anti-CD3 fragments were obtained by pepsin digestion. Briefly, anti-CD3 monoclonal antibody was dialyzed against citrate buffer (pH 3.5), and then 0.1 mg/ml pepsin dissolved in citrate was used at an enzyme/antibody ratio of 1:20 and incubated at 37°C for 3.5 hours, which was previously optimized. The reaction was stopped by adding 1:10 volume of 3 mol/L Tris base (pH 8.8). The pepsin-digested mAb preparation was immediately dialyzed against phosphate-buffered saline (pH 8.0, 4°C). A protein A-Sepharose CL-4B column was prepared, and the mixture was concentrated and loaded onto a prepared Sephacryl S-200 superfine column. Fractions, corresponding to a molecular weight of 110 kd were collected. The purity of anti-CD3 mAb F(ab′)2 was verified by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and the preparation was stored in phosphate-buffered saline at 4°C until use.
Antibody Treatment
The diabetic NOD mice, within 7 days of the onset of overt diabetes, were treated intravenously with anti-CD3 F(ab′)2 (40 μg/mouse) for 5 consecutive days. The untreated diabetic littermates were regarded as controls. The percentage of remission of diabetes was calculated as follows:
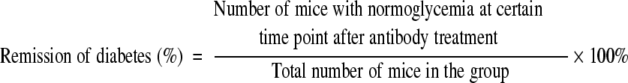 |
In some cases, NOD mice treated with anti-CD3 antibody received intraperitoneally antibody to interleukin (IL)-4 (11B11) and/or IL-10 (JES2A5) or rat IgG at a dose of 0.5 mg/mouse on days −1, 0, 2, 5, and then every 5 days. These antibodies were purchased from eBiosciences (San Diego, CA).
Lymphocyte Preparation
Single-cell suspension was prepared from spleen or pancreatic draining lymph nodes by passing them through nylon mesh. Lymphocytes from livers were isolated by 40%/80% Percoll solution.15
In Vitro Proliferation Assay
Lymphocytes from pancreatic draining lymph nodes were isolated from the anti-CD3-treated mice and from the control counterparts 3 weeks after antibody therapy. Proliferation assay was performed as described previously.16 Briefly, lymphocytes were suspended with RPMI 1640 culture medium supplement with 10% fetal calf serum and were seeded in 96-well plate in triplicate (4 × 105 cells/well) with 10 μg/ml glutamic acid decarboxylase 65 (GAD65), 10 μg/ml GAD500–585 (both prepared in our laboratory), 20 μg/ml porcine insulin, 15 μg/ml myelin basic protein, or 5 μg/ml concanavalin (all from Sigma Chemical Co, St. Louis, MO). On day 3 cultures were pulsed with 0.5 μCi of [3H]thymidine per well for the last 16 hours, and the cells were harvested and counted by standard liquid scintillation.
Enzyme-Linked Immunosorbent Assay of Cytokine Production
Fluorescence-activated cell sorted hepatic NKT cells (5 × 104) were incubated in 96-well flat-bottomed microtiter plates in the presence of 0.5 μg/ml anti-CD3 monoclonal antibodies. In some experiments, Splenocytes (5 × 105) were incubated in the presence of 20 μg/ml recombinant GAD500–585 protein. Supernatants were harvested after 48 hours. The levels of IL-4, interferon (IFN)-γ, and IL-10 were determined in triplicate in 0.1 ml of supernatant by sandwich enzyme-linked immunosorbent assay. The enzyme-linked immunosorbent assay kits used in this study were purchased from BD PharMingen (San Diego, CA).
Preparation for α-GalCer-Loaded CD1d:Ig Protein Staining Cocktail
α-Galactosylceramide (α-GalCer) (Toronto Research Chemicals Inc., Toronto, Canada) and CD1d:Ig (PharMingen) were mixed together in phosphate-buffered saline at appropriate ratio according to the manufacturer’s instructions and incubated at 37°C overnight. The staining cocktail was prepared by mixing α-GalCer-loaded CD1d:Ig protein with phycoerythrin-conjugated A85-1 mAb (PharMingen) at a ratio of 1:1 dimer to A85-1 mAb, and the mixture was incubated for 1 hour at room temperature in the dark.
Flow Cytometry
Antibodies used for flow cytometry analysis included fluorescein isothiocyanate-labeled anti-murine T cell receptor-β (H57-597) from eBiosciences and phycoerythrin-labeled anti-murine CD3ε (145 2C11) from PharMingen. Cells were stained in phosphate-buffered saline with 2% heat-inactivated fetal calf serum and 0.2% sodium azide and fixed using phosphate-buffered saline with 1% paraformaldehyde. The NKT cell population was identified as T cell receptor-β+α-GalCer-loading dimer+ cells. For the detection of peripheral blood CD3+ cells, whole blood was treated with fluorescence-activated cell sorting lysing solution (Becton Dickinson, Tokyo, Japan) to eliminate red blood cells after staining. Data collection and analysis were performed on a fluorescence-activated cell sorting Calibur flow cytometry using CellQuest software (Becton Dickinson, San Jose, CA).
Histopathology
NOD mice were sacrificed by cervical dislocation, and pancreas was immediately removed. Each pancreas was fixed with 10% buffered formalin, embedded in paraffin, sectioned at 4.5 μm, and stained with hematoxylin and eosin.17 Insulitis grade was scored as follows: 0, normal islet; 1, mononuclear infiltration, largely in the periphery, in <25% of the islet; 2, 25 to 50% of the islets showing mononuclear infiltration; 3, >50% of the islet showing mononuclear infiltration; 4, small, retracted islet with few mononuclear cells.
Adoptive Transfer of Diabetes
Purified NKT cells (2 × 105 cells) from the liver of anti-CD3-treated or control mice were mixed with splenocytes (1 × 107 cells) from untreated, acutely diabetic NOD mice and administered intravenously into the tail veins of 4- to 8-week-old NOD.scid mice. Age-matched NOD.scid mice receiving only 1 × 107 diabetic splenocytes were used as positive control. Recipients were tested every week for diabetes and diagnosed as described above.
Statistical Analysis
The Kaplan-Meier method was used to compare diabetes remission. Student’s t-test was used to compare mean values. Values of P < 0.05 were considered significant.
Results
Anti-CD3 F(ab′)2 mAbs Effectively Reversed New-Onset Diabetes in NOD Mice
From 10 weeks of age, NOD mice were monitored for fasting glycemia weekly. New onset diabetic mice were randomly divided into two groups and were transfused with anti-CD3 antibody or left untreated as described in Materials and Methods. Blood glucose were screened once a week. As shown in Figure 1A, 80% (16 of 20) of mice injected with anti-CD3 F(ab′)2 returned to normoglycemia by the fourth week after treatment. This effect was maintained over 40 weeks (Figure 1B), indicating that short-phase anti-CD3 F(ab′)2 treatment resulted in significant and durable remission of hyperglycemia to normoglycemia in NOD mice.
Figure 1.
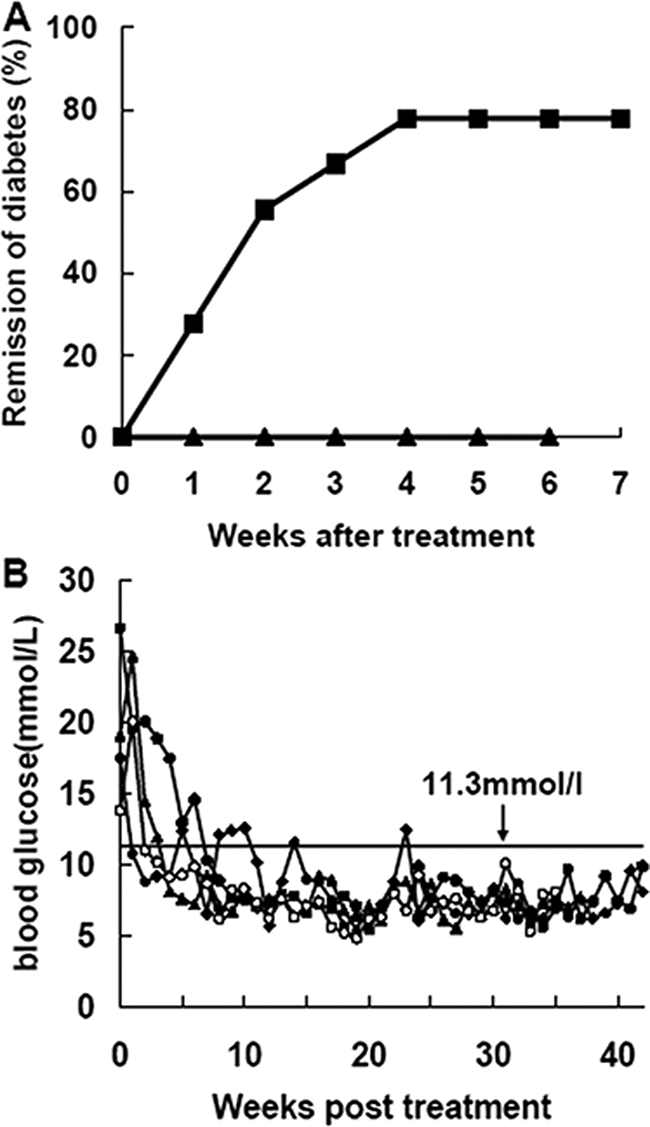
Anti-CD3 F(ab′)2 antibody efficiently reversed new-onset diabetes in NOD mice. Antibody preparation and injection are described in Materials and Methods. A: New-onset diabetic NOD mice were injected with anti-CD3 F(ab′)2. The diabetic untreated littermates were regarded as controls. Mice were screened for glucose levels every week after antibody treatment, and complete remission was defined as a return to normal glycemia. Results were analyzed statistically between the control group (▴, n = 15) and the antibody-treated group (▪, n = 20). B: Representative data of glucose control from four anti-CD3 F(ab′)2-treated NOD mice. Blood glucose was detected weekly after antibody treatment. A value less than 11.3 mmol/L was regarded as normal glycemic.
Anti-CD3 F(ab′)2 Treatment Diminished the Insulitis in NOD Mice
To determine whether reversal of diabetes following anti-CD3 F(ab′)2 treatment correlated with reduction and/or collapse of inflammatory infiltrates, we characterized the histology of pancreata from NOD mice 10 weeks and 20 weeks after antibody treatment. The results showed that, in contrast to the islet from the control diabetic mouse that showed much infiltration of numerous mononuclear cells (Figure 2A), anti-CD3 F(ab′)2 (Figure 2, B–D) treatment diminished the insulitis in NOD mice.
Figure 2.

Anti-CD3 F(ab′)2 treatment diminished insulitis in NOD mice. A: Representative islet from untreated diabetic mice (n = 4) with infiltration of numerous lymphocytes. B: Representative islets from mice (n = 6) 10 weeks after anti-CD3 F(ab′)2 treatment. C: Representative islets from mice (n = 5) 20 weeks after anti-CD3 F(ab′)2 treatment. D: Insulitis scores of pancreatic islets from anti-CD3 F(ab′)2-treated mice (n = 8) or controls (n = 6). Seventy-two islets for control group and 94 islets for treated group were examined, respectively. Magnifications: A and C, ×200; B, ×100. Arrows denote islets.
Anti-CD3 F(ab′)2 Treatment Had No Influence on the Number of CD3+ Cells in Vivo
Anti-CD3 antibodies function mainly by depleting CD3+ T cells in treating allograft rejection.18,19 To determine whether this was available to explain the effect of anti-CD3 F(ab′)2 on reversing established diabetes, the number of CD3+ cells after anti-CD3 F(ab′)2 treatment was examined. We found that anti-CD3 F(ab′)2 treatment did not result in significant increase or decrease in CD3+ cell numbers in peripheral blood (Figure 3A), spleen, or inguinal lymph nodes (Figure 3B). Therefore, reversal of diabetes by anti-CD3 F(ab′)2 treatment was not due to a change in the abundance of CD3+ cells.
Figure 3.
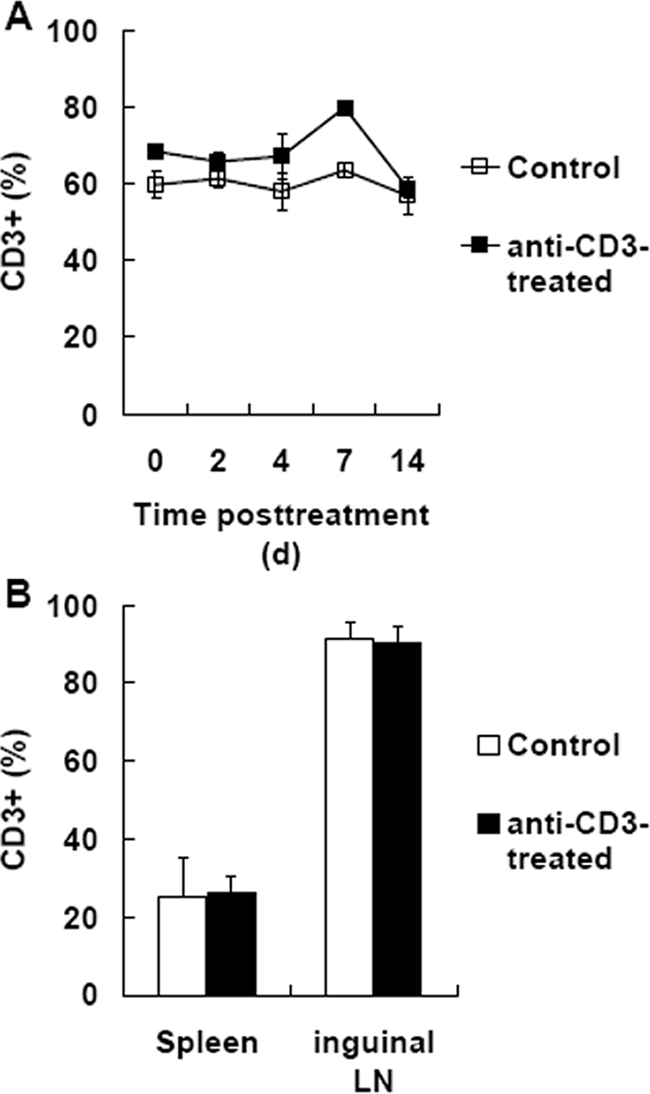
Anti-CD3 F(ab′)2 treatment did not affect CD3+ cell abundance. A: At different time after anti-CD3 F(ab′)2 treatment, the proportions of CD3+ cells were detected by PE-labeled anti-CD3ε antibody in peripheral blood. B: Two weeks after anti-CD3 F(ab′)2 treatment, cells were collected from spleen as well as inguinal lymph nodes of NOD mice and detected by flow cytometry. Data are shown as means ± SD, n = 4–6.
Anti-CD3 F(ab′)2 Treatment Induced Immune Tolerance to Pancreatic β-Cell Autoantigens
We next explored further the underlying mechanisms responsible for remission of diabetes by anti-CD3 F(ab′)2 treatment. To determine whether anti-CD3 F(ab′)2 administration diminished all T cell response or specifically inhibited the activation of autoantigen-specific T cells, we tested the T cell response to glutamic acid decarboxylase and insulin, two candidates of islet β-cell autoantigens,20 as well as some unrelated antigens such as myelin basic protein and concanavalin as stimulators. In addition, as it is documented that pancreatic draining lymph nodes are critical sites for priming effector cells through the pathogenesis of type 1 diabetes,21,22 we isolated mononuclear cells from pancreatic draining lymph nodes and examined the proliferation of these T cells in response to different antigens in vitro. When whole glutamic acid decarboxylase (GAD65) and GAD500–585, a peptide consisting of dominant epitopes of GAD65,23,24 as well as insulin were used as autoantigens in the in vitro T cell proliferation assay, we found that injection of anti-CD3 F(ab′)2 antibodies resulted in significant hyporesponsiveness of lymphocytes to GAD65, GAD500–585, and insulin compared to the control group (*P < 0.05 and **P < 0.01) (Figure 4). However, no statistical differences on the proliferative responses to concanavalin or myelin basic protein between the two groups were observed (P > 0.05). These results demonstrated that anti-CD3 antibody administration induced autoantigen-specific tolerance, which accounted for reversal of diabetes in NOD mice.
Figure 4.

Anti-CD3 F(ab′)2 administration diminished the T cell response in NOD mice. Lymphocytes isolated from pancreatic draining lymph nodes of anti-CD3 F(ab′)2-treated NOD mice or the controls 3 weeks after antibody treatment were reacted with recombinant mouse GAD65 protein (GAD65), 500–585 part of GAD65 (GAD500–585), porcine insulin, concanavalin, or myelin basic protein, and the cells were incubated with 0.5μCi of [3H]thymidine. Proliferation was determined by [3H]thymidine uptake. Data are expressed as stimulation indices (SI) ± SD of the mean from four mice with the background of 1249–3914 cpm, tested in triplicate.*P < 0.05, **P < 0.01 as compared with the control counterparts.
We have also examined in another study whether CD4+ T regulatory cells are involved in anti-CD3 F(ab′)2 antibody treatment-induced tolerance. Unexpectedly, both the abundance and the function of CD4+ T regulatory cells are comparable to untreated controls.25
CD1d-Restricted NKT Cells Were Involved in Anti-CD3 F(ab′)2 Antibody-Mediated Diabetes Reversion
Accumulative evidence has implicated CD1d-restricted NKT cells as a unique T lymphocyte sublineage in the regulation of immune responses associated with a broad range of diseases, including autoimmunity.26 Amplified activity of NKT cells contributed to manipulating autoimmunity aggression via controlling pathogenic cells in vivo through an IL-4-dependent pathway. Thus, to determine whether CD1d-restricted NKT cells were involved in restoring immune tolerance after CD3 antibody treatment, we first tested the abundance of NKT cells (T cell receptor-β+dimer+) in anti-CD3-treated mice versus the controls. At various time points after treatment, cells were collected from different lymphoid organs of NOD mice and analyzed by flow cytometry. The percentage and absolute number of NKT cells significantly increased after treatment in liver but not in spleen and mesenteric lymph nodes (Figure 5, A and B). This is consistent with the concept that the liver compartments harbor unique microarchitecture and vascularization in favor of preferential distribution of blood-borne anti-CD3ε antibodies to the liver.27 Furthermore, when hepatic NKT cells were isolated from remitting mice 4 weeks after antibody treatment, these NKT cells secreted increased amounts of IL-4 and IL-10 and lesser amounts of IFN-γ (Figure 5C), indicating a population of functionally immunomodulatory NKT cells occurred. In addition, we examined the level of IL-4 and IFN-γ in the supernatant of spleen cells from control or remitting mice 4 weeks after treatment. Consistent with Th2 phenotype of NKT cells, Th2 shift could also be detected in periphery (Figure 5D).
Figure 5.

Anti-CD3 administration expanded NKT cells and enhanced anti-inflammatory response. The proportion (A) and absolute number (B) of NKT cells in various lymphoid compartments of NOD mice after anti-CD3 treatment were determined by fluorescence-activated cell sorting analysis. Representative data of three experiments are shown. C: Hepatic NKT cells were isolated from control or anti-CD3 F(ab′)2-treated mice, respectively, and stimulated with anti-CD3 monoclonal antibodies (0.5 μg/ml) for 48 hours. The production of IL-4, IFN-γ, and IL-10 was determined in the supernatant by enzyme-linked immunosorbent assay. D: Splenocytes from antibody-treated or control mice 4 weeks after treatment were cultured in the presence of GAD500–585 at the concentration of 20 μg/ml for 48 hours, and then the supernatants were collected. The levels of IL-4 and IFN-γ were determined, and the ratio of IL-4 to IFN-γ is presented. Values are shown as means ± SD of four mice. *P < 0.05 as compared with control mice.
To determine a role of Th2 cytokines IL-4 and IL-10 in anti-CD3-mediated diabetes reversion, NOD mice treated with anti-CD3 were injected with antibodies specific for IL-4 and/or IL-10 to neutralize the bioactivity of these cytokines in vivo. In contrast to those only receiving isotype control antibodies or not, the mice receiving αIL-4, αIL-10, or combination of these two antibodies did not exhibit diabetes remission after anti-CD3 treatment (Figure 6), indicating Th2 cytokines IL-4 and IL-10 were critical for anti-CD3 effect.
Figure 6.

Neutralization of IL-4 and/or IL-10 prevented diabetes remission after anti-CD3 treatment. Diabetic NOD mice receiving CD3-specific antibody were injected intraperitoneally with neutralizing antibody to IL-4, IL-10, or a mixture of these two antibodies. As controls, anti-CD3-treated mice received isotypes or not. The remission of diabetes was evaluated for 5 weeks. N = 8–15 for each group.
To further address the protective potency of NKT cells, we fractionated NKT cells from the liver of antibody-treated mice and examined protective effect of these cells. Six- to 8-week-old NOD.scid recipients were transfused intravenously with splenocytes from diabetic NOD mice alone (diabetogenic cells) or mixed with NKT cells from antibody-treated or control NOD mice. In these experiments, 100% of mice injected with diabetogenic cells alone developed diabetes within 2 to 4 weeks of the transfer. In contrast, NKT cells from antibody-treated remitting mice significantly inhibited or delayed diabetes transfer into recipients. Whereas, no protection of NKT cells from control diabetic mice was observed (Figure 7). Taken together, these data indicated that the treatment with anti-CD3 antibodies induced a protective IL-4/IL-10-secreting subset of NKT cells.
Figure 7.
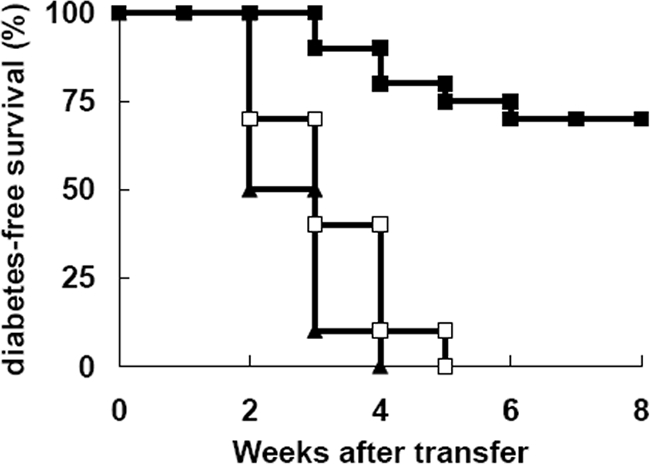
Expanded NKT cells suppressed the transfer of diabetes. NOD.scid females were injected with diabetic splenocytes (1 × 107 cells) alone (▴, n = 10) or mixed with isolated NKT cells (2 × 105 cells) from anti-CD3-treated (▪, n = 20) or untreated mice (□, n = 20). The development of diabetes in the recipients was monitored weekly. Data were pooled from three separate experiments.
Discussion
Nonmitogenic anti-CD3 antibodies have been used to treat established immunological diseases such as type 1 diabetes, multiple sclerosis, psoriatic arthritis, and arteriosclerosis.12,28,29,30 However, the underling mechanisms are largely unclear. Here our data showed that a short-term administration of CD3 antibodies was sufficient for reversing the new-onset diabetes for a long time, and the mechanisms involved the induction and maintenance of long-term immune tolerance, rather than a consequence of T cell depletion (Figures 1–4). The elements responsible for the specific tolerance were explored from different methods.
First we examined the role of CD4+CD25+ Tregs, a putative population of regulatory T cells,31 in anti-CD3 induced glycemia reversal and tolerance restoration. Not consistent with a previous report,14 which showed the involvement of CD4+CD25+ Tregs in CD3-antibody treated mice, neither increased number nor enhanced function of Foxp3+CD4+ T cells in NOD mice were found in our study, as depletion of CD4+CD25+ T cells using neutralizing anti-CD25 antibodies had no effect on the efficacy of anti-CD3 therapy.25 The discrepancy could be resolved because up-regulation of CD4+CD25+ T cells is substantially due to T-cell activation rather than induction/expansion of CD4+ Tregs. This prompted us to survey other pathways required for maintaining immune tolerance to self islets following anti-CD3 treatment.
In searching the T cell populations responsible for the immune tolerance in our model, we found that CD1d-restricted NKT cells, a dominant subset in mice,32 were significantly up-regulated in anti-CD3-treated mice. These cells secreted high amounts of IL-4/IL-10 and less IFN-γ as compared to those from untreated controls (Figure 5). Further, using a cotransfer model, these cells from antibody-treated remitting mice blocked diabetes transfer, suggesting a protective function of expanded NKT cells. This is in agreement with that NKT cell-deficient NOD mice displayed exacerbated diabetes33 and activation of NKT cells via repeated administration of CD1d ligands, for example α-GalCer, protected NOD mice from diabetes.34,35 In addition, these functionally regulatory NKT cells perhaps derive from expansion of remnant NKT cell pool.36
The mechanisms by which enhanced NKT cells affect the autoimmune process need to be determined. Here we focus on the alteration of Th1/Th2 profile in NKT cells and peripheral lymphocytes. NKT cells has been described as one of the immune regulators that work through secreting different cytokines, such as IFN-γ or IL-4, to promote different helper T cell response.37,38 Our study indicated that the cytokine pattern of NKT cells deviated to Th2 phenotype after anti-CD3 treatment with the characteristics of decreased production of IFN-γ and increased IL-4/IL-10 production, which might contribute to suppressing peripheral pathogenic Th1 response to islets in the progression of overt autoimmunity,39,40 although the details anti-CD3 treatment modulates NKT cell bioactivities need elucidation in the future experiments. Importantly, blockade of IL-4 and/or IL-10 activities in vivo led to abrogation of the diabetes-suppressive effect after anti-CD3 treatment, indicating a critical role of Th2 cytokines IL-4 and IL-10 in the NKT cell-mediated regulation of autoimmunity. These results are consistent with previous studies that underscore Th2 shift as a key mechanism of NKT-induced protection against diabetes.34,35,41 Notably, Th2 shift could also be detected in the cytokine production patterns of spleen cells from CD3 antibody-treated mice. So in our system, CD3 antibody administration might work through regulating the activity of NKT cells that resulted in up-regulated production of IL-4/IL-10 and decreased IFN-γ production; thereafter these cells drive effector cells toward a nonpathogenic Th2 phenotype in the periphery. In some cases, NKT cells could inhibit the IL-2 and IFN-γ production and later proliferation of the effector CD4+ T cells, resulting in an anergic phenotype, rather than blocking their initial activation and expansion42 or dampening T cell differentiation into effectors directly by cell contacts.43 Besides direct action on pathogenic T cells, the activation of NKT cells may influence other cell type, including dendritic cells, NK cells, and regulatory T cells.44,45,46,47 Activated NKT cells induce dendritic cell maturation and accumulation in pancreatic lymph nodes, where they subsequently recruit and tolerize pathogenic T cells.44 Once activated, NKT cells could also lead to subsequent activation of conventional NK cells.46
Collectively, these data have implied the involvement of CD1d-restricted NKT cells in anti-CD3 therapeutic effect on overt diabetes. Up-regulated NKT cells in anti-CD3-treated group express a Th2 shift on stimulation, which might affect the T cell balance in the peripheral system. Although the mechanisms in detail remain to be determined, our findings have important implications for understanding the mechanisms involved as well as potential modifications for the regimen of anti-CD3 therapy in the clinic.
Acknowledgments
We thank Lun Song for critical suggestions.
Footnotes
Address reprint requests to Prof. Yan Li, Department of Molecular Immunology, Institute of Basic Medical Sciences, Taiping Road, No. 27, Beijing, 100850 People’s Republic of China. E-mail: liyan62033@yahoo.com.cn.
Supported by grant 2007CB512406 from the National Key Basic Research Program of China and grant 30571732 from the National Natural Science Foundation of China.
G.C. and G.H. contributed equally to this work.
References
- Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell. 1996;85:291–297. doi: 10.1016/s0092-8674(00)81106-x. [DOI] [PubMed] [Google Scholar]
- Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med. 1999;5:601–604. doi: 10.1038/9442. [DOI] [PubMed] [Google Scholar]
- Knip M, Veijola R, Virtanen SM, Hyoty H, Vaarala O, Akerblom HK. Environmental triggers and determinants of type 1 diabetes. Diabetes. 2005;54:125–136. doi: 10.2337/diabetes.54.suppl_2.s125. [DOI] [PubMed] [Google Scholar]
- Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- Poulton LD, Smyth MJ, Hawke CG, Silveira P, Shepherd D, Naidenko OV, Godfrey DI, Baxter AG. Cytometric and functional analysis of NK and NKT cell deficiencies in NOD mice. J Immunol. 2001;13:887–896. doi: 10.1093/intimm/13.7.887. [DOI] [PubMed] [Google Scholar]
- Yang Y, Bao M, Yoon JW. Intrinsic defects in the T-cell lineage results in natural killer T-cell deficiency and the development of diabetes in the nonobese diabetic mouse. Diabetes. 2001;50:2691–2699. doi: 10.2337/diabetes.50.12.2691. [DOI] [PubMed] [Google Scholar]
- Kukreja A, Cost G, Marker J, Zhang C, Sun Z, Lin-Su K, Ten S, Sanz M, Exley M, Wilson B, Porcelli S, Maclaren N. Multiple immuno-regulatory defects in type 1 diabetes. J Clin Invest. 2002;109:131–140. doi: 10.1172/JCI13605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond KJL, Poulton LD, Palmisano LJ, Silveira PA, Godfrey DI, Baxter AG. α/β-T cell receptor (TCR)+CD4−CD8− (NKT) thymocytes prevent insulin-dependent diabetes mellitus in nonobese diabetic (NOD)/Lt mice by the influence of interleukin (IL)-4 and/or IL-10. J Exp Med. 1998;187:1047–1056. doi: 10.1084/jem.187.7.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepault F, Gagnerault MC. Characterization of peripheral regulatory CD4+ T cells that prevent diabetes onset in nonobese diabetic mice. J Immunol. 2000;164:240–247. doi: 10.4049/jimmunol.164.1.240. [DOI] [PubMed] [Google Scholar]
- Tang Q, Henriksen KJ, Bi M, Finger EK, Szot G, Ye J, Masteller EL, McDevitt H, Bonyhadi M, Bluestone JA. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med. 2004;199:1455–1465. doi: 10.1084/jem.20040139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronenberg M, Rudensky A. Regulation of immunity by self-reactive T cells. Nature. 2005;435:598–604. doi: 10.1038/nature03725. [DOI] [PubMed] [Google Scholar]
- Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci USA. 1994;91:123–127. doi: 10.1073/pnas.91.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, Gitelman SE, Harlan DM, Xu D, Zivin RA, Bluestone JA. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med. 2002;346:1692–1698. doi: 10.1056/NEJMoa012864. [DOI] [PubMed] [Google Scholar]
- Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-β-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med. 2003;9:1202–1208. doi: 10.1038/nm924. [DOI] [PubMed] [Google Scholar]
- Falcone M, Yeung B, Tucker L, Rodriguez E, Sarvetnick N. A defect in interleukin 12-induced activation and interferon γ secretion of peripheral natural killer T cells in nonobese diabetic mice suggests new pathogenic mechanisms for insulin-dependent diabetes mellitus. J Exp Med. 1999;190:963–972. doi: 10.1084/jem.190.7.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisch R, Wang B, Atkinson MA, Serreze DV, Friedline R. A glutamic acid decarboxylase 65-specific Th2 cell clone immunoregulates autoimmune diabetes in nonobese diabetic mice. J Immunol. 2001;166:6925–6936. doi: 10.4049/jimmunol.166.11.6925. [DOI] [PubMed] [Google Scholar]
- Yoon JW, Rodrigues MM, Currier C, Notkins AL. Long-term complications of virus-induced diabetes mellitus in mice. Nature. 1982;296:566–569. doi: 10.1038/296566a0. [DOI] [PubMed] [Google Scholar]
- Delaney VB, Campbell WG, Jr, Nasr SA, McCue PA, Warshaw B, Whelchel JD. Efficacy of OKT3 monoclonal antibody therapy in steroid-resistant, predominantly vascular acute rejection. Transplantation. 1988;45:743–748. doi: 10.1097/00007890-198804000-00016. [DOI] [PubMed] [Google Scholar]
- Janssen O, Wesselborg S, Kabelitz D. Immunosuppression by OKT3-induction of programmed cell death (apoptosis) as a possible mechanism of action. Transplantation. 1992;53:233–234. [PubMed] [Google Scholar]
- Lernmark A, Agardh CD. Immunomodulation with human recombinant autoantigens. Trends Immunol. 2005;26:608–612. doi: 10.1016/j.it.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Levisetti MG, Suri A, Frederick K, Unanue ER. Absence of lymph nodes in NOD mice treated with lymphotoxin-beta receptor immunoglobulin protects from diabetes. Diabetes. 2004;53:3115–3119. doi: 10.2337/diabetes.53.12.3115. [DOI] [PubMed] [Google Scholar]
- Jaakkola I, Jalkanen S, Hanninen A. Diabetogenic T cells are primed both in pancreatic and gut-associated lymph nodes in NOD mice. Eur J Immunol. 2003;33:3255–3264. doi: 10.1002/eji.200324405. [DOI] [PubMed] [Google Scholar]
- Song L, Wang J, Wang R, Yu M, Sun Y, Han G, Li Y, Qian J, Scott DW, Kang Y, Soukhareva N, Shen B. Retroviral delivery of GAD-IgG fusion construct induces tolerance and modulates diabetes: a role for CD4+ regulatory T cells and TGF-β? Gene Ther. 2004;11:1487–1496. doi: 10.1038/sj.gt.3302327. [DOI] [PubMed] [Google Scholar]
- Han G, Li Y, Wang J, Wang R, Chen G, Song L, Xu R, Yu M, Wu X, Qian J, Shen B. Active tolerance induction and prevention of autoimmune diabetes by immunogene therapy using recombinant adenoassociated virus expressing glutamic acid decarboxylase 65 peptide GAD500–585. J Immunol. 2005;174:4516–4524. doi: 10.4049/jimmunol.174.8.4516. [DOI] [PubMed] [Google Scholar]
- Chen G, Han G, Wang J, Wang R, Xu R, Shen B, Qian J, Li Y. Essential roles of TGF-β in anti-CD3 antibody therapy: reversal of diabetes in nonobese diabetic mice independent of Foxp3+CD4+ regulatory T cells. J Leukoc Biol. 2008;83:280–287. doi: 10.1189/jlb.0707498. [DOI] [PubMed] [Google Scholar]
- Laloux V, Beaudoin L, Jeske D, Carnaud C, Lehuen A. NKT cell-induced protection against diabetes in V alpha 14-J alpha 281 transgenic nonobese diabetic mice is associated with a Th2 shift circumscribed regionally to the islets and functionally to islet autoantigen. J Immunol. 2001;166:3749–3756. doi: 10.4049/jimmunol.166.6.3749. [DOI] [PubMed] [Google Scholar]
- Wingender G, Schumak B, Schurich A, Gessner JE, Endl E, Limmer A, Knolle PA. Rapid and preferential distribution of blood-borne αCD3εAb to the liver is followed by local stimulation of T cells and natural killer T cells. Immunology. 2006;117:117–126. doi: 10.1111/j.1365-2567.2005.02272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran GT, Carter N, He XY, Spicer TS, Plain KM, Nicolls M, Hall BM, Hodgkinson SJ. Reversal of experimental allergic encephalomyelitis with non-mitogenic, non-depleting anti-CD3 mAb therapy with a preferential effect on T(h)1 cells that is augmented by IL-4. Int Immunol. 2001;13:1109–1120. doi: 10.1093/intimm/13.9.1109. [DOI] [PubMed] [Google Scholar]
- Utset TO, Auger JA, Peace D, Zivin RA, Xu D, Jolliffe L, Alegre ML, Bluestone JA, Clark MR. Modified anti-CD3 therapy in psoriatic arthritis: a phase I/II clinical trial. J Rheumatol. 2002;29:1907–1913. [PubMed] [Google Scholar]
- Steffens S, Burger F, Pelli G, Dean Y, Elson G, Kosco-Vilbois M, Chatenoud L, Mach F. Short-term treatment with anti-CD3 antibody reduces the development and progression of atherosclerosis in mice. Circulation. 2006;114:1977–1984. doi: 10.1161/CIRCULATIONAHA.106.627430. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S. Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and non-self. Nat Immunol. 2005;6:345–352. doi: 10.1038/ni1178. [DOI] [PubMed] [Google Scholar]
- Matsuda JL, Naidenko OV, Gapin L, Nakayama T, Taniguchi M, Wang CR, Koezuka Y, Kronenberg M. Tracking the response of nature killer T cells to a glycolipid antigen using CD1d tetramers. J Exp Med. 2000;192:741–754. doi: 10.1084/jem.192.5.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi FD, Flodstrom M, Balasa B, Kim SH, Van Gunst K, Strominger JL, Wilson SB, Sarvetnick N. Germ line deletion of the CD1 locus exacerbates diabetes in the NOD mice. Proc Natl Acad Sci USA. 2001;98:6777–6782. doi: 10.1073/pnas.121169698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Wilson MT, Serizawa I, Wu L, Singh N, Naidenko OV, Miura T, Haba T, Scherer DC, Wei J, Kronenberg M, Koezuka Y, Van Kaer L. The natural killer T-cell ligand alpha-galactosylceramide prevents autoimmune diabetes in non-obese diabetic mice. Nat Med. 2001;7:1052–1056. doi: 10.1038/nm0901-1052. [DOI] [PubMed] [Google Scholar]
- Sharif S, Arreaza GA, Zucker P, Mi QS, Sondhi J, Naidenko OV, Kronenberg M, Koezuka Y, Delovitch TL, Gombert JM, Leite-De-Moraes M, Gouarin C, Zhu R, Hameg A, Nakayama T, Taniguchi M, Lepault F, Lehuen A, Bach JF, Herbelin A. Activation of natural killer T cells by alpha-galactosylceramide treatment prevents the onset and recurrence of autoimmune Type 1 diabetes. Nat Med. 2001;7:1057–1062. doi: 10.1038/nm0901-1057. [DOI] [PubMed] [Google Scholar]
- Wang B, Geng YB, Wang CR. CD1-restricted NKT cells protect nonobese diabetic mice from developing diabetes. J Exp Med. 2001;194:313–320. doi: 10.1084/jem.194.3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N, Hong S, Scherer DC, Serizawa I, Burdin N, Kronenberg M, Koezuka Y, Van Kaer L. Cutting edge: activation of NK T cells by CD1d and α-galactosylceramide directs conventional T cells to the acquisition of a Th2 phenotype. J Immunol. 1999;163:2373–2377. [PubMed] [Google Scholar]
- Cui J, Watanabe N, Kawano T, Yamashita M, Kamata T, Shimizu C, Kimura M, Shimizu E, Koike J, Koseki H, Tanaka Y, Taniguchi M, Nakayama T. Inhibition of T helper cell type 2 cell differentiation and immunoglobulin E response by ligand-activated Vα14 natural killer T cells. J Exp Med. 1999;190:783–792. doi: 10.1084/jem.190.6.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delovitch TL, Singh B. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity. 1997;7:727–738. doi: 10.1016/s1074-7613(00)80392-1. [DOI] [PubMed] [Google Scholar]
- Rabinovitch A. Immunoregulatory and cytokine imbalances in the pathogenesis of IDDM. Therapeutic intervention by immunostimulation? Diabetes. 1994;43:613–621. doi: 10.2337/diab.43.5.613. [DOI] [PubMed] [Google Scholar]
- Lehuen A, Lantz O, Beaudoin L, Laloux V, Carnaud C, Bendelac A, Bach JF, Monterio RC. Overexpression of natural killer T cells protects Valpha14-Jalpha281 transgenic nonobese diabetic mice against diabetes. J Exp Med. 1998;188:1831–1839. doi: 10.1084/jem.188.10.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudoin L, Laloux V, Novak J, Lucas B, Lehuen A. NKT cells inhibit the onset of diabetes by impairing the development of pathogenic T cells specific for pancreatic beta cells. Immunity. 2002;17:725–736. doi: 10.1016/s1074-7613(02)00473-9. [DOI] [PubMed] [Google Scholar]
- Novak J, Beaudoin L, Griseri T, Lehuen A. Inhibition of T cell differentiation into effectors by NKT cells require cell contacts. J Immunol. 2005;174:1954–1961. doi: 10.4049/jimmunol.174.4.1954. [DOI] [PubMed] [Google Scholar]
- Chen YG, Choisy-Rossi CM, Holl TM, Chapman HD, Besra GS, Porcelli SA, Shaffer DJ, Roopenian D, Wilson SB, Serreze DV. Activation NKT cells inhibit autoimmune diabetes through tolerogenic recruitment of dendritic cells to pancreatic lymph nodes. J Immunol. 2005;174:1196–1204. doi: 10.4049/jimmunol.174.3.1196. [DOI] [PubMed] [Google Scholar]
- Naumov YN, Bahjet KS, Gausling R, Abraham R, Exley MA, Koezuka Y, Balk SB, Strominger JK, Clare-Salzer M, Wilson SB. Activation of CD1d-restricted T cells protects NOD mice from developing diabetes by regulating dendritic cell subsets. Proc Natl Acad Sci USA. 2001;98:13838–13843. doi: 10.1073/pnas.251531798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa Y, Takeda K, Yagita H, Kakuta S, Iwakura Y, Van Kaer L, Saiki I, Okumura K. Critical contribution of IFN-gamma and NK cells, but not perforin-mediated cytotoxicity, to anti-metastatic effect of alpha-galactosylceramide. Eur J Immunol. 2001;31:1720–1727. [PubMed] [Google Scholar]
- Nakamura T, Sonoda KH, Faunce DE, Gumperz J, Yamamura T, Miyake S, Stein-Streilein J. CD4(+) NKT cells, but not conventional CD4(+) T cells, are required to generate efferent CD8(+) T regulatory cells following antigen inoculation in an immune-privileged site. J Immunol. 2003;171:1266–1271. doi: 10.4049/jimmunol.171.3.1266. [DOI] [PubMed] [Google Scholar]