

Abstract
Sulforaphane (SFN) is an isothiocyanate found in cruciferous vegetables, with particularly high levels detected in broccoli and broccoli sprouts. Over a decade ago, this phytochemical was identified as a likely chemopreventive agent based on its ability to induce Phase 2 detoxification enzymes, as well as to inhibit Phase 1 enzymes involved in carcinogen activation. Considerable attention has focused on SFN as a ‘blocking’ agent, with the ability to modulate the Nrf2/Keap1 pathway, but recent evidence suggests that SFN acts by numerous other mechanisms. SFN induces cell cycle arrest and apoptosis in cancer cells, inhibits tubulin polymerization, activates checkpoint 2 kinase, and inhibits histone deacetylase activity. The latter findings suggest that SFN may be effective during the post-initiation stages of carcinogenesis, as a ‘suppressing’ agent. Moreover, pharmacological administration of SFN may be a promising therapeutic approach to the treatment of cancers, including those characterized by increased inflammation and involving viral or bacterial-related pathologies. The present review discusses the more widely established chemoprotective mechanisms of SFN, but makes the case for additional work on mechanisms that might be of importance during later stages of carcinogenesis, beyond Keap1.
Keywords: Sulforaphane, Chemoprevention
1. Introduction
An estimated 1.4 million Americans will be diagnosed with cancer in 2005, with over 570,000 mortalities (American Cancer Society). Although advances in treatment have been made, there continues to be a need for intervention strategies, including compounds that act as primary protective agents by preventing, delaying, or reversing pre-neoplastic lesions, as well as those that act on secondary or recurrent cancers as therapeutic agents. Compounds found in the diet are of particular interest because of their accessibility to the general population, and ongoing research continues to identify novel candidates for use in cancer chemoprevention clinical trials.
Isothiocyanates (ITCs) are found in cruciferous vegetables such as broccoli, Brussels sprouts, cauliflower, and cabbage. Sulforaphane (SFN) is an ITC found at high levels in broccoli and broccoli sprouts [1,2]. Since it was first identified in 1992 as a potential chemopreventive agent [1], there has been a sharp increase in PubMed citations mentioning SFN (Fig. 1). Early research focused on Phase 2 enzyme induction by SFN, as well as inhibition of enzymes involved in carcinogen activation, but there has been growing interest in other mechanisms of chemoprotection by SFN.
Fig. 1.

Increasing interest in SFN, based on annual citations in PubMed.
Thus, in addition to its ability to act as a ‘blocking’ agent against early initiating events, recent evidence points to SFN as a ‘suppressing’ agent, helping to delay or reverse growth and/or survival of transformed cells (Fig. 2). The precise mechanism(s) that operate during the post-initiation phase are not well understood, and only a handful of suppressing agents have been studied in any detail. Nonetheless, this review considers the evidence that SFN might represent a multi-faceted chemopreventive agent, with the ability to act during blocking, suppressing and therapeutic stages. The various mechanisms will be discussed in turn, namely: (1) inhibition of Phase 1 enzymes, (2) modulation of Phase 2 enzymes, (3) eradication of infection, (4) inhibition of growth promoting signaling pathways, (5) alteration of signaling pathways, cell cycle arrest, and apoptosis, and (6) inhibition of recurrence (refer to Fig. 2).
Fig. 2.

SFN acts at various steps during multi-stage carcinogenesis. This figure illustrates the various ‘blocking’ and ‘suppressing’ mechanisms by which SFN inhibits multi-stage carcinogenesis. Each of the protective mechanisms (1–6) is described in further detail in the text. Adapted from a previous review [5].
2. Prevention of chemical carcinogenesis: carcinogen detoxification
Pathways that augment carcinogen detoxification and excretion can reduce reactive intermediate(s) that might otherwise interact with DNA, thereby effectively blocking initiating events early in carcinogenesis. Evidence suggests that SFN accomplishes this through two mechanisms, namely inhibition of Phase 1 enzymes and induction of Phase 2 enzymes (Fig. 2).
2.1. Inhibition of Phase 1 enzymes
Phase 1 enzymes can metabolically activate pro-carcinogens; thus, under certain circumstances, the inhibition of Phase 1 enzymes can be considered a preventive measure against chemically induced carcinogenesis. Activities of cytochromes P450 (CYPs) 1A1 and 2B1/2 were inhibited dose-dependently by SFN in rat hepatocytes, as determined by 7-ethoxyresorufin-O-deethylase (EROD) and pentoxyresorufin-O-dealkylase (PROD), respectively [3]. In human hepatocytes, SFN decreased mRNA levels of CYP3A4, and this translated to a decrease in CYP3A4 activity [3]. Furthermore, studies of p-nitro-phenol hydroxylase activity in acetone-induced rat liver microsomes established that SFN was a competitive inhibitor of CYP2E1 [4]. Subsequent work in mouse hepatocytes showed dose-dependent inhibition of unscheduled DNA synthesis induced by N-nitrosodimethylamine (NDMA), a carcinogen known to be activated by CYP2E1, and SFN exhibited antimutagenic effects against NDMA at nanomolar concentrations in the Salmonella assay. SFN had no effects on the mutagenicity of sodium azide, a direct-acting mutagen [4], suggesting that SFN indeed inhibited one or more enzymes in the S9 fraction. Interestingly, SFN was reported to have no effect against CYP1A2 [4], an enzyme involved in the metabolic activation of heterocyclic amines [5]. Nonetheless, SFN inhibited the S9-dependent mutagenic activities of several heterocyclic amines in the Salmonella assay, including 2-amino-3-methylimidazo[4,5-f]quinoline (IQ), 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP), and 3-amino-1,4-dimethyl-5H-pyridol[4,3-b] indole (Trp-P-1) [6]. Other mechanisms were not examined for SFN, such as complex formation with the carcinogen, degradation of the activated metabolites, or scavenging of electrophiles [5], which can serve to limit the extent to which initiating agents interact with DNA and form covalent adducts. Interestingly, SFN decreased the number of PhIP-DNA adducts by approximately 40% in human HepG2 cells pre-treated with 1 μM SFN followed by 10 ng PhIP; however, there was no decrease in DNA adduct formation with post-treatment of SFN, suggesting that SFN had no effect on PhIP DNA-adduct repair, but rather prevented PhIP interactions with DNA [7]. SFN was also shown to protect against benzo(a)pyrene (BaP)-induced single-strand DNA breaks in the comet assay, with concentrations as low as 5 μM SFN [8]. Using THLE cells that express CYP2E1 or CYP1A2, SFN also inhibited double-strand breaks initiated by NDMA and IQ, respectively [9]. In non-cancerous human mammary epithelial MCF-10F cells, SFN provided protection against DNA adduct formation by BaP and 1,6-dinitropyrene [10]. In the latter studies, protection was observed by concentrations as low as 0.1 μM SFN, which inhibited 68% of BaP-DNA adducts, with a maximum of 80% inhibition being detected at 2 μM SFN. Interestingly, an increase in NADPH:quinone reductase (NQO1) and glutathione-S-transferase P1 (GSTP1) protein expression was also reported in these cells [10]. Collectively, these reports provide evidence for protection by SFN against carcinogen-DNA damage in vitro and suggest a role for inhibition of certain Phase 1 enzymes, but they do not completely rule out the possibility that SFN operates via other blocking mechanisms.
2.2. Modulation of Phase 2 enzymes
An additional blocking mechanism, and indeed the first to be associated with SFN, was the potent induction of Phase 2 enzymes. SFN augments various pathways known to facilitate carcinogen detoxification and excretion. Using quinone reductase (QR) activity as a measure for inducer activity in Hepa 1c1c7 murine hepatoma cells, SFN was shown to double QR activity at concentrations on the order of 0.4–0.8 μM SFN [1]. Furthermore, mice treated by gavage with 15 μmol SFN/mouse per day for 5 days had an increase in QR and glutathione-S-transferase (GST) in the liver, forestomach, glandular stomach, proximal small intestine, and lungs [1], establishing that SFN also induces Phase 2 enzymes in vivo following oral ingestion.
Although many cell lines respond to SFN, there is some variability in the extent and type of Phase 2 enzyme induction, depending on cell type. In human HepG2 cells, for example, SFN increases mRNA of UDP-glucuronosyltranferase (UGT) 1A1 and GSTA1 [7], increases QR activity [11], and increases UGT1A1 protein as well as bilirubin glucuronidation [12]. An increase in GST and QR activities also was observed with SFN treatment in murine Hepa1c1c7 cells [11,13]. Whereas there was an apparent decrease in GST activity in HT29 cells, undifferentiated CaCo-2 cells demonstrated a dose- and time-dependent induction of GSTA1 and UGT1A1 upon SFN administration [14]. Human prostate cells responded to SFN with an increase in NQO1 and GSTA1 mRNA, as well as an increase in microsomal GST activity and QR activity [15]. Furthermore, SFN increased γ-glutamylcysteine synthetase light chain mRNA, resulting in an increase in cellular glutathione [15]. In primary rat hepatocytes, SFN has been reported to induce mRNA of GSTA1/2 and GSTP1 [3], whereas in human primary hepatocytes, SFN induced mRNA of GSTA1/2 and GSTM1 but decreased mRNA of CYP3A4 [3]. The net result of the aforementioned responses to SFN is to increase cellular defenses, leading to enhanced carcinogen detoxification and protection against potential mutagenic events.
Importantly, these effects of SFN in cell culture can be extrapolated to in vivo situations. As previously mentioned, induction of GST and QR activities was observed in tissues of mice given SFN orally, demonstrating that systemic uptake and bioavailability of SFN and/or its metabolites was feasible [1]. Confirming these observations, rats treated by gavage for 5 consecutive days demonstrated a dose-dependent (200–1000 μmol SFN/kg per day) increase in QR and GST activity in the liver, colon, and pancreas [13], and 40 μmol SFN/kg per day for 5 days by gavage increased QR and GST activities in the rat forestomach, duodenum, and bladder [16].
In recent years, two main mechanisms have been proposed to explain the induction of Phase 2 enzymes by SFN, namely disruption of Nrf2–Keap1 interactions and mitogen-activated protein kinase (MAPK) activation (Fig. 3). These mechanisms may act synergistically to induce genes that contain the so-called ‘antioxidant response element’ (ARE). In early experiments, SFN was shown to be the most potent inducer among several compounds tested in an ARE reporter assay [17]. The disruption of Nrf2–Keap1 interactions, the translocation of Nrf2 to the nucleus, and the induction ARE-containing genes has been extensively reviewed for SFN [18–21]. Recent evidence suggests that specific cysteine residues of Keap1, the anchor that prevents Nrf2 from entering the nucleus, act as ‘sensors’ that are modified by SFN and other Phase 2 enzyme-inducers [22]. In mice treated by gavage with 9 μmol SFN per day for 1 week, several classes of genes were identified as targets of SFN in a transcriptional microarray, including cellular NADPH regenerating enzymes, xenobiotic metabolizing enzymes, antioxidant enzymes, and biosynthetic enzymes of glutathione and glucuronidation conjugation pathways [23]. The importance of Nrf2 was illustrated through the use of Nrf2 knockout mice, in which the response to SFN in induction of the aforementioned classes of genes was abrogated. Further studies in which mice were fed broccoli seeds, which contain high levels of SFN, in the diet for 7 days resulted in an increase in protein expression of NQO1, GSTA1/2, GSTA3, and GSTM1/2 in the stomach, small intestine, and liver of wild-type mice [24]. Although the response in Nrf2 knockout mice was not as strong compared to wild-type mice, broccoli seeds nonetheless were able to increase GSTA1/2, GSTA3, and GSTP1 in the stomach and GSTA3 in the liver, hinting at other possible mechanisms.
Fig. 3.
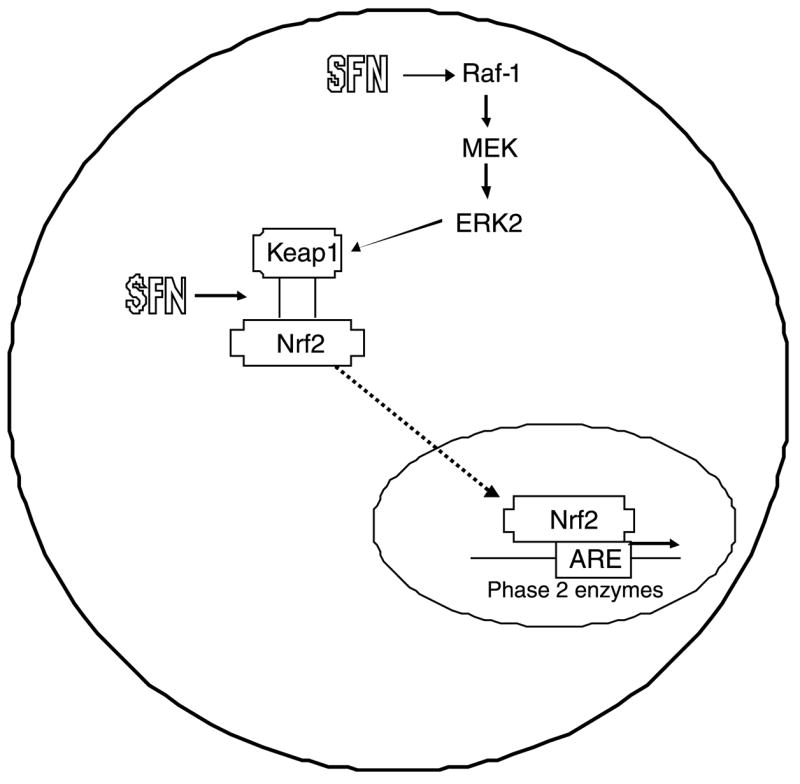
SFN induces ARE expression through disruption of the Keap1–Nrf2 complex. SFN can interact directly with sulfhydryl residues on Keap1, causing Nrf2 to be released. Alternatively, SFN can activate the MAPK pathway, causing phosphorylation of Keap1 and release of Nrf2. Once released, Nrf2 enters the nucleus, where it transactivates ARE-responsive genes.
An additional mechanism through which SFN might activate ARE-driven genes is via the mitogen activated protein kinase (MAPK) pathway (Fig. 3). SFN was shown to increase ERK2 activity, which is downstream from Raf-1 and MEK [25]. Use of a MAPK inhibitor attenuated activation of QR activity in HepG2 cells and ARE reporter activity, and SFN directly activated Raf-1 [25]. Subsequently, MAPK activation was observed in rat livers 2 h after rats were treated by gavage with 50 μmol SFN [26]. This, along with other data (reviewed in [27,28], suggests that MAPK activation may play a role in the activation of Nrf2, and that SFN may regulate ARE-mediated transcription both through its effects on Keap1–Nrf2 destabilization, as well as MAPK activation.
Blocking effects of SFN also have been reported in other studies, in vivo. Administration of 75 or 150 μmol SFN 3 days prior and 1 day after a single dose of DMBA dose-dependently decreased mammary tumor incidence and multiplicity in Sprague–Dawley rats [29]. Importantly, the latter study also showed a delay in onset of tumor formation. Hot water extracts from broccoli sprouts, containing high levels of the SFN precursor glucoraphanin, were inhibitory toward DMBA-induced mammary tumors [2]. SFN blocked BaP-initiated forestomach tumors in mice [30]. Interestingly, in a pre- versus post-initiation experiment, SFN and its metabolite SFN-N-acetyl-cysteine (SFN-NAC) demonstrated different outcomes in prevention of azoxymethane (AOM)-induced colonic aberrant crypt foci [31]. SFN administered both pre- and post-initiated effectively inhibited the total number of crypts and the number of rats with more than four crypts. However, SFN-NAC was active only post-initiation [31], suggesting that the parent compound, SFN, and the metabolite, SFN-NAC, may elicit different mechanisms of chemoprevention. The results of the latter study clearly suggest that SFN acts not only as a blocking agent, but also as a suppressing agent during later stages (Fig. 2).
2.3. Eradication of infection
Although many studies with SFN have focused on detoxification of chemical carcinogens, evidence also has been reported for protective effects against viral or bacterial pathogens (Fig. 3). For example, chronic infection with Helicobacter pylori (H. pylori) is associated with an increased risk of gastric cancer [32]. SFN was recently shown to inhibit several strains of H. pylori, including antibiotic-resistant strains, with a minimal inhibitory concentration of 4 μg/ml [30]. Moreover, SFN administered at 7.5 μmol per day for 5 days eradicated H. pylori from human gastric xenografts in nude mice [33]. The precise mechanism(s) through which SFN protected in these studies remains unknown.
2.3.1. Chemoprevention: beyond Keap1
Only recently has SFN been described as a chemopreventive and therapeutic agent that might act through suppressing mechanisms, i.e. post-initiation. Some studies reported decreased viability of neoplastically transformed cells as compared with the corresponding non-transformed cells or pre-neoplastic cells, suggesting some selectivity of SFN action. The downstream effects of SFN appear to be dependent on cell type, as well as the concentration and length of SFN treatment. While several studies have described effects on signaling pathways, cell cycle arrest and/or apoptosis (Table 1), relatively few mechanisms have been explored to explain precisely how SFN mediates these changes.
Table 1.
Responses to SFN treatment in various cell lines
| Cells | Dose (mM) | Treatment time (h) | Changes detected | References |
|---|---|---|---|---|
| HT-29 | 15 | 48 | Decreased cell viability; increased Bax, cyclin A, cyclin B1, p21, Rb phosphorylation, cdc2 kinase activities; cytochrome c release from mitochondria; G2/M arrest; roscovitine decreased SFN-induced apoptosis | [36–38] |
| HCT116 | 15 | 48 | Increased p21, acetylated histones H3 and H4 | [51] |
| CaCo-2 | 50 | 48 | Decreased cell viability | [36] |
| LnCaP | 9–50 | 30 | G1 arrest; sub-G1 peak; decreased DNA synthesis | [39] |
| DU-145 | 3–90 | 24–48 | G1 arrest; increased p21, TUNEL, caspase activity; Decreased cyclin D1 | [40] |
| PC-3 | 20–40 | 4–72 | G2/M arrest; increased sub-G1, increased Chk2 kinase activity, caspase activity, Bax; decreased cell viability, cyclin B1, Cdc25B, Cdc25C | [41,42] |
| F3II | 5 | 24 | IC50=5 μM; G2/M arrest | [43] |
| 100–300 | 1 | Interference with tubulin polymerization in vitro | ||
| 15 | 36 | Increased cdc2 kinase activity; decreased Bcl-2 | ||
| MCF-7 | 15 | 24 | G2/M arrest; increased cyclin B1; disruption of microtubule polymerization | [44,45] |
| 13.7 | 48 | IC50=13.7 μM by sulforhodamine B assay | ||
| MCF-12A | 40.5 | 48 | IC50=40.5 μM by sulforhodamine B assay | [45] |
| Lymphoblastoid | 3.9 | 48 | IC50=3.9 μM for loss of cell viability; increased apoptosis | [46] |
| 1 | 48 | |||
| HL-60 | 3.4 | 72 | IC50=3.4 mM for loss of cell viability | [47] |
| Jurkat | 30 | 48 | G2/M arrest; increased Bax, p53, apoptosis; decreased Bcl-2 | [48] |
| DAOY | 2 | 24 | Increased caspase activity | [49] |
| 10 | 18 | Increased TUNEL, cleaved PARP | ||
| 10 | 24 | Loss of cell viability (40% of controls) | ||
| MIA PaCa-2 | 10–40 | 24 | G2/M arrest; caspase-3 cleavage; decreased cell viability, GSH levels; no Chk1 activation | [50] |
| PANC-1 | 5 | 24 | G2/M arrest; decreased cell viability, GSH levels | [50] |
2.4. Inhibition of growth-promoting signaling pathways
During the various steps of multi-stage carcinogenesis (Fig. 2), tumor modulators can alter signaling pathways in a manner that promotes or inhibits cell growth. Whereas initiating events often constitutively activate survival and/or growth signaling pathways, chemopreventive agents can override the aberrant signaling events and thereby act as tumor suppressors. SFN interferes with two well-known signaling pathways, beyond Keap1. First, SFN acts as an anti-inflammatory agent through decreasing the DNA binding capability of nuclear factor κB (NFκB) [34]. In murine RAW macrophages, SFN decreased lipopolysaccharide (LPS)-induced nitric oxide (NO), prostaglandin E2, and tumor necrosis factor μ(TNFμ), as well as inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX2) protein expression and mRNA levels [34]. Second, SFN inhibited UVB-induced AP-1 activation in human keratinocytes at concentrations ranging from 1 to 10 μM [35]. It was shown that SFN interfered with AP-1 binding to DNA, and cysteine residues in AP-1 may be important for the inhibitory effects [35].
2.5. Induction of cell cycle arrest and/or apoptosis
The effects of SFN on cell viability were described in HT-29 human colon cancer cells, where 100 μM SFN for 24 or 48 h resulted in a decrease in cell viability to 10% of controls [36]. Further work established an IC50 of 15 μM SFN in HT-29 cells and 50 μM in CaCo-2 cells, suggesting possible selective effects toward undifferentiated versus differentiated cells, respectively [36]. Alterations in cell cycle progression also have been reported in HT-29 cells treated with SFN, with a decrease in the percentage of cells in G1 and an increase in G2/M, as well as increases in cyclin A and cyclin B1 protein levels [37], and hyperphosphorylation of the retinoblastoma tumor suppressor protein (Rb) [38]. Furthermore, there was an increase in Bax protein expression, loss of cytochrome c from mitochondria, and concomitant elevation of cytosolic cytochrome c [37]. Apoptotic markers also were observed upon treatment with SFN, such as phosphatidylserine translocation, nuclear condensation, membrane blebbing, and vesicle formation [37].
Similar responses have been observed in human prostate cancer cells after treatment with SFN. Androgen-dependent LnCaP cells exposed to 9 μM SFN underwent a G1 arrest, whereas increasing the dose to 50 μM caused the cells to become apoptotic, with activation of caspases and a dose-dependent decrease in DNA synthesis [39]. SFN caused androgen-independent DU-145 cells to also undergo G1 cell cycle arrest, with inhibition of Cdk4 activity, an increase in p21Cip1/Waf1 expression, and a decrease in cyclin D1 expression [40]. Furthermore, apoptosis was induced in these cells, as evidence by increased terminal deoxynucleotidyl transferase-mediated nick-end labeling (TUNEL), increased caspase-3 (cleaved), and decreased Bcl-2 expression [40]. While the latter studies with LnCaP and DU-145 cells reported a G1 arrest, SFN caused an apparent G2/M arrest in PC-3 cells [41]. The latter cells also underwent apoptosis, as evidenced by an increase in DNA fragmentation, cleavage of poly-ADP ribose polymerase (PARP), and increased expression of Bax and cleaved caspase-3 and caspase-9 [42].
SFN-mediated cell cycle arrest and apoptosis has been reported in several other cell lines. For example, a G2/M arrest was described in murine sarcomatid mammary F311 cells [43] and human MCF-7 mammary cancer cells [44], with cytotoxic effects also reported in MCF-7 cells [45]. Loss of cell viability was observed with concentrations as low as 1 μM SFN in lymphoblastoid cells [46], 4.3 μM SFN in HL60 cells [47], and 3 μM SFN in human T-cell leukemia cells [48]. Apoptosis induction in medulloblastoma cells occurred at 5 μM SFN, with an 8-fold increase in caspase activity at 10 μM SFN [49]. SFN also elicited caspase-3 cleavage and G2/M arrest in human pancreatic cancer cells, and cellular toxicity due to SFN was positively correlated with a decrease in cellular GSH levels [50]. Based on the numerous reports of cell cycle arrest and apoptotic activity induced by SFN in multiple cell types, SFN can potentially act against several types of cancer, and at early, intermediate and late stages (Fig. 2).
Apoptotic markers have also been observed with SFN in xenograft models. For example, in BALB/c mice implanted with murine mammary carcinoma cells and subsequently given 15 nmol SFN for 13 days by i.v. injection there was a 60% decrease in tumor mass, a decrease in proliferating cell nuclear antigen (PCNA) levels, and an increase in cleaved PARP [43]. PC-3 xenografts from mice treated by gavage with 5.6 μmol SFN three times per week for 3 weeks were approximately one-fourth the volume compared to controls, and the implants from SFN-treated mice exhibited an increase in TUNEL staining, and increases in the expression of pro-apoptotic proteins such as Bax and Bid [42].
Several mechanisms have been proposed to explain how SFN induces changes in cell cycle progression and apoptosis. First, two groups demonstrated that SFN maintains Cdc2 kinase in its active form, which was correlated with induction of apoptosis [38,43]. However, another study reported reduction of Cdc25C, a phosphatase responsible for activation of Cdc2; this reduction of Cdc25C by SFN was mediated via activation of checkpoint 2 kinase (Chk2) [41]. Reduction of Chk2 by siRNA slightly decreased SFN-mediated G2/M arrest, suggesting that Chk2-independent mechanisms also contribute to SFN-induced cell cycle arrest, but Chk2 protein levels may influence the sensitivity of cells to SFN [41]. Based on observations that SFN treatment resulted in condensed, unaligned chromatin at the metaphase plate in MCF-7 cells [44], and aberrant and mildly depolymerized spindles in F311 cells [43], it was hypothesized that SFN disrupts tubulin polymerization, resulting in cell cycle arrest during mitosis. SFN did decrease tubulin polymerization in vitro, albeit at non-physiological concentrations (100–300 μM) [43,44].
Finally, SFN was recently identified as a novel histone deacetylase (HDAC) inhibitor in human embryonic kidney 293 (HEK293) and in HCT116 human colon cancer cells [51]. HDAC inhibitors have been shown to induce cell cycle arrest and apoptosis [52], and they appear to have less effect on normal cells, perhaps due to induction of thioredoxin [53]. At physiologically relevant concentrations, SFN increased acetylated histones H3 and H4, and expression of p21Cip1/Waf1. Moreover, in a chromatin immunoprecipitation (ChIP) assay, SFN dose-dependently increased the amount of acetylated histone H4 associated with the P21 promoter. A metabolite of SFN, SFN–cysteine, was shown to be the likely inhibitor of HDAC activity [51]. These findings were recapitulated in benign prostate hyperplasia (BPH-1), LnCaP, and PC-3 human prostate cells, together with marked changes in cell cycle progression and apoptosis (M.C. Myzak et al., unpublished data). Importantly, the promoter region of the P21 gene appears to lack an ARE, supporting a Nrf2-independent mechanism by SFN. HDAC inhibition provides an important new avenue for future research on SFN, both as a tumor suppressing agent and as a chemotherapeutic agent during late stages of carcinogenesis.
3. Conclusions
Accumulating evidence suggests that SFN is a highly promising dietary preventive agent, due to its ability to confer chemoprotection through several distinct pathways and via multiple mechanisms of action. Early studies focused on the potent Phase 2 enzyme inducing activities of SFN, and alterations in Nrf2/Keap1 signaling. However, recent reports have identified SFN effects on cell cycle checkpoint controls, apoptosis mediators, and HDAC activity, suggesting the need to keep one eye beyond Keap1. Due to its ability to induce cell cycle arrest and apoptosis in various tumor cells, SFN has the potential to act against secondary and possibly recurrent cancers (Fig. 2), and the diversity of mechanisms makes SFN a strong candidate for possible therapeutic application, either alone or in combination with other agents that provide synergistic or additive protection. Epidemiological evidence suggests a reduced risk for cancers of the prostate [54–56], lung [57–59], breast [60], and colon [61,62] in people who consume cruciferous vegetables. Further studies will be required to determine to what extent these protective effects of whole vegetables can be attributed to SFN and/or its metabolites, and the various mechanisms discussed in the present review and outlined in Fig. 4.
Fig. 4.
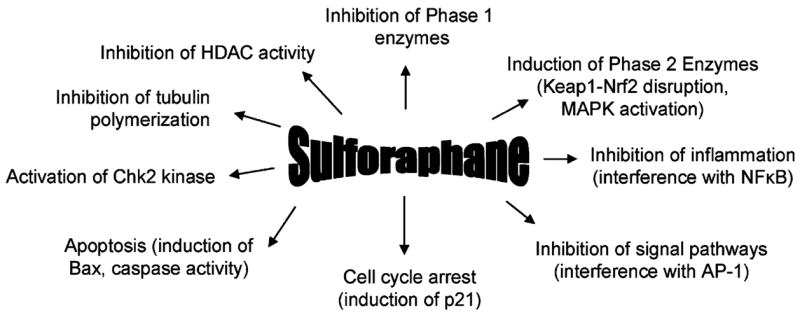
Multiple mechanisms of chemoprotection by SFN. This figure combines empirically tested as well as postulated activities of SFN, based largely on in vitro studies, plus in vivo findings where available. Each of the protective mechanisms is described in further detail in the text.
Acknowledgments
Grant Support: This work was supported in part by NIH grants CA66525 (RHD), CA80176 (RHD), CA90890 (RHD), and by the National Institute of Environmental Health Sciences Center grant P30 ES00210.
References
- 1.Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci USA. 1992;89:2399–2403. doi: 10.1073/pnas.89.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fahey JW, Zhang Y, Talalay P. Broccoli sprouts: an exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc Natl Acad Sci USA. 1997;94:10367–10372. doi: 10.1073/pnas.94.19.10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maheo K, Morel F, Langouet S, Kramer H, Le Ferrec E, Ketterer B, Guillouzo A. Inhibition of cytochromes P-450 and induction of glutathione S-transferases by sulforaphane in primary human and rat hepatocytes. Cancer Res. 1997;57:3649–3652. [PubMed] [Google Scholar]
- 4.Barcelo S, Gardiner JM, Gescher A, Chipman JK. CYP2E1-mediated mechanism of anti-genotoxicity of the broccoli constituent sulforaphane. Carcinogenesis. 1996;17:277–282. doi: 10.1093/carcin/17.2.277. [DOI] [PubMed] [Google Scholar]
- 5.Dashwood RH. Modulation of heterocyclic amine-induced mutagenicity and carcinogenicity: an ‘A-to-Z’ guide to chemopreventive agents, promoters, and transgenic models. Mutat Res. 2002;511:89–112. doi: 10.1016/s1383-5742(02)00005-4. [DOI] [PubMed] [Google Scholar]
- 6.Kaur Shishu IP. Inhibition of mutagenicity of food-derived heterocyclic amines by sulforaphane—a constituent of broccoli. Indian J Exp Biol. 2003;41:216–219. [PubMed] [Google Scholar]
- 7.Bacon JR, Williamson G, Garner RC, Lappin G, Langouet S, Bao Y. Sulforaphane and quercetin modulate PhIP-DNA adduct formation in human HepG2 cells and hepatocytes. Carcinogenesis. 2003;24:1903–1911. doi: 10.1093/carcin/bgg157. [DOI] [PubMed] [Google Scholar]
- 8.Bonnesen C, Eggleston IM, Hayes JD. Dietary indoles and isothiocyanates that are generated from cruciferous vegetables can both stimulate apoptosis and confer protection against DNA damage in human colon cell lines. Cancer Res. 2001;61:6120–6130. [PubMed] [Google Scholar]
- 9.Barcelo S, Mace K, Pfeifer AM, Chipman JK. Production of DNA strand breaks by N-nitrosodimethylamine and 2-amino-3-methylimidazo[4,5-f]quinoline in THLE cells expressing human CYP isoenzymes and inhibition by sulforaphane. Mutat Res. 1998;402:111–120. doi: 10.1016/s0027-5107(97)00288-1. [DOI] [PubMed] [Google Scholar]
- 10.Singletary K, MacDonald C. Inhibition of benzo[a]pyrene-and 1,6-dinitropyrene-DNA adduct formation in human mammary epithelial cells bydibenzoylmethane and sulforaphane. Cancer Lett. 2000;155:47–54. doi: 10.1016/s0304-3835(00)00412-2. [DOI] [PubMed] [Google Scholar]
- 11.Jiang ZQ, Chen C, Yang B, Hebbar V, Kong AN. Differential responses from seven mammalian cell lines to the treatments of detoxifying enzyme inducers. Life Sci. 2003;72:2243–2253. doi: 10.1016/s0024-3205(03)00101-2. [DOI] [PubMed] [Google Scholar]
- 12.Basten GP, Bao Y, Williamson G. Sulforaphane and its glutathione conjugate but not sulforaphane nitrile induce UDP-glucuronosyl transferase (UGT1A1) and glutathione transferase (GSTA1) in cultured cells. Carcinogenesis. 2002;23:1399–1404. doi: 10.1093/carcin/23.8.1399. [DOI] [PubMed] [Google Scholar]
- 13.Matusheskiand NV, Jeffery EH. Comparison of the bioactivity of two glucoraphanin hydrolysis products found in broccoli, sulforaphane and sulforaphane nitrile. J Agric Food Chem. 2001;49:5743–5749. doi: 10.1021/jf010809a. [DOI] [PubMed] [Google Scholar]
- 14.Svehlikova V, Wang S, Jakubikova J, Williamson G, Mithen R, Bao Y. Interactions between sulforaphane and apigenin in the induction of UGT1A1 and GSTA1 in CaCo-2 cells. Carcinogenesis. 2004;25:1629–1637. doi: 10.1093/carcin/bgh169. [DOI] [PubMed] [Google Scholar]
- 15.Brooks JD, Paton VG, Vidanes G. Potent induction of phase 2 enzymes in human prostate cells by sulforaphane. Cancer Epidemiol Biomarkers Prev. 2001;10:949–954. [PubMed] [Google Scholar]
- 16.Munday R, Munday CM. Induction of phase II detoxification enzymes in rats by plant-derived isothiocyanates: comparison of allyl isothiocyanate with sulforaphane and related compounds. J Agric Food Chem. 2004;52:1867–1871. doi: 10.1021/jf030549s. [DOI] [PubMed] [Google Scholar]
- 17.Prestera T, Talalay P. Electrophile and antioxidant regulation of enzymes that detoxify carcinogens. Proc Natl Acad Sci USA. 1995;92:8965–8969. doi: 10.1073/pnas.92.19.8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McMahon M, Itoh K, Yamamoto M, Chanas SA, Henderson CJ, McLellan LI, et al. The Cap’n’Collar basic leucine zipper transcription factor Nrf2 (NF-E2 p45-related factor 2) controls both constitutive and inducible expression of intestinal detoxification and glutathione biosynthetic enzymes. Cancer Res. 2001;61:3299–3307. [PubMed] [Google Scholar]
- 19.McMahon M, Itoh K, Yamamoto M, Hayes JD. Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J Biol Chem. 2003;278:21592–21600. doi: 10.1074/jbc.M300931200. [DOI] [PubMed] [Google Scholar]
- 20.Kwak MK, Wakabayashi N, Itoh K, Motohashi H, Yamamoto M, Kensler TW. Modulation of gene expression by cancer chemopreventive dithiolethiones through the Keap1–Nrf2 pathway Identification of novel gene clusters for cell survival. J Biol Chem. 2003;278:8135–8145. doi: 10.1074/jbc.M211898200. [DOI] [PubMed] [Google Scholar]
- 21.Kwak MK, Wakabayashi N, Greenlaw JL, Yamamoto M, Kensler TW. Antioxidants enhance mammalian proteasome expression through the Keap1–Nrf2 signaling pathway. Mol Cell Biol. 2003;23:8786–8794. doi: 10.1128/MCB.23.23.8786-8794.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, et al. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res. 2002;62:5196–5203. [PubMed] [Google Scholar]
- 24.McWalter GK, Higgins LG, McLellan LI, Henderson CJ, Song L, Thornalley PJ, et al. Transcription factor Nrf2 is essential for induction of NAD(P)H:quinone oxidoreductase 1, glutathione S-transferases, and glutamate cysteine ligase by broccoli seeds and isothiocyanates. J Nutr. 2004;134:3499S–3506S. doi: 10.1093/jn/134.12.3499S. [DOI] [PubMed] [Google Scholar]
- 25.Yu R, Lei W, Mandlekar S, Weber MJ, Der CJ, Wu J, Kong AT. Role of a mitogen-activated protein kinase pathway in the induction of phase II detoxifying enzymes by chemicals. J Biol Chem. 1999;274:27545–27552. doi: 10.1074/jbc.274.39.27545. [DOI] [PubMed] [Google Scholar]
- 26.Hu R, Hebbar V, Kim BR, Chen C, Winnik B, Buckley B, et al. In vivo pharmacokinetics and regulation of gene expression profiles by isothiocyanate sulforaphane in the rat. J Pharmacol Exp Ther. 2004;310:263–271. doi: 10.1124/jpet.103.064261. [DOI] [PubMed] [Google Scholar]
- 27.Kong AN, Yu R, Chen C, Mandlekar S, Primiano T. Signal transduction events elicited by natural products: role of MAPK and caspase pathways in homeostatic response and induction of apoptosis. Arch Pharm Res. 2000;23:1–16. doi: 10.1007/BF02976458. [DOI] [PubMed] [Google Scholar]
- 28.Kong AN, Owuor E, Yu R, Hebbar V, Chen C, Hu R, Mandlekar S. Induction of xenobiotic enzymes by the MAP kinase pathway and the antioxidant or electrophile response element (ARE/EpRE) Drug Metab Rev. 2001;33:255–271. doi: 10.1081/dmr-120000652. [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Kensler TW, Cho CG, Posner GH, Talalay P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proc Natl Acad Sci USA. 1994;91:3147–3150. doi: 10.1073/pnas.91.8.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fahey JW, Haristoy X, Dolan PM, Kensler TW, Scholtus I, Stephenson KK, et al. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc Natl Acad Sci USA. 2002;99:7610–7615. doi: 10.1073/pnas.112203099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung FL, Conaway CC, Rao CV, Reddy BS. Chemoprevention of colonic aberrant crypt foci in Fischer rats by sulforaphane and phenethyl isothiocyanate. Carcinogenesis. 2000;21:2287–2291. doi: 10.1093/carcin/21.12.2287. [DOI] [PubMed] [Google Scholar]
- 32.Forman D. Helicobacter pylori infection and cancer. Br Med Bull. 1998;54:71–78. doi: 10.1093/oxfordjournals.bmb.a011682. [DOI] [PubMed] [Google Scholar]
- 33.Haristoy X, Angioi-Duprez K, Duprez A, Lozniewski A. Efficacy of sulforaphane in eradicating Helicobacter pylori in human gastric xenografts implanted in nude mice. Antimicrob Agents Chemother. 2003;47:3982–3984. doi: 10.1128/AAC.47.12.3982-3984.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heiss E, Herhaus C, Klimo K, Bartsch H, Gerhauser C. Nuclear factor kappa B is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J Biol Chem. 2001;276:32008–32015. doi: 10.1074/jbc.M104794200. [DOI] [PubMed] [Google Scholar]
- 35.Zhu M, Zhang Y, Cooper S, Sikorski E, Rohwer J, Bowden GT. Phase II enzyme inducer, sulforaphane, inhibits UVB-induced AP-1 activation in human keratinocytes by a novel mechanism. Mol Carcinog. 2004;41:179–186. doi: 10.1002/mc.20052. [DOI] [PubMed] [Google Scholar]
- 36.Gamet-Payrastre L, Lumeau S, Gasc N, Cassar G, Rollin P, Tulliez J. Selective cytostatic and cytotoxic effects of glucosinolates hydrolysis products on human colon cancer cells in vitro. Anticancer Drugs. 1998;9:141–148. doi: 10.1097/00001813-199802000-00005. [DOI] [PubMed] [Google Scholar]
- 37.Gamet-Payrastre L, Li P, Lumeau S, Cassar G, Dupont MA, Chevolleau S, et al. Sulforaphane, a naturally occurring isothiocyanate, induces cell cycle arrest and apoptosis in HT29 human colon cancer cells. Cancer Res. 2000;60:1426–1433. [PubMed] [Google Scholar]
- 38.Parnaud G, Li P, Cassar G, Rouimi P, Tulliez J, Combaret L, Gamet-Payrastre L. Mechanism of sulforaphane-induced cell cycle arrest and apoptosis in human colon cancer cells. Nutr Cancer. 2004;48:198–206. doi: 10.1207/s15327914nc4802_10. [DOI] [PubMed] [Google Scholar]
- 39.Chiao JW, Chung FL, Kancherla R, Ahmed T, Mittelman A, Conaway CC. Sulforaphane and its metabolite mediate growth arrest and apoptosis in human prostate cancer cells. Int J Oncol. 2002;20:631–636. doi: 10.3892/ijo.20.3.631. [DOI] [PubMed] [Google Scholar]
- 40.Wang L, Liu D, Ahmed T, Chung FL, Conaway C, Chiao JW. Targeting cell cycle machinery as a molecular mechanism of sulforaphane in prostate cancer prevention. Int J Oncol. 2004;24:187–192. [PubMed] [Google Scholar]
- 41.Singh SV, Herman-Antosiewicz A, Singh AV, Lew KL, Srivastava SK, Kamath R, et al. Sulforaphane-induced G2/M phase cell cycle arrest involves checkpoint kinase 2-mediated phosphorylation of cell division cycle 25C. J Biol Chem. 2004;279:25813–25822. doi: 10.1074/jbc.M313538200. [DOI] [PubMed] [Google Scholar]
- 42.Singh AV, Xiao D, Lew KL, Dhir R, Singh SV. Sulforaphane induces caspase-mediated apoptosis in cultured PC-3 human prostate cancer cells and retards growth of PC-3 xenografts in vivo. Carcinogenesis. 2004;25:83–90. doi: 10.1093/carcin/bgg178. [DOI] [PubMed] [Google Scholar]
- 43.Jackson SJ, Singletary KW. Sulforaphane: a naturally occurring mammary carcinoma mitotic inhibitor, which disrupts tubulin polymerization. Carcinogenesis. 2004;25:219–227. doi: 10.1093/carcin/bgg192. [DOI] [PubMed] [Google Scholar]
- 44.Jacksonand SJ, Singletary KW. Sulforaphane inhibits human MCF-7 mammary cancer cell mitotic progression and tubulin polymerization. J Nutr. 2004;134:2229–2236. doi: 10.1093/jn/134.9.2229. [DOI] [PubMed] [Google Scholar]
- 45.Tseng E, Scott-Ramsay EA, Morris ME. Dietary organic isothiocyanates are cytotoxic in human breast cancer MCF-7 and mammary epithelial MCF-12A cell lines. Exp Biol Med (Maywood) 2004;229:835–842. doi: 10.1177/153537020422900817. [DOI] [PubMed] [Google Scholar]
- 46.Misiewicz I, Skupinska K, Kowalska E, Lubinski J, Kasprzycka-Guttman T. Sulforaphane-mediated induction of a phase 2 detoxifying enzyme NAD(P)H:quinone reductase and apoptosis in human lymphoblastoid cells. Acta Biochim Pol. 2004;51:711–721. [PubMed] [Google Scholar]
- 47.Zhang Y, Tang L, Gonzalez V. Selected isothiocyanates rapidly induce growth inhibition of cancer cells. Mol Cancer Ther. 2003;2:1045–1052. [PubMed] [Google Scholar]
- 48.Fimognari C, Nusse M, Cesari R, Iori R, Cantelli-Forti G, Hrelia P. Growth inhibition, cell-cycle arrest and apoptosis in human T-cell leukemia by the isothiocyanate sulforaphane. Carcinogenesis. 2002;23:581–586. doi: 10.1093/carcin/23.4.581. [DOI] [PubMed] [Google Scholar]
- 49.Gingras D, Gendron M, Boivin D, Moghrabi A, Theoret Y, Beliveau R. Induction of medulloblastoma cell apoptosis by sulforaphane, a dietary anticarcinogen from Brassica vegetables. Cancer Lett. 2004;203:35–43. doi: 10.1016/j.canlet.2003.08.025. [DOI] [PubMed] [Google Scholar]
- 50.Pham NA, Jacobberger JW, Schimmer AD, Cao P, Gronda M, Hedley DW. The dietary isothiocyanate sulforaphane targets pathways of apoptosis, cell cycle arrest, and oxidative stress in human pancreatic cancer cells and inhibits tumor growth in severe combined immunodeficient mice. Mol Cancer Ther. 2004;3:1239–1248. [PubMed] [Google Scholar]
- 51.Myzak MC, Karplus PA, Chung FL, Dashwood RH. A novel mechanism of chemoprotection by sulforaphane: inhibition of histone deacetylase. Cancer Res. 2004;64:5767–5774. doi: 10.1158/0008-5472.CAN-04-1326. [DOI] [PubMed] [Google Scholar]
- 52.Marks PA, Richon VM, Rifkind RA. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J Natl Cancer Inst. 2000;92:1210–1216. doi: 10.1093/jnci/92.15.1210. [DOI] [PubMed] [Google Scholar]
- 53.Ungerstedt JS, Sowa Y, Xu WS, Shao Y, Dokmanovic M, Perez G, et al. Role of thioredoxin in the response of normal and transformed cells to histone deacetylase inhibitors. Proc Natl Acad Sci USA. 2005;102:673–678. doi: 10.1073/pnas.0408732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kolonel LN, Hankin JH, Whittemore AS, Wu AH, Gallagher RP, Wilkens LR, et al. Vegetables, fruits, legumes and prostate cancer: a multiethnic case-control study. Cancer Epidemiol Biomarkers Prev. 2000;9:795–804. [PubMed] [Google Scholar]
- 55.Giovannucci E, Rimm EB, Liu Y, Stampfer MJ, Willett WC. A prospective study of cruciferous vegetables and prostate cancer. Cancer Epidemiol Biomarkers Prev. 2003;12:1403–1409. [PubMed] [Google Scholar]
- 56.Cohen JH, Kristal AR, Stanford JL. Fruit and vegetable intakes and prostate cancer risk. J Natl Cancer Inst. 2000;92:61–68. doi: 10.1093/jnci/92.1.61. [DOI] [PubMed] [Google Scholar]
- 57.Spitz MR, Duphorne CM, Detry MA, Pillow PC, Amos CI, Lei L, et al. Dietary intake of isothiocyanates: evidence of a joint effect with glutathione S-transferase polymorphisms in lung cancer risk. Cancer Epidemiol Biomarkers Prev. 2000;9:1017–1020. [PubMed] [Google Scholar]
- 58.Lewis S, Brennan P, Nyberg F, Ahrens W, Constantinescu V, Mukeria A, et al. Dietary intake of isothiocyanates: evidence of a joint effect with glutathione S-transferase polymorphisms in lung cancer risk. Cancer Epidemiol Biomarkers Prev. 2000;9:1017–1020. [PubMed] [Google Scholar]; Lewis S, Brennan P, Nyberg F, Ahrens W, Constantinescu V, Mukeria A, et al. Dietary intake of isothiocyanates: evidence of a joint effect with glutathione S-transferase polymorphisms in lung cancer risk. Cancer Epidemiol Biomark Prev. 2001;10:1105–1106. [PubMed] [Google Scholar]
- 59.Zhao B, Seow A, Lee EJ, Poh WT, Teh M, Eng P, et al. Dietary isothiocyanates, glutathione S-transferase -M1-T1 polymorphisms and lung cancer risk among Chinese women in Singapore. Cancer Epidemiol Biomarkers Prev. 2001;10:1063–1067. [PubMed] [Google Scholar]
- 60.Fowke JH, Chung FL, Jin F, Qi D, Cai Q, Conaway C, et al. Urinary isothiocyanate levels, brassica, and human breast cancer. Cancer Res. 2003;63:3980–3986. [PubMed] [Google Scholar]
- 61.Acquavella J, Cullen MR. Correspondence re: H. J. Lin et al., Glutathione transferase null genotype, broccoli, and lower prevalence of colorectal adenomas. Cancer Epidemiol Biomarkers Prev. 1998;7:647–652. [PubMed] [Google Scholar]; Acquavella J, Cullen MR, et al. Correspondence re: H. J. Lin et al., Glutathione transferase null genotype, broccoli, and lower prevalence of colorectal adenomas. Cancer Epidemiol Biomarkers Prev. 1999;8:947–949. [PubMed] [Google Scholar]
- 62.Seow A, Yuan JM, Sun CL, Van Den Berg D, Lee HP, Yu MC. Dietary isothiocyanates, glutathione S-transferase polymorphisms and colorectal cancer risk in the Singapore Chinese Health Study. Carcinogenesis. 2002;23:2055–2061. doi: 10.1093/carcin/23.12.2055. [DOI] [PubMed] [Google Scholar]