

Abstract
β-Catenin is a cadherin-binding protein involved in cell–cell adhesion, which also functions as a transcriptional activator when complexed in the nucleus with members of the T-cell factor (TCF)/lymphoid enhancer factor (LEF) family of proteins. There is considerable interest in mechanisms that down-regulate β-catenin, since this provides an avenue for the prevention of colorectal and other cancers in which β-catenin is frequently over-expressed. We show here that physiologically relevant concentrations of the tea polyphenol epigallocatechin-3-gallate (EGCG) inhibited β-catenin/TCF-dependent reporter activity in human embryonic kidney 293 cells transfected with wild type or mutant β-catenins, and there was a corresponding decrease in β-catenin protein levels in the nuclear, cytosolic and membrane-associated fractions. However, β-catenin accumulated as punctate aggregates in response to EGCG treatment, including in human colon cancer cells over-expressing β-catenin endogenously. Confocal microscopy studies revealed that the aggregated β-catenin in HEK293 cells was extra-nuclear and co-localized with lysosomes, suggesting that EGCG activated a pathway involving lysosomal trafficking of β-catenin. Lysosomal inhibitors leupeptin and transepoxysuccinyl-L-leucylamido(4-guanido)butane produced an increase in β-catenin protein in total cell lysates, without a concomitant increase in β-catenin transcriptional activity. These data provide the first evidence that EGCG facilitates the trafficking of β-catenin into lysosomes, presumably as a mechanism for sequestering β-catenin and circumventing further nuclear transport and activation of β-catenin/TCF/LEF signaling.
Keywords: β-Catenin/TCF/LEF signaling, EGCG, Colon cancer, Proteasomes, Lysosomes
1. Introduction
There is a great deal of current interest in β-catenin, adenomatous polyposis coli (APC), and other members of the Wnt signaling pathway and their role in the development of human cancers [1–4]. In colorectal cancers, the APC tumor suppressor gene is a common target for mutation [5], but colon tumors with wild type APC typically have genetic changes in CTNNB1, the gene for β-catenin [6–8]. β-Catenin is a cadherin-binding protein involved in cell–cell adhesion [9,10], but also functions as a transcriptional activator when complexed in the nucleus with members of the T-cell factor (TCF)/lymphoid enhancer factor (LEF) family of proteins [1–4]. As a consequence, there is considerable interest in mechanisms that down-regulate β-catenin and/or its target genes, since this provides an avenue for the prevention of colorectal and other cancers.
We recently reported on the inhibition of polyp formation by green tea and white tea in the Apcmin mouse, and observed an apparent decrease in β-catenin protein levels in the intestinal mucosa, along with attenuated expression of downstream β-catenin/Tcf targets, such as cyclin D1 and c-Jun [11]. Green tea and white tea, as well as physiologically relevant concentrations of the major tea polyphenol epigallocatechin-3-gallate (EGCG), also inhibited β-catenin/TCF transcriptional activity in human embryonic kidney 293 (HEK293) cells [12]. There was a corresponding decrease in β-catenin protein expression in total cell lysates, without changes in TCF4 levels. To investigate the mechanism in greater detail, we sought to determine the effects of EGCG on several mutant forms of β-catenin transfected into HEK293 cells, as well as on β-catenin that was expressed endogenously at high levels in colon cancer cells. We report here that β-catenin protein expression was diminished by EGCG in several cellular compartments, but there was an accumulation of β-catenin in lysosomes, without a concomitant increase in transcriptional activity. The results suggest that EGCG activated a pathway of β-catenin trafficking into lysosomes, thereby sequestering β-catenin and limiting the nuclear transport and further activation β-catenin/TCF target genes.
2. Materials and methods
2.1. Materials
HEK293 cells and human colorectal cancer cells (HT29 and HCT116) were obtained from sources described previously [12]. The following chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, USA); EGCG, transepoxysuccinyl-L-leucylamido(4-guanido)-butane (E64), and N-CBZ-Leu-Leu-Leu-AL (MG132). Leupeptin was purchased from Roche (Indianapolis, IN, USA). Minimum essential medium eagles (MEM), McCoys 5A media, horse serum and fetal bovine serum were purchased from Gibco BRL, Life Technologies/Invitrogen (Carlsbad, CA, USA). Effectene transfection reagent was from Qiagen (Valencia, CA, USA), whereas 4′,6-diamidine-2′-phenylindole dihydrochloride (DAPI), Lysotracker Red and Alexa Fluor 488 were purchased from molecular probes (Eugene, OR, USA).
2.2. Expression constructs
The β-catenin/TCF/LEF-responsive luciferase reporter ‘TOPflash’, and constructs for human TCF4 and wild type (WT) β-catenin were provided by Drs. Hans Clevers and Marc van de Wetering. Constructs expressing mutant D32G, D32N, D32Y, S33Y, and Δ45-β-catenin were generated by fragment switching, as described previously [13,14]. Green fluorescent protein (GFP)-WT-β-catenin and GFP-Δ45-β-catenin expression constructs were prepared by cloning the corresponding human β-catenin cDNA’s into an N-terminal GFP Fusion TOPO vector (Invitrogen). The expression vectors were then transformed into TOP10 Escherichia coli cells according to the manufacturer’s instructions.
2.3. Cell culture, transient transfections, and reporter assays
HEK293 cells were grown in MEM with 2 mM L-glutamine supplemented with 10% horse serum and 1 mM sodium pyruvate, whereas HT29 and HCT116 cells were maintained in McCoys 5A media with 10% fetal bovine serum, 100 units/ml penicillin and 100 μg/ml streptomycin (Sigma). Cells were maintained at 37 °C under 5% CO2. Transient co-transfections of HEK293 cells were performed, in triplicate, using effectene transfection reagent (Qiagen), as described elsewhere [12]. Briefly, 1 × 106 cells were seeded onto poly-D-lysine coated 60 mm plates the day before transfection with 0.5 μg each of β-catenin, TCF4, and TOPflash constructs. pSV-β-galactosidase (Promega, Madison, WI, USA) was included as a control for transfection efficiency, and empty vector was used to standardize for the amount of DNA. After 48 h, cells were lysed and reporter activities were determined as published [12,13]. In some experiments, cells were harvested after 48 h and cytoplasmic and nuclear fractions were isolated using NE-PER reagents (Pierce, Rockford, IL, USA). To isolate membrane-associated proteins [15], cells were lysed in 0.5% NP-40, 10 mM Tris–HCl, 2.5 mM MgCl2, and 1 mM phenylmethanesulfonyl fluoride (PMSF) on ice for 20 min. Cells were disrupted with a 21-gauge needle, vortexed, and centrifuged at 10,000 rpm for 3 min at 4 °C. The pellets were lysed in 25 mM NaH2PO4, 0.5 M NaCl, 1 mM EDTA, 0.5% Triton X-100, 10% glycerol, 5 mM MgCl2 and 1 mM PMSF on ice for 20 min with vortexing.
2.4. SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and immunodetection
Protein concentrations were determined as reported previously [16] for total cell lysates, whereas cytoplasmic, nuclear, and membrane-associated proteins were assayed according to manufacturer’s instructions using the Bradford kit (Biorad, Hercules, CA, USA). Equal amounts of protein were loaded onto Nupage 4–12% Bis–Tris gels (Invitrogen) and transferred to nitrocellulose membranes (Invitrogen). Equal loading and protein transfer were confirmed by staining blots with amido black (not shown). The primary antibody was mouse monoclonal anti-β-catenin (Transduction Laboratories, Lexington, KY, USA) or anti-myc tag (Cell Signaling Technology, Beverly, MA, USA), followed by anti-HRPx secondary antibody. Anti-β-actin (Sigma) was used as a loading control. Immunodetection was performed using Western Lightning Chemiluminescence Reagent Plus (PE Life Sciences, Torrance, CA, USA) coupled with image analysis and quantification on an AlphaInnotech photodocumentation system.
2.5. Expression of GFP-fusion proteins, and immunocytochemistry
HEK293 cells were seeded onto 2% gelatin-coated glass coverslips placed within multiwell (six-well) plates. Cells were transiently transfected with GFP-tagged WT- or Δ45-β-catenin using the effectene transfection reagent (Qiagen) and treated with 0 or 25 μM EGCG. After 24 or 48 h, cells were washed in PBS, fixed in 10% buffered neutral formalin, and nuclei were stained with DAPI. Cells were mounted and stored in the dark at −20 °C until viewed with a Zeiss LSM 510 Meta laser scanning confocal microscope. Alternatively, for detection of lysosomes, cells were incubated in 50 nM lysotracker red for 1 h prior to being fixed and stained, as mentioned above. HT29 and HCT116 cells also were seeded onto glass coverslips placed within six-well plates, and when cells reached 80% confluency EGCG or E64 was added to the media. 24 or 48 h later, cells were washed in PBS, fixed, permeabilized in methanol:acetone (1:1) for 10 min at −20 °C, and then stained with DAPI. Non-specific binding was blocked with 2% BSA–0.2% Tween 20 in PBS for 1 h at room temperature, followed by overnight incubation at 4 °C with a 1:2000 dilution of rabbit polyclonal anti-β-catenin antibody (Abcam, Cambridge, MA, USA). The secondary antibody, rabbit IgG conjugated to Alexa fluor 488 (molecular probes), was used at a dilution of 1:1000. Lysosomes were detected using a 1:1000 dilution of mouse monoclonal LAMP-1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA) conjugated to phycoerythrin.
2.6. Flow cytometry
Cells were treated at 80% confluency with 0 or 25 μM EGCG, and 24 h later the harvested cells were counted and fixed in 1% formaldehyde for 20 min at room temperature. Permeabilized cells were incubated in 96-well plates with a 1:1000 dilution of rabbit anti-β-catenin antibody (Abcam). Non-specific binding was blocked by pre-incubating cells with rat IgG, and appropriately labeled isotype controls were used routinely to correct for possible nonspecific fluorescence. Listmode data were collected on a Coulter Epics XL flow cytometer and analyzed using WinList software (Verity Software House Inc., Topsham, ME, USA).
3. Results
3.1. EGCG reduces β-catenin/TCF4-dependent reporter activity and β-catenin protein expression in HEK293 cells
Physiologically relevant concentrations of EGCG were shown previously to inhibit TOPflash reporter activity in HEK293 cells transfected with TCF4 plus WT-β-catenin [12]. To extend these findings, we transfected HEK293 cells with TCF4 plus various β-catenin mutants, and treated the cells with 0, 5, 10 or 25 μM EGCG (Fig. 1). Concentration-dependent inhibition of reporter activity was observed in cells transfected with TCF4 plus WT, Δ45, D32G, D32N, D32Y or S33Y β-catenin (Fig. 1A). The inhibition was most striking in cells transfected with TCF4 plus Δ45-β-catenin, but in all cases 25 μM EGCG lowered reporter activity to a level comparable with that seen for empty vector. Endogenous β-catenin protein was detected at low levels in the total cell lysates of HEK293 cells treated with no DNA or vector alone (Fig. 1B, left lanes, upper blot). β-Catenin levels were increased markedly in cells transfected with TCF4 plus S33Y-β-catenin (Fig. 1B, lane ‘0’), and there was a concentration-dependent decrease in β-catenin protein expression with 5, 10 and 25 μM EGCG treatment. This decrease in protein expression was seen with monoclonal anti-β-catenin antibody, which detects total (endogenous plus transfected) β-catenin, and also with anti-myc-tag antibody, which is specific for the myc tag in transfected β-catenins. These findings with S33Y-β-catenin were recapitulated in HEK293 cells treated with EGCG and WT, Δ45, D32G, D32N, or D32Y-β-catenins (data not shown).
Fig. 1.
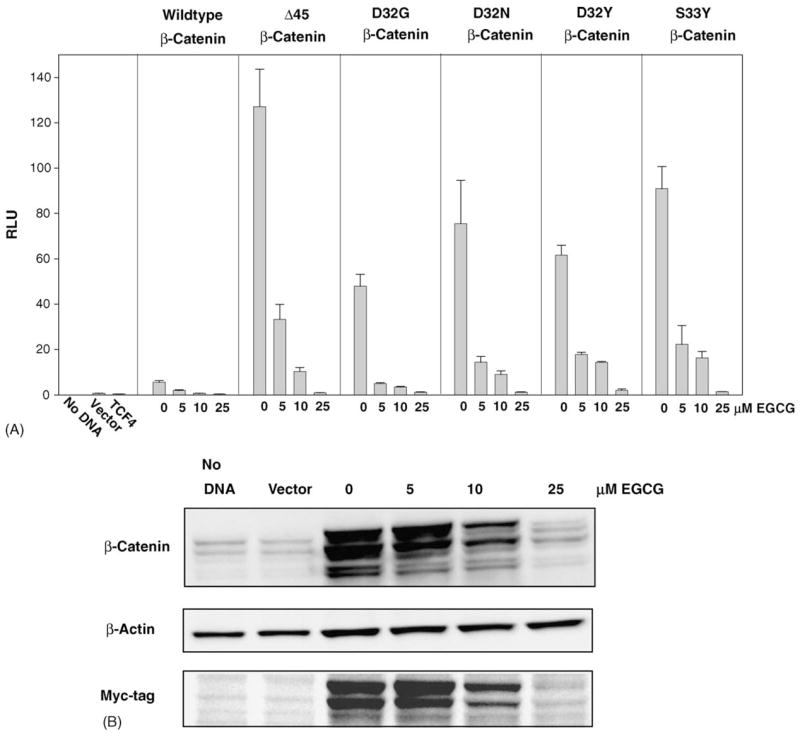
EGCG reduces β-catenin/TCF-dependent reporter activity and β-catenin protein expression in HEK293 cells. (A) Reporter assays of HEK293 cells transiently co-transfected with plasmids expressing wild-type or mutant β-catenin (Δ45, D32G, D32N, D32Y or S33Y), TCF4, and the reporter TOPflash, in the presence of increasing concentrations of EGCG (0, 5, 10, 25 μM). Cells were harvested after 48 h, and luciferase and β-galactosidase reporter assays were performed according to a published methodology [12]. Experiments were conducted three or more times, and the figure shows representative data (mean ± S.D.) from a single assay. (B) Representative immunoblot data of total cell lysates from cells transfected with S33Y-β-catenin and probed with anti-β-catenin, anti-β-actin, and anti-myc-tag antibodies.
3.2. EGCG decreases nuclear, cytoplasmic and membrane-associated β-catenin protein expression, as well as membrane-associated E-cadherin
Because the β-catenin protein levels were attenuated by EGCG in total cell lysates, we next investigated the effect of EGCG treatment on β-catenin expression in the nucleus, cytosol, and membrane-associated fractions of HEK293 cells. As expected, in cells transfected with TCF4 plus S33Y-β-catenin, cytoplasmic (cyt) and nuclear (nuc) β-catenin protein levels were strongly increased compared with cells transfected with empty vector (Fig. 2A). There was a concentration-dependent decrease in β-catenin protein expression both in the cytosol and nucleus of cells treated with 0, 5, 10 or 25 μM EGCG (Fig. 2A). In the membrane-associated fractions, EGCG also attenuated β-catenin expression levels, as well as E-cadherin (Fig. 2B). After normalizing to β-actin, densitometry analyses indicated that nuclear and membrane-associated β-catenin levels, as well as membrane-associated E-cadherin, were decreased by ~50% following treatment with 25 μM EGCG, whereas cytoplasmic β-catenin was reduced by ~25% (Fig. 2C). The immunoblotting data presented in Fig. 2 were from cells transfected with S33Y-β-catenin, but similar results were obtained with EGCG and all other β-catenin mutants studied here, as well as WT-β-catenin (data not shown).
Fig. 2.
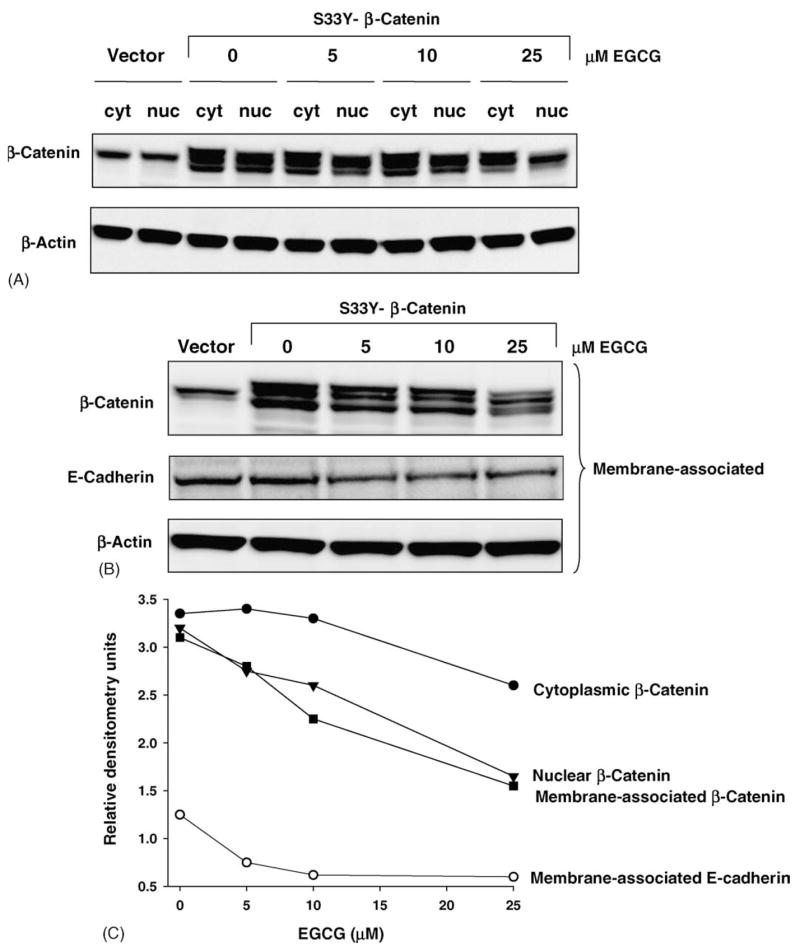
EGCG decreases nuclear, cytosolic, and membrane-associated β-catenin protein expression, as well as membrane-associated E-cadherin. (A) Representative immunoblot data of cytoplasmic and nuclear fractions of HEK293 cells transfected with S33Y-β-catenin and probed with anti-β-catenin and anti-β-actin antibodies. (B) Western blot of membrane-associated proteins from HEK293 cells transiently transfected with S33Y-β-catenin and probed with anti-β-catenin, anti-E-cadherin and anti-β-actin antibodies. (C) Densitometry data for results shown in (A) and (B), normalized to β-actin, showing relative expression levels of β-catenin and E-cadherin with increasing concentration of EGCG.
3.3. Punctate aggregates of β-catenin form in HEK293 cells treated with EGCG
Confocal microscopy studies were next performed in order to examine the loss of β-catenin protein expression in cells treated with EGCG. Transient transfection of HEK293 cells with GFP-WT-β-catenin produced high levels of green fluorescence in all cellular compartments, as expected, indicating that β-catenin was expressed throughout the cell (Fig. 3A, left panel). However, when cells were treated under the same conditions with 25 μM EGCG, fluorescent green ‘dots’ of various sizes were detected, which we refer to hereafter as punctate aggregates of β-catenin (Fig. 3A, right panel). These punctate aggregates were seen only after treatment with EGCG, and only in cells expressing high levels of WT or mutant β-catenin. For example, cells transfected with GFP-Δ45-β-catenin also had punctate aggregates after EGCG treatment, and these aggregates were frequently co-localized within the lysosomal compartment (Fig. 3B). Fluorescence intensities were determined separately for lysosomes and β-catenin in order to calculate the co-localization coefficients, for cells within an entire field of view (Fig. 3C). In cells treated with GFP-Δ45-β-catenin and 25 μM EGCG, 51.7% of the punctate aggregates of β-catenin were co-localized with lysosomes, and 98.3% of the lysosomes were co-localized with the β-catenin aggregates (quantified using Zeiss LSM software). These results suggested that EGCG induced a pathway involving the trafficking of β-catenin into lysosomes.
Fig. 3.

EGCG induces the formation of punctate aggregates in HEK293 cells transfected with GFP-β-catenin fusion proteins. (A) GFP-WT-β-catenin without EGCG treatment (green, left panel) and in the presence of 25 μM EGCG (right panel). (B) Punctate aggregates induced by 25 μM EGCG in cells transfected with GFP-Δ45-β-catenin (green, left panel), staining for lysosomes (red, middle panel), and merged image showing co-localization of β-catenin and lysosomes (in yellow, right panel). (C) Fluorescence intensity maps for lysosomes (left panel) and β-catenin (right panel) were quantified using LSM software (Zeiss) in order to calculate co-localization coefficients (see text).
3.4. Lysosomal trafficking and destruction of β-catenin
To further elucidate the role of lysosomes in the degradation of β-catenin, HEK293 cells were transfected with WT-β-catenin and treated with the lysosomal inhibitors E64 or leupeptin, or with the proteosomal inhibitor MG132. The latter compound resembled EGCG in its ability to inhibit TOPflash reporter activity (Fig. 4A), but unlike EGCG, MG132 strongly increased rather than decreased β-catenin protein levels in the total cell lysates (Fig. 4B, compare lane‘6’ with the corresponding vehicle control in lane ‘5’). Similar results were obtained with the proteasome inhibitor N-acetyl-Leu-Leu-norleucinal (ALLN) in studies that identified proteosomal degradation as a major pathway for β-catenin destruction [13,17]. In both cases, ALLN and MG132 accumulated β-catenin in a pool that exhibited diminished transcriptional activity (Fig. 4A and [13,17]), in contrast to the results obtained with lysosomal inhibitors E64 and leupeptin, which had no significant effect on TOPflash reporter activities. Nonetheless, both of the lysosomal inhibitors increased the β-catenin protein expression in total cell lysates at 48 h (Fig. 4B, compare lanes ‘7’ and ‘9’ for 0 and 50 μM E64, and lanes ‘4’ and ‘10’ for 0 and 50 μM leupeptin). The results indicated that, following transient transfection into HEK293 cells, a portion of the total cellular pool of β-catenin protein was trafficked into lysosomes, and that interference in this pathway caused an accumulation of β-catenin, without increasing the overall transcriptional activity. Control experiments revealed that lysosomal inhibitors had no apparent effect on β-catenin in non-transfected HEK293 cells (data not shown).
Fig. 4.
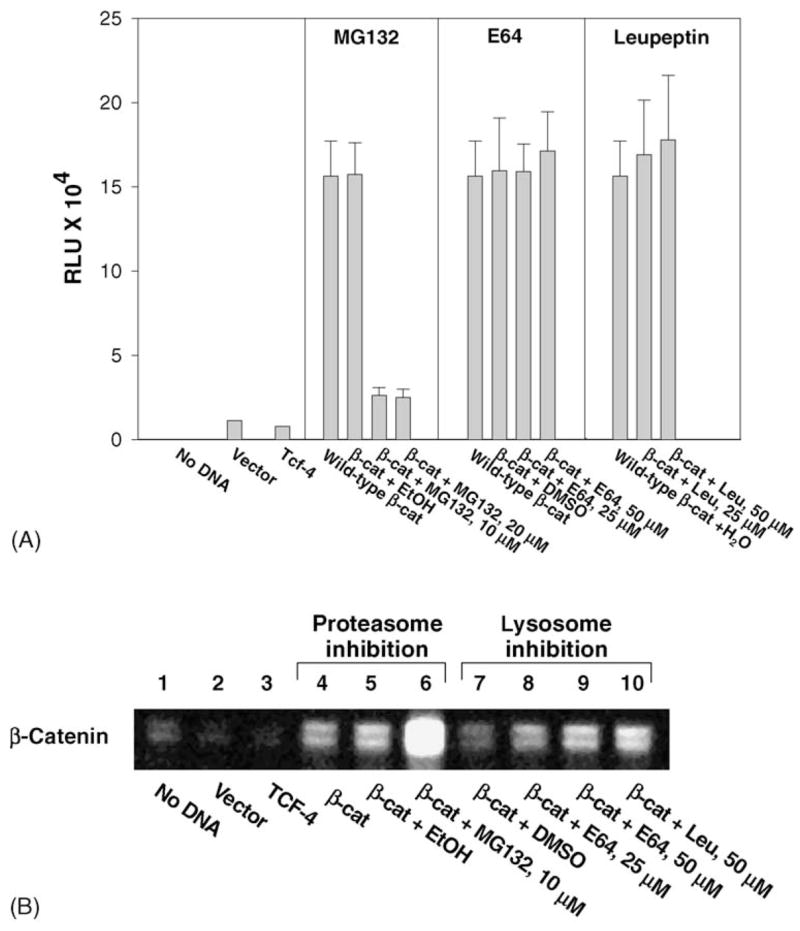
Lysosomal inhibitors induce accumulation of β-catenin without a concomitant increase in transcriptional activity. (A) TOPflash reporter activities in HEK293 cells transfected with TCF4 plus WT-β-catenin and treated with the proteasome inhibitor MG132, or the lysosomal inhibitors E64 or leupeptin. (B) Western blot of β-catenin in total cell lysates from cells transiently transfected with TCF4 plus WT-β-catenin and treated with MG132 or E64. The loading control was β-actin (not shown). EtOH, ethanol vehicle used with MG132; DMSO, vehicle used with E64; H2O was the vehicle control for leupeptin.
3.5. Punctate aggregates of β-catenin also form in human colon cancer cells treated with EGCG
The experiments described to this point involved over-expression of β-catenin by transient transfection into HEK293 cells, which normally express low endogenous levels of cytosolic β-catenin. We next sought to examine the effects of EGCG in cells containing high endogenous levels of β-catenin, namely human colon cancer cells. In HT29 cells, which over-express WT-β-catenin due to mutations in APC, immunocytochemical analyses revealed generalized staining of β-catenin in all cellular compartments, with particularly high levels in the plasma membrane (Fig. 5A, left panel). Punctate aggregates of β-catenin were seen rarely, if ever, in the absence of EGCG treatment. However, aggregates were clearly observed in the presence of 25 μM EGCG (Fig. 5A, right panel), and at higher magnification the accumulated β-catenin was detected almost exclusively outside the nucleus (inset). Fluorescence intensities were determined for lysosomes and β-catenin in the absence of EGCG treatment (Fig. 5B), and an increase was detected in the basal expression levels of both parameters after treatment with 25 μM EGCG (Fig. 5C). Co-localization coefficients indicated an increase in β-catenin within the lysosomal compartment in cells treated with 25 μM EGCG, although this was less marked than in HEK293 cells transfected with exogenous β-catenin. In the presence or absence of EGCG, the highest expression of β-catenin occurred at the cell periphery of HT29 cells (Fig. 5A). Flow cytometry studies revealed, surprisingly, a net increase in total β-catenin expression within the cell population following EGCG treatment (Fig. 5D), without a corresponding increase in the TOPflash activity, including in cells treated with E64 (Fig. 5E). The data presented in Fig. 5 were for HT29 cells treated with EGCG, but similar findings were obtained in HCT116 colon cancer cells, which have high endogenous levels of mutant Δ45-β-catenin (results not shown).
Fig. 5.

EGCG induces punctate aggregates of β-catenin in human colon cancer cells, and an overall accumulation of β-catenin, without a concomitant increase in transcriptional activity. (A) Immunocytochemical localization of endogenous β-catenin in HT29 cells, in the absence (left panel) and presence of 25 μM EGCG treatment (right panel). Green, β-catenin; blue, DAPI. At higher magnification, punctate aggregates of β-catenin were clearly identified as being outside the nucleus (inset, right panel). (B) and (C) 2.5-D images of HT29 cells in the absence and presence of 25 μM EGCG, respectively. Left panels show lysosomal fluorescence intensities, detected by LAMP-1 antibody conjugated to phycoerythrin, and right panels show β-catenin fluorescence intensities determined using anti-β-catenin antibody conjugated to Alexa Fluor 488. (D) Higher net expression of β-catenin in the cell population, 24 h after exposure of HT29 cells to 25 μM EGCG. Histograms depict intracellular staining for β-catenin, as determined by flow cytometry: blue line, isotype control; green line, HT29 cells in the absence of EGCG; red line, HT29 cells treated with 25 μM EGCG. Data shown in the figure are representative of results from three independent experiments. (E) Transcriptional activities in HT29 cells treated with EGCG or E64. Cells were transiently transfected with TOPflash, but no exogenous TCF4 or β-catenin. Data are given as mean ± S.D., n=3.
4. Discussion
The present work confirms and extends our previous studies showing that physiologically relevant concentrations of EGCG inhibited β-catenin/TCF4-dependent transcriptional activity in HEK293 cells transiently transfected with WT-β-catenin [12]. The inhibitory effects reported here for EGCG were seen with several β-catenin mutants (Fig. 1), and expression levels of β-catenin protein were decreased in the nucleus, cytosol, and membrane-associated fractions of HEK293 cells (Fig. 2). However, there was an unexpected increase in β-catenin within the lysosomal compartment, as evidenced by the formation of punctate aggregates and their co-localization with lysosomes (Fig. 3). Previous studies described the appearance of β-catenin aggregates and rod-like structures when GFP-β-catenin was transfected into MDCK and COS7 cells, but these accumulations were detected in the nucleus rather than in lysosomes [18]. In the latter report, punctate spots fused to form rods, and this was observed when total cellular levels of GFP-β-catenin were two to three times higher than the levels of endogenous β-catenin. No rodlike aggregates were seen in the present investigation, and punctate accumulations of β-catenin were detected only in cells treated with EGCG. To our knowledge, this is the first study to show that a natural chemopreventive agent can induce such lysosomal accumulations of β-catenin.
Several of the changes seen with EGCG in HEK293 cells transiently transfected with GFP-β-catenin were recapitulated in human colon cancer cells expressing high endogenous levels of β-catenin (Fig. 5). Thus, EGCG treatment led to the formation of punctate aggregates of β-catenin in HT29 cells, these aggregates were observed outside the nucleus, and there was increased co-localization with lysosomes, although this was less marked than in HEK293 cells transfected with exogenous β-catenin. A key difference between the results obtained in HEK293 cells and in HT29 cells was that, in the latter case, EGCG had no inhibitory effects on β-catenin expression in the total lysates (data not presented). Indeed, flow cytometry studies revealed a net increase in the expression of β-catenin after EGCG treatment in HT29 cells, but this was not associated with an increase in β-catenin/TCF-dependent transcriptional activity (Fig. 5D and E). Our interpretation of these findings is that, in colon cancer cells, EGCG is capable of activating several pathways for trafficking and/or sequestration of β-catenin.
Based on our initial experiments suggesting lysosomal trafficking of β-catenin, we tested the lysosomal inhibitors E64 and leupeptin and observed a consistent increase in β-catenin protein expression in total cell lysates, albeit less marked than in cells treated with the proteasome inhibitors ALLN [13] or MG132 (Fig. 4). As seen with EGCG, the increased expression of β-catenin protein in response to lysoso-mal inhibitors was not associated with a concomitant increase in TOPflash activities. These data suggested that β-catenin was sequestered and incapable of further activation of β-catenin/TCF/LEF target genes, including in HT29 and HCT116 colon cancer cells (Fig. 5, and data not shown).
The working hypothesis is that, under normal circumstances, the ubiquitin/proteasome pathway represents the major route for downregulating β-catenin in cells [17], but alternative pathways become increasingly important as β-catenin accumulates in the cytoplasm. Lysosomal trafficking, even in the short term, might provide an important mechanism by which chemopreventive agents limit β-catenin entry into the nucleus and activation of β-catenin/TCF/LEF target genes. In addition to lysosomal trafficking, other pathways may be activated by chemopreventive agents in order to down-regulate β-catenin expression. An interesting model has been proposed recently for the dynamic turnover of Adherins junctions, in which the novel ubiquitin ligase ‘Hakai’ facilitates the proteosomal degradation of β-catenin and at the same time shunts E-cadherin into lysosomes [19–21]. Based on the results presented here, it is interesting to speculate as to whether β-catenin also might be shunted into lyso-somes in response to chemopreventive agents that activate such a ‘remodeling’ process. We have reported that chlorophyllin and butyrate, which strongly induce E-cadherin expression in cancer cells, shift the pool of β-catenin from the nucleus to the cytoplasm and into the plasma membrane [22,23]. In the present study, there was an apparent decrease in membrane-associated E-cadherin and β-catenin in HEK293 cells treated with EGCG (Fig. 2C), but it remains to be determined whether this involves the activation of Adherins junction remodeling, and whether such a pathway occurs in cancer cells treated with EGCG.
In summary, the present investigation has shown that physiologically relevant concentrations of EGCG inhibited β-catenin/TCF-dependent reporter activity in HEK293 cells transfected with wild type or mutant β-catenins, and there was a corresponding decrease in β-catenin protein levels in the nucleus, cytosol and membrane-associated fractions. β-Catenin accumulated, however, as punctate aggregates in response to EGCG treatment, in cells over-expressing β-catenin endogenously or via transient transfection. The accumulated β-catenin co-localized to a higher extent within lysosomes, and lysosomal inhibitors increased β-catenin protein expression in total cell lysates without augmenting transcriptional activity. These data provide the first evidence for lysosomal trafficking/sequestering of β-catenin in response to EGCG, the major polyphenol in green tea and white tea [11,12]. It remains to be determined whether such a mechanism involves β-catenin/E-cadherin turnover at the plasma membrane via the Hakai pathway, and whether, in the long term, this helps to circumvent β-catenin entry into the nucleus, thereby limiting the activation of β-catenin/TCF/LEF target genes.
Acknowledgments
We thank Dr. Clevers and Dr. van de Wetering, University Hospital Utrecht, The Netherlands, for constructs. This research was supported in part by NIH grants CA65525, CA80176, and CA90890. Studies with the Coulter Epics XL flow cytometer and the Zeiss LSM 510 Meta laser scanning confocal microscope were performed, respectively, in the Cell Culture and Cell and Tissue Analyses Service Cores of the Environmental Health Sciences Center, supported by center grant P30 ES00210. Support for O.C. was provided, in part, by a postdoctoral fellowship, under grant number T32 ES07060 from the National Institute of Environmental Health Sciences.
Abbreviations
- ALLN
N-acetyl-Leu-Leu-norleucinal
- APC
adenomatous polyposis coli
- CTNNB1
HUMAN gene coding for β-catenin
- DAPI
4′,6-diamidine-2′-phenylindole dihydrochloride
- EGCG
epigallocatechin-3-gallate
- E64
transepoxysuccinyl-L-leucylamido(4-guanido)butane
- GFP
green fluorescent protein
- HEK293
human embryonic kidney 293
- LEF
lymphoid enhancer factor
- MEM
Minimum Essential Medium Eagles
- MG132
N-CBZ-Leu-Leu-Leu-AL
- TCF
T-cell factor
- WT
wild type
References
- 1.Polakis P. Wnt signaling and cancer. Genes Develop. 2000;14:1837–1851. [PubMed] [Google Scholar]
- 2.Oving IM, Clevers H. Molecular causes of colon cancer. Eur J Clin Invest. 2002;32:448–457. doi: 10.1046/j.1365-2362.2002.01004.x. [DOI] [PubMed] [Google Scholar]
- 3.Clements WM, Wang J, Sarnaik A, Kim OJ, MacDonald J, Fenoglio-Preiser C, Groden J, Lowry AM. β-Catenin mutation is a frequent cause of Wnt pathway activation in gastric cancer. Cancer Res. 2002;62:3503–3506. [PubMed] [Google Scholar]
- 4.Lustig B, Behrens J. The Wnt signaling pathway and its role in tumor development. J Cancer Res Clin Oncol. 2003;129:199–221. doi: 10.1007/s00432-003-0431-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kinzler KW, Vogelstein B. Cancer-susceptibility genes, gatekeepers and caretakers. Nature. 1997;386:761–763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- 6.Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW. Activation of β-catenin-TCF signaling in colon cancer by mutations in β-catenin or APC. Science. 1997;275:1787–1790. doi: 10.1126/science.275.5307.1787. [DOI] [PubMed] [Google Scholar]
- 7.Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. β-Catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci USA. 1997;94:10330–10334. doi: 10.1073/pnas.94.19.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/β-catenin/TCF pathway in colorectal cancer. Cancer Res. 1998;58:1130–1134. [PubMed] [Google Scholar]
- 9.Hajra KM, Fearon ER. Cadherin and catenin alterations in human cancer. Genes Chromosomes Cancer. 2002;34:255–268. doi: 10.1002/gcc.10083. [DOI] [PubMed] [Google Scholar]
- 10.Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. The cadherincatenin adhesion system in signaling and cancer. J Clin Invest. 2002;109:987–991. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Orner GA, Dashwood WM, Blum CA, Diaz GD, Li Q, Dashwood RH. Suppression of tumorigenesis in the Apcmin mouse: down-regulation of β-catenin signaling by a combination of tea plus sulindac. Carcinogenesis. 2003;24:263–267. doi: 10.1093/carcin/24.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dashwood WM, Orner GA, Dashwood RH. Inhibition of β-catenin/Tcf activity by white tea, green tea, and epigallocatechin-3-gallate (EGCG): minor contribution of H2O2 at physiologically relevant EGCG concentrations. Biochem Biophys Res Commun. 2002;296:584–588. doi: 10.1016/s0006-291x(02)00914-2. [DOI] [PubMed] [Google Scholar]
- 13.Al-Fageeh M, Li Q, Dashwood WM, Myzak MC, Dashwood RH. Phosphorylation and ubiquitination of oncogenic mutants of β-catenin containing substitutions at Asp32. Oncogene. 2004;23:4839–4846. doi: 10.1038/sj.onc.1207634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Porfiri E, Rubinfeld B, Albert I, Hovanes K, Waterman M, Polakis P. Induction of a β-catenin/LEF-1 complex by Wnt-1 and transforming mutants of β-catenin. Oncogene. 1997;15:2833–2839. doi: 10.1038/sj.onc.1201462. [DOI] [PubMed] [Google Scholar]
- 15.Balajee AS, Geard CR. Induction of DNA repair and signal transduction proteins triggered by ionizing radiation in ‘bystander’ cells. http://cpmcnet.columbia.edu/dept/radoncology/crr/reports2001/b10.htm.
- 16.Blum CA, Orner GA, Fong AT, Bailey GS, Stoner GD, Horio DT, Dashwood RH. β-Catenin mutation in rat colon tumors initiated by 1,2-dimethylhydrazine and 2-amino-3-methylimidazo[4,5-f]quinoline, and the effect of post-initiation treatment with chlorophyllin and indole-3-carbinol. Carcinogenesis. 2001;22:315–320. doi: 10.1093/carcin/22.2.315. [DOI] [PubMed] [Google Scholar]
- 17.Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. β-Catenin is a target for the ubiquitin-proteosome pathway. EMBO J. 1997;16:3797–3804. doi: 10.1093/emboj/16.13.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giannini AL, Vivanco MdM, Kypta RM. Analysis of β-catenin aggregation and localization using GFP fusion proteins: nuclear import of β-catenin by the α-catenin/Tcf complex. Exp Cell Res. 2000;255:207–220. doi: 10.1006/excr.1999.4785. [DOI] [PubMed] [Google Scholar]
- 19.Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, Sommer T, Birchmeier W. Hakai a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol. 2002;4:222–231. doi: 10.1038/ncb758. [DOI] [PubMed] [Google Scholar]
- 20.Pece S, Gutkind JS. E-cadherin and Hakai: signaling, remodeling, or destruction? Nat Cell Biol. 2002;4:E72–E74. doi: 10.1038/ncb0402-e72. [DOI] [PubMed] [Google Scholar]
- 21.Dashwood RH. Adherins junction remodeling – a novel target for cancer chemo-prevention and chemotherapy? Int J Cancer Prev. 2004;1:15–18. [Google Scholar]
- 22.Diaz GD, Li Q, Dashwood RH. Caspase-8 and apoptosis-inducing factor mediate a cytochrome c-independent pathway of apoptosis in human colon cancer cells induced by the dietary phytochemical chlorophyllin. Cancer Res. 2003;63:1254–1261. [PubMed] [Google Scholar]
- 23.Carter O, Bailey GS, Dashwood RH. The dietary phytochemical chlorophyllin alters E-cadherin and β-catenin expression in human colon cancer cells. J Nutr. 2004;134:3441S–3444S. doi: 10.1093/jn/134.12.3441S. [DOI] [PMC free article] [PubMed] [Google Scholar]