

Abstract
Phospholipase D2 (PLD2), one of the two mammalian members of the PLD family, has been implicated in cell proliferation, transformation, tumor progression and survival. However, as precise mechanistic details are still unknown, we investigated here if the PLD2 isoform would signal through the PI3K/AKT pathway. Transient expression of PLD2 in COS7 cells with either the WT or with a Y179F mutant, resulted in an increased basal phosphorylation of AKT in residues T308 and S473, in a PI3K-dependent manner. Transfection of PLD2-Y179F (but not the wild type) caused an increased (>2-fold) DNA synthesis even in the absence of extracellular stimuli. Other signaling mechanisms downstream such PLD/PI3K dependence (that might lead to DNA synthesis regulation), were further studied. PLD2-Y179F caused an increase in phosphorylation of p42/p44ERK and in the expression of G0/G1 phase transition markers, p21CIP and PCNA, and these effects, too, were dependent on PI3K. Interestingly, Akt once activated, enabled the phosphorylation of PLD2 on residue T175, an effect that was inhibited by LY296004. Lastly, if PLD2-Y179F is further mutated in residue K758 (PLD2 Y179F-K758R), which renders inactive a catalytic site, DNA synthesis is then abrogated, indicating that the activity of the enzyme (i.e. synthesis of PA) is necessary for the observed effects. In conclusion, the unavailability of residue Y179 on PLD2 to become phosphorylated leads to an augmentation of DNA synthesis concomitantly with MEK and AKT phosphorylation, in a process that is dependent on PI3K and independent of any extracellular stimuli. This might be critical for the maintenance of the PLD2-regulated proliferative status.
Keywords: PLD2, PI3K, AKT, DNA synthesis, phosphorylation
INTRODUCTION
Phospholipase D (PLD) hydrolyzes phosphatidylcholine to produce phosphatidic acid (PA) and choline. PA is subsequently converted to lyso-PA by phospholipase A2 or to diacylglycerol (DAG) by phosphatidate phosphohydrolase [1]. Both PA and lyso-PA are mitogenic intracellular messengers, and DAG, via direct stimulation of PKC, is also associated with cell proliferation [2]. PLD plays a role in growth factor-induced cellular proliferation [3–6] and facilitates oncogenic transformation [7–10]. Endogenous PLD activity is significantly increased in several types of cancers [11–15], as well as in cell lines transformed with oncogenes [4, 5, 8, 10, 16–19]. PLD may participate in the activation of p42/44ERK via Raf-1 translocation to the plasma membrane [20–23], as well as in the survival signaling mediated by PI3K/AKT activation [24]. Although the precise mechanisms involving the isoform PLD2 in cell proliferation and survival are still unknown, recent evidence suggests that residues Y169 and Y179 serve to recruit the Grb2/Sos complex and that PLD2-Y169 is involved in Ras/MAPK activation [25, 26].
PI3Ks phosphorylate cell membrane phosphatidylinositols to generate PIP3. PIP3 then recruits AKT to the membrane where it is sequentially phosphorylated on T308 and S473 [27, 28]. Once activated, AKT modulates downstream effectors relevant to the regulation of cell growth (i.e. Raf/MEK, mTOR/p70S6K), cell cycle progression and cell proliferation (i.e. p21CIP1/PCNA), or apoptosis (i.e. Bad, caspase 9, Mdm2) [29–32]. Interestingly, most of these effectors can also be regulated by PLD [16, 21, 22, 25, 33–36]. Furthermore, AKT and bacterial PLD interact [37] and PLD plays a critical role in the PI3K-dependent activation of AKT [38–42]. The addition of exogenous bacterial PLD to CHO cells results in AKT activation in a PA- and PI3K-dependent manner [43].
Transient expression of PLD inhibits drug-induced apoptosis via the activation of PI3K/AKT and extracellular signal-regulated kinase (ERK) survival signaling pathways [36, 41, 44]. The present study was undertaken to investigate the molecular mechanisms implicated in PLD2-mediated de novo DNA synthesis and to find out whether or not they involve the PI3K/AKT signaling cascade and cell cycle progression, independently of extracellular stimuli.
MATERIAL & METHODS
Materials, cells and antibodies
COS-7 and RAW-264.7 cells and media were from ATCC (Manassas, VA); antibodies against T308 or S473-phosphorylated AKT, T202/Y204-phosphorylated p42/44ERK, AKT, p42/44ERK, and myc were from Cell Signaling Inc. (Beverly, MA). Anti-PCNA and p21CIP1 antibodies were from Biolegend (San Diego, CA). Anti-myc coupled to agarose beads was from Santa Cruz Biotech (Santa Cruz, CA). DMEM was from Sigma (St. Louis, MO); Lipofectamine, Plus reagent, Opti-MEM reduced serum medium were from Invitrogen (Carlsbad, CA); reagents for nucleofection were from Amaxa (Gaithersburg, MD); Scintiverse II scintillation cocktail was purchased from Fisher (Pittsburgh, PA); electrophoresis chemicals were from Bio-Rad Laboratories (Richmond, CA). Precast 4–20% polyacrylamide gels were from Pierce Biotechnology, Inc. (Rockford, IL). LY294002 and PD98059 were from Calbiochem (San Diego, CA).
Mutagenesis of pcDNA-mycPLD2
The pcDNA-mycPLD2 construct was used as a template to create point mutants following the QuickChange II Site-Directed mutagenesis protocol (Stratagene, La Jolla, CA). MycPLD2-Y179F was created by using the following sense primer: 5′-TC TTG ACC ATG TCT TTC TTT CGA AAC TAC CAT GCC-3′. Note the simultaneous introduction of a BstB I restriction site by silent mutation (underlined) together with the desired missense mutation for screening purposes. Restriction enzyme analysis of the PLD2-Y179F mutant was performed by PCR amplification of the region containing the expected mutation (sense: 5′-GCT TGA CTC ACG GCG ACT TTT C-3′, antisense: 5′-CAC CTC TTG GAC CAG CGA TAA CA-3′). The single transition of adenine (A) in position 536 to thymidine (T) (A536T) of the PLD2 cDNA (which introduces the Y179F mutation and the BstB I site. Positive digestion of the PCR fragment from Y179F cDNA inidcates the presence of the mutation. The catalytically inactive double mutant Y179F-K758R was created using pcDNA-mycPLD2 Y179F as a template and the K758R mutagenic primer 5′-TC TAC ATC CAC AGC AGG GTG CTC ATC GCA G-3′. All oligos and their reverse complements were PAGE/HPLC purified (Integrated DNA Technologies, Coralville, IA). Molecular identity of pcDNA-mycPLD2 mutants were confirmed by PCR, restriction digestion and direct sequence analysis (Cleveland Genomics, Cleveland OH).
Transient transfection of COS7 or RAW-267.4 cells
COS7 were routinely transfected following lipofectamine-based procedures as described in detail elsewhere [45]. RAW cells were nucleofected with the Amaxa-based (Gaithersburg, MD) electroporation cuvette with 2×106 cells/100 µl in nucleofector solution V. Cells were "zapped" in program D-32 (as per the manufacturer’s instructions) after which they were resuspended in 1 ml DMEM + 20% FBS and immediately plated into six-well plates. Cells were incubated at 37 °C for 48 hr, then harvested and used for the experiments described herein.
Myc immunoprecipitation
Transiently or mock-transfected COS7 cells were lysed in lysis buffer [50 mM Tris-HCl pH 7.5, 1 mM EDTA, 1 mM EGTA, 0.5 mM sodium orthovanadate, 0.1% β-mercaptoethanol, 1% Triton X-100, 1% deoxycholic acid, 50 mM sodium fluoride, 5 mM sodium pyrophosphate, 10 mM sodium β-glycerolphosphate plus freshly added protease/phosphatase inhibitors (0.1 mM PMSF, 1 µg/ml aprotinin, 1 µg/ml pepstatin, 1 µg/ml leupeptin, 1 µM microcystin)] and about 500 µg of protein were incubated overnight with 50 µg anti-myc/Agarose in a final volume of 500 µl lysis buffer/inhibitors with gentle rotation. Agarose beads were spun-down at 12,000×g and washed 5 times with ice-cold lysis buffer/inhibitors, resuspended in loading buffer, boiled 5, subjected to SDS-PAGE and transferred to PDVF membranes. Western blots were developed by the ECL method (Amersham, UK).
ERK and AKT phosphorylation and general immunodetection
Specific p42/44ERK and AKT phosphorylation was analyzed by immuno-detecting the phosphorylation state of residues T202/Y204 or T308/S473, respectively. Transiently transfected COS7 cells were lysed in lysis buffer (plus protease and phosphatase inhibitors). About 75 µg of protein were resolved in 4–20% SDS-PAGE. Gels were transfered to PDVF membranes and developed by the ECL method (Amersham, UK).
[3H]-Thymidine incorporation
Newly synthezised (de novo) DNA was estimated by [3H]-thymidine incorporation into cellular DNA as described recently. Briefly, mycPLD2-transfected COS7 cells growing in 6 well plates were treated with 0.6 µCi/ml/well [3H]-thymidine for ~16 h. Cells were washed twice with ice-cold PBS and precipitated with 5% TCA. Precipitated cellular DNA was washed twice with 5% TCA and solubilized by adding 550 µl/well of 12.5 N NaOH. Precipitable [3H]-DNA was measured by scintillation counting.
Immunofluorescence
Expression of pcDNA-mycPLD2 (WT or Y179F), were analyzed by immunofluorescence following procedures as described in detail elsewhere [26]. Cells were treated with 2 µg/ml anti-myc antibodies and with 0.5 µg/ml FITC-conjugated secondary antibody and, lastly, with DAPI for nuclear staining.
Statistical analysis
The analysis of multiple intergroup differences in each experiment was conducted by one-way analysis of variance followed by Student test. A p<0.05 was used as the criterion of statistical significance.
RESULTS
PLD2 regulates basal AKT phosphorylation
In order to investigate if PLD2 would modulate cell survival and proliferation through the AKT/PI3K pathway, we transiently transfected COS7 cells with PLD2 Y179F, a mutant known to increase DNA synthesis in this cell line [25, 26]. The expression levels and localization of PLD2 WT did not differ from PLD2 Y179F, as shown in Figure 1A. Further, the level of endogenous PLD2 in COS-7 cells is very low as compared to the overexpressed counterpart (Fig. 1B). When COS7 cells were transfected with either the WT or Y179F constructs, we observed that AKT phosphorylation increased in residues T308 and S473, as evidenced by the slow migrating AKT form (band "b") detected with two anti-phospho-AKT antibodies (Figure 1C,F). Of note, this was observed without growth factor or serum stimulation of the cells. Basal and PLD2-driven AKT phosphorylation in residues T308/S473 was completely eliminated when COS7 cells were incubated in the presence of the PI3K inhibitor LY294002 (Figure 1D,G), suggesting that PLD2-WT increases AKT phosphorylation in a PI3K-dependent manner. The blocking effect of the PI3K inhibitor was not due to decreased expression of the constructs in COS7 cells treated with the inhibitor, since mycPLD2 (either WT or Y179F) were expressed at comparable levels in whole cell lysates. They did not affect cell viability either, that remained >90% as assayed by the trypan blue exclusion test (not shown). Figure 1E,H show positive controls for Akt phosphorylation that are comparable to those presented for COS-7 cells.
Figure 1. PLD2-Y179F increases AKT phosphorylation.
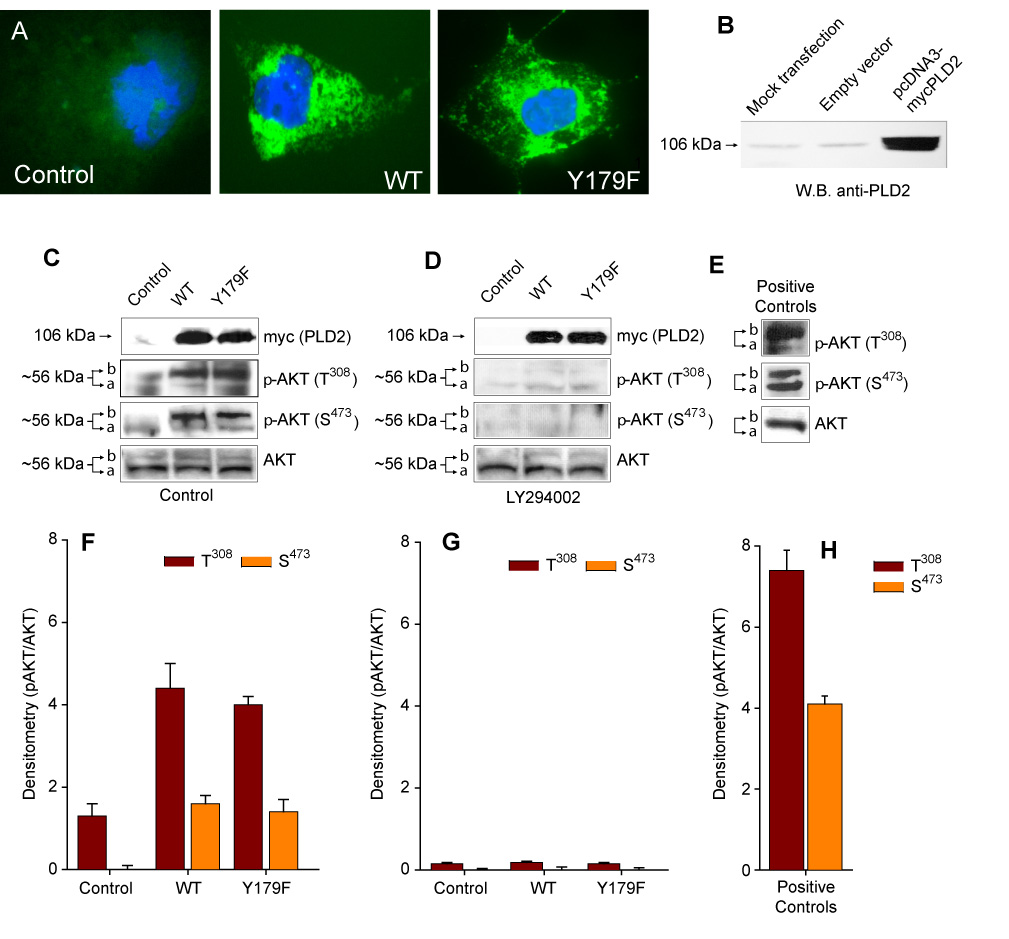
(A). Immunofluorescence detection of PLD2 WT or Y179F expression in COS7 cells using a FITC-conjugated anti-myc antibody against the myc-tag. (B). Western blot showing endogenous and overexpressed proteins, developed with anti-PLD2 antibodies. (C–E). PLD2-dependent increase of AKT phosphorylation. Shown are representative experiments obtained in lysates from COS7 cells Control (mock-transfected) and transiently expressing PLD2 (WT or Y179F) incubated overnight in the absence (C) or presence (D) of 25 uM LY294002. Cell lysates were subjected to SDS-PAGE, transfered to PDVF membranes and the expression levels of mycPLD2, AKT and p-AKT (AKT phosphorylated in T308 or S473) analyzed by using specific antibodies. Positive Controls (Calyculin A-treated total lysates of Jurkat cell grown in 10% FBS) were also included (E). Arrows refer to the mobility shift in AKT phosphorylation (a = dephospho form, b = phospho form). (F–H). Densitometric analysis in arbitrary units expressed as mean ± SEM (n=3) of the optical density of the immunoblots normalized to total AKT signal (pAKT/AKT) for Control (F), LY294002-treated (G) or Positive Controls (H).
PLD2 Y179F regulates DNA synthesis in a PI3K/AKT-dependent manner
PLD2 has been described to be located upstream of PI3K/AKT signaling [24]. Thus, basal PI3K activity may be necessary for PLD2 Y179F-induced de novo DNA synthesis. As shown in Figure 2A, COS7 cells overexpressing PLD2 Y179F have increased DNA synthesis when compared to WT-transfected cells, as demonstrated previously [25]. However, LY294002 or wortmannin blocked PLD2 Y179F-induced [3H]-thymidine incorporation, without affecting de novo DNA synthesis (Figs. 2B,C), suggesting that AKT activation and PLD2 Y179F functionality are PI3K-dependent. The blocking effect of both PI3K inhibitors was not due to decreased expression of the constructs in COS7 cells treated with the inhibitors, since mycPLD2 WT or Y179F were expressed at comparable levels in whole cell lysates (Figure 2D). They did not affect cell viability either, as assayed by the trypan blue exclusion test (not shown). The effect of PLD2-Y179F on DNA synthesis was observed in cells other than COS-7. Fig. 2E shows an increase thymidine incorporation with PLD2-Y179F in cells overexpressing the protein (Fig. 2F), making the initial observation more general. Together, these results suggest that AKT may regulate the cell fate (survival vs. DNA synthesis) via PLD2 Y179F. The possibility that PLD2 Y179F may be a substrate for AKT was investigated next.
Figure 2. PLD2-Y179F increases DNA synthesis in a PI3K-dependent manner.
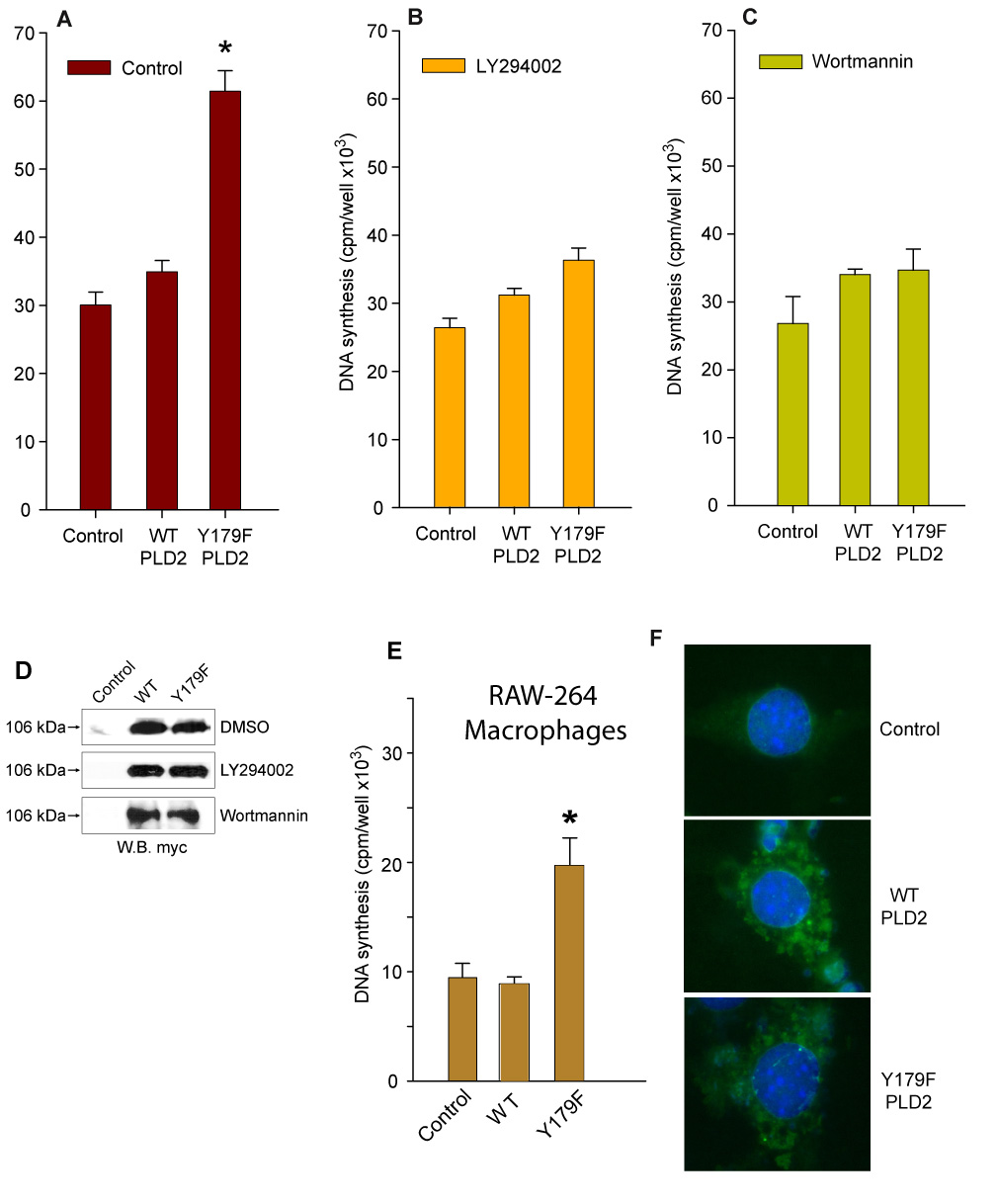
(A, B and C). PLD2 Y179F-dependent increase of de novo DNA synthesis. COS7 cells transiently expressing pcDNA-mycPLD2 WT or Y179F were incubated in serum-free media 16 hs in the presence of 0.6 uCi/ml [3H]-thymidine (A), 25 uM LY294002 (B) or Wortmannin (C). [3H]-thymidine incorporation into newly synthesized DNA is expressed as cpm/well (mean ± SEM, n=5, *p<0.001 vs. untreated Control). (D). Expression levels of PLD2 WT or Y179F are maintained in spite of inhibitors treatments. COS7 cells were transiently transfected with mycPLD2 WT or Y179F and incubated with vehicle (DMSO) or in the presence of 25 uM LY294002 or wortmannin. Shown are representative Western blots demonstrating comparable expression levels of the plasmids. (E–F). Study in RAW-264 macrophages of expression of PLD2 (wt and mutant) showing thymidine incorporation (E) and immunofluorescence pattern of protein intracellular expression, developed with TRITC-anti-myc antibodies (nuclei stained with DAPI).
PLD2 as a putative AKT substrate
To ascertain whether PLD2 could be a substrate for PLD2-driven phosphorylation of AKT, Western blotting experiments were performed with a polyclonal phospho-(S/T)-AKT kinase substrate antibody. This antibody recognizes peptides and proteins containing phospho-(S/T) preceded by R at position n-3 and/or n-5 [RxRxx(pS/pT)], the suggested consensus site for AKT kinase phosphorylation (Fig. 3A) [46–48]. As shown in Figure 3B, the AKT substrate antibody detected a band corresponding to immunoprecipitated mycPLD2 WT (~106 kDa), suggesting that PLD2 WT is phosphorylated in S/T within a consensus site for AKT kinase phosphorylation. Although mycPLD2 WT and Y179F were recognized by the antibody, mycPLD2 Y179F provided a stronger signal (Figure 3B). This PLD2 phosphorylation appears to be dependent on the PI3K/AKT activation, since LY294002 completely abolished AKT kinase substrate phosphorylation of mycPLD2 WT or Y179F (Fig. 3B, left panel). These results suggest that PLD2 is a substrate of activated AKT kinase and that mycPLD2 Y179F is the better substrate of the two.
Figure 3. MycPLD2 T175 is a substrate of AKT and mycPLD2 T175 mutants cannot increase de novo DNA synthesis.
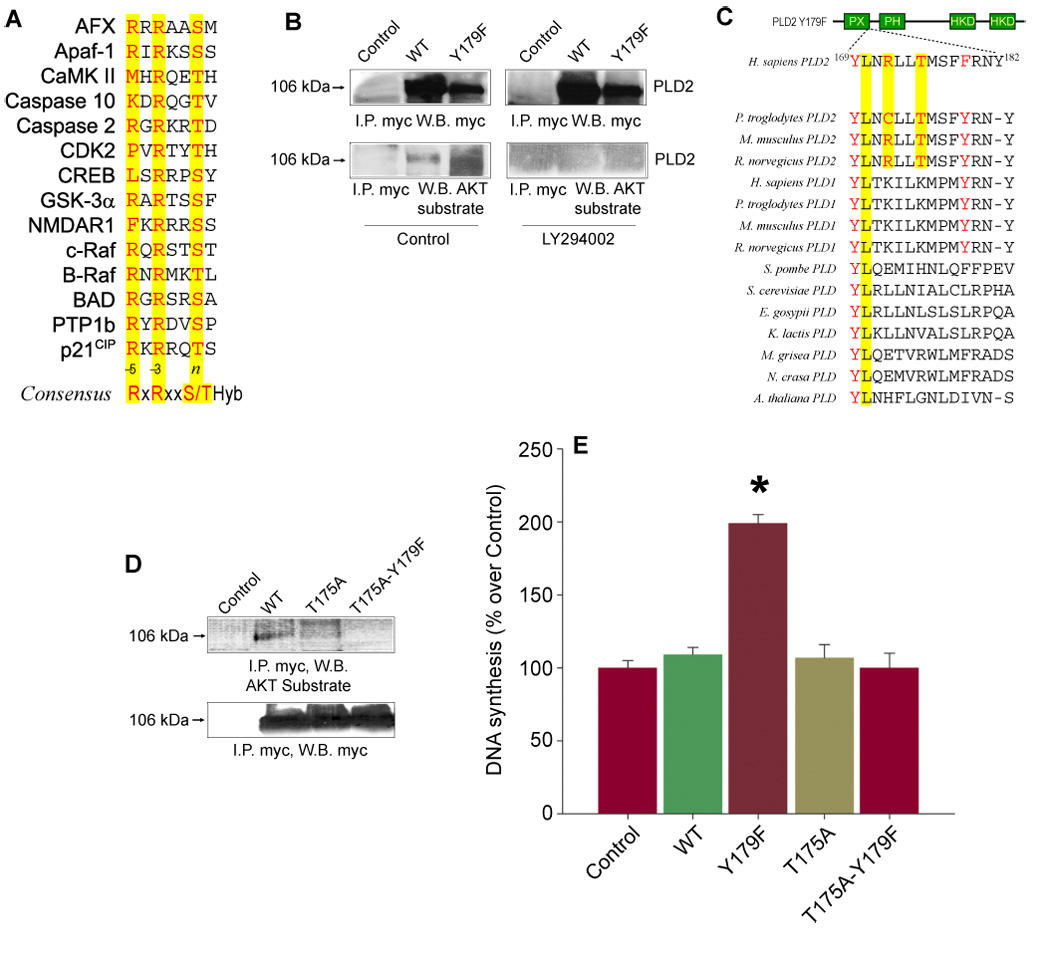
(A). Protein sequence alignment of known AKT substrates showing the sequence neighborhood of the AKT consensus phosphorylation site RxRxx(S/T)-Hyb, where Hyb stands for any hydrophobic residue. (B). Immunoprecipitation of mycPLD2 WT and Y179F. COS7 cells transiently expressing mycPLD2 WT or Y179F were incubated in the presence of vehicle (DMSO) or 25 M LY294002 overnight. Whole lysates were obtained, immunoprecipitated using a myc antibody coupled to agarose beads and resolved by SDS-PAGE. The immunological presence of mycPLD2 and phosphorylated AKT kinase substrate in mycPLD2 was revealed by using an anti-myc antibody or a polyclonal antibody directed against the sequence RxRxx(pS/pT), respectively. Shown is a representative experiment done in triplicate. (C). Protein sequence alignment of eukaryotic PLDs showing mammalian PLD2 conservation of residue T175 within the context LxRxxT. (D). AKT-dependent phosphorylation of mycPLD2 WT, T175A and T175A-Y179F. COS7 cells transiently expressing mycPLD2 WT or T175 mutants were incubated in the presence of vehicle (DMSO) overnight. Whole lysates were obtained, immunoprecipitated as indicated above and resolved by SDS-PAGE. The immunological presence of mycPLD2 and phosphorylated AKT kinase substrate in mycPLD2 was revealed by using an anti-myc antibody or a polyclonal antibody directed against the sequence RxRxx(pS/pT), respectively. Shown is a representative experiment done in triplicate. (E). [3H]-thymidine incorporation into newly synthesized DNA in COS7 cells Control (mock transfected) and transiently expressing PLD2 WT, T175A, Y179F or T175A-Y179F. Results are expressed as mean ± SEM [n=5, *p<0.001 vs. Control (30,200 ± 1,894 cpm/well)].
Manual analysis of eukaryotic PLD protein sequences revealed that T175, conserved only in mammalian PLD2s (Figure 3C), is encoded within the context 170LnRllT175, a potential target for activated AKT [48]. Based on this, residue T175 in pcDNA-mycPLD2 WT or Y179F was mutated to A175 by site-directed mutagenesis and transfected into COS7 cells. As shown in Figure 3D, the anti-AKT kinase substrate antibody did not detect immunoprecipitated mycPLD2 T175 mutants, providing immunological evidence that PLD2-stimulated AKT is in turn phosphorylating PLD2 Y179F in residue T175, and it does so more efficiently than mycPLD2 WT.
Both mycPLD2 T175 mutants were also tested for their ability to increase [3H]-thymidine incorporation in COS7 cells. As shown in Figure 3E, transient expression of mycPLD2 WT or T175 mutants did not stimulate de novo DNA synthesis when compared to mycPLD2 Y179F. These results suggest that residue T175, encoded within the potential consensus site for AKT-mediated phosphorylation (170LnRllT175), is a key participant of mycPLD2 Y179F-induced de novo DNA synthesis.
PLD2-Y179F regulates DNA synthesis via Mek activation
We next set out to determine the signaling mechanism by which PLD2-Y179F might regulate DNA synthesis. AKT activates the Raf/Mek signaling cascade and regulates the equilibrium between cell cycle arrest and proliferation [29] and overexpression of PLD2 Y179F phosphorylates p42/44ERK inducing DNA synthesis [25]. Hence, we tested the impact of LY294002-induced PI3K/AKT inhibition on mycPLD2 Y179F-mediated p42/44ERK phosphorylation. Transient expression of PLD2 Y179F resulted in increased phosphorylation of p42/44ERK (Figs. 4A,C). However, this effect was not observed in the presence of LY294002 (Figs. 4B,D), suggesting that mycPLD2-Y179F phosphorylates and activates p42/44ERK in a PI3K-dependent manner. Both constructs were expressed at comparable levels in whole cell lysates and did not affect cell viability (not shown).
Figure 4. PI3K-regulation of p42/44ERK phosphorylation and Mek-mediated regulation of PLD2-Y179F-induced de novo DNA synthesis.
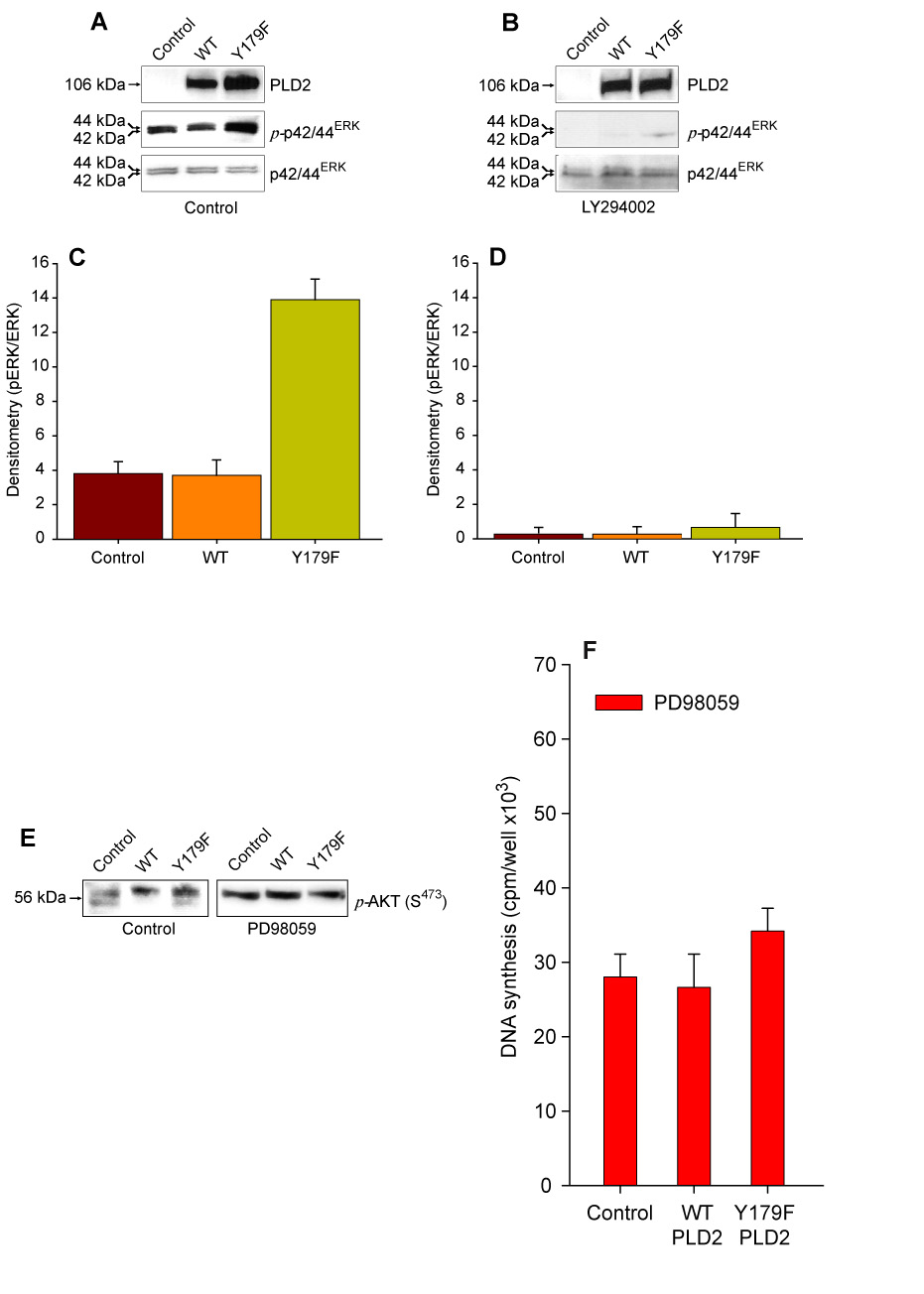
(A–D). Phosphorylation of p42/44ERK is PI3K-dependent. Mock- (Control) or mycPLD2 WT- and Y179F-transfected COS7 cells were incubated in the absence (A) or presence (B) of 25 uM LY294002. Whole lysates were obtained, resolved by SDS-PAGE and transferred to PDVF membranes. The immunological presence of mycPLD2, phosphorylated p42/44ERK and total p42/44ERK were analyzed by using specific antibodies. Shown is a representative experiment done in triplicate. (C and D). Densitometric analysis in arbitrary units expressed as mean ± SEM (n=3) of the optical density of the immunoblots normalized to total p42/44ERK signal (pp42/44ERK/p42/44ERK) for Control (C) or LY294002-treated cells (D). (E). Lack of an effect of Mek inhibitor PD98059 on PLD-induced AKT phosphorylation in residue S473. (F). [3H]-thymidine incorporation into newly synthesized DNA in COS7 cells Control (mock transfected) and transiently expressing PLD2 WT or Y179F. Results are expressed as mean ± SEM [n=3, *p<0.001 vs. Control].
On the other hand, PD98059 (Mek inhibitor) treatment of COS7 failed to inhibit mycPLD2 Y179F-induced basal phosphorylation of AKT in S473 (Figure 4E), but inhibited PLD2 Y179F-induced DNA synthesis (Figure 4F). These results suggest that AKT activation is located upstream of Mek and that Mek participates in PLD2 Y179F-induced DNA synthesis.
PLD2 Y179F regulates p21CIP1 and PCNA expression in a PI3K-dependent manner
To continue analyzing the mechanism of PLD2-Y179F regulation of DNA synthesis, we based our next series experiments in the known fact that AKT promotes cell survival through regulation of key effectors that directly or indirectly regulate the G0/G1 to S phase transition, such as p21CIP1 or PCNA [30–32, 49, 50]. Moreover, overexpression of PLD2 Y179F in COS7 cells correlated with increase expression of PCNA [25], a marker of cells committed to the S phase of the cell cycle [51]. Therefore, the role of PLD2 Y179F in the regulation of p21CIP1 and PCNA expression was investigated. As shown in Figures 5A,C, mock- or mycPLD2 WT-transfected COS7 cells did not significantly change the steady state levels of p21CIP1 or PCNA expression under basal conditions. Conversely, COS7 cells transiently expressing mycPLD2-Y179F produced an increase in p21CIP1 or PCNA expression. However, LY294002 completely blocked the mycPLD2 Y179F-induced effects (Figs. 5B,D). These results suggest that mycPLD2-Y179F promotes S phase entry and that mycPLD2 WT promotes cell survival, probably by arresting cells in G0/G1 through regulation of the cyclin-dependent kinase inhibitor p21CIP1/PCNA in a PI3K/AKT dependent manner.
Figure 5. PLD2-Y179F expression results in the releasing of the cell cycle arrest.
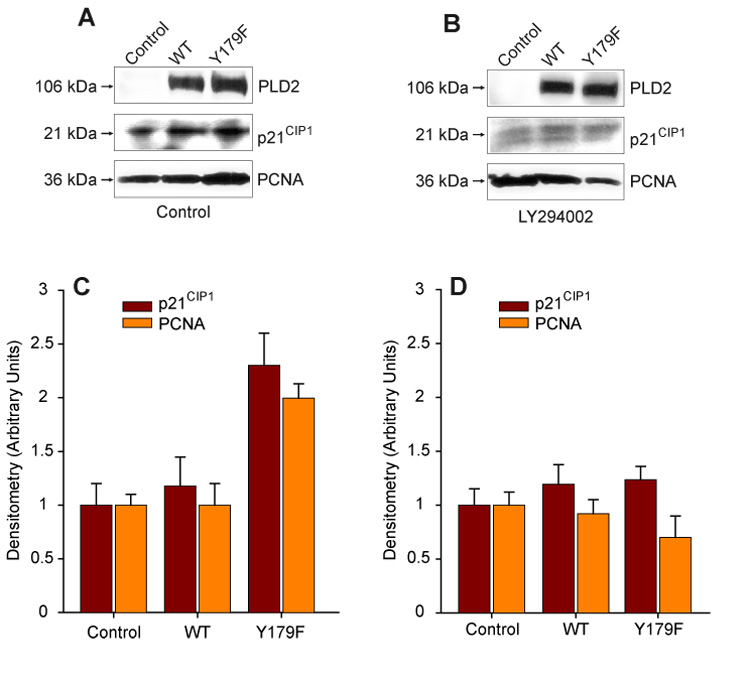
PLD2 WT- or Y179F-mediated regulation of p21CIP1 or PCNA under basal conditions (DMSO-treated) (A) or incubated in the presence of 25 uM LY294002 (B). COS7 cells Control (mock-transfected) and transiently expressing PLD2 (WT or Y179F) were incubated overnight in the presence or absence of the inhibitor. Cell lysates were subjected to SDS-PAGE, transferred to PDVF membranes and the expression levels of mycPLD2, p21CIP1 or PCNA simultaneously analyzed by Western blot using specific antibodies. Shown is a representative experiment carried out in triplicate. (C,D). Densitometric analysis in arbitrary units expressed as mean ± SEM (n=3) of the optical density of the immunoblots shown in A and B.
PLD2 Y179F-induced DNA synthesis is PA-dependent
Lastly, we wanted to ascertain if the effect of PLD2-Y179F on DNA synthesis would require the activity of the enzyme (i.e. catalysis of PC and PA generation). The following observations prompted us to investigate the role of PLD on PLD2 Y179F-induced DNA synthesis: i) PLD2 Y179F increases AKT phosphorylation and DNA synthesis and PCNA expression in a PI3K-dependent manner; ii) PLD2 Y179F-mediated phosphorylation of p42/44ERK is not dependent on PI3K; iii) PA binds the catalytic subunit of PI3K [52]; and iv) PA increases DNA synthesis when added exogenously [53]. Accordingly, the impact of PA generation on DNA synthesis and AKT phosphorylation was tested in COS7 cells transiently expressing PLD2-Y179F, is a fully active enzyme [25] or PLD2 Y179F/K758R, a catalytically inactive mutant. As shown in Figure 5, transient expression of mycPLD2 Y179F-K758R resulted in neither increased de novo DNA synthesis, AKT phosphorylation in S473, nor PCNA upregulation. Since PLD2-WT and -Y179F induce phosphorylation of AKT in a PI3K-dependent manner, but only PLD2 Y179F promotes DNA synthesis, these results indicate that PLD2-generated PA is involved in the PI3K-mediated activation of AKT.
DISCUSSION
Conclusive evidence implicating PI3K in PLD2 signaling has remained elusive. We present here data indicating that there is a link between PLD2 and PI3K-mediated signaling, that further affects cell cycle progression and DNA synthesis. The relevance of PLD2 in cell proliferation has been reported earlier [reviewed in [2, 43]]. Transient expression of PLD2 WT per se does not increase de novo DNA synthesis [3, 25], but provides survival signals by phosphorylation of AKT in a PI3K-dependent manner [24, 38–42] thus preventing uncontrolled cell proliferation [32]. We show here that transient expression of PLD2 WT or Y179F in COS7 cells resulted in increased AKT phosphorylation in residues T308/S473 in a PI3K-dependent manner. However, PLD2-Y179F, but not PLD2-WT, led to an increase in DNA synthesis. This prompted us to study its mechanisms and relevance.
Most of the AKT-dependent phosphorylations in response to PI3K activation occur in S or T within the consensus sequence RxRxx(S/T) followed by any hydrophobic amino acid (i.e. M, I, L, V, F, W or C) [48]. The requirement for R residues at position n-3 is very stringent, whereas R at n-5 appears not to be so stringent [46–48]. The sequence 170LnRllT175 located in the PX domain of PLD2 thus conforms a putative consensus site for AKT kinase phosphorylation. The results presented here provide evidence that PLD2 T175 is a substrate of activated AKT kinase.
MycPLD2 Y179F appears to be a better substrate for AKT since a stronger signal with this antibody was derived experimentally. Interestingly, residue T175 in PLD2 is located in the sequence 169YLNRLLTMSFYRNY182 which overlaps with one of the two demonstrated binding sites for the Grb2/Sos complex (underlined). Since residue Y169 in PLD2 is involved in Ras/MAPK activation and de novo DNA synthesis [25], together, these information suggest that mutation of Y179 may allow AKT kinase activity to phosphorylate T175 more efficiently, thus regulating PLD2 mitogenic potential via PLD2 Y169-mediated engagement of the Grb2/Sos/Ras/MAPK signaling cascade.
Transient expression of mycPLD2 Y179F increases p42/44ERK phosphorylation [25, 26] and AKT crosstalk with Raf modulating its function as a Mek activator [29, 54, 55]. MycPLD2 Y179F-mediated activation of p42/44ERK was dependent on PI3K activity suggesting that PI3K/AKT is located upstream of p42/44ERK. Moreover, Mek activity also appears to be required for PLD2 Y179F-induced DNA synthesis since PD98059-mediated Mek inhibition blocked this effect, but not PLD2-mediated AKT phosphorylation. Hence, these results indicate that the PI3K/AKT signaling cascade activated in response to PLD2 Y179F expression is in turn regulating Mek/MAPK signaling and de novo DNA synthesis. The opposite situation seems to be unlikely, since PLD2 Y179F-induced phosphorylation of AKT in residues T308/S473 was not affected by Mek inhibition.
AKT's targets known to regulate cell proliferation and transition from G0/G1 to S phase of the cell cycle are in turn regulated by PLD2 Y179F expression. AKT regulates S phase entry by direct phosphorylation of p21CIP1, preventing the formation of the complex p21CIP1/PCNA, which results in an inhibition of de novo DNA synthesis [56]. Also PI3K activation negatively regulates expression of p27KIP1 inducing DNA synthesis [57].
Exogenously added PA increases [3H]-thymidine incorporation [53]. In a phospholipid binding assay that uses unsaturated fatty acids containing PA, it was demonstrated that PA binds to the PI3K catalytic subunit, p110γ[52]. In neutrophils, PA also promotes tyrosine phosphorylation of p85, the regulatory subunit of PI3K [58]. Moreover, the addition of exogenous bacterial PLD to mammalian cells results in AKT activation in a PA- and PI3K-dependent manner [37, 43]. In agreement to these, our results indicate that PLD2-stimulated PI3K/AKT activation is PA-dependent. Indeed, PLD2 Y179F-induced DNA synthesis, phosphorylation of S473 in AKT and PCNA expression were not detected in COS7 cells expressing the catalytically inactive mutant PLD2 Y179F-K758R.
As PLD2-generated PA may serve to integrate AKT activation and DNA synthesis, we would like to propose the model depicted in Figure 7. In it, AKT is activated by PLD2 Y179F via PA-mediated activation of PI3K resulting in a PLD2-mediated increase in DNA synthesis via PLD2 Y169-mediated recruitment the Grb2/Sos complex, as demonstrated [25]. However, in the presence of functional Y169 and Y179 residues in PLD2, AKT may not efficiently phosphorylate T175 due to the binding of the second Grb2/Sos complex. This model may provide a short-loop feedback mechanism in which AKT-mediated phosphorylation of T175 in PLD2 may result in the regulation of Y179-mediated recruitment of the Grb2/Sos complex resulting in the modulation of DNA synthesis vs. survival signals.
Figure 7. Proposed model of PLD2/PI3K/AKT functional interaction.
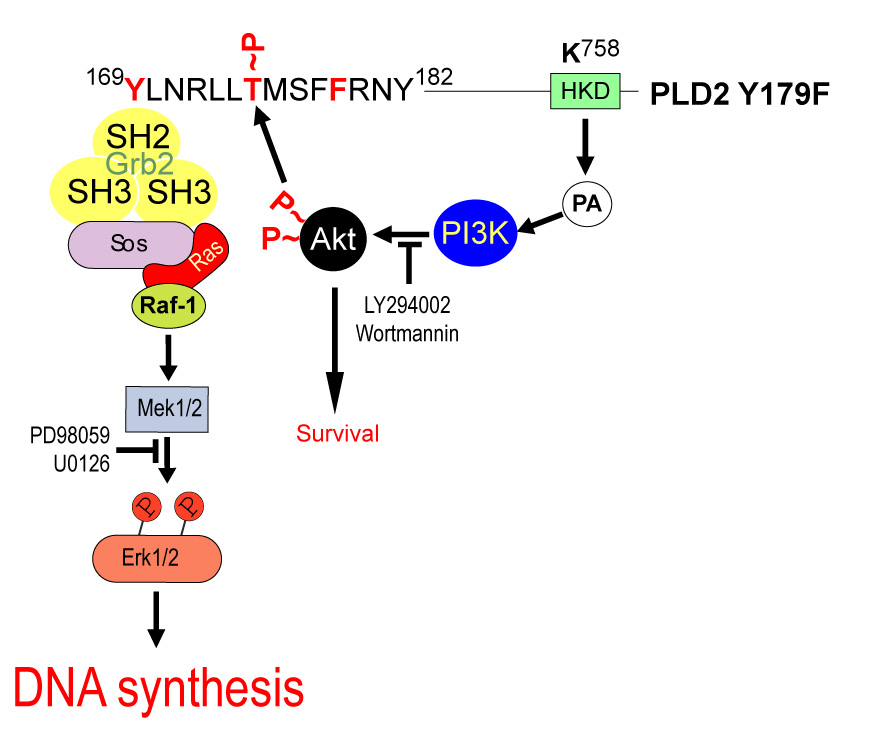
DNA synthesis induced by PI3K-mediated AKT activation and regulation of the Mek signaling cascade is driven by PLD2 Y179F overexpression. The figure shows a PLD2 region encompassing the protein region involved in this study (amino acids Y169-Y182). Also, a critical PLD2 residue for catalysis (K758) is depicted in the green box. As PA is produced by PLD2-Y179F, it induces ERK and AKT activation, which are mediated by PI3K, all leading to an increase in DNA synthesis. It is possible AKT would cross-talk to MEK (but not vice versa), through phosphoryaltion of PLD2 on residue T175 as explained in the text.
Figure 6. PLD2-Y179F-induced de novo DNA synthesis, AKT phosphorylation and PCNA expression depends on PLD2 catalysis.
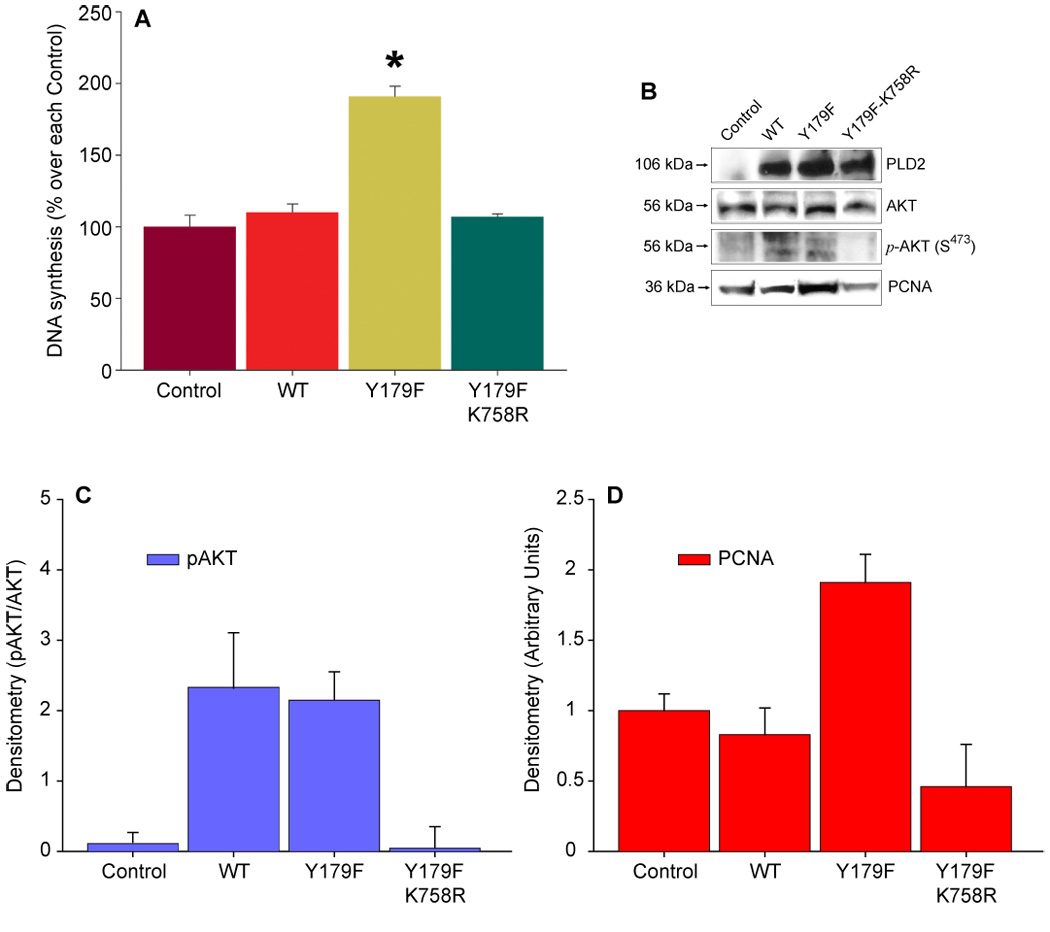
(A). Mock-transfected COS7 cells (Control) or transiently expressing PLD2 WT, Y179F or the catalytically inactive mutant Y179F-K758R were incubated in the presence of 0.6 uCi/ml [3H]-thymidine for 16 h. Precipitable DNA counts were then analyzed by scintillation counting. Results are expressed as % of Control (29,789 ± 2,366 cpm/well) without transfection as mean ± SEM, n=3, *p<0.001. (B). Cell lysates of COS7 cells transiently expressing PLD2 WT, Y179F or Y179F-K758R were obtained, resolved by SDS-PAGE and transferred to PDVF membranes. The immunological presence of mycPLD2, phosphorylated AKT (S473) and PCNA were analyzed by using specific antibodies. Shown is a representative experiment done in triplicate. (C,D). Densitometric analysis in arbitrary units expressed as mean ± SEM (n=3) of the optical density of the immunoblots shown in A and B.
Acknowledgements
This work has been supported by grants from the National Institutes of Health (HL056653), Research Incentive Program (Wright State University, School of Medicine) grant 229082 and Research Challenge (Ohio Board of Regents) grant 666761 to J.G.-C.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Exton JH. Biochim Biophys Acta. 1994;1212(1):26–42. doi: 10.1016/0005-2760(94)90186-4. [DOI] [PubMed] [Google Scholar]
- 2.Foster DA, Xu L. Mol Cancer Res. 2003;1(11):789–800. [PubMed] [Google Scholar]
- 3.Ahn BH, Kim SY, Kim EH, Choi KS, Kwon TK, Lee YH, Chang JS, Kim MS, Jo YH, Min do S. Mol Cell Biol. 2003;23(9):3103–3115. doi: 10.1128/MCB.23.9.3103-3115.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lucas L, del Peso L, Rodriguez P, Penalva V, Lacal JC. Oncogene. 2000;19(3):431–437. doi: 10.1038/sj.onc.1203323. [DOI] [PubMed] [Google Scholar]
- 5.del Peso L, Lucas L, Esteve P, Lacal JC. Biochem J. 1997;322(Pt 2):519–528. doi: 10.1042/bj3220519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu Z, Hornia A, Joseph T, Sukezane T, Frankel P, Zhong M, Bychenok S, Xu L, Feig LA, Foster DA. Mol Cell Biol. 2000;20(2):462–467. doi: 10.1128/mcb.20.2.462-467.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang H, Luo JQ, Urano T, Frankel P, Lu Z, Foster DA, Feig LA. Nature. 1995;378(6555):409–412. doi: 10.1038/378409a0. [DOI] [PubMed] [Google Scholar]
- 8.Jiang YW, Song J, Zang Q, Foster DA. Biochem Biophys Res Commun. 1994;203(2):1195–2003. doi: 10.1006/bbrc.1994.2309. [DOI] [PubMed] [Google Scholar]
- 9.Aguirre-Ghiso JA, Frankel P, Farias EF, Lu Z, Jiang H, Olsen A, Feig LA, de Kier Joffe EB, Foster DA. Oncogene. 1999;18(33):4718–4725. doi: 10.1038/sj.onc.1202850. [DOI] [PubMed] [Google Scholar]
- 10.Joseph T, Bryant A, Frankel P, Wooden R, Kerkhoff E, Rapp UR, Foster DA. Oncogene. 2002;21(22):3651–3658. doi: 10.1038/sj.onc.1205380. [DOI] [PubMed] [Google Scholar]
- 11.Uchida N, Okamura S, Kuwano H. Anticancer Res. 1999;19(1B):671–675. [PubMed] [Google Scholar]
- 12.Uchida N, Okamura S, Nagamachi Y, Yamashita S. J Cancer Res Clin Oncol. 1997;123(5):280–285. doi: 10.1007/BF01208639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshida M, Okamura S, Kodaki T, Mori M, Yamashita S. Oncol Res. 1998;10(8):399–406. [PubMed] [Google Scholar]
- 14.Zhao Y, Ehara H, Akao Y, Shamoto M, Nakagawa Y, Banno Y, Deguchi T, Ohishi N, Yagi K, Nozawa Y. Biochem Biophys Res Commun. 2000;278(1):140–143. doi: 10.1006/bbrc.2000.3719. [DOI] [PubMed] [Google Scholar]
- 15.Nozawa S, Ohno T, Banno Y, Dohjima T, Wakahara K, Fan DG, Shimizu K. J Biol Chem. 2005 doi: 10.1074/jbc.M411626200. [DOI] [PubMed] [Google Scholar]
- 16.Fang Y, Park IH, Wu AL, Du G, Huang P, Frohman MA, Walker SJ, Brown HA, Chen J. Curr Biol. 2003;13(23):2037–2044. doi: 10.1016/j.cub.2003.11.021. [DOI] [PubMed] [Google Scholar]
- 17.Lucas L, Penalva V, Ramirez de Molina A, Del Peso L, Lacal JC. Int J Oncol. 2002;21(3):477–485. [PubMed] [Google Scholar]
- 18.Frankel P, Ramos M, Flom J, Bychenok S, Joseph T, Kerkhoff E, Rapp UR, Feig LA, Foster DA. Biochem Biophys Res Commun. 1999;255(2):502–507. doi: 10.1006/bbrc.1999.0234. [DOI] [PubMed] [Google Scholar]
- 19.Song JG, Pfeffer LM, Foster DA. Mol Cell Biol. 1991;11(10):4903–4908. doi: 10.1128/mcb.11.10.4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Slaaby R, Du G, Altshuller YM, Frohman MA, Seedorf K. Biochem J. 2000;351(Pt 3):613–619. [PMC free article] [PubMed] [Google Scholar]
- 21.Rizzo MA, Shome K, Vasudevan C, Stolz DB, Sung TC, Frohman MA, Watkins SC, Romero G. J Biol Chem. 1999;274(2):1131–1139. doi: 10.1074/jbc.274.2.1131. [DOI] [PubMed] [Google Scholar]
- 22.Rizzo MA, Shome K, Watkins SC, Romero G. J Biol Chem. 2000;275(31):23911–23918. doi: 10.1074/jbc.M001553200. [DOI] [PubMed] [Google Scholar]
- 23.Hong JH, Oh SO, Lee M, Kim YR, Kim DU, Hur GM, Lee JH, Lim K, Hwang BD, Park SK. Biochem Biophys Res Commun. 2001;281(5):1337–1342. doi: 10.1006/bbrc.2001.4517. [DOI] [PubMed] [Google Scholar]
- 24.Banno Y, Takuwa Y, Akao Y, Okamoto H, Osawa Y, Naganawa T, Nakashima S, Suh PG, Nozawa Y. J Biol Chem. 2001;276(38):35622–35628. doi: 10.1074/jbc.M105673200. [DOI] [PubMed] [Google Scholar]
- 25.Di Fulvio M, Lehman N, Lin X, Lopez I, Gomez-Cambronero J. Oncogene. 2006;25(21):3032–3040. doi: 10.1038/sj.onc.1209340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Di Fulvio M, Frondorf K, Henkels KM, Lehman N, Gomez-Cambronero J. J Mol Biol. 2007;367(3):814–824. doi: 10.1016/j.jmb.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Datta K, Bellacosa A, Chan TO, Tsichlis PN. J Biol Chem. 1996;271(48):30835–30839. doi: 10.1074/jbc.271.48.30835. [DOI] [PubMed] [Google Scholar]
- 28.Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P. Oncogene. 1998;17(3):313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- 29.Zimmermann S, Moelling K. Science. 1999;286(5445):1741–1744. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]
- 30.Song G, Ouyang G, Bao S. J Cell Mol Med. 2005;9(1):59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chan TO, Rittenhouse SE, Tsichlis PN. Annu Rev Biochem. 1999;68:965–1014. doi: 10.1146/annurev.biochem.68.1.965. [DOI] [PubMed] [Google Scholar]
- 32.Vivanco I, Sawyers CL. Nat Rev Cancer. 2002;2(7):489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 33.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Science. 2001;294(5548):1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 34.Lehman N, Ledford B, Di Fulvio M, Frondorf K, McPhail LC, Gomez-Cambronero J. Faseb J. 2007 doi: 10.1096/fj.06-6652com. PubMed 17242159. [DOI] [PubMed] [Google Scholar]
- 35.Kwun HJ, Lee JH, Min do S, Jang KL. FEBS Lett. 2003;544(1–3):38–44. doi: 10.1016/s0014-5793(03)00446-0. [DOI] [PubMed] [Google Scholar]
- 36.Hui L, Abbas T, Pielak RM, Joseph T, Bargonetti J, Foster DA. Mol Cell Biol. 2004;24(13):5677–5686. doi: 10.1128/MCB.24.13.5677-5686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edwards JL, Apicella MA. Cell Microbiol. 2006;8(8):1253–1271. doi: 10.1111/j.1462-5822.2006.00707.x. [DOI] [PubMed] [Google Scholar]
- 38.Banno Y, Ohguchi K, Matsumoto N, Koda M, Ueda M, Hara A, Dikic I, Nozawa Y. J Biol Chem. 2005;280(16):16319–16324. doi: 10.1074/jbc.M410903200. [DOI] [PubMed] [Google Scholar]
- 39.Li F, Zhang C, Schaefer S, Estes A, Malik KU. Am J Physiol Heart Circ Physiol. 2005;289(6):H2592–H2601. doi: 10.1152/ajpheart.00450.2005. [DOI] [PubMed] [Google Scholar]
- 40.Li F, Malik KU. J Pharmacol Exp Ther. 2005;312(3):1043–1054. doi: 10.1124/jpet.104.076588. [DOI] [PubMed] [Google Scholar]
- 41.Yamada M, Banno Y, Takuwa Y, Koda M, Hara A, Nozawa Y. Biochem J. 2004;378(Pt 2):649–656. doi: 10.1042/BJ20031398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lim HK, Choi YA, Park W, Lee T, Ryu SH, Kim SY, Kim JR, Kim JH, Baek SH. J Biol Chem. 2003;278(46):45117–45127. doi: 10.1074/jbc.M303789200. [DOI] [PubMed] [Google Scholar]
- 43.Nozawa Y. Biochim Biophys Acta. 2002;1585(2–3):77–86. doi: 10.1016/s1388-1981(02)00327-x. [DOI] [PubMed] [Google Scholar]
- 44.Kim J, Lee YH, Kwon TK, Chang JS, Chung KC, Min do S. Cancer Res. 2006;66(2):784–793. doi: 10.1158/0008-5472.CAN-05-1316. [DOI] [PubMed] [Google Scholar]
- 45.Horn J, Lopez I, Miller MW, Gomez-Cambronero J. Biochem Biophys Res Commun. 2005;332(1):58–67. doi: 10.1016/j.bbrc.2005.04.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Obata T, Yaffe MB, Leparc GG, Piro ET, Maegawa H, Kashiwagi A, Kikkawa R, Cantley LC. J Biol Chem. 2000;275(46):36108–36115. doi: 10.1074/jbc.M005497200. [DOI] [PubMed] [Google Scholar]
- 47.Yaffe MB, Leparc GG, Lai J, Obata T, Volinia S, Cantley LC. Nat Biotechnol. 2001;19(4):348–353. doi: 10.1038/86737. [DOI] [PubMed] [Google Scholar]
- 48.Alessi DR, Caudwell FB, Andjelkovic M, Hemmings BA, Cohen P. FEBS Lett. 1996;399(3):333–338. doi: 10.1016/s0014-5793(96)01370-1. [DOI] [PubMed] [Google Scholar]
- 49.Li Y, Dowbenko D, Lasky LA. J Biol Chem. 2002;277(13):11352–11361. doi: 10.1074/jbc.M109062200. [DOI] [PubMed] [Google Scholar]
- 50.Ahn JY, Liu X, Liu Z, Pereira L, Cheng D, Peng J, Wade PA, Hamburger AW, Ye K. Embo J. 2006;25(10):2083–2095. doi: 10.1038/sj.emboj.7601111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moldovan GL, Pfander B, Jentsch S. Cell. 2007;129(4):665–679. doi: 10.1016/j.cell.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 52.Kirsch C, Wetzker R, Klinger R. Biochem Biophys Res Commun. 2001;282(3):691–696. doi: 10.1006/bbrc.2001.4623. [DOI] [PubMed] [Google Scholar]
- 53.Moolenaar WH, Kruijer W, Tilly BC, Verlaan I, Bierman AJ, de Laat SW. Nature. 1986;323(6084):171–173. doi: 10.1038/323171a0. [DOI] [PubMed] [Google Scholar]
- 54.Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K, Moelling K, Yancopoulos GD, Glass DJ. Science. 1999;286(5445):1738–1741. doi: 10.1126/science.286.5445.1738. [DOI] [PubMed] [Google Scholar]
- 55.Guan KL, Figueroa C, Brtva TR, Zhu T, Taylor J, Barber TD, Vojtek AB. J Biol Chem. 2000;275(35):27354–27359. doi: 10.1074/jbc.M004371200. [DOI] [PubMed] [Google Scholar]
- 56.Rossig L, Jadidi AS, Urbich C, Badorff C, Zeiher AM, Dimmeler S. Mol Cell Biol. 2001;21(16):5644–5657. doi: 10.1128/MCB.21.16.5644-5657.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mahimainathan L, Ghosh-Choudhury N, Venkatesan BA, Danda RS, Choudhury GG. Am J Physiol Renal Physiol. 2005;289(1):F72–F82. doi: 10.1152/ajprenal.00277.2004. [DOI] [PubMed] [Google Scholar]
- 58.Siddiqui RA, English D. Biochim Biophys Acta. 2000;1483(1):161–173. doi: 10.1016/s1388-1981(99)00172-9. [DOI] [PubMed] [Google Scholar]