

Abstract
Human cytomegalovirus (CMV) infection is a major cause of morbidity in immunosuppressed individuals, and congenital CMV infection is a leading cause of birth defects in newborns. Infection with pathogenic viral strains alters cell-cell and cell-matrix interactions, affecting extracellular matrix remodeling and endothelial cell migration. The multifunctional cytokine transforming growth factor (TGF)-β1 regulates cell proliferation, differentiation, and extracellular matrix remodeling. Secreted as a latent protein complex, TGF-β1 requires activation before binding to receptors that phosphorylate intracellular effectors. TGF-β1 is activated by integrin αvβ6, which is strongly induced in the epithelium by injury and inflammation but has not previously been found in endothelial cells. Here, we report that CMV infection induces integrin αvβ6 expression in endothelial cells, leading to activation of TGF-β1, signaling through its receptor ALK5, and phosphorylation of its intracellular effector Smad3. Infection of endothelial cells was also found to stimulate collagen synthesis through a mechanism dependent on both TGF-β1 and integrin αvβ6. Immunohistochemical analysis showed integrin αvβ6 up-regulation in capillaries proximal to foci of CMV infection in lungs, salivary glands, uterine decidua, and injured chorionic villi of the placenta, demonstrating both its induction in endothelium and up-regulation in epithelium in vivo. Our results suggest that activation of TGF-β1 by integrin αvβ6 contributes to pathological changes and may impair endothelial cell functions in tissues that are chronically infected with CMV.
Human cytomegalovirus (CMV) is a ubiquitous pathogen that infects more than half the population of the United States. On primary infection, latency is established in granulocyte-macrophage progenitors.1,2 High levels of CD8+ antigen-specific cytotoxic T lymphocytes reactive with CMV peptides indicate that reactivated infections require sustained immunosurveillance.3,4 CMV replicates in epithelial and endothelial cells in salivary glands, lungs, kidney, uterus, and placenta.5,6,7,8,9,10 Because cytopathic alterations, characteristic of a late stage of infection, were found in these cells, they are likely to reflect biologically relevant mechanisms operative during in vivo infection. Clinical studies indicate that chronic subclinical CMV infection and immune rejection in organ transplant recipients are risk factors for graft failure.11,12,13,14
Transforming growth factor-β1 (TGF-β1), a multifunctional cytokine, plays a central role in cell proliferation, migration, and synthesis of extracellular matrix (ECM) in the endothelium.15 In most cell types, TGF-β1 signals through the type I receptor activin receptor-like kinase 5 (ALK5). In addition to expressing ALK5, endothelial cells express a second TGF-β1 receptor, the type I receptor ALK1. When activated, ALK1 induces phosphorylation of the nuclear effectors Smad1 and Smad5, which promote endothelial cell proliferation and migration.16 In contrast, activated ALK5 induces Smad2 and Smad3 phosphorylation, leading to the inhibition of endothelial cell proliferation. TGF-β1 is secreted as an inactive, noncovalent complex with latency-associated peptide and requires activation before it can bind to its receptors. Reported mechanisms of TGF-β1 activation include cleavage by metalloproteinases or plasmin and binding to thrombospondin 1 or either of the integrins αvβ6 and αvβ8.15,17,18,19,20,21 One of the in vivo activators of TGF-β1 is integrin αvβ6.17,18 This activation model is particularly interesting because integrin αvβ6 is expressed principally on epithelial cells, which are very sensitive to TGF-β1-mediated growth inhibition. Integrin αvβ6 is strongly up-regulated at sites of epithelial repair and inflammation in lung and kidney,22 and also because of the overlap of the phenotypes of TGF-β1- and integrin β6 subunit-deficient mice. Mice lacking the β6 subunit show increased inflammation and decreased fibrosis, both of which processes are strongly regulated by TGF-β1.18,23,24
Recent work has provided evidence for the induction of TGF-β1 in a variety of cells and tissues on CMV infection. TGF-β1 was released in increasing amounts from splenocytes infected with rat CMV in vitro.25 TGF-β1 protein was increased in alveoli and stromal cells in rat lungs, spleen, and liver after radiation-induced immune suppression of CMV-infected rats.25 Furthermore, CMV-infected murine astrocytes increased TGF-β1 transcription and protein levels.26 In human kidney allografts, CMV proteins and DNA were associated with locally increased TGF-β1 in tubuli and arterial endothelium long after viral clearance from the blood.27 Brain biopsy specimens from AIDS patients with CMV encephalitis were found to contain viral inclusions that co-localized with TGF-β1 protein in cells with astrocyte-specific glial filaments.26 In addition, TGF-β1 induction in human fibroblasts has been shown to involve the transactivation of its promoter by immediate-early 2 protein through an Egr-1 consensus site by binding the zinc finger domain of Egr-1.28,29 Although the evidence suggests that TGF-β1 may be directly involved in CMV pathogenesis, little is known about the cellular proteins involved in virus-mediated TGF-β1 activation, or what specific functional role it plays in vivo. In recent experiments, we found that a subpopulation of freshly isolated human cytotrophoblasts from term placentas expressed integrin αvβ6, which activates TGF-β1 in vitro.30 We hypothesized that CMV infection could increase not only the production of latent TGF-β1 but also its activation, causing TGF-β1-mediated cellular responses.
Here we report that CMV-infected endothelial cells from pulmonary, uterine, and placental blood vessels activate TGF-β1 through the induction of the epithelial integrin αvβ6, promoting signaling through ALK5 and Smad3. This signaling pathway plays a fundamental role in mediating profibrotic responses at later times after infection. In our studies, immunohistochemical analysis of CMV-infected tissues showed integrin αvβ6 expression in both epithelial and endothelial cells proximal to infected foci and sites of injury. These results suggest that integrin αvβ6-mediated TGF-β1 activation could be relevant to the development of fibrosis in persistent infection.
Materials and Methods
Cell Culture, Virus, and Infection
Human microvascular endothelial cells-lung (HMVEC-L) and uterine (UtMVECs) from two donors and human umbilical vein endothelial cells (HUVECs) from two donors were maintained in EBM-2 medium supplemented with EGM-2-MV and EGM-2 Singlequots (Cambrex BioScience Walkersville, Inc., Walkersville, MD). Endothelial cells from different sources, 98% positive by immunofluorescence staining with a rabbit antiserum to von Willebrand factor complex (Novocastra Laboratories Ltd., Newcastle upon Tyne, UK), were used between passages 4 and 6. Cells were infected with the pathogenic clinical CMV strain VR181431 at 1 PFU/cell and cultured up to 15 days.
Serological and Other Reagents
The following antibodies to integrins were purified from hybridoma supernatants: mouse anti-human β5 mAb ALULA32; mouse anti-human αv mAb L 230,33 mouse anti-αvβ6 CSβ6,34 and rabbit anti-αvβ6 mAb 4B5.35 Mouse anti-human α5 mAb P3D10 and mouse anti-human β1 monoclonal antibody (mAb) P5D236 were generous gifts of Dr. Elizabeth Wayner (Fred Hutchinson Cancer Center, Seattle, WA). Mouse anti-αvβ6 mAbs 3G9 and 2A1 were gifts from Drs. Shelia Violette and Paul Weinreb (Biogen Idec Inc., Cambridge, MA),37 and mouse anti-human αvβ8 mAb 14E5 was a gift from Dr. Stephen Nishimura.19 The following antibodies were purchased: mouse anti-human β3 mAb (clone VI-PL2; BD Biosciences, San Diego, CA), mouse anti-TGF-β1, -β2, and -β3 mAb (clone 1D11), chicken anti-human-TGF-β polyclonal antibody, anti-human ALK1 goat polyclonal antibody and mouse anti-human endoglin (CD105) mAb (R&D Systems, Minneapolis, MN); rabbit polyclonal antiserum to von Willebrand factor complex (Novocastra Laboratories; and DakoCytomation, Carpinteria, CA); mouse anti-Smad2/3 mAb, and mouse anti-human Grb2 mAb (BD Biosciences); rabbit anti-TGF-β mAb (clone 56E4), rabbit polyclonal anti-Smad1, anti-Smad5, anti-pSmad1/5/8, which specifically recognizes phosphorylated Smad1 (Ser463/465), Smad5 (Ser163/465), and Smad8 (Ser426/428), rabbit anti-pSmad3 mAb, which specifically recognizes phosphorylated Smad3 (Ser423/425) (clone C25A9) (Cell Signaling Technology, Beverly, MA); anti-TGF-β receptor I (TGF-β RI, H-100; Santa Cruz Biotechnology, Santa Cruz, CA), anti-human type IV collagen goat polyclonal antibody (Southern Biotechnology, Birmingham, AL), mouse monoclonal anti-human CMV-infected-cell protein p76 and DNA binding protein UL44 (clone DDG9 and CCH2) (DakoCytomation), and mouse anti-actin mAb (Sigma-Aldrich, St. Louis, MO). Mouse mAbs to CMV glycoprotein B (gB) (CH112-2, UL55) and immediate-early (IE1 and IE2) nuclear proteins (CH160, UL122 and UL123), produced in the Pereira laboratory, were used as described.10,30 CH112-2 was conjugated with fluorescein isothiocyanate (eBioscience, San Diego, CA). TGF-β receptor I ALK5 kinase inhibitor SB431542 was purchased from Tocris Bioscience (Ellisville, MO).
Flow Cytometry
Cells were harvested using cell dissociation buffer (Invitrogen, Carlsbad, CA). For analysis of surface antigen expression, cells were blocked with normal goat or donkey serum, washed with phosphate-buffered saline (PBS), incubated with primary antibodies or isotype-matched control IgG for 1 hour at 4°C, and detected with phycoerythrin-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA). Stained cells were analyzed by FACScan and CellQuest software (BD Biosciences). Propidium iodide was used to gate out contributions from dead cells. Intracellular expression was analyzed after fixation with 4% formaldehyde for 10 minutes at room temperature and permeabilization with 0.01% Triton X-100 for 5 minutes at 4°C.
Immunoblot Analysis
Cells were lysed in buffer [50 mmol/L Tris-HCl, pH 7.6, 100 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid, 1% Triton X-100, 0.1% sodium dodecyl sulfate (SDS), 20 mmol/L NaF, 1 mmol/L Na3VO4, and protease inhibitor cocktail (Sigma-Aldrich)] and then clarified by centrifugation. Proteins were separated by SDS-polyacrylamide gel electrophoresis (PAGE), transferred to a nitrocellulose membrane, and blocked for 1 hour in PBS containing 5% skim milk and 0.05% Tween 20. After incubation with primary antibody for 16 hours at 4°C, and then with peroxidase-conjugated secondary antibody for 1 hour, blots were developed with the ECL Western blotting detection system (GE Health Care, Piscataway, NJ).
TGF-β Reporter Cell Assay
Mink lung epithelial cells (TMLCs), which are TGF-β reporter cells stably expressing a truncated portion of the plasminogen activator inhibitor-1 promoter fused to the luciferase gene (a gift from Daniel Rifkin, New York University, New York, NY), were used to quantify TGF-β activation.38 Exposure to active TGF-β, either produced by activating cell types or present in cell-free supernatants to TMLCs, resulted in a dose-dependent increase in luciferase activity in the cell lysates.18,19,38 Briefly, TMLCs were co-cultured with uninfected control HUVECs and VR1814-infected HUVECs (5 × 104 cells) at 3, 7, or 10 days for 16 to 24 hours; cell lysates were then analyzed for luciferase activity.18,38 Cells infected for 10 days were co-cultured with TMLCs with or without anti-pan-TGF-β blocking antibody (1D11, mouse IgG1) or anti-αvβ6 function-blocking antibody (3G9, mouse IgG1). As negative controls, mouse IgG1 isotype control antibody and isotype-matched non-function-blocking anti-αvβ6 antibody (CSβ6, mouse IgG1) were used in the same concentrations as the neutralizing antibodies. All assays were performed in duplicate.
Quantification of TGF-β1 by Enzyme-Linked Immunosorbent Assay
Levels of secreted TGF-β1 were measured by determining its concentration in conditioned medium using a commercial sandwich enzyme-linked immunosorbent assay (Quantikine TGF-β1 immunoassay; R&D Systems). Cells were grown in six-well plates; conditioned medium was harvested on day 1 and then at 2-day intervals thereafter, cleared by centrifugation, and stored at −80°C. Conditioned medium was acid-activated and directly assayed by visualization with tetramethylbenzidine on an enzyme-linked immunosorbent assay plate reader at 450 nm according to the manufacturer’s instructions. Protein concentrations were calculated from a standard curve with twofold serial dilutions and a high standard of 2000 pg/ml.
Immunohistochemical Analysis
Lung and Salivary Gland Specimens
Samples from 11 lungs and 1 salivary gland were obtained from 12 patients with CMV infection with histological evidence of nuclear inclusion bodies. These were obtained from families at autopsy with informed consent from the Tokyo Women’s Medical University, Tokyo, Japan. Tissue sections were stained with hematoxylin and eosin and immunostained using the avidin-biotin complex method on formalin-fixed and paraffin-embedded tissues. Tissue sections were deparaffinized, and antigen retrieval was performed by incubating slides in a pepsin solution at 37°C or by microwave treatment in a 10 mmol/L citrate buffer (pH 6.0). Endogenous peroxidase activity was blocked by incubation in a solution with 3% hydrogen peroxide for 10 minutes. Nonspecific binding was blocked with PBS containing 5% normal donkey serum (1 hour) before incubation with a primary antibody overnight (4°C). Biotinylated anti-rabbit or anti-mouse IgG (Vector Laboratories Inc., Burlingame, CA) was applied to the slides, which were subsequently treated using a Vectastain ABC kit (Vector Laboratories). Slides were visualized with a solution containing 0.01% (w/v) 3,3′-diaminobenzidine tetrahydrochloride, 0.02% (v/v) hydrogen peroxide, and 50 mmol/L Tris-HCl (pH 7.6) counterstained with hematoxylin, dehydrated, cleared, and mounted.
Decidual and Placental Biopsy Specimens
Approval for the use of human patients was obtained from the institutional review board of the University of California, San Francisco, San Francisco, CA. Detection of CMV replication and virion structural proteins in the biopsy specimens was reported.10,39 Tissue sections were processed for immunohistochemistry as described.9 Briefly, tissues were fixed in 3% paraformaldehyde, infiltrated with 5 to 15% sucrose followed by embedding in optimal-cutting-temperature compound, and frozen in liquid nitrogen. For double staining, tissue sections were simultaneously incubated with primary antibodies from different species and secondary antibodies labeled with fluorescein isothiocyanate or tetramethyl rhodamine isothiocyanate. Nuclei were counterstained with TO-PRO-3 iodide (Invitrogen).
Statistical Analysis
Data are expressed as mean ± SE. Student’s t-test was used to analyze the difference in expression between control and CMV-infected cells. A P value of <0.05 was considered significant.
Results
CMV-Infected HUVECs Express αvβ6
Previous investigators reported that human fibroblasts infected with a laboratory CMV strain expressed TGF-β1 transcripts and protein, but they did not examine activation of the latent protein.28,29 The propeptide of TGF-β1, latency-associated peptide-β1, contains an Arg-Gly-Asp (RGD) motif that is recognized by a subset of integrins having in common the integrin αv subunit18,19,40,41,42 and α5β1.43 Furthermore, the integrins αvβ6 and αvβ8 have been shown to activate TGF-β1 in vivo.18,19 To examine whether CMV infection alters the expression level of αv integrin β subunit partners and integrin α5, we infected HUVECs with VR1814, a pathogenic clinical CMV strain, and quantified the surface expression of integrins β1, β3, β5, β6, β8, and α5 by flow cytometry at 10 days after infection. Level of infectivity was evaluated by immunofluorescence staining and flow cytometric analysis of CMV gB expression at the cell surface. The results showed nuclear immunofluorescence of CMV IE1 and IE2 proteins and cytoplasmic gB staining in >90% of infected cells. Flow cytometry detected surface expression of gB in 60.8 ± 6.3% of infected cells. In control uninfected HUVECs, integrin subunits β1, β3, β5, and α5 were expressed abundantly, but there was no expression of β6 and only minimal expression of integrin β8 (Figure 1A). Integrin β6, whose expression is considered restricted to epithelial cells, was strongly induced in CMV-infected HUVECs, whereas levels of integrins β1, β3, β5, β8, and α5, as well as αv (data not shown), were unchanged. An analysis of the kinetics of integrin β6 induction in infected HUVECs showed that the protein was increasingly detected from 5 to 10 days after infection (Figure 1B). Expression of integrin β6 was confirmed at 10 days by immunoblot analysis (Figure 1C). These data suggested that integrin αvβ6, aberrantly expressed in infected HUVECs, participates in TGF-β1 activation. Our subsequent investigations focused on assessing integrin αvβ6 function in infected HUVECs.
Figure 1.

CMV strain VR1814-infected HUVECs induce integrin αvβ6 expression at late times. A: Flow cytometric analysis of integrin subunits β1, β3, β5, β6, β8, and α5 in HUVECs at 10 days after infection. Experiments were repeated at least five times. Typical histograms from control (cont.) and infected (inf.) HUVECs are shown. Shaded areas represent expression of specific proteins. Lines represent isotype control. B: Flow cytometric analysis of integrin αvβ6 in HUVECs at 3, 5, 7, and 10 days after infection (dpi) and control (cont). Typical histograms are shown. Shaded areas represent expression of specific proteins. Lines represent isotype control. Experiments were repeated at least four times. C: Cell lysates (100 μg) from control or infected HUVECs at 10 days after infection were immunoblotted with an anti-integrin αvβ6 (2A1) and anti-actin antibodies as a loading control. Molecular mass (kDa) is shown on the left.
CMV Induces Integrin β6-Dependent TGF-β1 Activation
In the next series of experiments, we assessed whether integrin αvβ6 in HUVECs induced by CMV activates TGF-β1. First, we quantified the level of TGF-β1 released into the medium from CMV-infected HUVECs and uninfected control cells. After day 1, conditioned medium from infected and control cells was collected on alternate days and frozen. To quantify TGF-β1 by enzyme-linked immunosorbent assay, conditioned medium was acid-treated to convert the latent TGF-β1 to the immunoreactive form. Increasing amounts of TGF-β1 were secreted from HUVECs as early as 3 days after infection (Figure 2A). Significantly more TGF-β1 was released from cells at 5 to 7 days after infection. In contrast, control cells did not show any increase in the amounts of soluble TGF-β1 in a comparable culture period (Figure 2A).
Figure 2.

Integrin αvβ6-dependent TGF-β1 activation in CMV-infected HUVECs. A: TGF-β1 production by infected HUVECs. Conditioned medium was collected from control (open circles) and infected (filled circles) HUVECs at 1 to 9 days, and TGF-β1 was quantified by enzyme-linked immunosorbent assay. Results are the mean (±SE) of three experiments done in duplicate. Asterisks indicate the amount of TGF-β1 in infected HUVECs as compared with uninfected controls (*P < 0.05, **P < 0.01). B: Surface expression of TGF-β1 in HUVECs was analyzed by flow cytometry at 3, 7, and 10 days after infection and controls (cont). Typical histograms are shown. Shaded areas represent expression of specific proteins. Lines represent isotype control. Experiments were repeated at least three times. C: Total TGF-β1 was analyzed by flow cytometry using permeabilized cells at 3, 7, and 10 days after infection (inf.) and controls (cont). Left: Typical histograms at 10 days are shown. Shaded areas represent expression of specific proteins. Lines represent isotype control. Right: Results are the mean fluorescence intensity (±SE) of three experiments. Asterisks indicate expression in infected HUVECs as compared with uninfected controls (*P < 0.05). D: TGF-β bioassay of active TGF-β produced by infected HUVECs. Equal numbers of TMLC TGF-β reporter cells, and control (cont.) or infected HUVECs (inf.) were cultured for 16 to 24 hours at 3, 7, and 10 days after infection. Relative luciferase activity in cell lysates was defined as the measured activity divided by TMLC baseline activity. Results are the mean (±SE) from 6 to 11 experiments done in duplicate. Asterisks indicate the TGF-β1 activity in infected HUVECs as compared with uninfected controls (*P < 0.05, **P < 0.001). E: Inhibition of luciferase activity in TGF-β bioassay by anti-integrin αvβ6. HUVECs infected for 10 days were co-cultured with TMLCs with anti-TGF-β neutralizing antibody (1D11); function-blocking anti-αvβ6 antibody (3G9); isotype-matched, non-function-blocking anti-αvβ6 antibody (CSβ6); or mouse IgG1 isotype control antibody (control Ab). Results are the mean (±SE) from three to five experiments done in duplicate. Asterisks indicate inhibition of TGF-β1 activation relative to untreated infected HUVECs (*P < 0.05, **P < 0.01, ***P < 0.001).
Because secretion of TGF-β1 is increased by infection, we then asked how much surface (ie, bound) and total cellular TGF-β1 was present by flow cytometry. Surface expression of TGF-β1 on infected cells was increased at 7 to 10 days after infection, whereas no change was observed in uninfected control cells (Figure 2B). Expression of total TGF-β1 in infected cells was significantly increased at 10 days after infection (Figure 2C).
To determine whether CMV activates TGF-β1, we co-cultured HUVECs with TMLCs. At 3, 7, and 10 days after infection, control HUVECs or infected cells were trypsinized and then co-cultured with TMLCs for 16 to 24 hours before measurement of luciferase activity in cell lysates. We found a dramatic increase in luciferase activity, indicating TGF-β1 activation, in 7- to 10-day-infected HUVECs co-cultured with TMLCs (Figure 2D). Little luciferase activity was observed in control HUVECs co-cultured with TMLCs (Figure 2D). We then tested whether the increased luciferase activity is dependent on TGF-β1 or integrin αvβ6. HUVECs infected for 10 days were co-cultured with TMLCs, with or without function-blocking antibodies against either TGF-β (1D11) or αvβ6 (3G9). Negative controls included isotype-matched, non-function-blocking antibodies with either unrelated specificity or non-function-blocking specificity against αvβ6 (CSβ6). The increase in luciferase activity was partly abrogated by function-blocking anti-TGF-β (1D11) and anti-β6 (3G9) but not by control antibodies (CSβ6 or isotype control) (Figure 2E), indicating that TGF-β1 activation after CMV infection is at least integrin αvβ6-dependent. Although the inhibition of luciferase activity by neutralizing antibodies was dose-dependent, even very high concentrations of anti-TGF-β were able to reduce luciferase activity by only ∼50% compared with untreated cells, suggesting that CMV may also activate the plasminogen activator-1 promoter through a mechanism not dependent on TGF-β1.
CMV-Infected HUVECs Undergo ALK5/Smad3 Signaling
Activated TGF-β1 can bind the type I receptors ALK1 and ALK5, which then phosphorylate the transcriptional activators Smad1/5 and Smad2/3, respectively.44 To determine which of these TGF-β1 signaling pathways is activated in CMV-infected HUVECs, we analyzed Smad1/5 and Smad3 phosphorylation by immunoblotting with antibodies specific to Smads and their phosphorylated forms (Figure 3A). Smad3 phosphorylation was strongly detected in 7- and 10-day-infected cells. In contrast, only weak staining for phosphorylated Smad1/5 was observed, and this level either did not change or was decreased at 10 days after infection. Phosphorylated Smad1/5 was also weakly detected in the control. Protein levels of Smad1, Smad5, and Smad2/3 were the same in both infected and control cells.
Figure 3.

A: CMV-infected HUVECs induce Smad3 phosphorylation. Cell lysates from control (cont.) or infected (inf.) HUVECs at 3, 7, and 10 days after infection were fractionated by 10% SDS-PAGE and blotted on nitrocellulose. Phosphorylation of Smad3 (pSmad3) and Smad1/5 (pSmad1/5) was analyzed by immunoblotting using phospho-specific Smad3 and Smad1/5/8 (pSmad1/5) antibodies. Equal loading of the gels was confirmed using Smad2/3, Smad1, and Smad5 protein levels. B: Effects of anti-TGF-β antibody, anti-αvβ6 antibody (3G9), and ALK5 kinase inhibitor on Smad3 phosphorylation. Infected HUVECs were cultured without antibody (untreated) or with anti-TGF-β neutralizing antibody (1D11, 40 μg/ml), function-blocking anti-αvβ6 antibody (3G9, 80 μg/ml), mouse IgG1 isotype control antibody (control Ab, 80 μg/ml), the ALK5 kinase inhibitor SB431542 (2.5 μmol/L), or the vehicle DMSO for 8 days. Lysates were fractionated by 10% SDS-PAGE and blotted. Filters were incubated with antibodies to phosphorylated Smad3 (pSmad3), phosphorylated Smad1/5/8 (pSmad1/5), and Grb2 (loading control). Results are representative of at least four independent experiments.
To determine the relative contributions of TGF-β1 and αvβ6 to the observed ALK5 and Smad3 signaling, we performed function-blocking experiments using anti-TGF-β (1D11) and anti-αvβ6 (3G9) antibodies in 8-day-infected HUVECs. Both neutralizing antibodies blocked Smad3 phosphorylation, whereas the isotype control antibody had little effect (Figure 3B). Treatment of infected cells with the ALK5 kinase inhibitor SB431542 also prevented Smad3 phosphorylation (Figure 3B). Phosphorylation of Smad1/5 was not blocked by treatment with these neutralizing antibodies, suggesting that the activation of Smad1/5 depends on a separate pathway. Together the results of these experiments show that CMV-infected HUVECs release increasing amounts of TGF-β1 and activate TGF-β1 through an integrin αvβ6-mediated mechanism that stimulates ALK5 signaling and downstream Smad3 phosphorylation.
Induction of Integrin β6 Requires TGF-β/ALK5 Signaling and Viral DNA Replication
We next assessed how integrin β6 was induced on CMV infection in HUVECs. It has been reported that TGF-β1 induces de novo synthesis of integrin β6 in normal human keratinocytes45 and strongly up-regulates its expression in primary cultures of human airway epithelial cells.46 Having found increased secretion of TGF-β1 in infected cells as early as 3 days after infection (Figure 2A), we then investigated the effect of TGF-β1 on induction of integrin β6. As expected, expression of integrin β6 was greatly reduced (by ∼70%) by treatment with the anti-TGF-β neutralizing antibody (Figure 4A). In addition, the ALK5 kinase inhibitor SB431542 (0.1 μmol/L to 1 μmol/L) was able to increasingly block the induction of integrin β6 with increasing inhibitor concentrations and nearly abolish it at high concentrations, whereas the control solution, containing the same concentration of the solvent dimethyl sulfoxide had no effect (Figure 4A). Next, we investigated whether soluble factors participate in the induction of integrin β6. After day 1, conditioned medium from infected cells was collected on alternate days and frozen. HUVECs were cultured with the filtered conditioned medium for 8 days, and expression of integrin β6 was analyzed. No integrin β6 expression was observed in cells cultured with conditioned medium from any time point (data not shown), even though the secretion of TGF-β1, which could be mostly present in an inactive form, from infected cells increased throughout time. We then asked whether viral late gene expression is required for the up-regulation of integrin β6 in infected cells because the expression was observed only at late times after infection. HUVECs were infected and cultured in the presence of the viral polymerase inhibitors Foscarnet (400 μmol/L) or phosphonoacetic acid (100 μg/ml). Both viral polymerase inhibitors blocked induction of integrin β6 (Figure 4B) and strongly suppressed induction of TMLC luciferase activity (Figure 4C). The remaining luciferase activity was further reduced by the addition of an anti-TGF-β antibody, but not by an anti-integrin β6 neutralizing antibody (3G9) (data not shown), indicating that increased luciferase activity was not attributable to integrin αvβ6-mediated TGF-β1 activation. Together, these results indicate that TGF-β1/ALK5 signaling and viral DNA replication are important factors for the induction of integrin β6 in HUVECs.
Figure 4.

Induction of integrin αvβ6 expression requires TGF-β signaling and viral DNA replication A: Infected HUVECs were cultured with or without chicken anti-TGF-β polyclonal antibody (20 μg/ml), chicken IgY isotype control antibody (control Ab, 20 μg/ml), the ALK5 kinase inhibitor SB431542 (0.5 μmol/L), or the vehicle DMSO for 7 days, and surface expression of integrin αvβ6 was analyzed by flow cytometric analysis. Typical histograms are shown. Shaded areas represent expression of specific proteins. Lines represent isotype control. Experiments were repeated at least two times. B: Surface expression of integrin αvβ6 was analyzed by flow cytometric analysis at 7 days after infection with or without viral DNA polymerase inhibitors, Foscarnet, and phosphonoacetic acid (PAA). Typical histograms are shown. Shaded areas represent expression of specific proteins. Lines represent isotype control. Experiments were repeated six times. C: Active TGF-β was not produced by infected HUVECs in the presence of viral DNA polymerase inhibitors. Equal numbers of TMLC TGF-β reporter cells and control (cont.) or infected HUVECs were cultured for 16 to 24 hours at 7 days after infection. Relative luciferase activity in cell lysates was defined as the measured activity divided by TMLC baseline activity. Representative data (mean ± SE) are from four experiments done in triplicate.
CMV-Infected HUVECs Dysregulate ALK1 and ALK5 Protein Levels
Endothelial cells express ALK1, which stimulates Smad1/5 phosphorylation during angiogenesis and counterbalances TGF-β1/ALK5 signaling.47,48 The ALK1 signaling pathway involves an accessory receptor, endoglin, which is highly expressed in endothelial cells, and indirectly inhibits TGF-β1/ALK5 signaling. Preferential phosphorylation of Smad3 in CMV-infected HUVECs suggested that the ratio of ALK1 and ALK5 receptors on the cell surface might be altered. By flow cytometry, we found that uninfected HUVECs expressed ALK1, endoglin, and ALK5 (Figure 5A). Intensities of both ALK1 and ALK5 changed appreciably in infected HUVECs at late time points, with a significant decrease in ALK1 and endoglin expression and a significant increase in ALK5 expression as compared with uninfected cells (Figure 5A, Table 1). Immunoblot analysis revealed the same pattern of changes in expression levels (Figure 5B). Interestingly, the shift in receptor expression occurred even when cells were treated with anti-integrin αvβ6, anti-TGF-β neutralizing antibody, or the ALK5 kinase inhibitor (data not shown), indicating that this change was independent of αvβ6-mediated TGF-β1 activation. Then we investigated the possibility that soluble factors mediate the observed changes in the expression of ALK1, endoglin, and ALK5. After day 1, conditioned medium from infected cells was collected on alternate days and frozen. HUVECs were cultured with the filtered conditioned medium for 8 days, and the surface expression of the receptors was analyzed by flow cytometry. Expression of ALK1 was decreased in cells cultured with conditioned medium from all time points. Expression of ALK5 increased in cells cultured with the conditioned medium from 5, 7, and 9 days after infection. Expression of endoglin was not much affected by conditioned medium from any time point (Figure 5C).
Figure 5.

CMV-infected HUVECs increase expression of ALK5 and reduce ALK1. A: Surface expression of ALK1, endoglin, and ALK5 was analyzed by flow cytometric analysis at 7 days after infection in the absence or presence of viral DNA polymerase inhibitors. Typical histograms from control (cont.) and infected (inf.) HUVECs are shown. Shaded areas represent expression of specific proteins. Lines represent isotype control. Numbers represent mean fluorescence intensity. The experiments were repeated at least four times. B: Cell lysates from control (cont.) or infected (inf.) HUVECs at 3 and 10 days after infection were fractionated by 10% SDS-PAGE and blotted on nitrocellulose. Filters were incubated with antibodies to ALK1, endoglin, ALK5, and Grb2 (loading control). C: Surface expression of ALK1, endoglin, and ALK5 was analyzed by flow cytometric analysis at 7 days of culture with conditioned medium (CM) from infected HUVECs. Relative surface expression as expressed by mean fluorescence intensity was normalized for control HUVECs in the same experiment. Results are the mean (±SE) from three to seven experiments. Asterisks indicate relative expression level of receptors in HUVECs cultured with conditioned medium as compared with controls (*P < 0.05, **P < 0.01).
Table 1.
CMV-Infected Endothelial Cells Increase Expression of ALK5 and Reduce ALK1/Endoglin
| ALK1
|
Endoglin
|
ALK5
|
||||
|---|---|---|---|---|---|---|
| Control | Infected | Control | Infected | Control | Infected | |
| HMVEC-L | 84.6 ± 21.6 | 14.9 ± 13.9* | 2399.0 ± 376.3 | 1190.7 ± 137.8† | 23.7 ± 9.4 | 180.5 ± 83.1* |
| UtMVECs | 516.9 ± 76.1 | 297.0 ± 61.5* | 4640.9 ± 465.0 | 2564.3 ± 523.2† | 216.4 ± 56.3 | 338.9 ± 90.8 |
| HUVECs | 138.8 ± 20.0 | 49.2 ± 9.2‡ | 2479.9 ± 355.2 | 426.8 ± 87.2‡ | 81.8 ± 23.7 | 547.0 ± 107.5‡ |
Surface expression of ALK1, endoglin, and ALK5 was analyzed by flow cytometry late in infection. Numbers represent mean fluorescence intensity (MFI) (mean ± SE) of 3 to 11 experiments. Asterisks and symbols indicate significantly changed MFI in infected cells compared with uninfected control cells
(P < 0.05;
P < 0.01;
P < 0.001). HMVEC-L: ALK1 (n = 3), endoglin (n = 5), ALK5 (n = 4); UtMVECs: ALK1 (n = 3), endoglin (n = 5), ALK5 (n = 6); HUVECs: ALK1 (n = 10), endoglin (n = 5), ALK5 (n = 11).
Finally, viral polymerase inhibitors were able to partially block the change in ALK1 and ALK5 expression (Figure 5A), suggesting that a part of those changes may be mediated through immediate-early or early genes. In contrast, expression of endoglin was not changed by infection while in the presence of a viral DNA polymerase inhibitor (Figure 5A), indicating that viral replication is required for change in endoglin expression. Together, these results indicate that there are both direct effects of viral infection on receptor expression and indirect effects that depend on secreted molecules. Our results confirmed that CMV-infected HUVECs reduce ALK1 and endoglin expression, whereas they increase ALK5 expression. Increased availability of ALK5 for TGF-β1 binding, in conjunction with reduced levels of ALK1 and endoglin in infected HUVECs, could explain preferential Smad3 phosphorylation and possible downstream signaling events.
Integrin αvβ6-Mediated TGF-β Activation Increases ECM Production in CMV-Infected Cell Cultures
TGF-β1 is a potent fibrotic factor responsible for the synthesis of ECM, and profibrotic TGF-β1 responses are induced primarily via ALK5/Smad3 signal transduction in normal fibroblasts.49 TGF-β1 also potently promotes the synthesis and deposition of ECM in endothelial cells.50 In microarray analysis, HUVECs infected with recombinant adenovirus carrying a constitutively active form of ALK5 up-regulate ECM genes, whereas ALK1 either does not exhibit a significant effect or causes down-regulation of these genes.51 Therefore, we investigated whether CMV-activated TGF-β1 could increase ECM production and whether blocking TGF-β1 activation could prevent the effect. Surface expression of type IV collagen, analyzed by flow cytometry, was significantly increased in infected HUVECs at late time points (Figure 6, A and B). Immunoblot analysis also showed an increased production of type IV collagen in infected cells (Figure 6C). To evaluate the effect of inhibition of activation of TGF-β1 on CMV-induced profibrotic response, we treated infected cells with anti-TGF-β (1D11) and anti-αvβ6 (3G9) antibodies for 7 days. The results showed that these neutralizing antibodies prevented CMV-induced elevation of type IV collagen expression and that 40 μg/ml of either antibody almost completely abolished the effect (Figure 6D). Immunoblot analysis revealed that neutralizing antibodies reduced the production of type IV collagen in infected cells and had no effect on uninfected control cells (Figure 6E). Furthermore, the ALK5 kinase inhibitor SB431542 had an inhibitory effect on surface expression of type IV collagen in infected cells in a dose-dependent manner (data not shown). A similar effect was seen in control cells, indicating that the ALK5 kinase inhibitor blocked the basal level of TGF-β more efficiently than blocking antibodies and had a greater effect on inhibition of type IV collagen synthesis. In addition, surface expression of fibronectin was increased at late times after infection, and was reduced by the ALK5 kinase inhibitor (data not shown). Taken together, these results indicate that ECM production is increased by integrin αvβ6-mediated TGF-β1 activation in infected HUVECs.
Figure 6.

Increased type IV collagen synthesis by CMV infection was blocked by anti-TGF-β and anti-αvβ6 neutralizing antibodies. A: Surface expression of type IV collagen was analyzed by flow cytometric analysis at 10 days after infection. Typical histograms from control (cont.) and infected (inf.) HUVECs are shown. Shaded areas represent expression of specific proteins. Lines represent isotype control. B: The results represent the mean fluorescence intensity of type IV collagen (mean ± SE) from three to seven experiments. Asterisks indicate surface expression in infected HUVECs as compared with uninfected controls (*P < 0.01). C: Cell lysates from control (cont.) or infected (inf.) HUVECs at 3, 7, and 10 days after infection were fractionated by 8% SDS-PAGE and blotted on nitrocellulose. Filters were incubated with anti-type IV collagen (Col IV) and anti-actin (loading control) antibodies. D: Surface expression of type IV collagen was analyzed by flow cytometric analysis at 7 days without antibody (untreated) or with anti-TGF-β neutralizing antibody (1D11), function-blocking anti-αvβ6 antibody (3G9), or mouse IgG1 isotype control antibody (control Ab). Relative surface expression as expressed by mean fluorescence intensity was normalized for control HUVECs in the same experiment. Results are the mean (±SE) from three experiments. Treatment with neutralizing antibodies significantly decreased surface expression of type IV collagen compared with infected cells (*P < 0.01). E: Effects of anti-TGF-β antibody and anti-αvβ6 antibody on type IV collagen production. Control and infected HUVECs were cultured without antibody (untreated) or with anti-TGF-β neutralizing antibody (1D11, 40 μg/ml), function-blocking anti-αvβ6 antibody (3G9, 40 μg/ml), or mouse IgG1 isotype control antibody (control Ab, 40 μg/ml) for 7 days. Lysates were fractionated by 8% SDS-PAGE and blotted. Filters were incubated with specific antibodies. Results are representative of at least four independent experiments.
CMV-Infected Microvascular Endothelial Cell Types Induce Integrin αvβ6 and Switch TGF-β Receptor Expression
To determine whether CMV infection altered integrin αvβ6 expression in other endothelial cell types, we analyzed VR1814-infected HMVEC-L and UtMVECs for surface expression of integrin αvβ6 at 10 days after infection and compared it with surface expression in infected HUVECs. Integrin αvβ6 was induced in both microvascular endothelial cell types after infection (Figure 7). Interestingly, integrin αvβ6 was present in uninfected UtMVECs, but the induction level at late times after infection was not different from that of infected HUVECs. In addition, we compared the levels of the repertoire of TGF-β receptors expressed by HMVEC-L and UtMVECs (Table 1). All endothelial cells expressed high levels of ALK1 and endoglin and lower levels of ALK5. After infection, ALK1 and endoglin expression were significantly decreased, and ALK5 was significantly increased, as was observed in infected HUVECs. Interestingly, levels of TGF-β receptor expression on the surface of infected cells differed according to the vascular beds from which the endothelial cells were obtained.
Figure 7.
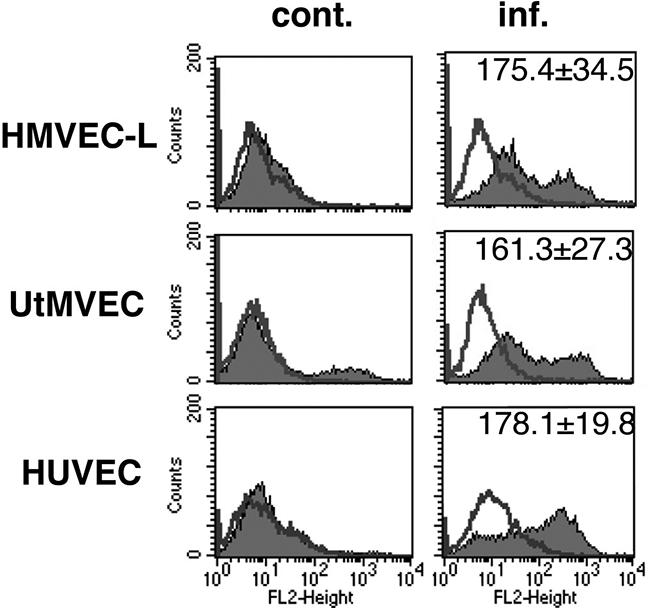
Different CMV-infected endothelial cell types induce different levels of integrin αvβ6. Flow cytometric analysis of integrin αvβ6 in HMVEC-L, UtMVECs, and HUVECs at 10 days after infection with VR1814. Typical histograms from control (cont.) and infected (inf.) cells are shown. Shaded areas represent expression of specific proteins. Lines represent isotype control. Numbers represent mean fluorescence intensity (mean ± SE). The experiments were repeated at least three times.
Up-Regulated Integrin αvβ6 in Blood Vessels of CMV-Infected Organs
Having found that the pathogenic CMV strain VR1814 induces integrin αvβ6, which initiates TGF-β1/ALK5 signaling in infected endothelial cells in vitro, we examined specimens from salivary gland, lung, uterus, and placenta with natural infection to ascertain whether expression occurs in vivo. We performed immunohistochemical analysis on tissues with confirmed histological evidence of cytomegalic cells (ie, sites of viral replication and active infection). In submandibular glands, islands of integrin αvβ6-positive cells were detected among much larger areas of nonexpressing cells (Figure 8, A, C, and D). Expression of integrin αvβ6 was found in infected cytomegalic cells (owl’s eye appearance) (Figure 8, B and C) and was up-regulated in nearby epithelium (Figure 8D). In infected lungs, strong integrin αvβ6 induction was seen in endothelial cells (Figure 8E). However, induction was infrequent (2 of 11 lung samples), and only focal expression of integrin αvβ6 was found. Analysis of serial sections from infected lungs showed a vascular staining pattern for von Willebrand factor (Figure 8F) proximal to infected endothelial cells (Figure 8G) that induced integrin αvβ6 expression (Figure 8H). Interestingly, integrin αvβ6-specific antibodies showed that the protein was present in blood vessels immediately adjacent to CMV-infected cells, but no staining was observed in distal capillaries (Figure 8F).
Figure 8.

CMV-infected tissues induce integrin αvβ6 expression in epithelium and vascular endothelium in vivo. Samples (1 submandibular gland and 11 lung) obtained from 12 patients with CMV infection with histological evidence of nuclear inclusion bodies were evaluated for integrin β6 expression A–D: Integrin αvβ6 immunostaining in CMV-infected cells and gland epithelium in submandibular gland. A and B: Serial sections of infected submandibular gland immunostained with antibodies to integrin αvβ6 (A) and CMV replication proteins in infected cells (B). C and D: Integrin αvβ6 was strongly up-regulated in epithelial cells of submandibular glands proximal to cytomegalic cells (foci of viral replication). E: Integrin αvβ6 immunostaining in vascular endothelium of CMV-infected lung. Expression of integrin αvβ6 in blood vessels was found in two samples. F–H: Serial sections of infected lung immunostained with antibodies to von Willebrand factor (vWF) (F), CMV replication proteins in infected cells (G), and integrin αvβ6 induction (H). Black arrowheads, integrin αvβ6-positive cytomegalic cells; white arrowheads, glandular epithelium; black arrows, integrin αvβ6-positive endothelial cells. BV, blood vessels. Original magnifications: ×20 (A, B); ×40 (C–H).
We reported that CMV replicates at the uterine-placental interface, transmitting virus from infected capillaries to decidual cells and cytotrophoblast progenitor cells of epithelial origin in the adjacent placenta.10,52 In the present study, we found that infected UtMVECs induce integrin αvβ6 expression, suggesting that the same induction could occur in utero. We therefore examined three paired decidual and adjacent placental biopsy specimens naturally infected with CMV in early gestation, and eight placentas from healthy deliveries at term. In the decidua, immunostaining for CMV virion gB revealed areas with infected decidual cells (Figure 9A). Nearby, an infected capillary showed up-regulated integrin αvβ6 expression in an overall diffuse staining pattern (Figure 9A). When glandular epithelia were infected, integrin αvβ6 was induced in proximal blood vessels (Figure 9B). At times, marked expression was found in endothelial cells without evidence of viral proteins in surrounding tissue (Figure 9C). Occasionally, endothelial cells were infected, but capillaries showed little or no integrin αvβ6 staining (Figure 9D). In the placenta, immunostaining revealed clusters of cytotrophoblast progenitor cells with intense membrane expression of integrin αvβ6 in chorionic villi where syncytiotrophoblasts had signs of local damage (Figure 10). For example, we found intense surface membrane staining on cytotrophoblast progenitors underneath syncytial knotting (Figure 10A) and in the vicinity of blood clots adhering to villi in contact with maternal blood (Figure 10B). Occasional cytotrophoblasts contained scattered cytoplasmic vesicles with CMV gB, a pattern suggesting virion uptake in caveolar vesicles without replication.53,54 In contrast, integrin αvβ6 was not expressed by cytotrophoblasts when CMV virion gB accumulated in villus core macrophages, and syncytiotrophoblasts were undamaged (Figure 10C). Similar patterns of expression were seen in the other placenta.
Figure 9.

Integrin αvβ6 induction in blood vessels of CMV-infected decidua in early gestation. A: Immunostaining of integrin αvβ6 expression (green) in infected blood vessel (BV) proximal to infected decidual cells immunostained for CMV glycoprotein B (gB) (red). B: Integrin αvβ6 expression (green) in blood vessel proximal to infected glandular epithelium (red). C: Integrin αvβ6 expressed in blood vessel of the same tissue (decidua 16) in an area without viral proteins. D: Integrin αvβ6-negative BV. Expression of integrin αvβ6 was found in two of three decidual biopsy specimens. Original magnifications, ×400.
Figure 10.

Up-regulated integrin αvβ6 expression in villus cytotrophoblast progenitor cells, epithelial cells of the placenta. A–C: CMV-infected early gestation placenta. D–F: Uninfected placenta at term. Cytotrophoblasts broadly induced integrin αvβ6 (green) proximal to sites of damage, syncytial knotting (A), and adherent blood clots (B), but not in healthy chorionic villi with macrophage (Mφ) uptake of CMV virion gB proteins (C) of the same tissue (placenta 10). Expression of integrin αvβ6 in cytotrophoblasts was found in two of three placental biopsy specimens. D and E: Cytotrophoblasts contiguous with fibrinotic deposits (ECM accumulation) on the villous surface strongly up-regulate integrin αvβ6 (green). F: Integrin αvβ6-negative villus in healthy villus in the same tissue (placenta 24). Similar patterns were found in five of eight term placenta. CTB, cytotrophoblast; STB, syncytiotrophoblast; VC, villus core; BV, blood vessel. Original magnifications, ×400.
Immunostaining of a placenta at term (five of eight) revealed high integrin αvβ6 induction in cytotrophoblast progenitor cells located next to fibrinoids, which are large ECM deposits formed on the surface of chorionic villi in contact with maternal blood (Figure 10, D and E). In areas with undamaged chorionic villi, cytotrophoblasts showed little or no detectable integrin αvβ6 expression (Figure 10F). Together these results confirm and extend our in vitro findings and show that integrin αvβ6 is up-regulated in diverse infected tissues. However, not all endothelial cells adjacent to the infected cells expressed integrin αvβ6, suggesting a requirement for additional cellular factors or a special environment.
Discussion
To our knowledge, the data presented here are the first reported evidence for virus-induced expression of an epithelium-specific integrin, αvβ6, in endothelial cells. Furthermore, although induction of TGF-β1 in endothelial cells by CMV infection has been reported previously, these data provide evidence for a mechanism by which CMV may cause the activation of latent TGF-β1 and downstream signaling that could alter vascular function or otherwise contribute the fibrotic and vascular components of CMV pathology. However, we cannot rule out the possibility of small contributions from other TGF-β1 activation mechanisms, such as those involving metalloproteinases, plasmin, thrombospondin 1, and integrin αvβ8.15,17,18,19,20,21 Furthermore the absolute contribution of αvβ6 to TGF-β1 activation in vivo remains unclear. Nonetheless, our data suggest that integrin αvβ6 is the main activator of TGF-β1 in CMV-infected endothelial cells in vitro (Figure 2E) and further suggest that this is a major pathway for TGF-β1 activation in vivo. The mechanisms underlying integrin αvβ6 induction in infected vasculature are still unclear. Integrin αvβ6 is a TGF-β-inducible integrin expressed at sites of epithelial inflammation and remodeling in diverse organs and binds to the RGD site in TGF-β latency-associated peptide.18,22 This complex is tethered by a disulfide linkage to the latent TGF-β binding protein, resulting in a conformational change in the latent complex and allowing cell-associated active TGF-β to interact with TGF-β receptors on immediately adjacent cells without releasing free active TGF-β.55,56 This indicates that cell-to-cell contact is required for integrin αvβ6-mediated activation of TGF-β. Our immunohistological analysis suggested that integrin αvβ6 in endothelial cells is induced by infection or up-regulated through contact with adjacent infected cells (Figures 8 and 9). Although TGF-β1 induces the de novo synthesis of integrin αvβ6 in normal human keratinocytes,45 TGF-β1 secreted by infected HUVECs (mostly in an inactive form) (Figure 2A) is less likely to play a role in the induction, because conditioned medium from infected cells did not induce expression of integrin αvβ6. Our data indicate that viral DNA replication may be necessary for the de novo synthesis of integrin αvβ6 in endothelial cells, as demonstrated by the lack of induction of integrin αvβ6 when viral late gene expression is blocked (Figure 4B). In addition, TGF-β1/ALK5 signaling plays an important role in the induction of integrin αvβ6, because anti-TGF-β neutralizing antibody and the ALK5 kinase inhibitor strongly suppressed the expression of the integrin (Figure 4A). Once integrin αvβ6 is expressed, it may act to continuously process TGF-β1 into its active from. Active TGF-β1, locally bound to the ECM, could serve as an amplifier for the induction of integrin αvβ6 in adjacent cells by cell-to-cell contact. The activation of TGF-β1 and expression of αvβ6 could then be involved in a positive feedback loop45,46 that amplifies the response within endothelial cells.
However, we cannot exclude the possibility that endothelial cells might normally express trace amounts of integrin αvβ6 that become dramatically up-regulated by some other mechanism. Integrin αvβ6 binds an RGD motif, the most common integrin binding sequence,57 also contained in several CMV proteins. When present in the plasma membrane of infected cells, these proteins could serve as integrin αvβ6 receptors through an RGD motif and activate signaling pathways. CMV protein UL148, and CMV-specific transmembrane proteins UL30 and US2358,59 each contain an RGD motif. It is intriguing that UL148, encoded by pathogenic CMV strains, confers tropism for endothelial cells and leukocytes60,61 and could theoretically mediate functional changes associated with pathogenesis.
Integrin αvβ6 is expressed at low levels on uninjured epithelia of healthy tissues but dramatically increases in response to epithelial injury and wound healing during subclinical, acute, or chronic inflammation.22 In a mouse model of lung injury and edema induced by bleomycin treatment, integrin αvβ6-mediated activation of TGF-β1 induces early lung edema and late lung fibrosis in normal mice, whereas mice with an interruption in the β6 integrin gene show exaggerated inflammation in the lungs and skin but are protected from pulmonary fibrosis and edema.18,23,62 This combination of effects suggests a localized deficiency of active TGF-β1 resulting from loss of β6 integrin.23 Likewise, lack of the β6 integrin gene protects against tubulointerstitial renal fibrosis in mice with unilateral urethral obstruction63 and blockade of integrin αvβ6 protects against renal fibrosis in a murine model of Alport’s syndrome.24 In transgenic mice overexpressing human integrin β6 in the epithelium, chronic ulcers with areas of severe fibrosis develop, and TGF-β1 expression significantly increases in lesions as compared with normal skin.64 These various lines of evidence suggest that integrin-αvβ6-mediated TGF-β activation is of general importance in the development of fibrosis in multiple epithelial organs, and the same mechanism may well contribute to fibrosis and other pathological outcomes in CMV disease.
Episodes of immune rejection, ischemia, and CMV infection are among the risk factors for chronic allograft nephropathy with interstitial inflammation, glomerular lesions, and interstitial fibrosis.13,65 In a rat CMV model of chronic renal allograft rejection, remarkable fibrosis, combined with glomerular and tubular damage, was found within weeks after graft transplantation.66 In human kidney allografts, persistent CMV infection without acute rejection was associated with increased TGF-β1 expression in arterial endothelium and tubular epithelial cells.67 Patients with ongoing CMV infection had significantly increased urinary excretion of TGF-β1 and developed interstitial fibrosis in the kidney 6 months after transplantation.68 Although CMV-infected transplants were not examined in our study, integrin αvβ6-mediated TGF-β1 activation could be central to the pathology associated with viral replication and interstitial fibrosis in diverse tissues.
Another interesting and potentially relevant phenomenon is that vascular intimal thickening is increased in biopsy specimens from kidney allografts with persistent CMV infection.67 Intimal thickening could involve an endothelial-to-mesenchymal transition caused by TGF-β, as observed in cardiac fibrosis in mouse models,69 as well as during embryonic development of the heart.70 In addition, in vitro studies have shown that embryonic endothelial cells changed to an epithelioid phenotype corresponding with the down-regulation of von Willebrand factor-related antigen.71 In the present study, we found that CMV-infected endothelial cells express epithelial integrin αvβ6 in vitro (Figures 1 and 7) and in vivo (Figures 8 and 9), switch expression levels of TGF-β receptors (Figure 5, Table 1), and down-regulate endothelial-specific proteins, including VE-cadherin, von Willebrand factor, and PECAM-1 (T.T. and L.P., unpublished). Taken together, these results suggest that CMV-infected endothelial cells undergo a phenotypic change to a nonendothelial cell type, a transition that could be associated with CMV pathogenesis.
In pregnancies affected by congenital CMV infection, substantial evidence of virus-initiated pathology is provided by inflammation, leukocytic infiltration, edema, and fibrinotic deposits that occlude blood vessels in the villus core.72,73 Except in cases of severe symptomatic CMV disease, evidence of ongoing viral replication in the placenta is seldom detected. Here we determined that integrin αvβ6 is up-regulated in blood vessels in early gestation decidua with focal sites of viral replication and in villus cytotrophoblasts in placentas containing viral DNA (Figures 9 and 10). Remarkably strong induction was observed in cytotrophoblasts near blood clots adhering to damaged chorionic villi and in cells contiguous with fibrinoids composed of fibronectin, laminin, and collagen IV, suggesting that integrin-mediated TGF-β1 activation contributes to pathology in the uterine and fetal compartment. Purified villus cytotrophoblasts isolated from placentas at term that contain CMV DNA, and virion proteins without active replication express integrin αvβ6 that activates TGF-β1.30 Deposition of ECM protein by integrin αvβ6-mediated activation of TGF-β1 (Figure 6), impairment of ECM degradation by down-regulation of matrix metalloproteinase 2 activity by CMV-encoded viral interleukin-10,74 and increased production of the tissue inhibitor of metalloproteinases 1, which is independent of TGF-β1 activation (T.T. and L.P., unpublished), could explain the marked pathology at the uterine-placental interface in congenital infection.
In most cell types, TGF-β1 binds to the ubiquitously expressed ALK5 receptor, which activates Smad2 and Smad3. In endothelial cells, TGF-β1 can also bind to ALK-1 in the presence of functional ALK5, resulting in phosphorylation of both Smad1 and Smad5.75 However, it has been suggested that TGF-β1 may not represent the physiological ligand for the ALK1 receptor, since Goumans and colleagues75 investigated binding of TGF-β1 to ALK1 under specific experimental conditions. We found that phosphorylation of Smad1/5 was present during the course of infection and that the levels of phosphorylation either did not change or decreased at late times after infection (Figure 3A). In addition, phosphorylated Smad1/5 was not blocked by treatment with either anti-TGF-β or anti-αvβ6 neutralizing antibody (Figure 3B). This raises the possibility that another ligand(s) could bind to ALK1 and/or endoglin and activate Smad1/5. Bone morphogenetic protein (BMP)-9 and BMP-10 have been shown to bind to ALK1, inducing phosphorylation of Smad1/5 in human dermal microvascular endothelial cells, and could have an effect on angiogenesis.76 In addition, overexpression of endoglin increases the BMP-9 response.76 Endoglin has been shown to interact with TGF-β1, TGF-β3, activin A, BMP-2, and BMP-7 in the presence of type I or type II receptor.77 Although the function of endoglin in endothelial cells is still unclear, it has been shown to antagonize TGF-β1-induced ALK5/Smad3 signaling and enhance the BMP-7/Smad1/5 pathway.78 Phosphorylation of Smad1/5 could be attributable to a response to BMP(s). It will be interesting to find out whether CMV infection induces BMP/Smad1/5 signaling. If so, it will be important to ask what specific functional role it plays in vivo.
Although our results provide a better understanding of the regulation of TGF-β1 in CMV-infected endothelial cells, much remains to be learned. For one, TGF-β1 activation could occur through other mechanisms not dependent on integrin αvβ6, and these could be important contributors to TGF-β effects in vivo. In addition, analysis of infected tissues suggests that up-regulation of αvβ6 in the endothelium could be limited and require additional cellular factor(s) and/or a special environment, suggested by the patchy distribution of integrin αvβ6 in blood vessels (Figures 8 and 9). Although animal models for the study of human CMV pathogenesis do not exist because of strict species specificity, studies of integrin αvβ6 knockout mice infected with murine CMV may provide clues about the physiological role of integrin αvβ6 induction in infected tissues and vascular beds.
Acknowledgments
We thank Drs. Akihiro Matsuura, Yasuhiro Tsutsui, and Isao Kosugi for biopsy specimens; Drs. Stephen Nishimura, Elizabeth Wayner, Shelia Violette, and Paul Weinreb for providing antibodies; Hsin-Ti Chang for valuable technical assistance; Dr. Dusko Ilic and members of the Pereira and Sheppard laboratories for insightful discussions; and Mary McKenney and Dr. Matthew Petitt for editing the manuscript.
Footnotes
Address reprint requests to Lenore Pereira, University of California San Francisco, 513 Parnassus Ave., San Francisco, CA 94143-0640. E-mail: lenore.pereira@ucsf.edu.
Supported by the National Institutes of Health (grants AI46657 and AI53782 to L.P., HL53949 to D.S., DK74538 to T.S.), the Thrasher Research Fund (no. 02821-7 to L.P.), the University of California San Francisco Academic Senate (to T.T.), and the American Heart Association (to H.K.).
T.T. and H.K. contributed equally to this study.
We dedicate this article to Dr. Hisaaki Kawakatsu who died during this study.
References
- Hahn G, Jores R, Mocarski ES. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc Natl Acad Sci USA. 1998;95:3937–3942. doi: 10.1073/pnas.95.7.3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves MB, MacAry PA, Lehner PJ, Sissons JG, Sinclair JH. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci USA. 2005;102:4140–4145. doi: 10.1073/pnas.0408994102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillespie GM, Wills MR, Appay V, O’Callaghan C, Murphy M, Smith N, Sissons P, Rowland-Jones S, Bell JI, Moss PA. Functional heterogeneity and high frequencies of cytomegalovirus-specific CD8(+) T lymphocytes in healthy seropositive donors. J Virol. 2000;74:8140–8150. doi: 10.1128/jvi.74.17.8140-8150.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn HS, Haney DJ, Ghanekar SA, Stepick-Biek P, Lewis DB, Maecker HT. Dynamics of CD4 and CD8 T cell responses to cytomegalovirus in healthy human donors. J Infect Dis. 2002;186:15–22. doi: 10.1086/341079. [DOI] [PubMed] [Google Scholar]
- Plachter B, Sinzger C, Jahn G. Cell types involved in replication and distribution of human cytomegalovirus. Adv Virus Res. 1996;46:195–261. doi: 10.1016/s0065-3527(08)60073-1. [DOI] [PubMed] [Google Scholar]
- Sinzger C, Grefte A, Plachter B, Gouw AS, The TH, Jahn G. Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J Gen Virol. 1995;76:741–750. doi: 10.1099/0022-1317-76-4-741. [DOI] [PubMed] [Google Scholar]
- Sinzger C, Plachter B, Stenglein S, Jahn G. Immunohistochemical detection of viral antigens in smooth muscle, stromal, and epithelial cells from acute human cytomegalovirus gastritis. J Infect Dis. 1993;167:1427–1432. doi: 10.1093/infdis/167.6.1427. [DOI] [PubMed] [Google Scholar]
- Sinzger C, Müntefering H, Löning T, Stöss H, Plachter B, Jahn G. Cell types infected in human cytomegalovirus placentitis identified by immunohistochemical double staining. Virchows Arch A Pathol Anat Histopathol. 1993;423:249–256. doi: 10.1007/BF01606887. [DOI] [PubMed] [Google Scholar]
- Fisher S, Genbacev O, Maidji E, Pereira L. Human cytomegalovirus infection of placental cytotrophoblasts in vitro and in utero: implications for transmission and pathogenesis. J Virol. 2000;74:6808–6820. doi: 10.1128/jvi.74.15.6808-6820.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira L, Maidji E, McDonagh S, Genbacev O, Fisher S. Human cytomegalovirus transmission from the uterus to the placenta correlates with the presence of pathogenic bacteria and maternal immunity. J Virol. 2003;77:13301–13314. doi: 10.1128/JVI.77.24.13301-13314.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman JA, Rubin RH. Infection in organ-transplant recipients. N Engl J Med. 1998;338:1741–1751. doi: 10.1056/NEJM199806113382407. [DOI] [PubMed] [Google Scholar]
- Kroshus TJ, Kshettry VR, Savik K, John R, Hertz MI, Bolman RM., III Risk factors for the development of bronchiolitis obliterans syndrome after lung transplantation. J Thorac Cardiovasc Surg. 1997;114:195–202. doi: 10.1016/S0022-5223(97)70144-2. [DOI] [PubMed] [Google Scholar]
- Helantera I, Koskinen P, Tornroth T, Loginov R, Gronhagen-Riska C, Lautenschlager I. The impact of cytomegalovirus infections and acute rejection episodes on the development of vascular changes in 6-month protocol biopsy specimens of cadaveric kidney allograft recipients. Transplantation. 2003;75:1858–1864. doi: 10.1097/01.TP.0000064709.20841.E1. [DOI] [PubMed] [Google Scholar]
- Potena L, Holweg CT, Chin C, Luikart H, Weisshaar D, Narasimhan B, Fearon WF, Lewis DB, Cooke JP, Mocarski ES, Valantine HA. Acute rejection and cardiac allograft vascular disease is reduced by suppression of subclinical cytomegalovirus infection. Transplantation. 2006;82:398–405. doi: 10.1097/01.tp.0000229039.87735.76. [DOI] [PubMed] [Google Scholar]
- Lebrin F, Deckers M, Bertolino P, Ten Dijke P. TGF-beta receptor function in the endothelium. Cardiovasc Res. 2005;65:599–608. doi: 10.1016/j.cardiores.2004.10.036. [DOI] [PubMed] [Google Scholar]
- Chen YG, Massague J. Smad1 recognition and activation by the ALK1 group of transforming growth factor-beta family receptors. J Biol Chem. 1999;274:3672–3677. doi: 10.1074/jbc.274.6.3672. [DOI] [PubMed] [Google Scholar]
- Rifkin DB. Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. J Biol Chem. 2005;280:7409–7412. doi: 10.1074/jbc.R400029200. [DOI] [PubMed] [Google Scholar]
- Munger JS, Huang X, Kawakatsu H, Griffiths MJ, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D. The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell. 1999;96:319–328. doi: 10.1016/s0092-8674(00)80545-0. [DOI] [PubMed] [Google Scholar]
- Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. The integrin alpha(v) beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- Breuss JM, Gallo J, DeLisser HM, Klimanskaya IV, Folkesson HG, Pittet JF, Nishimura SL, Aldape K, Landers DV, Carpenter W. Expression of the beta 6 integrin subunit in development, neoplasia and tissue repair suggests a role in epithelial remodeling. J Cell Sci. 1995;108:2241–2251. doi: 10.1242/jcs.108.6.2241. [DOI] [PubMed] [Google Scholar]
- Huang XZ, Wu JF, Cass D, Erle DJ, Corry D, Young SG, Farese RV, Jr, Sheppard D. Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin. J Cell Biol. 1996;133:921–928. doi: 10.1083/jcb.133.4.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahm K, Lukashev ME, Luo Y, Yang WJ, Dolinski BM, Weinreb PH, Simon KJ, Chun Wang L, Leone DR, Lobb RR, McCrann DJ, Allaire NE, Horan GS, Fogo A, Kalluri R, Shield CF, III, Sheppard D, Gardner HA, Violette SM. Alphav beta6 integrin regulates renal fibrosis and inflammation in Alport mouse. Am J Pathol. 2007;170:110–125. doi: 10.2353/ajpath.2007.060158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haagmans BL, Teerds KJ, van den Eijnden-van Raaij AJ, Horzinek MC, Schijns VE. Transforming growth factor beta production during rat cytomegalovirus infection. J Gen Virol. 1997;78:205–213. doi: 10.1099/0022-1317-78-1-205. [DOI] [PubMed] [Google Scholar]
- Kossmann T, Morganti-Kossmann MC, Orenstein JM, Britt WJ, Wahl SM, Smith PD. Cytomegalovirus production by infected astrocytes correlates with transforming growth factor-beta release. J Infect Dis. 2003;187:534–541. doi: 10.1086/373995. [DOI] [PubMed] [Google Scholar]
- Helanterä I, Loginov R, Lautenschlager I, Helantera I, Koskinen P, Tornroth T, Gronhagen-Riska C. The role of cytomegalovirus infection in chronic allograft nephropathy. Transplantation. 2005;79:379. doi: 10.1097/01.tp.0000141359.21175.29. [DOI] [PubMed] [Google Scholar]
- Michelson S, Alcami J, Kim SJ, Danielpour D, Bachelerie F, Picard L, Bessia C, Paya C, Virelizier JL. Human cytomegalovirus infection induces transcription and secretion of transforming growth factor beta 1. J Virol. 1994;68:5730–5737. doi: 10.1128/jvi.68.9.5730-5737.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo YD, Chiou CJ, Choi KS, Yi Y, Michelson S, Kim S, Hayward GS, Kim SJ. The IE2 regulatory protein of human cytomegalovirus induces expression of the human transforming growth factor beta1 gene through an Egr-1 binding site. J Virol. 1996;70:7062–7070. doi: 10.1128/jvi.70.10.7062-7070.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata T, McDonagh S, Kawakatsu H, Pereira L. Cytotrophoblasts infected with a pathogenic human cytomegalovirus strain dysregulate cell-matrix and cell-cell adhesion molecules: a quantitative analysis. Placenta. 2007;28:527–537. doi: 10.1016/j.placenta.2006.05.006. [DOI] [PubMed] [Google Scholar]
- Revello GM, Baldanti F, Percivalle E, Sarasini A, De-Giuli L, Genini E, Lilleri D, Labo N, Gerna G. In vitro selection of human cytomegalovirus variants unable to transfer virus and virus products from infected cells to polymorphonuclear leukocytes and to grow in endothelial cells. J Gen Virol. 2001;82:1429–1438. doi: 10.1099/0022-1317-82-6-1429. [DOI] [PubMed] [Google Scholar]
- Neurohr C, Nishimura SL, Sheppard D. Activation of transforming growth factor-beta by the integrin alphavbeta8 delays epithelial wound closure. Am J Respir Cell Mol Biol. 2006;35:252–259. doi: 10.1165/rcmb.2006-0013OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinacker A, Chen A, Agrez M, Cone RI, Nishimura S, Wayner E, Pytela R, Sheppard D. Role of the integrin alpha v beta 6 in cell attachment to fibronectin. Heterologous expression of intact and secreted forms of the receptor. J Biol Chem. 1994;269:6940–6948. [PubMed] [Google Scholar]
- Huang X, Wu J, Spong S, Sheppard D. The integrin alphavbeta6 is critical for keratinocyte migration on both its known ligand, fibronectin, and on vitronectin. J Cell Sci. 1998;111:2189–2195. doi: 10.1242/jcs.111.15.2189. [DOI] [PubMed] [Google Scholar]
- Huang X, Wu J, Zhu W, Pytela R, Sheppard D. Expression of the human integrin beta6 subunit in alveolar type II cells and bronchiolar epithelial cells reverses lung inflammation in beta6 knockout mice. Am J Respir Cell Mol Biol. 1998;19:636–642. doi: 10.1165/ajrcmb.19.4.3293. [DOI] [PubMed] [Google Scholar]
- Dittel BN, McCarthy JB, Wayner EA, LeBien TW. Regulation of human B-cell precursor adhesion to bone marrow stromal cells by cytokines that exert opposing effects on the expression of vascular cell adhesion molecule-1 (VCAM-1). Blood. 1993;81:2272–2282. [PubMed] [Google Scholar]
- Weinreb PH, Simon KJ, Rayhorn P, Yang WJ, Leone DR, Dolinski BM, Pearse BR, Yokota Y, Kawakatsu H, Atakilit A, Sheppard D, Violette SM. Function-blocking integrin alphavbeta6 monoclonal antibodies: distinct ligand-mimetic and nonligand-mimetic classes. J Biol Chem. 2004;279:17875–17887. doi: 10.1074/jbc.M312103200. [DOI] [PubMed] [Google Scholar]
- Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct. Anal Biochem. 1994;216:276–284. doi: 10.1006/abio.1994.1042. [DOI] [PubMed] [Google Scholar]
- McDonagh S, Maidji E, Chang H-T, Pereira L. Patterns of human cytomegalovirus infection in term placentas: a preliminary analysis. J Clin Virol. 2006;35:210–215. doi: 10.1016/j.jcv.2005.08.011. [DOI] [PubMed] [Google Scholar]
- Munger JS, Harpel JG, Giancotti FG, Rifkin DB. Interactions between growth factors and integrins: latent forms of transforming growth factor-beta are ligands for the integrin alphavbeta1. Mol Biol Cell. 1998;9:2627–2638. doi: 10.1091/mbc.9.9.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Munger JS, Steadele M, Busald C, Tellier M, Schnapp LM. Integrin alpha8beta1 mediates adhesion to LAP-TGFbeta1. J Cell Sci. 2002;115:4641–4648. doi: 10.1242/jcs.00145. [DOI] [PubMed] [Google Scholar]
- Ludbrook SB, Barry ST, Delves CJ, Horgan CM. The integrin alphavbeta3 is a receptor for the latency-associated peptides of transforming growth factors beta1 and beta3. Biochem J. 2003;369:311–318. doi: 10.1042/BJ20020809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano Y, Ihn H, Yamane K, Jinnin M, Mimura Y, Tamaki K. Involvement of alphavbeta5 integrin-mediated activation of latent transforming growth factor beta1 in autocrine transforming growth factor beta signaling in systemic sclerosis fibroblasts. Arthritis Rheum. 2005;52:2897–2905. doi: 10.1002/art.21246. [DOI] [PubMed] [Google Scholar]
- ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Zambruno G, Marchisio PC, Marconi A, Vaschieri C, Melchiori A, Giannetti A, De Luca M. Transforming growth factor-beta 1 modulates beta 1 and beta 5 integrin receptors and induces the de novo expression of the alpha v beta 6 heterodimer in normal human keratinocytes: implications for wound healing. J Cell Biol. 1995;129:853–865. doi: 10.1083/jcb.129.3.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Yokosaki Y, Ferrando R, Balmes J, Sheppard D. Differential regulation of airway epithelial integrins by growth factors. Am J Respir Cell Mol Biol. 1996;15:664–672. doi: 10.1165/ajrcmb.15.5.8918373. [DOI] [PubMed] [Google Scholar]
- Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S, Li E. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci USA. 2000;97:2626–2631. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M, Mummery C, Arthur HM, ten Dijke P. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal transduction. EMBO J. 2004;23:4018–4028. doi: 10.1038/sj.emboj.7600386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida W, Mori Y, Lakos G, Sun L, Shan F, Bowes S, Josiah S, Lee WC, Singh J, Ling LE, Varga J. Intracellular TGF-beta receptor blockade abrogates Smad-dependent fibroblast activation in vitro and in vivo. J Invest Dermatol. 2006;126:1733–1744. doi: 10.1038/sj.jid.5700303. [DOI] [PubMed] [Google Scholar]
- Pepper MS. Transforming growth factor-beta: vasculogenesis, angiogenesis, and vessel wall integrity. Cytokine Growth Factor Rev. 1997;8:21–43. doi: 10.1016/s1359-6101(96)00048-2. [DOI] [PubMed] [Google Scholar]
- Ota T, Fujii M, Sugizaki T, Ishii M, Miyazawa K, Aburatani H, Miyazono K. Targets of transcriptional regulation by two distinct type I receptors for transforming growth factor-beta in human umbilical vein endothelial cells. J Cell Physiol. 2002;193:299–318. doi: 10.1002/jcp.10170. [DOI] [PubMed] [Google Scholar]
- McDonagh S, Maidji E, Ma W, Chang HT, Fisher S, Pereira L. Viral and bacterial pathogens at the maternal-fetal interface. J Infect Dis. 2004;190:826–834. doi: 10.1086/422330. [DOI] [PubMed] [Google Scholar]
- Maidji E, McDonagh S, Genbacev O, Tabata T, Pereira L. Maternal antibodies enhance or prevent cytomegalovirus infection in the placenta by neonatal fc receptor-mediated transcytosis. Am J Pathol. 2006;168:1210–1226. doi: 10.2353/ajpath.2006.050482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maidji E, Genbacev O, Chang HT, Pereira L. Developmental regulation of human cytomegalovirus receptors in cytotrophoblasts correlates with distinct replication sites in the placenta. J Virol. 2007;81:4701–4712. doi: 10.1128/JVI.02748-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annes JP, Chen Y, Munger JS, Rifkin DB. Integrin alphaVbeta6-mediated activation of latent TGF-beta requires the latent TGF-beta binding protein-1. J Cell Biol. 2004;165:723–734. doi: 10.1083/jcb.200312172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keski-Oja J, Koli K, von Melchner H. TGF-beta activation by traction? Trends Cell Biol. 2004;14:657–659. doi: 10.1016/j.tcb.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Ruoslahti E, Pierschbacher MD. New perspectives in cell adhesion: RGD and integrins. Science. 1987;238:491–497. doi: 10.1126/science.2821619. [DOI] [PubMed] [Google Scholar]
- Chambers J, Angulo A, Amaratunga D, Guo H, Jiang Y, Wan JS, Bittner A, Frueh K, Jackson MR, Peterson PA, Erlander MG, Ghazal P. DNA microarrays of the complex human cytomegalovirus genome: profiling kinetic class with drug sensitivity of viral gene expression. J Virol. 1999;73:5757–5766. doi: 10.1128/jvi.73.7.5757-5766.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigoutsos I, Novotny J, Huynh T, Chin-Bow ST, Parida L, Platt D, Coleman D, Shenk T. In silico pattern-based analysis of the human cytomegalovirus genome. J Virol. 2003;77:4326–4344. doi: 10.1128/JVI.77.7.4326-4344.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerna G, Percivalle E, Baldanti F, Revello MG. Lack of transmission to polymorphonuclear leukocytes and human umbilical vein endothelial cells as a marker of attenuation of human cytomegalovirus. J Med Virol. 2002;66:335–339. doi: 10.1002/jmv.2150. [DOI] [PubMed] [Google Scholar]
- Hahn G, Revello MG, Patrone M, Percivalle E, Campanini G, Sarasini A, Wagner M, Gallina A, Milanesi G, Koszinowski U, Baldanti F, Gerna G. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J Virol. 2004;78:10023–10033. doi: 10.1128/JVI.78.18.10023-10033.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittet JF, Griffiths MJ, Geiser T, Kaminski N, Dalton SL, Huang X, Brown LA, Gotwals PJ, Koteliansky VE, Matthay MA, Sheppard D. TGF-beta is a critical mediator of acute lung injury. J Clin Invest. 2001;107:1537–1544. doi: 10.1172/JCI11963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma LJ, Yang H, Gaspert A, Carlesso G, Barty MM, Davidson JM, Sheppard D, Fogo AB. Transforming growth factor-beta-dependent and -independent pathways of induction of tubulointerstitial fibrosis in beta6(−/−) mice. Am J Pathol. 2003;163:1261–1273. doi: 10.1016/s0002-9440(10)63486-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häkkinen L, Koivisto L, Gardner H, Saarialho-Kere U, Carroll JM, Lakso M, Rauvala H, Laato M, Heino J, Larjava H. Increased expression of beta6-integrin in skin leads to spontaneous development of chronic wounds. Am J Pathol. 2004;164:229–242. doi: 10.1016/s0002-9440(10)63113-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul LC. Chronic allograft nephropathy: an update. Kidney Int. 1999;56:783–793. doi: 10.1046/j.1523-1755.1999.00611.x. [DOI] [PubMed] [Google Scholar]
- Lautenschlager I, Soots A, Krogerus L, Kauppinen H, Saarinen O, Bruggeman C, Ahonen J. Effect of cytomegalovirus on an experimental model of chronic renal allograft rejection under triple-drug treatment in the rat. Transplantation. 1997;64:391–398. doi: 10.1097/00007890-199708150-00003. [DOI] [PubMed] [Google Scholar]
- Helantera I, Loginov R, Koskinen P, Tornroth T, Gronhagen-Riska C, Lautenschlager I. Persistent cytomegalovirus infection is associated with increased expression of TGF-{beta}1. PDGF-AA and ICAM-1 and arterial intimal thickening in kidney allografts. Nephrol Dial Transplant. 2005;20:790–796. doi: 10.1093/ndt/gfh714. [DOI] [PubMed] [Google Scholar]
- Helanterä I, Teppo AM, Koskinen P, Tornroth T, Gronhagen-Riska C, Lautenschlager I. Increased urinary excretion of transforming growth factor-beta(1) in renal transplant recipients during cytomegalovirus infection. Transpl Immunol. 2006;15:217–221. doi: 10.1016/j.trim.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Zeisberg EM, Tarnavski O, Zeisberg M, Dorfman AL, McMullen JR, Gustafsson E, Chandraker A, Yuan X, Pu WT, Roberts AB, Neilson EG, Sayegh MH, Izumo S, Kalluri R. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat Med. 2007;13:952–961. doi: 10.1038/nm1613. [DOI] [PubMed] [Google Scholar]
- Nakajima Y, Yamagishi T, Hokari S, Nakamura H. Mechanisms involved in valvuloseptal endocardial cushion formation in early cardiogenesis: roles of transforming growth factor (TGF)-beta and bone morphogenetic protein (BMP). Anat Rec. 2000;258:119–127. doi: 10.1002/(SICI)1097-0185(20000201)258:2<119::AID-AR1>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Arciniegas E, Parada D, Graterol A. Mechanically altered embryonic chicken endothelial cells change their phenotype to an epithelioid phenotype. Anat Rec A Discov Mol Cell Evol Biol. 2003;270:67–81. doi: 10.1002/ar.a.10177. [DOI] [PubMed] [Google Scholar]
- Garcia AG, Fonseca EF, Marques RL, Lobato YY. Placental morphology in cytomegalovirus infection. Placenta. 1989;10:1–18. doi: 10.1016/0143-4004(89)90002-7. [DOI] [PubMed] [Google Scholar]
- Benirschke K, Kaufmann P. New York: Springer,; Pathology of the Human Placenta. 2000 [Google Scholar]
- Yamamoto-Tabata T, McDonagh S, Chang H-T, Fisher S, Pereira L. Human cytomegalovirus interleukin-10 downregulates matrix metalloproteinase activity and impairs endothelial cell migration and placental cytotrophoblast invasiveness in vitro. J Virol. 2004;78:2831–2840. doi: 10.1128/JVI.78.6.2831-2840.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goumans MJ, Valdimarsdottir G, Itoh S, Lebrin F, Larsson J, Mummery C, Karlsson S, ten Dijke P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol Cell. 2003;12:817–828. doi: 10.1016/s1097-2765(03)00386-1. [DOI] [PubMed] [Google Scholar]
- David L, Mallet C, Mazerbourg S, Feige JJ, Bailly S. Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood. 2007;109:1953–1961. doi: 10.1182/blood-2006-07-034124. [DOI] [PubMed] [Google Scholar]
- Barbara NP, Wrana JL, Letarte M. Endoglin is an accessory protein that interacts with the signaling receptor complex of multiple members of the transforming growth factor-beta superfamily. J Biol Chem. 1999;274:584–594. doi: 10.1074/jbc.274.2.584. [DOI] [PubMed] [Google Scholar]
- Scherner O, Meurer SK, Tihaa L, Gressner AM, Weiskirchen R. Endoglin differentially modulates antagonistic transforming growth factor-beta1 and BMP-7 signaling. J Biol Chem. 2007;282:13934–13943. doi: 10.1074/jbc.M611062200. [DOI] [PubMed] [Google Scholar]