

Abstract
Background
There have been increasing concerns regarding the safety and efficacy of neuroleptics in people with dementia, but there are very few long-term trials to inform clinical practice. The aim of this study was to determine the impact of long-term treatment with neuroleptic agents upon global cognitive decline and neuropsychiatric symptoms in patients with Alzheimer disease.
Methods and Findings
Design: Randomised, blinded, placebo-controlled parallel two-group treatment discontinuation trial.
Setting: Oxfordshire, Newcastle and Gateshead, London and Edinburgh, United Kingdom.
Participants: Patients currently prescribed the neuroleptics thioridazine, chlorpromazine, haloperidol trifluoperazine or risperidone for behavioural or psychiatric disturbance in dementia for at least 3 mo.
Interventions: Continue neuroleptic treatment for 12 mo or switch to an identical placebo.
Outcome measures: Primary outcome was total Severe Impairment Battery (SIB) score. Neuropsychiatric symptoms were evaluated with the Neuropsychiatric Inventory (NPI).
Results: 165 patients were randomised (83 to continue treatment and 82 to placebo, i.e., discontinue treatment), of whom 128 (78%) commenced treatment (64 continue/64 placebo). Of those, 26 were lost to follow-up (13 per arm), resulting in 51 patients per arm analysed for the primary outcome. There was no significant difference between the continue treatment and placebo groups in the estimated mean change in SIB scores between baseline and 6 mo; estimated mean difference in deterioration (favouring placebo) −0.4 (95% confidence interval [CI] −6.4 to 5.5), adjusted for baseline value (p = 0.9). For neuropsychiatric symptoms, there was no significant difference between the continue treatment and placebo groups (n = 56 and 53, respectively) in the estimated mean change in NPI scores between baseline and 6 mo; estimated mean difference in deterioration (favouring continue treatment) −2.4 (95% CI −8.2 to 3.5), adjusted for baseline value (p = 0.4). Both results became more pronounced at 12 mo. There was some evidence to suggest that those patients with initial NPI ≥ 15 benefited on neuropsychiatric symptoms from continuing treatment.
Conclusions
For most patients with AD, withdrawal of neuroleptics had no overall detrimental effect on functional and cognitive status. Neuroleptics may have some value in the maintenance treatment of more severe neuropsychiatric symptoms, but this benefit must be weighed against the side effects of therapy.
Trial registration: Cochrane Central Registry of Controlled Trials/National Research Register (#ISRCTN33368770).
In a randomized trial of patients with dementia, Clive Ballard and colleagues show that withdrawal of neuroleptics had no overall detrimental effect, and by some measures improved, functional and cognitive status.
Editors' Summary
Background
The number of people with dementia (currently 25 million worldwide) is expected to increase by 5 million each year. The risk of dementia, including Alzheimer disease, increases sharply with age: Alzheimer's Disease International estimates that 1.4% of people 65–69 have dementia, whereas almost a full quarter of those over the age of 85 years are affected. Almost all older dementia patients will experience, along with the cognitive and functional decline typical of the illness, some neuropsychiatric symptoms. These symptoms can include agitation, aggression, and psychosis, and are often devastating for the older patient and his or her family and caregiver. Managing these symptoms is often a prime concern for health-care providers and families. Neuroleptics (sometimes called antipsychotics) are the class of drugs often used to manage or control neuropsychiatric problems, but there have been questions about their safety and appropriateness. Safety concerns involve risk of stroke, parkinsonism, sedation, edema, and chest infections but also include a worsening of cognitive decline with prolonged use of neuroleptics.
Why Was the Study Done?
Previous studies on the effectiveness and safety of neuroleptics in older people have been short term. Ballard and colleagues wanted to study over a longer period of time the impact of neuroleptic drugs on elderly patients with dementia. Specifically, they wanted to know if being on a neuroleptic was associated with more cognitive decline than coming off the drug. They also wanted to investigate whether discontinuing the drug exacerbated any neuropsychiatric symptoms, Parkinson disease-like symptoms, or other functional, language, and cognition difficulties frequently associated with dementia.
What Did the Researchers Do and Find?
The researchers recruited older patients with Alzheimer disease from across England who had been on neuroleptics for at least three months. They randomised patients to one of two groups: the first group continued taking the same neuroleptic at the same dosage level while the second group was switched to an identical-looking placebo. The researchers assessed the patients' cognitive status and neuropsychiatric symptoms upon their entry into the study. Six and 12 months later the researchers assessed any cognitive decline and the level of neuropsychiatric and other problems that patients were experiencing.
At both 6 and 12 months, the researchers found that there were no differences between the two groups (continued treatment and placebo) in terms of cognitive decline. The placebo group may have had less cognitive decline, but this was not statistically significant. They also found no overall differences between the two groups in the change in the number of neuropsychiatric symptoms over these time periods. Patients with severe neuropsychiatric problems at the outset of the trial did better on continued neuroleptic therapy, but this advantage was not statistically significant. There was a significant decline on the verbal fluency language tests among the patients who continued on their neuroleptic.
What Do these Findings Mean?
The researchers report perhaps the first trial of this duration on continued versus withdrawn neuroleptic treatment among older dementia patients. The findings do not indicate any benefit of continuing neuroleptic therapies in older patients on either cognitive or neuropsychiatric outcomes. The researchers conclude that neuroleptics, with their known safety issues, should not be used as first-line treatment to manage problems such as agitation or aggression. For older dementia patients whose neuropsychiatric symptoms are not remedied by nonpharmaceutical treatments, the researchers advise caution. More studies are urgently needed to find better solutions to help older patients with dementia who have agitation, aggression, and psychosis.
Additional Information
Please access these Web sites via the online version of this summary at http://dx.doi.org/10.1371/journal.pmed.0050076.
Alzheimer's Disease International is an umbrella organisation of organisations worldwide
The Alzheimer's Research Trust in the UK is a charity funding research to cure or prevent dementias
The US National Institutes of Aging has information on Alzheimer Disease in English and Spanish
Two governmental regulatory agencies—the Medicines and Healthcare Products Regulatory Agency in the UK and the Food and Drug Administration in the US—offer information about antipsychotics in people with dementia
Introduction
Worldwide, there are 25 million people with dementia [1], the majority of whom have Alzheimer disease (AD). It is a devastating illness that results in a progressive decline in cognitive ability and functional capacity, causes immense distress to patients, their carers, and families, and has an enormous societal impact. Currently the most frequent treatment issue for people with AD presenting to clinical services remains the management of neuropsychiatric symptoms, such as aggression, agitation, and psychosis. Over 90% of people with dementia develop these symptoms at some point during their illness [2]. The symptoms are frequently distressing for the patients who experience them [3] and problematic for their caregivers [4], in whom they are associated with clinically significant depression [5]. In addition, they are often the precipitant for institutional care [6].
Neuroleptics are widely used as the first-line pharmacological approach to treat these neuropsychiatric symptoms. Efficacy has been examined in eight randomized, placebo-controlled trials with typical neuroleptics [7,8] and 18 placebo-controlled trials with atypical neuroleptics [9–11]. The strongest evidence of efficacy is for risperidone, for which there are five published trials indicating a modest but significant diminution in aggression compared to placebo, but limited evidence of benefit for other neuropsychiatric symptoms [9–11]. However, given that in the US and Europe, up to 60% of people with dementia residing in care facilities are prescribed neuroleptics (e.g., [12,13]) for median periods of greater than a year [14,15], the pivotal question is whether longer-term therapy with atypical neuroleptics confers any treatment benefit. There are only two placebo-controlled trials of a neuroleptic for more than 14 wk, and neither showed significant efficacy of neuroleptic treatment for neuropsychiatric symptoms [16,17]. Similarly, longitudinal cohort studies [18] and placebo-controlled neuroleptic withdrawal studies [14,15,19] do not indicate benefit from neuroleptic therapy. However, all of the withdrawal studies continued for 3 mo or less, leaving some uncertainty regarding long-term symptom outcome. In addition, the largest study did suggest a benefit for treatment with atypical neuroleptics in people with scores greater than 14 on the Neuropsychiatric Inventory (NPI) [15].
Any beneficial effects of neuroleptics in people with AD must be weighed against the short- and long-term adverse effects which, according to meta-analyses, include parkinsonism, sedation, oedema, chest infections, stroke (odds ratio 2.5–3) and mortality (odds ratio 1.5–1.7) [7–11,20–22]. Additional evidence has also highlighted accelerated cognitive decline as an important potential negative consequence of prolonged use of neuroleptics [16,23]. A meta-analysis [17] has confirmed this observation, indicating 0.7 of a Mini Mental State Examination (MMSE) point (95% confidence interval [CI] 0.38 to 1.09) greater decline over 6–12 wk in neuroleptic-treated patients compared to those treated with placebo, which appears modest but represents a doubling in the expected rate of cognitive deterioration over this period.
In the US the Food and Drug Administration [22] has warned about the risk of increased mortality and stroke with neuroleptics in people with dementia, and most practice guidelines recommend nonpharmacological approaches as the first-line treatment for agitation and other neuropsychiatric symptoms (e.g., [24]). However, there is still considerable debate as to the place of neuroleptics in the management of severe and distressing symptoms that are intractable to other treatment approaches, especially when there is potential risk to the patient or to others.
The main aim of the trial was to determine whether ongoing treatment with neuroleptics accelerates cognitive decline in people with AD. We additionally sought to determine whether ongoing treatment with neuroleptics confers any benefit for the long-term maintenance treatment of neuropsychiatric symptoms in people with AD.
Methods
Participants
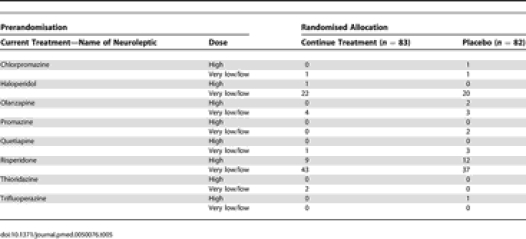
Participants were patients in Oxfordshire, South Birmingham, Newcastle and Gateshead, London and Edinburgh prescribed the neuroleptics thioridazine, chlorpromazine, haloperidol, trifluoperazine or risperidone for behavioural or psychiatric disturbance in dementia for at least 3 months.
Participants had to meet all inclusion criteria. The inclusion criteria were (a) patient lived in a nursing or residential home; (b) patient fulfilled the NINCDS/ADRDA criteria for possible or probable AD [25]; (c) patient had either a MMSE [26] score > 6 or a Severe Battery Impairment [27] score > 30; and (d) patient was taking at least 10 mg chlorpromazine equivalents (CPZe) of a typical neuroleptic or at least 0.5 mg daily of risperidone.
The exclusion criteria were (a) patient was unable to complete primary outcome measures at baseline assessment; (b) clinician responsible for care or study clinician considered that the patient suffered from any physical condition—including marked extrapyramidal disorder—that would have made participation in the trial distressing or likely to increase suffering; (c) patient was currently taking thioridazine and showing a prolonged QTc on electrocardiogram [28,29]; (d) the patient was likely to be unable to take capsules.
Ethics
Two caregivers of people with Alzheimer's disease were closely involved in the development of the protocol, which was peer reviewed through the auspices of the Alzheimer's Research Trust (Cambridge, UK). As this was a multicentre trial, the study was in the first instance reviewed and approved by a properly constituted Multi-Centre Research Ethics Committee (the North of England MREC). Subsequent site-specific approval was then granted by properly constituted Local Research Ethics Committees at each of the participating centres. All Ethics Committees were conducted under the auspices of the Central Organization of Research Ethics Committees (COREC, now the National Research Ethics Service UK, http://www.nres.npsa.nhs.uk/).
Consent
Potentially suitable individuals residing in care facilities, their next of kin, and staff within the care facilities were provided with comprehensive information about the study. Those wishing to take part were invited to participate. If the potential participant had adequate capacity, the individual him- or herself was asked to complete the written study consent procedures. In these circumstances, the next of kin (or if no family members were in contact, an appropriate guardian, usually the manager of the care facility) was also asked to provide written assent to participation. If participants did not have adequate capacity, then written assent from the next of kin and agreement as far as could be ascertained from the potential participant were obtained and considered appropriate to enable participation. This procedure was fully approved by the MREC, and was standard procedure in clinical trials involving vulnerable adults in the UK, until subsequent legislative changes (Mental Capacity Act 2005), introduced after the current study, enabled consent to be provided by a caregiver.
Interventions
Participants were randomised in equal numbers either to continue neuroleptic treatment for 12 mo or to switch to placebo. Three fixed dosages, named respectively (a) very low; (b) low; and (c) high, were chosen for each of the permitted neuroleptic drugs to correspond as near as possible to the dose the patient was being prescribed prior to trial entry (Table 1).
Table 1.
Fixed Dosage Regimens for the Respective Neuroleptics
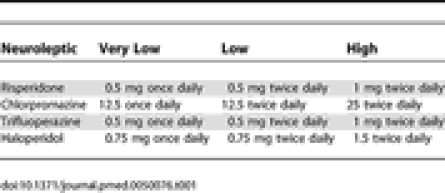
Each of the neuroleptics was overencapsulated to conceal the identity of the contents. Placebo capsules were identical to the overencapsulated neuroleptics, but contained only inert filler. The respective treatments were maintained at the same fixed dose throughout the 12 mo treatment period of the trial.
Objectives
The primary aim of this study was to determine whether treatment with neuroleptic agents is associated with an accelerated rate of cognitive decline in dementia. Secondary objectives were: (a) to examine the impact of neuroleptics on function and other cognitive outcomes; (b) to determine whether discontinuing neuroleptics was associated with an exacerbation of neuropsychiatric symptoms, both overall and in people with NPI scores above and below 14 [15]; (c) to examine the impact on parkinsonism; and (d) to determine the impact on global clinician rated outcome.
Outcomes
Primary outcomes.
The primary outcome was the total SIB score [27] (change from baseline to 6 mo). This is a well-validated instrument designed to evaluate global cognitive functioning in individuals who are too impaired to complete standard neuropsychological tests. There are 40 questions in the SIB, assessing social interaction, memory, orientation, language, attention, praxis, visuospatial ability, construction, and orienting to name. A total score is obtained by summing all questions and ranged from 0 to 100. A higher score indicates higher cognitive ability.
Secondary outcomes.
1. Standardised Mini Mental State Examination (SMMSE) [26]: A widely used instrument for assessing cognitive mental status. It assesses orientation, attention, immediate and short-term recall, language, and the ability to follow simple verbal and written commands. The maximum total score is 30. A higher total score indicates higher cognitive function. A standardized approach to the administration has been published and was adopted in the current study [26].
2. FAS test of Verbal Fluency [30]: A verbal fluency test for which the total score is presented as the sum of all acceptable words generated.
3. Bristol Activities of Daily Living Scale (BADLS) [31]: A 20-item questionnaire to measure daily living abilities, specifically in patients with dementia. The maximum score is 60.
4. Sheffield Test for Acquired Language Disorders (STALD) [32]: Developed as a nonspecialist clinical aid to help identify dysphasia. The test assesses receptive and expressive skills. It gives a total score ranging from 0 to 26.
5. NPI [33]: A caregiver-administered questionnaire that assesses 12 behaviours of patients on the basis of frequency and severity: delusions, hallucinations, agitation/aggression, depression/dysphoria, anxiety, elation/euphoria, apathy/indifference, disinhibition, irritability, aberrant motor behaviour, night-time behaviours, and appetite/eating behaviours. Each behaviour is scored by multiplying the frequency and severity (i.e., frequency × severity); the higher the score, the greater the neuropsychiatric impairment. A total score can be calculated by summing the scores of all behaviours (range 1 to 144). Lower scores indicate less frequent/severe.
6. Functional Assessment Staging (FAST) [34]: Has seven main stages that consist of physical and instrumental active daily living, which are intended to project the progression of loss of function in patients with dementia.
7. Modified Unified Parkinson's Disease Rating Scale (M-UPDRS): A modification of the full UPDRS, to focus only the items that were independent of cognitive function [35]. A score of 8 or more indicates significant parkinsonism [35].
8. Clinician's Global Impression of Change (CGIC): A widely used and validated rating scale [36] based on the health-care provider's “general clinical impressions” with or without the informant input (i.e., family members). It evaluates global function and is scored from 1 (very much improved) to 7 (very much worsened).
For scales requiring an informant, the information was provided by a nurse or professional caregiver who had regular contact with the individual, usually the key worker. As far as possible, the same informant provided information for subsequent assessments. The outcome assessment schedule is summarised in Table 2.
Table 2.
Outcome Assessment Schedule
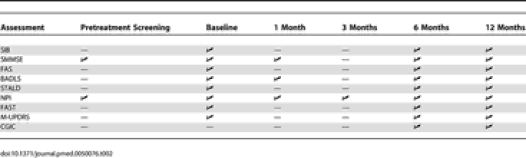
Although the study was of 12 mo treatment (or discontinuation), our primary focus was the progression of cognitive impairment at the 6 mo assessment. This schedule was predetermined in view of the frailty and predicted high mortality of this patient population.
Sample Size
The planned sample size of this trial was 110 patients per treatment group. In the absence of data from randomized clinical trials to inform power calculations at the time the protocol was developed, this calculation was undertaken using two different approaches. First, based upon the occurrence of clinically significant cognitive decline (defined as decline greater than the mean expected cognitive decline of four points per year on the MMSE), in 50% of patients continuing on neuroleptics versus 30% on placebo, allowing for a dropout rate of 15%–20%. Second, using a generic approach [37], the same sample size gives 80% power to the 5% level of significance to detect an effect size of 0.43 (regarded conservatively as a medium effect size). As we expected the SIB to be a more sensitive measure than MMSE, it was anticipated that the sample size would have more than sufficient power to detect a clinically important difference.
Randomisation
Randomisation was performed centrally at the Centre for Statistics in Medicine in Oxford (CSMO), using dedicated computer software (MINIM). The clinician responsible for randomisation of a patient faxed a randomisation form to the CSMO (or sent e-mail in exceptional circumstances) and provided details appropriate and sufficient for establishing eligibility. If a patient was eligible and informed consent/assent had been obtained and baseline assessments had been completed, the patient was randomised by the statistician either to continue taking medication or to discontinue (placebo group). The statistician directly communicated the allocation to the relevant trial pharmacy, ensuring concealment. The randomisation programme included a minimisation algorithm to ensure balanced allocation of participants across the intervention groups for the following important prognostic factors: presence or absence of extrapyramidal signs (EPS); visual hallucinations and delusions; use of cholinesterase inhibitors (y/n); SMMSE score (<6/≥6); and current neuroleptic medication (atypical/typical). The first 22 patients (10% of the target sample size) were allocated randomly to avoid predictability at the outset. These allocations were computer generated using block randomisation (block sizes of two and four) using Stata version 7 software (ralloc.ado v3.2.3 subroutine) [38]. Subsequently, the minimisation algorithm was applied with an allocation ratio that was not fully deterministic. The statistician carrying out the randomisation had no direct contact with patients and allocation was, therefore, totally independent of patient recruitment.
Blinding
The clinicians, those administering the trial medication, the caregivers, the relatives, the patients themselves, and those assessing the outcomes were all blinded to treatment allocation.
Statistical Methods
We used SPSS (release 12.0.1) to enter and manage data, and Stata (release 9.2) for analysis. Demographic factors and clinical characteristics were summarised with counts (percentages) for categorical variables, mean (standard deviation [SD]) for normally distributed continuous variables, or median (interquartile [IQR] or entire range) for other continuous variables. We restricted comparative analysis to those patients who started allocated treatment and had at least one assessment after randomisation, and participants were analysed in the groups to which they were allocated. Primary analysis was performed on patients with complete data at both baseline and week 26, including those who did not adhere to the protocol (e.g., those who never started treatment but for whom we had complete data). As the trial was conducted with masking of treatments, knowledge of allocation could not have contributed to drop-outs before or after treatment, so that exclusion of such patients did not impart bias. For the primary analysis, we summarised the change in the severe impairment battery (i.e., SIB at 6 mo − SIB at baseline) score from baseline to 6 mo using the mean and SD. To establish the magnitude and direction of the treatment effect, we used analysis of covariance (ANCOVA) to compare the two groups, giving the mean difference (in change in SIB from baseline to 6 mo) between groups (plus 95% CIs and p-values) with adjustment for baseline value. An additional ANCOVA was performed with adjustment for the minimisation factors, as well as geographical centre, in addition to the treatment contrast and baseline values. Changes in secondary outcomes were summarised and compared similarly.
For non-normally distributed continuous outcomes, we performed nonparametric Wilcoxon rank-sum tests to investigate differences between the treatment groups.
Prespecified sensitivity analyses were carried out to examine the robustness of conclusions to different assumptions about departures from randomised policies, dependent on the availability of data and the particular set of circumstances, namely:
1. Imputation is a method of “filling in” missing data with plausible values to give a completed dataset. Multiple imputation replaces each missing value with a set of plausible values that represent the uncertainty about the right value to impute. The procedure involves filling in missing data m times to generate m complete datasets. Then the m complete datasets are analysed by using standard statistical analyses, and finally, the results from the m complete datasets are combined to produce inferential results. Variables that were included in the imputation model were all assessments of SIB, NPI, FAS, SMMSE, BADLS, STALD-receptive, STALD-expressive, FAST, and CGIC collected at baseline, 6, and 12 mo. Other baseline covariates that were also included in the imputation model were allocated treatment group, sex, centre, presence of significant EPS, visual hallucinations or delusions, age at randomisation, type of neuroleptic drug at baseline, and whether the participant was taking cholinesterase inhibitors at trial entry.
2. In order to test the robustness of the SIB result, we limited a sensitivity analysis to those patients for whom the risk of possible floor and ceiling effects was smallest, i.e., SIB baseline cut-off values ≥ 40 but ≤ 90. Prespecified subgroup analyses were carried out on change in SIB and change in NPI. We performed the test of interaction, i.e., examined whether the treatment effects were consistent across subgroups. The subgroups investigated were (a) type of neuroleptic (atypical versus typical)—it made clinical sense to examine whether the type of neuroleptic (atypical versus typical) at baseline made a difference since, if one type was more harmful than the other, then discontinuation would benefit those patients more; (b) baseline NPI (≤ 14 versus ≥ 15)—we intended to look at consistency of treatment effect in relation to baseline NPI (≤ 14 versus 15 or more) in an attempt to replicate Ballard et al. 2004 [15]; and (c) centre effects (Newcastle versus Oxford versus London/Edinburgh). London and Edinburgh were grouped together because only a small number of participants were recruited from Edinburgh, and all were assessed by the London-based researchers.
Study Governance
A three-person independent data-monitoring committee (DMC) was charged with overseeing patient safety. Its remit included prompt review of serious adverse events and a comprehensive review of all adverse events based upon interim data reports. The remit also required advice regarding any new or emerging information on the safety of the study treatments. If required, the DMC would make recommendations to the trial steering group and the sponsor about the safe continuation of the trial and any issues of concern. These decisions relied upon the independence and expertise of the DMC members and did not follow any strict “stopping rules.”
Results
Participant Flow
The first patient was randomised in October 2001 and the last in December 2004. 165 patients were randomised (83 to continue treatment and 82 to placebo, i.e., discontinue treatment), of whom 128 (78%) commenced treatment (64 continue/64 placebo) (Figure 1). Of those, 26 were lost to follow-up (13 per arm), resulting in 51 patients per arm analysed for the primary outcome. Of those (102 total), 77 remained on allocated treatment at 6 mo follow-up (40 continue/37 placebo). A further 22 participants withdrew between 6 and 12 mo follow-up (Figure 1). Over the full 12 mo of the study 47 people did not complete follow-up. Of these individuals 21 died and a further 26 (55%) stopped the allocated treatment. Baseline demographic and clinical characteristics were evenly balanced across the two groups (Tables 3 and 4). The majority of the participants were taking risperidone/placebo risperidone (n = 101) or haloperidol/placebo haloperidol (n = 43) at baseline. A full breakdown of the neuroleptic drugs used in the study is given in Table 5.
Figure 1. Flow of Participants through the Trial.

Table 3.
Demographic and Clinical Characteristics and Assessments at Baseline (All Patients Randomized)
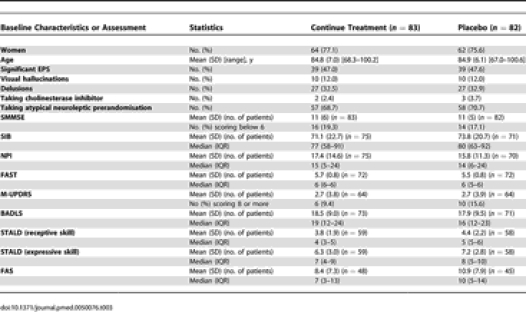
Table 4.
Demographic and Clinical Characteristics and Assessments at Baseline (Analysis Population)
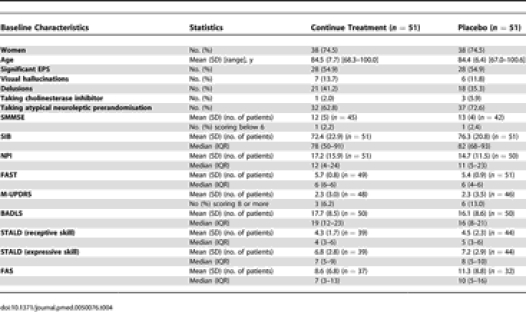
Table 5.
Details of Type and Dose of Treatment before Randomisation by Allocated Treatment Group
Primary Outcome—Severe Impairment Battery
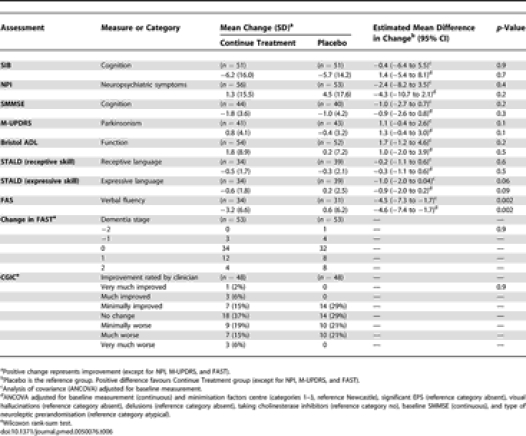
There was no significant difference between the continue treatment and placebo groups (n = 51 in both arms) in the estimated mean change in SIB scores between baseline and 6 mo (Figure 2; Table 6), 5.7 points (SD 14.2) deterioration for the placebo group, and a 6.2 (SD 16.0) deterioration for the continue treatment group; estimated mean difference in deterioration (favouring placebo) −0.4 (95% CI −6.4 to 5.5), adjusted for baseline value (p = 0.9).
Figure 2. Change in Total SIB Score (Baseline to 6 Months) by Treatment Group.

Table 6.
Summary of Change from Baseline to 6 Months for Main Outcomes plus Comparative Statistics
Secondary Outcomes
Neuropsychiatric symptoms.
For the NPI, there was no significant difference between the continue treatment and placebo groups (n = 56 and 53, respectively) in the estimated mean change in NPI scores between baseline and 6 mo, 4.5 points (SD 17.6) deterioration for the placebo group compared to a 1.3 (SD 15.5) deterioration for the continue treatment group; estimated mean difference in deterioration (favouring continue treatment) −2.4 (95% CI −8.2 to 3.5), adjusted for baseline value (p = 0.4). In patients with NPI scores ≤ 14 (Figure 3) the change in NPI over 6 mo was very similar in the neuroleptic and placebo groups (estimated difference 0.49, 95% CI −5.63 to 6.60), whereas in the people with NPI scores > 14 there was a five-point, albeit nonsignificant, advantage for people continuing to receive neuroleptics (estimated difference −5.33, 95% CI −15.82 to 5.17).
Figure 3. Subgroup Analysis (Change in NPI from Baseline to 6 Months).

Cognition, language, and function.
For the SMMSE, there was no significant difference between the continue treatment and placebo groups (n = 44 and 40, respectively) in the estimated mean change in SMMSE scores between baseline and 6 mo (Table 6), 1.0 points (SD 4.2) deterioration for the placebo group, and a 1.8 (SD 3.6) deterioration for the continue treatment group; estimated mean difference in deterioration (favouring placebo) −1.0 (95% CI −2.7 to 0.7), adjusted for baseline value (p = 0.2). For the BADLS, likewise there was no significant difference between the continue treatment and placebo groups (n = 54 and 52, respectively) in the estimated mean change in BADLS scores between baseline and 6 mo, 0.2 points (SD 7.2) worsening for the placebo group, and a 1.8 (SD 8.9) worsening for the continue treatment group; estimated mean difference in improvement (favouring placebo) 1.7 (95% CI −1.2 to 4.6), adjusted for baseline value (p = 0.2).
For the STALD (receptive skills), once again there was no significant difference between the continue treatment and placebo groups (n = 34 and 39 respectively) in the estimated mean change in STALD (receptive skills) scores between baseline and 6 mo, 0.3 points (SD 2.1) deterioration for the placebo group, and a 0.5 (SD 1.7) deterioration for the continue treatment group; estimated mean difference in deterioration (favouring placebo) −0.2 (95% CI −1.1 to 0.6), adjusted for baseline value (p = 0.6). For the STALD there was no significant difference between the continue treatment and placebo groups (n = 34 and 39, respectively) in the estimated mean change in receptive language scores between baseline and 6 mo. For expressive language there was no significant difference of placebo compared to the treatment group; estimated mean difference (favouring placebo) −1.0 (95% CI −2.0 to 0.04), adjusted for baseline value (p = 0.06).
However, for the FAS, there was strong evidence, i.e., a highly significant difference between the continue treatment and placebo groups (n = 34 and 31, respectively) in the estimated mean change in FAS totals between baseline and 6 mo. There was 0.6 points (SD 6.2) improvement for the placebo group compared to a 3.2 (SD 6.6) deterioration for the continue treatment group; estimated mean difference (favouring placebo) −4.5 (95% CI −7.3 to −1.7), adjusted for baseline value (p = 0.002).
Parkinsonism.
For the M-UPDRS, there was a slight but nonsignificant difference between the continue treatment and placebo groups (n = 41 and 43, respectively) in the estimated mean change in M-UPDRS scores between baseline and 6 mo, 0.4 points (SD 3.2) improvement for the placebo group compared to a 0.8 (SD 4.1) deterioration for the continue treatment group; estimated mean difference (favouring placebo) 1.1 (95% CI −0.4 to 2.6), adjusted for baseline value (p = 0.1).
Global Outcome
For the change in FAST and CGIC, there was no evidence whatsoever of any differences between the continue treatment and placebo groups (Wilcoxon rank-sum test p = 0.9 for both).
Prespecified Sensitivity Analyses
Imputed values were included for 12 participants, six in each treatment arm. These individuals had similar characteristics to the overall trial population, but as would be expected for participants unable to complete the trial, at baseline they were more cognitively impaired (SIB ± SD, 68.1 ± 22.3 versus 74.4 ± 21.9) and had higher levels of neuropsychiatric symptoms (NPI ± SD, 22.2 ± 14.8 versus 16.0 ± 13.9) than patients who subsequently completed the 6 month assessments. The results for SIB (mean difference −2.1, 95% CI −8.8 to 4.6, p = 0.5), NPI, SMMSE, UPDRS, BADLS, STALD, and FAS all remained consistent when missing data were replaced using multiple imputation techniques. In addition, the result for sensitivity analysis of SIB excluding participants whose baseline SIB was < 40 or > 90, i.e., excluding patients unlikely to benefit from treatment (mean difference −0.2, 95% CI −9.2 to 8.9, p = 0.9) was again consistent with the primary result.
Prespecified subgroup analysis.
There was no evidence of any interaction between treatment group and the various subgroups (Figures 3 and 4).
Figure 4. Subgroup Analysis (Change in SIB from Baseline to 6 Months).

Post-Hoc Additional Exploratory Sensitivity Analysis
Given that a substantial proportion of patients did not actually start allocated treatment (∼22%), we decided to examine the robustness of results by performing an analysis with all data available. In addition, according to the protocol, all patients with SMMSE < 6 at pretrial screening were not required to complete SMMSE and FAS at any subsequent assessments; however, some measures were collected during the follow-up. Therefore, additional sensitivity analysis was performed on SMMSE and FAS to assess if results remained consistent when these measures were included. Once again, there were no substantive differences between the primary and sensitivity analyses, i.e., the results are very robust.
Although not specified in the original analysis plan, to avoid omitting potentially important clinical differences, additional descriptive data were also obtained regarding emergent delusions and agitation in participants who did not have these symptoms at baseline. The pattern of emergent symptoms appeared similar in the two treatment arms at 1, 3, and 6 mo assessments, respectively, for both delusions (neuroleptics 14%, 16%, 5% versus placebo 5%, 8%, 13%) and agitation (neuroleptics 17%, 23%, 32% versus placebo 23%, 26%, 34%).
12 Month Assessment Data
Analysis at 12 mo was limited to the two main outcomes, cognitive function and neuropsychiatric features, given the large amount of missing data. There was no significant difference between the continue treatment and placebo groups (n = 28 and 27, respectively) in the estimated mean change in SIB scores between baseline and 12 mo, although there was a clinically important but not statistically significant numerical advantage of 8 points for the placebo group—8.5 points (SD 13.4) deterioration in the placebo group, and a 16.5 (SD 23.1) deterioration in the continue treatment group; estimated mean difference in deterioration (favouring placebo) −8.4 (95% CI −18.6 to 1.7), adjusted for baseline value (p = 0.1).
For the NPI, there was, however, a significant difference between the continue treatment and placebo groups (n = 28 and 31 respectively) in the estimated mean change in NPI scores between baseline and 12 mo, with 11.4 points (SD 17.7) deterioration for the placebo group compared to a 1.4 (SD 22.1) deterioration for the continue treatment group; estimated mean difference in deterioration (favouring continue treatment) −10.9 (95% CI −20.1 to −1.7), adjusted for baseline value (p = 0.02). A cut-off ≤ 14 on the NPI was again helpful in interpreting the results. For participants with baseline NPI scores below this threshold, there was no significant difference between treatment groups; estimated mean difference in deterioration (favouring continue treatment) −5.2 (95% CI −15.8 to 5.4), whereas for individuals with higher NPI scores there was a significant −16.9 point “advantage” for the group who continued neuroleptics (95% CI −32.5 to −1.2). However, the test of interaction, although underpowered, was not significant (p = 0.2); therefore there is no evidence of a statistical interaction.
Discussion
Because of difficulties in identifying people with Alzheimer disease in nursing homes who were taking neuroleptics and were able to complete the rigorous cognitive assessments, the recruitment target sample size based on the power calculation was not attained. However, despite this drawback, we report the largest and longest-duration randomized placebo controlled trial of neuroleptic discontinuation. To our knowledge this is the first study of this type to evaluate outcome over 6 mo and beyond.
Treatment with neuroleptics was not associated with significantly greater decline in global cognitive function than placebo, although there were numerical advantages for the placebo-treated group on the SIB and the SMMSE (1 point decrease on SMMSE, 0.4 point decrease on SIB overall, ∼3 point decrease on SIB in people with NPI scores ≤ 14) at 6 mo, which became more pronounced by month 12, at which point there was an 8 point advantage on the SIB for the placebo-treated group, equivalent to approximately 6 mo of average expected cognitive decline. The failure of these differences to attain statistical significance may be because of limited statistical power (a type II error), as the magnitude of difference in change in global cognition between neuroleptics and placebo at 6 mo was consistent with the effect size identified in a recent meta-analysis [11], and became more substantial over further follow-up. It would be more usual in a study of this type to hypothesise equivalence or the absence of an advantage to the treatment that is being removed, and there is certainly no evidence at all from these results suggesting any cognitive advantage favouring antipsychotics.
On secondary cognitive outcomes, there was a significant deterioration in verbal fluency for patients taking neuroleptics compared to people receiving placebo, and there was a nonsignificant numerical advantage for the placebo-treated group on the BADLS. There were nonsignificant numerical advantages for the placebo group with respect to the severity of parkinsonism.
There was a marginal nonsignificant 2.4 point advantage on the total NPI score for continuing neuroleptic treatment over the first 6 mo of treatment. Using a baseline NPI threshold ≤14, previously reported to be predictive of outcome in a 3 mo neuroleptic withdrawal trial [15], the change in NPI did not differ between the treatment groups. Participants with baseline NPI scores > 14 had an almost 5 point advantage (albeit nonsignificant) if they remained on neuroleptics, whereas there was no benefit for people with NPI scores below this threshold. For patients with more severe neuropsychiatric symptoms, there were modest benefits at 6 mo and more substantial advantages at 12 mo, which have to be weighed against the potential for serious adverse events. Some of the changes in NPI score are likely to be related to natural symptom course, or a Hawthorne effect, or regression to the mean, although there should be no imbalance in these factors between groups. There were no differences between groups for global clinician-rated outcome, and in an additional descriptive evaluation there appeared to be no difference in emergent delusions or agitation between groups.
In the post-hoc analysis there was no indication of a difference between people taking typical or atypical neuroleptics. The majority of individuals were taking risperidone or haloperidol, and the number of people taking other drugs was too small to enable any meaningful comparison. In particular, it will be important in further work to determine whether neuroleptics with more prominent antimuscarinic properties have a more potent impact on cognition in patients with dementia.
Several studies have demonstrated that psychological management approaches can replace neuroleptic therapy without any significant worsening of neuropsychiatric symptoms [14,39]; and evidence is emerging that cholinesterase inhibitors [40] or memantine [41] may be safer, effective alternatives for some symptoms. The authors of the recent CATIE study [17], a large, pragmatic, 36-wk placebo-controlled trial of atypical neuroleptics in AD, concluded that the modest benefits were not sufficient to justify therapy in the presence of the increased risk of serious adverse events. Clinicians should certainly try to replace atypical neuroleptics with safer management approaches. Taking into consideration CATIE, the results of 6- to 12-wk placebo-controlled trials, and our own data, we would suggest that there is, however, a limited place for atypical neuroleptics in the maintenance treatment of severe neuropsychiatric manifestations (particularly aggression) in AD when there is tangible risk or severe distress, and the symptoms have been refractory to other treatment approaches.
The magnitude of the impact of neuroleptics upon cognition, although consistent with a recent meta-analysis, was considerably less marked than reported in a recent 6 mo placebo-controlled trial of quetiapine [16]. There are numerous possible explanations for this difference, although it may be noted that unlike quetiapine, none of the atypical antipsychotics used in the current study had substantial antimuscarinic properties, and it is possible that antimuscarinic properties may exacerbate the impact of neuroleptics upon cognition. Although this is speculative, it is consistent with a study comparing cognition in patients with AD treated with either risperidone or olanzapine, where olanzapine treatment was associated with greater impairment of attentional and executive performance related to anticholinergic activity [42]. Within the current study there was a significant detrimental impact upon expressive language function, an important skill to enable social communication and maintain quality of life in people with AD residing in care facilities. Further work is needed to examine the effects on different aspects of cognitive function, to clarify the differential impact of individual neuroleptic drugs, and to determine whether the impact upon cognition is sufficient to interfere with everyday activities.
The results of the current study must be interpreted within the context of a number of limitations. In particular, the sample size was much smaller than intended, conferring limited statistical power, and the number of deaths and withdrawals precluded meaningful analysis of data beyond the 6 mo follow-up. In addition a sizeable proportion of patients did not start their allocated treatment for a variety of reasons, mainly related to frailty and concurrent illnesses. The sample size achieved was short of the 220 target due mainly to problems identifying eligible patients, which in turn led to slow recruitment, bringing on more centres, and ultimately curtailing recruitment due to a lack of resources. Given the vulnerability of the study population, a substantial number of deaths and withdrawals are an almost inevitable problem to contend with. Probably the only solution would be to exclude people with profound dementia or with a certain degree of physical frailty, but this would then diminish the validity of drawing more general conclusions from the results. Although it is difficult, therefore, to see how this problem could have been avoided, the high number of drop-outs must be considered when interpreting the results.
In addition, the reason for the initial prescription of neuroleptic drugs was unclear in the majority of instances. Most prescriptions had been instigated by primary care physicians and, as a number of the individuals had changed their primary care physician, or been admitted to a care facility, or changed care facility since neuroleptics were first prescribed, the clinical indication for the original prescription was often lost. Although in many ways this lack of information is unsatisfactory, it does reflect real clinical practice, so that the population studied and the information available were representative of what faces clinicians in their routine practice. As few individuals were under specialist care, it is unlikely that treatment-refractory symptoms were a reason for neuroleptic use in most of the participants. The data regarding outcome and the baseline severity of symptoms do, however, provide a useful basis for clinical decisions. It is possible that the profile of the original symptoms for which the neuroleptics were prescribed may have influenced outcome, but this possibility could practically be investigated only in people with neuroleptic prescriptions of shorter duration, for which the presenting symptoms would be easier to ascertain.
Conclusion
For most patients with AD, withdrawal of neuroleptics had no overall detrimental effect on functional and cognitive status and by some measures improved functional and cognitive status. Neuroleptics may have some value in the maintenance treatment of more severe neuropsychiatric symptoms, but this possibility must be weighed against the unwanted effects of therapy. The current study helps to inform a clinical management strategy for current practice, but the considerable risks of maintenance therapy highlight the urgency of further work to find, develop, and implement safer and more effective treatment approaches for neuropsychiatric symptoms in people with AD.
Supporting Information
(44 KB PDF)
Acknowledgments
Other DART AD investigators: Timothy Smith, Ruth Elvish, Claire Maddison, Carol Bannister.
Data Monitoring and Ethics Committee: Clive Holmes (Chair), John Geddes and Robert Clarke.
DART-AD Pharmacists: (North East) Vranda Barbieri, Kay Carson, Gavin Mankin; (Oxfordshire) Michael Marven, Kim Gray; (London) Charlotte Gibbs.
Abbreviations
- AD
Alzheimer disease
- ANCOVA
analysis of covariance
- BADLS
Bristol Activities of Daily Living Scale
- CGIC
Clinician's Global Impression of Change
- CI
confidence interval
- DMC
data-monitoring committee
- EPS
extrapyramidal signs and symptoms
- FAS
Verbal Fluency Task
- FAST
Functional Assessment Staging
- IQR
interquartile range
- NPI
Neuropsychiatric Inventory
- SD
standard deviation
- SIB
Severe Impairment Battery
- (S)MMSE
(Standardised) Mini Mental State Examination
- STALD
Sheffield Test for Acquired Language Disorders
- UPDRS
Unified Parkinson's Disease Rating Scale
Footnotes
Author contributions. CB, RMS, RJ, and EJ designed the experiments/the study. MML, MT, SD, and KK collected data or did experiments for the study. RMS, LY, and EJ analyzed the data. MML, MT, SD, and RJ enrolled patients. CB and EJ wrote the first draft of the paper. SD, RMS, RJ, and EJ contributed to the writing of the paper. MT collected all the data for the Oxfordshire patients over the first two years. SD managed the trial in Newcastle for the majority of its running time, collecting data and also setting up the trial in London. SD also entered data and helped to track down missing data prior to analysis, proof read the manuscript and offered suggestions for changes. RJ contributed to the writing of the grant application and was one of the holders of the grant from the Alzheimer's Research Trust. RJ was also a principal investigator for the study and supervised the collection of the clinical data in the Oxford (UK) patients. KK identified patients, collected data, and wrote the follow-up protocol for the study. LY critically reviewed the manuscript. EJ was the trial statistician and part of the trial management group which met every 2 months. EJ presented regular interim analyses to the Data Monitoring Committee, and co-authored the annual report to the funder. EJ co-wrote the statistical analysis plan and was senior reviewer of the final statistical report.
Funding: The DART-AD project was made possible by a grant from The Alzheimer's Research Trust, Cambridge, UK (http://www.alzheimers-research.org.uk) to Profs Ballard and Jacoby and to RM. The peer review process undertaken by the funder did result in some modifications to the study design. The funder has no other role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests: CB has received honoraria from Novartis, Pfizer, Shire, Lundbeck, Myriad, Janssen, Astra Zeneca, and Servier pharmaceutical companies and research grants from Novartis, Lundbeck, Astra-Zeneca, and Janssen pharmaceuticals. The remaining authors have declared that they have no competing interests.
References
- Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, et al. Alzheimer's Disease International. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–7. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard C, Ayre G, Gray A. Psychotic symptoms and behavioral disturbances in dementia: A review. Rev Neurol. 1999;155:44–52. [PubMed] [Google Scholar]
- Gilley DW, Whalen ME, Wilson RS, Bennett DA. Hallucinations and associated factors in Alzheimer's disease. J Neuropsych Clin Neurosci. 1991;3:371–376. doi: 10.1176/jnp.3.4.371. [DOI] [PubMed] [Google Scholar]
- Rabins PV, Mace NL, Lucas MJ. The impact of dementia on the family. J Am Med Soc. 1982;248:333–335. [PubMed] [Google Scholar]
- Donaldson C, Tarrier N, Burns A. The impact of the symptoms of dementia on caregivers. Brit J Psychiatr. 1997;170:62–68. doi: 10.1192/bjp.170.1.62. [DOI] [PubMed] [Google Scholar]
- Steele C, Rovner B, Chase GA. Psychiatric symptoms and nursing home placement of patients with Alzheimer's disease. Am J Psych. 1990;147:1049–51. doi: 10.1176/ajp.147.8.1049. [DOI] [PubMed] [Google Scholar]
- Schneider LS, Pollock VE, Lyness SA. A metaanalysis of controlled trials of neuroleptic treatment in dementia. J Am Geriatrics Soc. 1990;38:553–63. doi: 10.1111/j.1532-5415.1990.tb02407.x. [DOI] [PubMed] [Google Scholar]
- Lonergan E, Luxenberg J, Colford J, Birks J. Haloperidol for agitation in dementia. Cochrane Database Syst Rev 4. Art. No.: CD002852. 2005. doi: 10.1002/14651858.CD002852. [DOI] [PubMed]
- Ballard C, Howard R. Neuroleptic drugs in dementia: benefits and harm. Nat Rev Neurosci. 2006;7:492–500. doi: 10.1038/nrn1926. [DOI] [PubMed] [Google Scholar]
- Ballard C, Waite J. The effectiveness of atypical antipsychotics for the treatment of aggression and psychosis in Alzheimer's disease. Cochrane Database Syst Rev 1: Art. No.: CD003476. 2006. doi: 10.1002/14651858.CD003476.pub2. [DOI] [PMC free article] [PubMed]
- Schneider LS, Dagerman K, Insel PS. Efficacy and adverse effects of atypical antipsychotics for dementia: meta-analysis of randomized, placebo-controlled trials. Am J Geriatr Psychiatry. 2006;14:191–210. doi: 10.1097/01.JGP.0000200589.01396.6d. [DOI] [PubMed] [Google Scholar]
- McGrath AM, Jackson GA. Survey of prescribing in residents of nursing homes in Glasgow. BMJ. 1996;314:611–612. doi: 10.1136/bmj.312.7031.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margallo-Lana M, Swann A, O'Brien J, Fairbairn A, Reichelt K, et al. Prevalence and pharmacological management of behavioral and psychological symptoms amongst dementia sufferers living in care environments. Int J Geriatric Psych. 2001;16:39–44. doi: 10.1002/1099-1166(200101)16:1<39::aid-gps269>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Cohen-Mansfield J, Lipson S, Werner P, Billig N, Taylor L, et al. Withdrawal of haloperidol, thioridazine, and lorazepam in the nursing home: a controlled, double-blind study. Arch Int Med. 1999;159:1733–40. doi: 10.1001/archinte.159.15.1733. [DOI] [PubMed] [Google Scholar]
- Ballard CG, Thomas A, Fossey J, Lee L, Jacoby R, et al. A 3-month, randomized, placebo-controlled, neuroleptic discontinuation study in 100 people with dementia: the neuropsychiatric inventory median cutoff is a predictor of clinical outcome. J Clin Psych. 2004;65:114–119. doi: 10.4088/jcp.v65n0120. [DOI] [PubMed] [Google Scholar]
- Ballard C, Margallo-Lana M, Juszczak E, Douglas S, Swann A, et al. Quetiapine and rivastigmine and cognitive decline in Alzheimer's disease: randomised double blind placebo controlled trial. BMJ. 2005;330:874. doi: 10.1136/bmj.38369.459988.8F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider LS, Tariot PN, Dagerman KS, Davis SM, Hsiao JK, et al. CATIE-AD Study Group. Effectiveness of atypical antipsychotic drugs in patients with Alzheimer's disease. New Engl J Med. 2006;355:1525–38. doi: 10.1056/NEJMoa061240. [DOI] [PubMed] [Google Scholar]
- Ballard C, Margallo-Lana ML, Fossey J, Reichelt K, Mynt P, et al. A one year follow-up study of behavioral and psychological symptoms in dementia among people in care environments”. J Clin Psych. 2001;62:631–636. doi: 10.4088/jcp.v62n0810. [DOI] [PubMed] [Google Scholar]
- Bridges-Parlet S, Knopman D, Steffes S. Withdrawal of neuroleptic medications from institutionalised dementia patients, results of a double-blind baseline-treatment-controlled pilot study. J Geriatric Psych Neurol. 1997;10:119–126. doi: 10.1177/089198879701000306. [DOI] [PubMed] [Google Scholar]
- Schneider LS, Dagerman KS, Insel P. Risk of death with atypical antipsychotic drug treatment for dementia: Meta-analysis of randomized placebo-controlled trials. JAMA. 2005;294:1934–1943. doi: 10.1001/jama.294.15.1934. [DOI] [PubMed] [Google Scholar]
- Committee for the Safety of Medicines (2004 March 9) Message from Chairman, Committee on Safety of Medicines to medical contacts: Atypical antipsychotic drugs and stroke [memo] Edinburgh (Scotland): The Scottish Government; Available: http://www.scotland.gov.uk/Publications/2004/03/19074/34348. Accessed 21 February 2008. [Google Scholar]
- FDA. Deaths with antipsychotics in elderly patients with behavioral disturbances. Washington (D.C): U.S: Food and Drug Administration, FDA Public Health Advisory, Centre for Drug Evaluation and Research 13-7-2005; 2005. [Google Scholar]
- McShane R, Keene J, Gedling K, Fairburn C, Jacoby R, et al. Do neuroleptic drugs hasten cognitive decline in dementia? Prospective study with necropsy follow up. BMJ. 1997;314:266–270. doi: 10.1136/bmj.314.7076.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard R, Ballard C, O'Brien J, Burns A. UK and Ireland Group for Optimization of Management in dementia. Guidelines for the management of agitation in dementia. Int J Geriatric Psych. 2001;16:714–7. doi: 10.1002/gps.418. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurol. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Molloy DW, Alemayehu E, Roberts R. Reliability of a standardized Mini-Mental State Examination compared with the traditional Mini-Mental State Examination. Am J Psych. 1991;148:102–5. doi: 10.1176/ajp.148.1.102. [DOI] [PubMed] [Google Scholar]
- Schmitt FA, Ashford W, Ernesto C, Saxton J, Schneider LS, et al. The severe impairment battery: concurrent validity and the assessment of longitudinal change in Alzheimer's disease. The Alzheimer's disease cooperative study. Alzheimer Dis Assoc Disord. 1997;11:51–56. [PubMed] [Google Scholar]
- Committee on Safety of Medicines. Thioridazine: Restricted indications and new warnings on cardiotoxicity [memo] London: Department of Health, National Health Service; 2000 December 11. CEM/CMO/2000/18. Available: http://www.info.doh.gov.uk/doh/embroadcast.nsf/f011981a95f31f4180256c07003d34a0/fc5d1f63a16da86b80256dad00480816?OpenDocument. Accessed: 21 February 2008. [Google Scholar]
- Reilly JG, Ayis SA, Ferrier IN, Jones SJ, Thomas SH. QTc-interval abnormalities and psychotropic drug therapy in psychiatric patients. Lancet. 2000;355:1048–52. doi: 10.1016/s0140-6736(00)02035-3. [DOI] [PubMed] [Google Scholar]
- Benton AL, Hamsher K, Sivan AB. Multilingual Aphasia Examination. 3rd edition. Iowa City (Iowa): AJA Associates; 1983. [Google Scholar]
- Byrne LM, Wilson PM, Bucks RS, Hughes AO, Wilcock GK. The sensitivity to change over time of the Bristol Activities of Daily Living Scale in Alzheimer's disease. Int J of Ger Psych. 2000;15:656–61. doi: 10.1002/1099-1166(200007)15:7<656::aid-gps163>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Syder D, Levy C. Wanting to talk: Counselling case studies in communication disorders. Philadelphia (Pennsylvania): Whurr Publishers; 1998. pp. 256–288. [Google Scholar]
- Cummings JL, Mega M, Gray K, Rosenburg-Thomas S, Carusi DA. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurol. 1994;44:2308–2314. doi: 10.1212/wnl.44.12.2308. [DOI] [PubMed] [Google Scholar]
- Sclan SG, Reisberg B. Functional assessment staging (FAST) in Alzheimer's disease: reliability, validity, and ordinality. Int Psychogeriatrics. 1992;1:55–69. doi: 10.1017/s1041610292001157. [DOI] [PubMed] [Google Scholar]
- Ballard C, McKeith I, Burn D, Harrison R, O'Brien J, et al. The UPDRS scale as a means of identifying extrapyramidal signs in patients suffering from dementia with Lewy bodies. Acta Neurol Scand. 1997;96:366–371. doi: 10.1111/j.1600-0404.1997.tb00299.x. [DOI] [PubMed] [Google Scholar]
- Schneider LS, Olin JT, Doody RS, Clark CM, Morris JC, et al. Validity and reliability of the Alzheimer's Disease Co-operative Study-Clinical Global Impression of Change. The Alzheimer's Disease Co-operative Study. Alzheimer Dis Assoc Disord. 1997;11:22–32. doi: 10.1097/00002093-199700112-00004. [DOI] [PubMed] [Google Scholar]
- Cohen J. A power primer. Psychol Bull. 1992;112:155–159. doi: 10.1037//0033-2909.112.1.155. [DOI] [PubMed] [Google Scholar]
- [No author listed] Stata Statistical Software release 7.0. College Station (Texas): Stata Corporation; 2002. [Google Scholar]
- Fossey J, Ballard C, Juszczak E, James I, Alder N. Effect of enhanced psychosocial care on antipsychotic use in nursing home residents with severe dementia: cluster randomised trial. BMJ. 2006;332:756–761. doi: 10.1136/bmj.38782.575868.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier S, Feldman H, Hecker J, Vellas B, Ames D, et al. Donepezil MSAD Study Investigators Group. Efficacy of donepezil on behavioral symptoms in patients with moderate to severe Alzheimer's disease. Int Psychogeriatrics. 2002;14:389–404. doi: 10.1017/s104161020200858x. [DOI] [PubMed] [Google Scholar]
- Cummings JL, Schneider E, Tariot PN, Graham SM. Behavioral effects of memantine in Alzheimer's disease patients receiving donepezil treatment. Neurology. 2006;67:57–63. doi: 10.1212/01.wnl.0000223333.42368.f1. [DOI] [PubMed] [Google Scholar]
- Mulsant BH, Gharabawi GM, Bossie CA, Mao L, Martinez RA. Correlates of anticholinergic activity in patients with dementia and psychosis treated with risperidone or olanzapine. J Clin Psych. 2004;65:1708–14. doi: 10.4088/jcp.v65n1217. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(44 KB PDF)