

Abstract
The prostanoid synthesizing enzyme cyclooxygenase-2 (COX-2) is involved in the mechanisms of cerebral ischemia, an effect mediated by prostaglandin E2 through activation of EP1 receptors. Thus, inhibition of EP1 receptors is neuroprotective in models of ischemic stroke, but the molecular mechanisms of the effect have not been fully elucidated. We used oxygen glucose deprivation (OGD) in hippocampal slices as an injury model to investigate whether the neuroprotection afforded by EP1 receptor inhibition involves the PI3K/AKT survival pathway. EP1 receptor inhibition with SC51089 or SC51322 reduced the hippocampal damage produced by ODG by 28 ± 2% and 32 ± 3%, respectively (p<0.05). OGD induced a transient reduction of AKT activity that was partly counteracted by SC51089. LY294002 blocked the increase in phospho-AKT evoked by SC51089 and abolished the associated protective effect. The AKT activation induced by SC51089 was associated with phosphorylation of PTEN, the phosphatase that negatively regulates AKT. Furthermore, SC51089 attenuated the mitochondrial translocation of the proapoptotic protein BAD. These data indicate that EP1 receptor inhibition improves the survival of hippocampal slices by preventing the attenuation in AKT activity induced by OGD, and by reducing the mitochondrial translocation of BAD. The findings provide evidence for a link between EP1 receptors and the PI3K/AKT survival pathway and shed light on the molecular mechanisms of the prosurvival effect of EP1 receptor inhibition.
Keywords: hippocampal slice culture, oxygen glucose deprivation, AKT, PTEN, neuroprotection, BAD, EP1 receptors
INTRODUCTION
Cyclooxygenase-2 (COX-2) is a rate limiting enzyme for the production of prostanoids (Breyer et al. 2001; Turini and DuBois 2002). In brain, COX-2 expression is constitutive but is upregulated rapidly by NMDA receptor activation and by injuries such as ischemic stroke (Collaco-Moraes et al. 1996; Miettinen et al. 1997; Nogawa et al. 1997). It is well established that COX-2 activation contributes to ischemic brain injury. Thus, COX-2 gene inactivation or pharmacological inhibition attenuates the infarct and neurological dysfunction in mice subjected to focal cerebral ischemia (Nogawa et al. 1997; Iadecola et al. 2001; Sasaki et al. 2003). The mediator of the neurotoxic effect of COX-2 in cerebral ischemia is prostaglandin E2 (PGE2), and not superoxide, which is also produced by COX-2 (Kawano et al. 2006; Kunz et al. 2007).
PGE2 exerts its biological actions through specific G-protein coupled transmembrane receptors (Breyer et al. 2001). Recently, we showed that EP1 receptors are the downstream effectors of COX-2-derived PGE2. Inhibition or genetic inactivation of EP1 receptors counteracts the Ca2+ dysregulation induced by NMDA receptor overactivation and induces neuroprotection (Kawano et al. 2006). However, the downstream molecular events linking the restoration of Ca2+ homeostasis with neuroprotection have not been defined.
The serine/theronine kinase AKT/PKB (protein kinase B) is a key component in the survival signaling pathway transducing growth stimuli from growth factors (Manning and Cantley 2007). In the central nervous system, decreased AKT activity has been linked to the neuronal death induced by NMDA receptor activation, focal ischemia, or hypoxia (Luo et al. 2003; Hirai et al. 2004). On the other hand, increased AKT activity contributes to the neuroprotection induced by hypothermia (Zhao et al. 2005) and to the protection of human cerebral endothelial cells induced by hypoxic preconditioning (Zhang et al. 2007). The activity of AKT depends on the availability of phosphoinisitidylinositol-3,4,5-triphosphate (PIP3), which is generated by the enzyme phosphatidylinositol 3-kinase (PI3K) (Foster et al. 2003). The levels of PIP3 are determined by the activity of a lipid phosphatase, PTEN (phosphatase and tensin homologue deleted on chromosome 10) (Maehama and Dixon 1998), which dephosphorylates PIP3 and converts it back to PIP2. Therefore, the biological effects of AKT are determined by the balance between the activity of PI3K and PTEN, although the influence of PTEN can be more dominant (Seo et al. 2005).
After activation, AKT phosphorylates target proteins involved in cell growth, metabolism and survival (Manning and Cantley 2007). For example, AKT phosphorylates the pro-apoptotic protein BAD, preventing it from binding to and inactivating Bcl-xL in mitochondria (Datta et al. 1997). In turn, Bcl-xL exerts its anti-apoptotic effect and contributes to cell survival. In cerebral ischemia, BAD has been shown to be a key molecule regulating the balance between cell survival and death signals (Kamada et al. 2007).
Considering the key role that AKT has in cell survival in models of neurotoxicity, we sought to determine whether AKT is involved in the neuroprotective effect of EP1 inhibition. Using oxygen-glucose deprivation (OGD) as a model of ischemia in mouse hippocampal slice cultures, we found that EP1 receptor inhibition increases AKT phosphorylation, an event mediated by PTEN phosphorylation. EP1 receptor inhibition counteracts the suppression of AKT induced by OGD and reduces the translocation of BAD to mitochondria. The data suggest a previously unrecognized link between EP1 receptors and intrinsic pathways regulating cell survival and death.
MATERIALS AND METHODS
Materials and reagents
Hank's balanced salt solution (HBSS), Eagle's Basal Medium, Propidium iodide (PI) and other tissue culture supplies were from Invitrogen (Carlsbad, CA). Millicell CM membrane insert and polyvinylidene difluoride (PVDF) membrane for western blot were from Millipore (Bedford, MA). Horse serum and other chemicals were from Sigma (St. Louis, MO). Antibodies for AKT, phospho-AKT (p-AKT), PTEN, phospho-PTEN (p-PTEN) are from Cell Signaling (Danvers, MA). Horse radish peroxidase conjugated fluorescence secondary antibodies were from Jackson ImmunoResearch (West Grove, PA).
Organotypic mouse hippocampal slice culture
All procedures involving new born mice for hippocampal slices cultures were approved by and performed according to the guidelines of the Weill Cornell Medical College Institutional Animal Care and Use Committee. Hippocampal slice cultures were prepared according to the procedure of Muller et al (Muller et al. 2001) as previously described (Kawano et al. 2006). Briefly, hippocampi from 5−6 day old mouse pups were dissected out aseptically and coronal slices of 350 μM in thickness were obtained using a tissue chopper (McILwan tissue chopper, Vibratome Company, St. Louis, MO). Slices in good shape were transferred to the Millicell CM membrane insert (30 MM, 0.4 μm) placed in a 6-well plates filled with 1 ml medium (25% horse serum, 50% Eagle's Basal Medium, 25% HBSS, 5 mg/ml glucose). Six slices were seeded in each well. The slice cultures were maintained in a humidified chamber at 37°C with 5% CO2 and medium changed twice a week. The slices were cultured for 13 days before being used for experiments.
Oxygen and glucose deprivation and neuronal cell death measurement in hippocampal slices
Slices were first rinsed twice with warm OGD buffer (125 mM NaCl, 5 mM KCl, 1.2 mM Na2PO4, 26 mM NaHCO3, 1.8 mM CaCl2, 0.9 mM MgCl2, 10 mM Hepes, pH 7.4) plus 10 mM glucose and incubated for 30 min with the same rinsing buffer. The slices were then rinsed with OGD buffer twice and transferred to an OGD chamber (Billups-Rothenberg, San Diego, CA) and flushed with anoxic gas (95% N2 and 5% CO2) at 21 L/min for 5 min. The chamber was then sealed and placed into 37°C incubator for 1 hr. Controls were rinsed with OGD buffer but incubated in a normoxia conditions. After OGD treatment, the slices were transferred to normal culture medium and incubated for 23 hrs. Before fluorescence images were taken, propidium iodide (PI) was added to medium to 5 μM and incubated for 1 hr.
Measurement of neuronal cell death in slices was achieved by PI staining of the dead cells and quantitatively analyzing the mean PI fluorescence in CA1 region in each slice. Briefly, slices in culture medium containing PI (5 μg/ml) before and after OGD were imaged under a Nikon inverted fluorescence microscope equipped with a digital camera operated by a Mac computer using IPLab software (BD Biosciences, Rockville, MD). The slices were then treated with 100 μM NMDA and PI for 24 more hrs and images were recorded and served as maximum fluorescence intensity in slices. The same camera settings (slice position, exposure time, filter setting, gain) were used throughout experiments.
To calculate the percentage of cell death after OGD in CA1 region, digital images of PI staining at three different time points (before and after OGD, and after NMDA treatment) were outlined to define the same CA1 region and mean fluorescence intensity in obtained with corresponding time points: Fbasal, FOGD and Fmax. The percent cell death is calculated from the formula (FOGD - Fbasal)/(Fmax - Fbasal)×100% (Kawano et al. 2006).
Western blotting
Cultured slices after specified treatment were homogenized gently in detergent lysis buffer (25 mM Hepes, pH 7.4, 50 mM NaCl, 1 mM EDTA, 1% Triton X-100, protease inhibitor cocktail and phosphatase inhibitors) and incubated on ice for 10 min. The lysates were centrifuged 14,000×g for 10 min at 4°C. After protein concentration was determined for the supernatant, equal amount of proteins were gel-separated and transferred to membranes. Membranes were incubated with mouse-specific antibodies (dilution 1:1000) after blocking with 5% dry milk in PBS for 1 hour at room temperature. The membranes were washed 3 times with PBST (PBS + 0.1% Tween-20) and incubated with horse radish peroxidase conjugated anti-mouse secondary antibodies (dilution 1:2500) for one hour, washed 3 times with PBST, and developed by the enhanced chemiluminescence system (ECL, Amersham, Arlington Heights, IL). Protein band images were captured by using a Kodak Digital Imaging Station 2000R (Kodak, Rochester, NY).
Immunofluorescence staining of hippocampal slice
The treated slices were fixed in 3.25% acrine, 2% paraformaldehyde for 15 min. The slices were then permeablized with 1% TBST followed by incubation in 3% BSA for 1 hr. After primary antibody addition (1:200) and incubation overnight at room temperature, the slices were washed 3 time with PBST and incubated with diluted secondary antibody conjugated with F448 (Jackson ImmunoResearch, West Grove, PA). The nuclei were counterstained with 0.5 μg/ml DAPI (Molecular Probe) for 10 min at room temperature. BAD immunostaining was used to assess BAD translocation after OGD. The images were captured with a Leica confocal microscope using 63× lens.
RESULTS
Inhibition of COX-2 or EP1 receptors is neuroprotective in hippocampal slices
We first examined whether COX-2 and EP1 receptors contribute to the hippocampal injury caused by OGD. Hippocampal slices subjected to OGD showed substantial injury in the CA1 region (Fig 1A). Treatment with the COX-2 inhibitor NS398 and with the EP1 receptor antagonists SC51089 or SC51322, reduced the injury (Fig. 1A, B). These observations establish that the COX-2 and EP1 receptors contribute to the cell death in hippocampal slices subjected to OGD.
Figure 1.

EP1 receptor inhibition is neuroprotective in an hippocampal slice culture model of OGD induced cell death. (A) Fluorescence images showing PI staining in hippocampal slices subjected to OGD for one hour. Images were taken before OGD (Base), 24 hrs after OGD (OGD) and after NMDA incubation (MAX). Scale bar=1mm. (B) Quantitative measurements of cell death in CA1. Compared to sham treated controls, the cell death in the CA1 region measured as PI intensity was reduced by NS398 (5μM), SC51089 (10μM), or SC51322 (10μM). *p<0.05 (t-test), n=18−56 slices from 3−7 individual experiments. Data are presented as mean ± SEM.
AKT phosphorylation is modulated by EP1 receptors
Next, we examined the role of AKT phosphorylation in the protection exerted by EP1 receptor inhibition. In normoxic conditions, a basal level of p-AKT was observed (Fig. 2A, lane 1), as previously described by others (Hirai et al. 2004), and treatment with SC51089 increased the basal level of p-AKT (Fig. 2A. lane 2). Immediately after OGD, p-AKT was markedly attenuated, an effect partially counteracted by SC51089 (Fig. 2A lanes 3 and 4). One hour after OGD, p-AKT increased above basal levels, but the increase was more marked in slices treated with SC51089 (Fig. 2A, lane 5 and 6). Three hours after OGD, the increase in p-AKT by EP1 receptor inhibition was reduced to levels similar to those of vehicle-treated slices (Fig. 2B). Levels of p-AKT returned to baseline 6 hrs after OGD (ratio of p-AKT/t-AKT, vehicle 0.42 ± 0.23; SC-51089 0.29 ± 0.8. p>0.05). These observations suggest that EP1 receptors modulate AKT activity after OGD.
Figure 2.

EP1 inhibition increases AKT phosphorylation. Hippocampal slices were treated with one hr of OGD in the absence or presence of SC51089 (10 μM). The level of phosphorylation on AKT protein was assessed by immunoblotting with AKT and p-AKT specific antibodies. (A) The effect of SC51089 on p-AKT level after OGD. Slices were either sham treated or treated with SC51089 (10μM) and removed at indicated time points. Proteins from slices were probed separately with p-AKT and t-AKT (total-AKT) specific antibodies. Samples for lanes 1 and 2 were not treated with OGD (normoxia). Each lane was loaded with 25μg of protein. One representative gel image is shown from a total of 6 independent experiments. (B) Protein band intensity measurement. Each membrane was reprobed with β-actin antibody for sample loading adjustment. The p-AKT and AKT band intensities were then normalized against that of β-actin before the ratio of p-AKT/AKT was obtained. Absolute values of p-AKT/t-AKT ratio are presented. #p<0.05, *p<0.01, n=6. (C) The effect of SC51089 on 17-pt-PGE2 induced p-AKT level changes without OGD. Slices were pre-treated with NS398 (5μM) and SC-560 (10μM) followed by incubation with 17-pt-PGE2 (5μM) with or without SC51089 (10μM). After probing with p-AKT antibodies, the membrane was stripped and reprobed with AKT antibody. Each lane contained 25μg of proteins and one representative image from 3 independent experiments is shown here.
To further verify that the AKT phosphorylation is modulated by EP1 receptors, we added the stable analog of PGE2 17-phenyl-trinor-PGE2 (17-pt-PGE2) to slice cultures without OGD treatment. Endogenous PGE2 production was blocked by pre-treating the slices with the COX-1 inhibitor SC560 and COX-2 inhibitor NS398. In the absence of SC51089, 17-pt-PGE2 decreased p-AKT levels (Fig. 2C, lane 2). Co-incubation of 17-pt-PGE2 with SC51089 restored p-AKT levels (Fig. 2C, lane 3), indicating that inhibition of EP1 receptors is linked to the increase in AKT activity and demonstrating the specificity of the induction of p-AKT by EP1 receptor inhibition. This result also suggests that the presence of a constitutive negative regulation of AKT by EP1 receptors. Although p-AKT was decreased by treatment with 17-pt-PGE2, the viability of the slice was not affected (data not shown).
The increase in AKT phosphorylation resulting from EP1 receptor inhibition contributes to neuroprotection
To determine whether the increase in p-AKT contributes to the viability enhancement induced by EP1 receptor inhibition, we used the PI3K inhibitor LY294002 to lower the p-AKT level. As shown in Fig. 3A (lane 3 and 10), LY294002 blocked AKT phosphorylation both before and after OGD. In the presence of LY294002, SC51089 was unable to increase p-AKT after OGD (Fig 3A, lane 4), suggesting that the effect by EP1 is upstream of AKT phosphorylation. Importantly, LY294002 abolished the neuroprotective effect of EP1 receptor or COX-2 inhibition (Fig 3B). Similar results were obtained with the other PI3K inhibitor Wortmannin (data not shown). LY294002 by itself did not induce tissue damage in normal condition, although the basal level of AKT phosphorylation was reduced (Fig 3A, lane 10). In addition, LY294002 did not enhance the damage after OGD (Fig 3B), suggesting that in normal conditions, other survival pathways may work in concert to compensate for the loss of AKT activity. Similarly, the increase in p-AKT and the enhanced viability evoked by the COX-2 inhibitor NS398 was abolished by LY294002 (Fig. 3A, lane 8 and Fig. 3B).
Figure 3.

The AKT phosphorylation evoked by EP1 receptor inhibition increases cell viability. Hippocampal slices were treated with LY294002 (LY, 20μM), SC51089 (SC, 10μM), and NS398 (NS, 5μM), or in combinations as indicated followed by 1 hr of OGD. Immediately after OGD, one set of samples was taken out and lysates made for immunoblotting and another set of samples was used for viability measurement 24 hrs later. (A) Immunoblotting showing that increase in p-AKT by SC51089 and NS398 is abolished by LY294002. t-AKT images were obtained after stripping the membrane and reprobing it with t-AKT antibodies. One set of gel image from three experiments with similar results is shown. Each lane was loaded with 25μg of protein. (B) Quantitative analyses of cell viability in the CA1 region of slices that were treated in the same way as in (A). Note LY294002 alone does not increase cell death. Values for bar graphs were from three independent experiments and at least 18 slices were counted for each data point. *p<0.05, n=3/group.
EP1 receptor inhibition enhances PTEN phosphorylation
Since LY294002 abolished the upregulation in p-AKT and the neuroprotection afforded by SC51089, the effect of EP1 receptors on AKT phosphorylation is likely to lie upstream of AKT phosphorylation. Because PTEN negatively regulates AKT activity (Maehama and Dixon 1998) and phosphorylation of PTEN inactivates PTEN, we examined the phosphorylation state of PTEN. As shown in Fig. 4A, p-PTEN was reduced immediately after OGD (Fig. 4A, lanes 1 and 2), correlating with the reduction in p-AKT (Fig. 2). As observed for p-AKT, this early reduction in PTEN phosphorylation was counteracted by SC51089 (Fig. 4A, lane 3). SC51089 did not affect p-PTEN at 1 or 3 hrs after OGD (Fig. 4A, B). Therefore, EP1 receptor inhibition upregulates PTEN phosphorylation after OGD.
Figure 4.
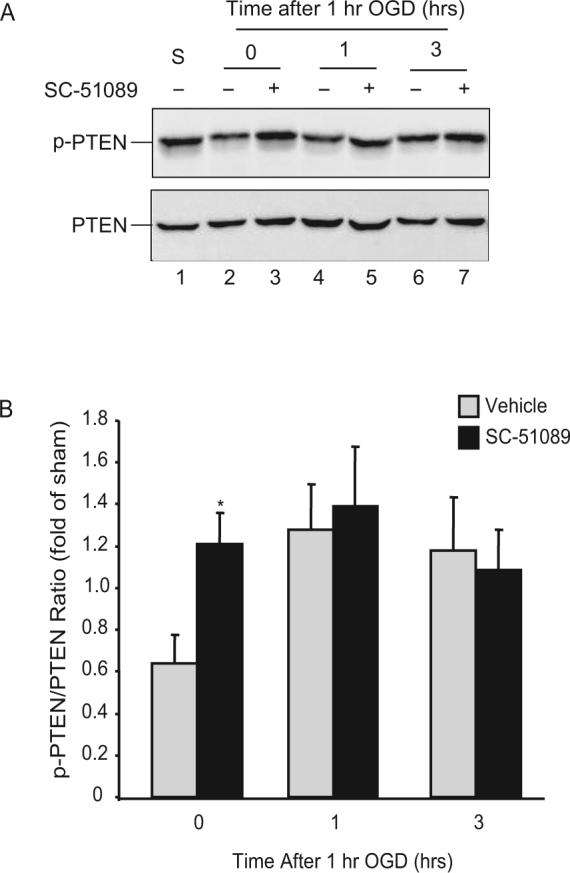
EP1 receptor inhibition increases phosphorylation of PTEN. Hippocampal slices were treated with SC51089 (10μM) and subjected to 1 hr OGD. Protein lysates were made from the slices and the phosphorylation status of PTEN was assessed by immunoblotting. Total PTEN was obtained from the same membrane after stripping and reprobing with PTEN antibodies. (A) The effect of SC51089 on PTEN phosphorylation after OGD. S; sham treated samples. Each lane contains 25μg of protein. One representative blot is shown from 3 separate experiments. (B) Analysis of p-PTEN/PTEN ratio change at time points indicated. Note the increase in p-PTEN after OGD by SC51089 treatment. *p<0.05, n=3/group.
BAD translocation after OGD is attenuated by EP1 inhibition
One mechanism by which p-AKT promotes cell viability is by preventing the translocation of the pro-apoptotic protein BAD into mitochondria (Datta et al. 1997). Therefore, we examined whether the increase in AKT phosphorylation mediated by EP1 receptor inhibition could interfere with the mitochondrial translocation of BAD after OGD. First, we sought to confirm that the OGD-induced cell death in hippocampal slices involves BAD translocation from cytosol to mitochondria. Using Mitotracker Red as a mitochondrial marker we found that BAD immunostaining did not co-localize with mitochondria in sham-treated slices (Fig. 5, panels A-C). Following OGD, BAD immunoreactivity was co-localized with Mitotracker Red (Fig 5, panels D-F), suggesting mitochondrial targeting of BAD immediately after the injury. Furthermore, the mitochondrially targeted BAD appeared as punctate staining surrounding the nuclei (Fig. 5F).
Figure 5.

Colocalization of BAD protein with the mitochondrial marker Mitotracker Red detected by confocal microscope. Hippocampal slices were treated with 1 hr of OGD followed by incubation with Mitotracker Red dye (3 μM) for 15 min. The slices were then fixed after 45 min in normoxia and labeled with BAD specific antibodies. Note the colocalization of BAD (green) with Mitotracker Red (red) after OGD (panel F). Arrows indicate the cells exhibiting both BAD and Mitotracker Red. Scale bar = 10μm.
We then examined the effect of SC51089 on the distribution of BAD immunoreactivity after OGD. Immediately after OGD, BAD immunoreactivity showed a punctate pattern (Fig. 6B), reflecting its mitochondrial localization (Fig. 5). Treatment with SC-51089 resulted in a more diffuse cytoplasmic localization of BAD (Fig. 6C), resembling the pattern observed in sham-treated slices (Fig. 6A). The PI3K inhibitor LY-294002 counteracted the effect of SC51089 on the BAD immunostaining pattern (Fig. 6D, arrows), suggesting that the inhibition of translocation BAD by SC-51089 is AKT dependent. Five hours after OGD, the punctate pattern of BAD was more pronounced and was associated with shrinkage of nuclei, consistent with development of cell death (Fig. 6 E, H) (Henshall et al. 2002; Jiang et al. 2006). In SC51089-treated slices nuclear shrinkage was not as marked as in OGD-treated slices, indicating improved slice viability (Fig. 6 F, I). LY294002 counteracted the effect of SC-51089 on nuclear shrinkage (Fig. 6H, L).
Figure 6.

EP1 receptor inhibition attenuates BAD protein translocation. Hippocampal slices were treated with 1 hr of OGD in the presence or absence of SC51089 (OGD/SC, SC:10μM) or in combination with LY294002 (OGD/SC/LY, LY:20μM). The treated slices were then fixed, processed for BAD immunostaining (green) and counterstained with the nuclear dye DAPI (blue) 1 hr and 5 hrs after1 hr of OGD. Top panel: Confocal images showing the effect of EP1 receptor inhibition on BAD translocation one hour after OGD. Note that the nuclei were not shrunken at this time point (1 hr after 1hr OGD), but that the BAD staining pattern became punctate and perinuclear (A, B). Treatment with SC51089 (C) partially reversed this change and the staining pattern became more similar to sham treated slices (A). LY294002 abolished the reversal of the staining pattern produced by SC51089 (arrows). Bottom panel: effect of EP1 receptor inhibition on BAD translocation five hours after OGD. Five hrs after 1 hr OGD, the cell death process is mostly complete as shown by their uniform shrunken nuclei (F and J). Note the difference between F and G. Treatment with SC51089 partially prevented the unclear shrinkage caused by OGD (G, arrows) and preserved the diffuse pattern of BAD staining. Treatment with LY294002 after SC51089 reversed the protective change induced by SC51089 (H and L). Scale bar = 20 μm for all images.
DISCUSSION
We used an in vitro model of ischemic brain injury to investigate the molecular mechanisms by which activation of EP1 receptors contributes to the brain damage produced by acute cerebral ischemia. We found that hippocampal slices treated with OGD exhibit dephosphorylation of AKT at early stages of the cell death process, and that such loss of phosphorylation could be counteracted by EP1 receptor inhibition. The preserved p-AKT levels induced by EP1 receptor inhibition contribute to the neuroprotection because inhibition of AKT activation with LY294002 blocked AKT phosphorylation and decreased the viability of the slices. Since AKT activity is largely dependent on the levels of PIP3, we examined the possibility that PTEN is involved in the process. Consistent with this prediction, we found that PTEN is rapidly dephosphorylated after OGD and its dephosphorylation is reversed by EP1 receptor inhibition. Unlike AKT, PTEN phosphorylation results in its inactivation, not activation. So a rapid dephosphorylation of PTEN increases PTEN activity and reduces PIP3 availability leading to dephosphorylation of AKT. Therefore, it is likely that the inhibition of EP1 receptors causes PTEN to lose its activity and, in turn, increase AKT activity. Downstream, the sustained p-AKT levels reduced the translocation of BAD from cytosol to mitochondria, a well known phenomenon in BAD mediated apoptotic cell death.
Another finding of the present study is that EP1 receptor inhibition increases AKT phosphorylation even in normal conditions (Fig 2A, lane 2). One possible explanation for this finding is that there is a constitutive interaction between EP1 receptors and the PTEN/AKT pathway and that EP1 receptors negatively regulate AKT phosphorylation. Inhibition of EP1 receptors interferes with this negative interaction, leading to an increase in AKT phosphorylation. Thus, EP1 inhibition could potentiate the AKT pathway and prepare the cells to better withstand stressful events.
The increase in AKT phosphorylation after OGD induced by EP1 receptor inhibition contributes to the increased cell viability, since LY294002 abolished AKT phosphorylation and prevented the increase in cell viability produced by EP1receptor inhibition. Thus, the neuroprotection against OGD mediated by EP1 receptors inhibition is dependent on AKT phosphorylation. The p-AKT level increase mediated by SC51089 cannot be the result of non-specific interactions between SC51089 and the PTEN/AKT pathway, because the PGE2 analog17-pt-PGE2 decreases p-AKT levels, an effect blocked by SC51089. Thus, the increase in p-AKT is a specific consequence of EP1 receptor inhibition.
Once activated, AKT phosphorylates several downstream proteins involved in cell metabolism and cell fate (Manning and Cantley 2007). One of the well known targets is the Bcl-2 family member BAD. BAD is a BH3 domain protein and it exerts its pro-apoptotic activity by binding and neutralizing the function of anti-apoptotic BCl-xL (Yang et al. 1995). In normal conditions, BAD is phosphorylated at serine-136 by p-AKT (Datta et al. 1997). Serine-136 phosphorylation makes it possible for BAD to bind to 14−3−3 proteins and to be sequestered in the cytosol (Datta et al. 2000). Following cell stress, the loss of AKT activity leads to BAD dephosphorylation and translocation to mitochondria, where it binds to BCl-xL. The reduced availability of BCL-xL leads to the formation of BAX homodimers and to activation of the mitochondrial cell death pathway (Zha et al. 1996). Our data demonstrate that OGD causes BAD protein translocation to mitochondria, an effect prevented in part by inhibition of EP1 receptors leading to improved cell survival. Thus, the neuroprotection mediated by EP1 receptor inhibition in hippocampal slices involves the activation of the AKT pathway, which in turn prevents the translocation of BAD into mitochondria.
We observed that the PGE2 analog 17-pt-PGE2 is not neurotoxic. This phenomenon was also observed in our earlier study in which 17-pt-PGE2 did not cause brain injury in vivo (Manabe et al. 2004). Similarly, in neuronal cell cultures, 17-pt-PGE2 did not induce cell death unless NMDA was added to the culture (Carlson 2003). Therefore, PGE2 is toxic only in the setting of NMDA receptor activation. One possible explanation for this finding is that, because PGE2 alters Ca2+ homeostasis via effects of EP1 receptors on the Na+/Ca2+ exchanger (Kawano et al. 2006), the deleterious actions of this prostaglandin are not expressed unless the Na+/Ca2+ exchanger is activated during excitotoxicity.
Based on the observations that EP1 receptors are predominantly present in neurons (Kawano et al. 2006) and that OGD-induced cell death in CA1 region involves neurons, we would anticipate that the effect of SC51089 is mainly neuronal. However, interactions with other cells cannot be ruled out at this time. Further studies are needed to identify the cell type involved in the effect of SC51089 in brain slices.
In summary, we have demonstrated that EP1 receptor inhibition increases AKT phosphorylation and leads to neuroprotection in an in vitro model of ischemic brain injury produced by OGD. The increase in p-AKT results from increased phosphorylation of PTEN and leads to a reduction in the mitochondrial translocation of the proapoptotic protein BAD. The findings unveil a previously unrecognized link between EP1 receptors and signaling pathway controlling cell survival, and provide further insight into the mechanisms through which EP1 receptor inhibition exerts its beneficial effect on the injured brain.
ACKNOWLEDGEMENTS
The Authors wish to thank Raymond Delnicki for technical assistance and Dr. Josef Anrather for useful discussion and suggestions.
This work was supported by NIH grant (NS35806).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Breyer RM, Bagdassarian CK, Myers SA, Breyer MD. Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol. 2001;41:661–690. doi: 10.1146/annurev.pharmtox.41.1.661. [DOI] [PubMed] [Google Scholar]
- Carlson NG. Neuroprotection of cultured cortical neurons mediated by the cyclooxygenase-2 inhibitor APHS can be reversed by a prostanoid. J Neurosci Res. 2003;71:79–88. doi: 10.1002/jnr.10465. [DOI] [PubMed] [Google Scholar]
- Collaco-Moraes Y, Aspey B, Harrison M, de Belleroche J. Cyclo-oxygenase-2 messenger RNA induction in focal cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:1366–1372. doi: 10.1097/00004647-199611000-00035. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- Datta SR, Katsov A, Hu L, Petros A, Fesik SW, Yaffe MB, Greenberg ME. 14−3−3 proteins and survival kinases cooperate to inactivate BAD by BH3 domain phosphorylation. Mol Cell. 2000;6:41–51. [PubMed] [Google Scholar]
- Foster FM, Traer CJ, Abraham SM, Fry MJ. The phosphoinositide (PI) 3-kinase family. J Cell Sci. 2003;116:3037–3040. doi: 10.1242/jcs.00609. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Araki T, Schindler CK, Lan J-Q, Tiekoter KL, Taki W, Simon RP. Activation of Bcl-2-Associated Death Protein and Counter-Response of Akt within Cell Populations during Seizure-Induced Neuronal Death. J. Neurosci. 2002;22:8458–8465. doi: 10.1523/JNEUROSCI.22-19-08458.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai K, Hayashi T, Chan PH, Zeng J, Yang GY, Basus VJ, James TL, Litt L. PI3K inhibition in neonatal rat brain slices during and after hypoxia reduces phospho-Akt and increases cytosolic cytochrome c and apoptosis. Brain Res Mol Brain Res. 2004;124:51–61. doi: 10.1016/j.molbrainres.2004.02.009. [DOI] [PubMed] [Google Scholar]
- Iadecola C, Niwa K, Nogawa S, Zhao X, Nagayama M, Araki E, Morham S, Ross ME. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proc Natl Acad Sci U S A. 2001;98:1294–1299. doi: 10.1073/pnas.98.3.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang P, Du W, Heese K, Wu M. The Bad Guy Cooperates with Good Cop p53: Bad Is Transcriptionally Up-Regulated by p53 and Forms a Bad/p53 Complex at the Mitochondria To Induce Apoptosis. Mol. Cell. Biol. 2006;26:9071–9082. doi: 10.1128/MCB.01025-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada H, Nito C, Endo H, Chan PH. Bad as a converging signaling molecule between survival PI3-K/Akt and death JNK in neurons after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2007;27:521–533. doi: 10.1038/sj.jcbfm.9600367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano T, Anrather J, Zhou P, Park L, Wang G, Frys KA, Kunz A, Cho S, Orio M, Iadecola C. Prostaglandin E2 EP1 receptors: downstream effectors of COX-2 neurotoxicity. Nat Med. 2006;12:225–229. doi: 10.1038/nm1362. [DOI] [PubMed] [Google Scholar]
- Kunz A, Anrather J, Zhou P, Orio M, Iadecola C. Cyclooxygenase-2 does not contribute to postischemic production of reactive oxygen species. J Cereb Blood Flow Metab. 2007;27:545–551. doi: 10.1038/sj.jcbfm.9600369. [DOI] [PubMed] [Google Scholar]
- Luo HR, Hattori H, Hossain MA, Hester L, Huang Y, Lee-Kwon W, Donowitz M, Nagata E, Snyder SH. Akt as a mediator of cell death. Proc Natl Acad Sci U S A. 2003;100:11712–11717. doi: 10.1073/pnas.1634990100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Manabe Y, Anrather J, Kawano T, Niwa K, Zhou P, Ross ME, Iadecola C. Prostanoids, not reactive oxygen species, mediate COX-2-dependent neurotoxicity. Ann Neurol. 2004;55:668–675. doi: 10.1002/ana.20078. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen S, Fusco FR, Yrjanheikki J, Keinanen R, Hirvonen T, Roivainen R, Narhi M, Hokfelt T, Koistinaho J. Spreading depression and focal brain ischemia induce cyclooxygenase-2 in cortical neurons through N-methyl-D-aspartic acid-receptors and phospholipase A2. Proc Natl Acad Sci U S A. 1997;94:6500–6505. doi: 10.1073/pnas.94.12.6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D, Toni N, Buchs P, Parisi L, Stoppini L. Interface organotypic hippocamlpal slice cultures. In: Fedoroff S, Richardson A, editors. Protocols for Neural Cell Culture. 3rd Edition Humana Press; 2001. pp. 13–27. [Google Scholar]
- Nogawa S, Zhang F, Ross ME, Iadecola C. Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci. 1997;17:2746–2755. doi: 10.1523/JNEUROSCI.17-08-02746.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Kitagawa K, Sugiura S, Omura-Matsuoka E, Tanaka S, Yagita Y, Okano H, Matsumoto M, Hori M. Implication of cyclooxygenase-2 on enhanced proliferation of neural progenitor cells in the adult mouse hippocampus after ischemia. J Neurosci Res. 2003;72:461–471. doi: 10.1002/jnr.10595. [DOI] [PubMed] [Google Scholar]
- Seo JH, Ahn Y, Lee SR, Yeol Yeo C, Chung Hur K. The major target of the endogenously generated reactive oxygen species in response to insulin stimulation is phosphatase and tensin homolog and not phosphoinositide-3 kinase (PI-3 kinase) in the PI-3 kinase/Akt pathway. Mol Biol Cell. 2005;16:348–357. doi: 10.1091/mbc.E04-05-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turini ME, DuBois RN. Cyclooxygenase-2: a therapeutic target. Annu Rev Med. 2002;53:35–57. doi: 10.1146/annurev.med.53.082901.103952. [DOI] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ. Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell. 1995;80:285–291. doi: 10.1016/0092-8674(95)90411-5. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ. Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell. 1996;87:619–628. doi: 10.1016/s0092-8674(00)81382-3. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Park TS, Gidday JM. Hypoxic preconditioning protects human brain endothelium from ischemic apoptosis by Akt-dependent survivin activation. Am J Physiol Heart Circ Physiol. 2007;292:H2573–2581. doi: 10.1152/ajpheart.01098.2006. [DOI] [PubMed] [Google Scholar]
- Zhao H, Shimohata T, Wang JQ, Sun G, Schaal DW, Sapolsky RM, Steinberg GK. Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J Neurosci. 2005;25:9794–9806. doi: 10.1523/JNEUROSCI.3163-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]