

Abstract
Plasma membrane calcium pumps (PMCAs) expel Ca2+ from eukaryotic cells to maintain overall Ca2+ homeostasis and to provide local control of intracellular Ca2+ signaling. Recent work indicates functional versatility among PMCA isoforms, with specific pumps being essential for cochlear hair cell function, sperm motility, feedback signaling in the heart, and pre- and post-synaptic Ca2+ regulation in neurons. The functional versatility of PMCAs is due to differences in their regulation by calmodulin, kinases and other signaling proteins, as well as to their differential targeting and retention in defined plasma membrane domains. The basis for this is the structural diversity of PMCAs. In mammals, four genes encode PMCA isoforms 1-4, and each of these has multiple variants generated by alternative RNA splicing. The alternatively spliced regions are intimately involved in the regulatory interactions and differential membrane localization of the pumps. The alternatively spliced C-terminal tail acts as auto-inhibitory domain by interacting with the catalytic core of the pump. The degree of inhibition and the kinetics of interaction with the major activator calmodulin differ between PMCA variants. This translates into functional differences in how PMCAs handle Ca2+ signals of different magnitude and frequency. Accumulating evidence thus demonstrates how structural diversity provides functional versatility in the PMCAs.
Keywords: Ca2+-ATPase, Ca2+ regulation, Ca2+ signaling, isoforms, PMCA, splice variant
PMCAs are important parts of the toolkit for cellular Ca2+ regulation
Ca2+ is a universal regulator of cellular processes ranging from fertilization to programmed cell death [1, 2]. Signaling by Ca2+ requires an elaborate “toolkit” of proteins to control the influx, efflux, and buffering of Ca2+ in and between different cellular compartments and among different cells [3]. High sensitivity and excellent signal to noise ratios are achieved by keeping cytosolic [Ca2+] very low (≤100 nM) during the resting state. This is accomplished by Ca2+-buffering proteins and by integral membrane transport systems that can remove Ca2+ from the cytosol even against large concentration gradients. Specialized transporters such as ion gradient-dependent Na+/Ca2+ exchangers and ATP-driven pumps are responsible for this “uphill” transport of Ca2+ across biological membranes [4]. The plasma membrane Ca2+ ATPases (PMCAs) are the major high-affinity Ca2+ handling system dedicated to the expulsion of Ca2+ from the cytosol. Due to their ubiquitous expression and low transport capacity, they have long been assumed to be a housekeeping system responsible for setting and maintenance of the normally low cytosolic [Ca2+] [5]. However, the discovery of multiple isoforms and alternative splice variants, and recent results on naturally occurring PMCA mutants and genetically engineered PMCA “knockout” mice demonstrate that the PMCAs play distinct roles in tissue- and cell-specific dynamic Ca2+ handling [6-8]. The range of functions for the PMCAs is therefore much wider than initially envisioned: PMCAs are not merely “sump pumps” needed to remove “excess” Ca2+, but instead are versatile tools needed to shape global and local Ca2+ signals with high spatial and temporal resolution.
Structural diversity among PMCAs
The PMCAs are P-type ion-transporting ATPases (so named for the formation of an obligatory phosphorylated intermediate during the reaction cycle [9]) and are members of the type IIB subfamily within the large superfamily of these ATPases [10]. In humans and other mammals, four major PMCA isoforms (PMCA 1, 2, 3, 4) are encoded by separate genes. In the human genome, these genes are localized on chromosomes 12q21.3, 3p25.3, Xq28, and 1q32.1. The encoded proteins range from 1115 to 1258 amino acid residues in length, with calculated molecular masses between ∼125 and 140 kDa. Although their global structures are very similar - with intracellular N- and C-terminal tails, 10 membrane-spanning segments, and two major cytosolic loops (see Figure 1) - the average sequence identity between PMCA isoforms amounts to only about 80% [11]. The sequence differences are not evenly distributed; rather regions of high sequence divergence are interspersed with regions of high sequence conservation. The regions with high sequence variability (indicated by thick black lines in Figure 1) are responsible for the structural diversity of different PMCAs. Of note, the N- and C-terminal tails are among the least conserved regions in PMCAs. As shown in Figure 1, the tail sequences are involved in numerous protein-protein interactions of functional importance, including binding of the major regulator calmodulin (CaM) to the C-terminal tail.
Figure 1.
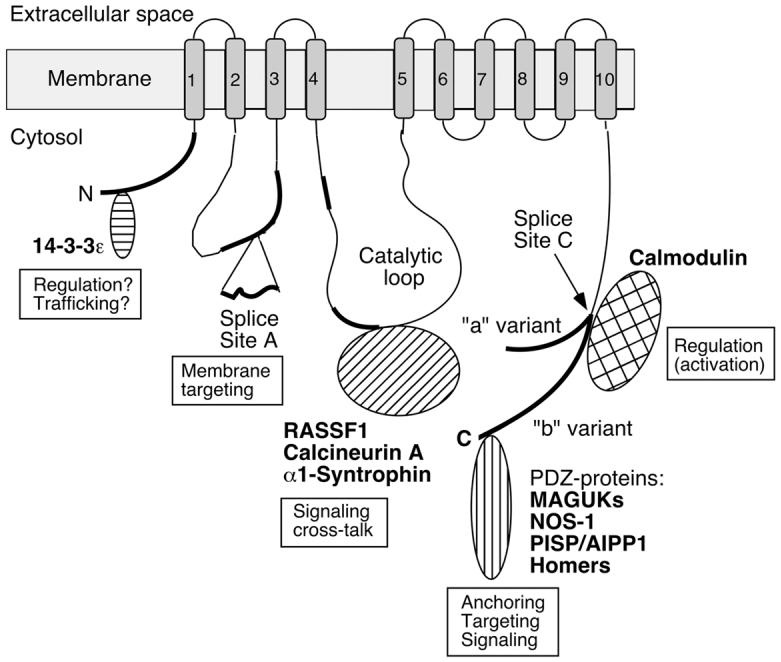
Scheme of the PMCA emphasizing regions of structural diversity among isoforms and sites of protein-protein interactions. Membrane-spanning regions are numbered 1-10 and shown as shaded boxes. The amino- (N) and carboxyl-terminal (C) ends are labeled, and the position of the large catalytic loop is indicated. Regions of significant sequence divergence among isoforms are shown as thick black lines. “Splice Site A” and “Splice Site C” denote the regions affected by alternative splicing. At site A the insertion of a peptide segment encoded by alternatively spliced exon(s) is indicated; at site C the two major splice variants (“a” and “b”) are shown with separate tails to indicate their divergent reading frames. Various PMCA-interacting proteins are schematically shown near the domain of the PMCA where they bind, and their known or suspected roles in providing functional diversity are indicated. The PMCA is represented in its activated state with calmodulin bound to the C-tail. For details, see the text.
The diversity of the PMCA family is further enhanced by alternative RNA splicing affecting two major “hotspots” called sites A and C (Fig. 1). The combinatorial use of alternative splice options at these two sites results in the generation of over 20 PMCA variants [12]. These splice variants differ in the length of the first intracellular loop (splicing at site A) and in the C-terminal tail (site C splicing). Table I lists the major PMCA isoforms and splice variants in humans. Many of these PMCAs show developmental-, tissue-, and cell-specific patterns of expression, suggesting their functional adaptation to the specific Ca2+ handling needs of a given cell type [12]. Alternative splicing adds structural diversity to the PMCAs: splicing at site A alters the size of the first cytosolic loop by inserting up to 45 “extra” amino acids (in PMCA2w splice variants). Although no high-resolution structural information is as yet available for any PMCA, the variation in size of the second intracellular loop likely leads to significant local (and global?) structural differences. In support of this notion, the w-insert at splice site A directs PMCA2 to the apical membrane in polarized cells, whereas pumps with the shorter x-insert are targeted to the lateral membrane [13, 14]. Splicing at site C leads to even more dramatic structural changes resulting from a reading frame shift caused by differential exon usage. Consequently, splice variants “a” and “b” of each PMCA differ in the length and amino acid sequence of their C-terminal tail (see Fig. 1). These differences underlie the divergent intra-molecular interactions of the a- and b-splice variants with the catalytic domain of the PMCA (important for auto-inhibition), and result in different interactions with external regulatory and signaling proteins. In turn, these structural differences lead to functional differences that enable different PMCA isoforms to fulfill tissue- and cell-specific roles in Ca2+ handling.
Table I.
Major human PMCA isoforms and gene deletion phenotypes
| Isoform | Splice variantsa | Length in amino acids | Tissue distributiona | Gene knockout phenotype (mouse)b |
|---|---|---|---|---|
| PMCA1 | PMCA1x/a PMCA1x/b PMCA1x/c PMCA1x/d |
1176 1220 1249 1258 |
Brain, nervous tissue Ubiquitous Skeletal muscle, heart Skeletal muscle, brain (rare) |
Embryonic lethal (homozygotes); vascular smooth muscle apoptosis (heterozygotes in PMCA4 null background); altered smooth muscle Ca2+ regulation (heterozygotes) |
| PMCA2 | PMCA2w/a PMCA2x/a PMCA2z/a PMCA2w/b PMCA2x/b PMCA2z/b |
1199 1168 1154 1243 1212 1198 |
Brain, vestibular and cochlear hair cells Brain (rare) Brain (more abundant than 2x/a) Brain, lactating mammary gland, pancreas Brain/excitable tissue Brain/excitable tissue |
Profoundly deaf, severe ataxia, reduced milk Ca2+, motor neuron loss [28], altered cerebellar synapse plasticity [27] (homozygotes); age-related and noise-induced hearing loss (heterozygotes) |
| PMCA3 | PMCA3x/a PMCA3z/a PMCA3x/b PMCA3z/b PMCA3x/f PMCA3z/f |
1173 1159 1220 1206 1129 1115 |
Brain Brain, pancreas (β-cells) Brain Brain (Fast) skeletal muscle, brain (rare) Brain (rare) |
Not determined |
| PMCA4 | PMCA4x/a PMCA4z/a PMCA4x/b PMCA4z/b |
1170 1158 1205 1193 |
Brain, heart, stomach Heart, testis, pancreas (islet of Langerhans) Ubiquitous Heart |
Male infertility due to sperm motility defect (homozygotes); smooth muscle apoptosis and altered Ca2+ regulation (homozygotes in PMCA1+/- background) |
Functional diversity among PMCAs
In the resting state, the PMCAs are auto-inhibited by a mechanism that involves binding of their C-terminal tail to the two major intracellular loops. Activation requires binding of Ca2+-CaM to the C-terminal tail and a conformational change that displaces the auto-inhibitory tail from the catalytic domain [15, 16]. PMCA activity is also affected by acidic phospholipids, phosphorylation of specific residues (both Ser/Thr and Tyr) in the C-terminal tail, partial proteolysis, and dimerization via the C-terminal tail [15, 17, 18]. Because PMCA isoforms and especially their C-terminal splice variants vary in sequence and - likely - in structure, their functional properties are also different. For example, the degree of auto-inhibition (determined as basal activity in the absence of CaM) differs between a- and b-splice variants due to the different strength of interaction of their C-terminal tails with the catalytic domain. Differences in the binding kinetics and affinity for CaM lead to very different activation profiles of these splice variants, and this translates into different functional properties (reviewed in [6, 12]).
Recent studies indicate that PMCA isoforms are differentially distributed in membrane sub-domains such as sphingolipid-rich membrane “rafts” [19, 20]. The differential partitioning of PMCA isoforms likely reflects structural differences, e.g., in the length and local arrangement of membrane-spanning sequences that favor specific lipid interactions. The lipid environment also impacts the functional properties of the PMCAs [21, 22]. The enrichment of distinct membrane domains in locales such as apical stereocilia of hair cells or pre-synaptic nerve terminals may lead to the local concentration of specific PMCAs and a concomitant effect on local Ca2+ handling properties. Alternative splicing at sites A and C influences the targeting and trafficking of the PMCAs (Fig. 1), underlining the importance of structural variations for the differential deployment and functional specialization of the pumps.
The functional properties of individual PMCAs are relevant to how the pumps handle a cellular Ca2+ signal. Studies on the kinetics of activation of different PMCAs reveal significant differences in their ability to respond to a sudden Ca2+ rise and to decode the frequency of repetitive Ca2+ spikes [23]. Comparing splice variants PMCA4a and 4b, which differ only in the length and sequence of the C-terminal tail showed that the rate for CaM activation in PMCA4b is much slower (t1/2 ∼ 1 min at 0.5 μM Ca2+) than in PMCA4a (t1/2 ∼ 20 sec). The inactivation rate of PMCA4b is even slower (t1/2 ∼ 20 min), suggesting that this isoform is geared towards handling slow, tonic Ca2+ changes [24]. On the other hand, the higher basal activity and faster activation by CaM allow PMCA4a to be more effective than PMCA4b in reducing an agonist-evoked intracellular Ca2+ pulse. Because PMCAs detect changes in the sub-plasmalemmal Ca2+ level, their “reaction time” to Ca2+ rises must be optimal for the desired outcome of Ca2+ signaling. Detailed kinetic studies show that PMCA isoforms such as PMCA3f and PMCA2b expressed in cells with fast Ca2+ responses (skeletal muscles, neurons) are generally “fast”, i.e., they are activated quickly by a rise in [Ca2+]i [25-27]. The different time course of inactivation (by CaM release) also results in striking differences in the “memory” of PMCA isoforms for their recent activation [23]. Thus, in cells with frequent Ca2+ spikes, PMCA2b will remain “pre-activated” and respond almost immediately to the next Ca2+ signal.
The functional versatility of different PMCAs is enhanced by specific protein interactions (see Fig. 1). These interactions serve to connect specific PMCAs to particular signaling pathways, to recruit them to multi-protein complexes linked to the actin cytoskeleton, or to alter their regulatory properties [7]. Many of these protein interactions are isoform- and/or splice variant-specific. Accordingly, most of the interacting partners bind to the structurally diverse regions of the PMCAs, including the alternatively spliced C-tail and the N-terminal tail.
Conclusion: Structural diversity underlies the functional versatility of PMCAs
Plasma membrane Ca2+ ATPases (PMCAs) are essential components of the cellular toolkit to regulate and fine-tune cytosolic Ca2+ concentrations. Work over the last 20 years has revealed a perplexing multitude of PMCA isoforms and alternative splice variants in mammalian tissues, raising questions about the specific role of different pumps in Ca2+ handling. Studies on the regulation and kinetics of individual isoforms suggest that PMCAs are functionally diverse and optimized to participate in dynamic Ca2+ handling according to tissue- and cell-specific demands. Different PMCAs help shape slow, tonic Ca2+ signals in some cells and provide rapid, efficient Ca2+ clearance in others. Underlying this functional diversity are structural differences among PMCAs. These structural differences are concentrated in the C-terminal alternatively spliced tail and in parts of specific intracellular regions of the pump involved in differential protein-protein interactions. Thus, while maintaining the overall structural framework necessary to function as membrane-bound high-affinity Ca2+-transporting ATPases, individual PMCA isoforms show significant local structural diversity in sub-domains involved in regulation of activity, membrane targeting, and signaling cross-talk. Future work will be directed at understanding the functional differences among PMCAs based on structural differences at the atomic resolution level.
Acknowledgments
This work was supported in part by NIH grants GM28835 and NS51769.
Abbreviations
- CaM
calmodulin
- MAGUKs
membrane-associated guanylate kinases
- NOS
nitric oxide synthase
- PDZ
postsynaptic density-95/disc-large/zona occludens
- PISP/AIPP
PMCA-interacting single-PDZ protein/ATPase-interacting PDZ protein
- PMCA
plasma membrane calcium pump
- RASSF
Ras association domain family
References
- 1.Berridge MJ, Bootman MD, Lipp P. Nature. 1998;395:645–648. doi: 10.1038/27094. [DOI] [PubMed] [Google Scholar]
- 2.Carafoli E, Santella L, Branca D, Brini M. Crit. Rev. Biochem. Molec. Biol. 2001;36:107–260. doi: 10.1080/20014091074183. [DOI] [PubMed] [Google Scholar]
- 3.Berridge MJ, Bootman MD, Roderick HL. Nature Rev. Mol. Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 4.Carafoli E. Annu. Rev. Biochem. 1987;56:395–433. doi: 10.1146/annurev.bi.56.070187.002143. [DOI] [PubMed] [Google Scholar]
- 5.Carafoli E. Physiol. Rev. 1991;71:129–153. doi: 10.1152/physrev.1991.71.1.129. [DOI] [PubMed] [Google Scholar]
- 6.Strehler EE, Treiman M. Curr. Mol. Med. 2004;4:323–335. doi: 10.2174/1566524043360735. [DOI] [PubMed] [Google Scholar]
- 7.Strehler EE, Caride AJ, Filoteo AG, Xiong Y, Penniston JT, Enyedi Á. Ann. N. Y. Acad. Sci. 2007;1099:226–236. doi: 10.1196/annals.1387.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prasad V, Okunade G, Liu L, Paul RJ, Shull GE. Ann. N. Y. Acad. Sci. 2007;1099:276–286. doi: 10.1196/annals.1387.029. [DOI] [PubMed] [Google Scholar]
- 9.Pedersen PL, Carafoli E. Trends Biochem. Sci. 1987;12:146–150. [Google Scholar]
- 10.Axelsen KB, Palmgren MG. J. Mol. Evol. 1998;46:84–101. doi: 10.1007/pl00006286. [DOI] [PubMed] [Google Scholar]
- 11.Strehler EE. J. Membr. Biol. 1991;120:1–15. doi: 10.1007/BF01868586. [DOI] [PubMed] [Google Scholar]
- 12.Strehler EE, Zacharias DA. Physiol. Rev. 2001;81:21–50. doi: 10.1152/physrev.2001.81.1.21. [DOI] [PubMed] [Google Scholar]
- 13.Chicka MC, Strehler EE. J. Biol. Chem. 2003;278:18464–18470. doi: 10.1074/jbc.M301482200. [DOI] [PubMed] [Google Scholar]
- 14.Hill JK, Williams DE, LeMasurier DE, Dumont RA, Strehler EE, Gillespie PG. J. Neurosci. 2006;26:6172–6180. doi: 10.1523/JNEUROSCI.0447-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carafoli E. FASEB J. 1994;8:993–1002. [PubMed] [Google Scholar]
- 16.Penniston JT, Enyedi A. J. Membr. Biol. 1998;165:101–109. doi: 10.1007/s002329900424. [DOI] [PubMed] [Google Scholar]
- 17.Dean WL, Chen D, Brandt PC, Vanaman TC. J. Biol. Chem. 1997;272:15113–15119. doi: 10.1074/jbc.272.24.15113. [DOI] [PubMed] [Google Scholar]
- 18.Pászty K, Antalffy G, Penheiter AR, Homolya L, Padányi R, Iliás A, Filoteo AG, Penniston JT, Enyedi Á. Biochem. J. 2005;391:687–692. doi: 10.1042/BJ20051012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sepúlveda MR, Berrocal-Carrillo M, Gasset M, Mata AM. J. Biol. Chem. 2006;281:447–453. doi: 10.1074/jbc.M506950200. [DOI] [PubMed] [Google Scholar]
- 20.Jiang L, Fernandes D, Mehta N, Bean JL, Michaelis ML, Zaidi A. J. Neurochem. 2007 doi: 10.1111/j.1471-4159.2007.04480.x. OnlineEarly Articles, doi:10.111/j.1471-4159.2007.04480.x. [DOI] [PubMed] [Google Scholar]
- 21.Pang Y, Zhu H, Wu P, Chen J. FEBS Lett. 2005;579:2397–2403. doi: 10.1016/j.febslet.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 22.Tang D, Dean WL, Borchman D, Paterson CA. Cell Calcium. 2006;39:209–216. doi: 10.1016/j.ceca.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 23.Caride AJ, Penheiter AR, Filoteo AG, Bajzer Z, Enyedi Á, Penniston JT. J. Biol. Chem. 2001;276:39797–39804. doi: 10.1074/jbc.M104380200. [DOI] [PubMed] [Google Scholar]
- 24.Caride AJ, Elwess NL, Verma AK, Filoteo AG, Enyedi Á, Bajzer Z, Penniston JT. J. Biol. Chem. 1999;274:35227–35232. doi: 10.1074/jbc.274.49.35227. [DOI] [PubMed] [Google Scholar]
- 25.Caride AJ, Filoteo AG, Penheiter AR, Pászty K, Enyedi Á, Penniston JT. Cell Calcium. 2001;30:49–57. doi: 10.1054/ceca.2001.0212. [DOI] [PubMed] [Google Scholar]
- 26.Scheuss V, Yasuda R, Sobczyk A, Svoboda K. J. Neurosci. 2006;26:8183–8194. doi: 10.1523/JNEUROSCI.1962-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Empson RM, Garside ML, Knöpfel T. J. Neurosci. 2007;27:3753–3758. doi: 10.1523/JNEUROSCI.0069-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kurnellas MP, Nicot A, Shull GE, Elkabes S. FASEB J. 2005;19:298–300. doi: 10.1096/fj.04-2549fje. [DOI] [PMC free article] [PubMed] [Google Scholar]