

Abstract
The tumor suppressor protein p53 activates growth arrest and pro-apoptotic genes in response to DNA damage. It is known that negative feedback by p21Cip1/Waf1/Sdi1 represses p53-dependent transactivation of PUMA. The current study investigates PUMA feedback on p53 during oxidative stress from hyperoxia and the subsequent effects on cell survival mediated through p21 and Bcl-XL. Deletion of PUMA in HCT116 colon carcinoma cells increased levels of p53 and p21 resulting in a larger G1 population during hyperoxia. P21-dependent increase in Bcl-XL levels protected PUMA-deficient cells against hyperoxic cell death. Bax and Bak were both able to promote hyperoxic cell death. Bcl-XL protection against hyperoxic death was lost in cells lacking Bax, not PUMA, suggesting that Bcl-XL acts to inhibit Bax-dependent death. These results indicate PUMA exerts negative feedback on p53 and p21, leading to p21-dependent growth suppressive and survival changes. Enhanced survival was associated with increased Bcl-XL to block Bax activated cell death during oxidative stress.
Keywords: Free radicals, cell death
INTRODUCTION
Reactive oxygen species (ROS) play an important role as secondary messengers in signaling pathways and are generated through aerobic respiration or by the radiolysis of water. Since aerobic organisms must tolerate aberrant ROS formation due to electron leakage along the respiratory chain, anti-oxidant systems have developed to detoxify ROS thus preventing oxidative injury to the cell. Oxidation of intracellular macromolecules occurs when anti-oxidant defenses are overwhelmed by ROS production. Oxidative stress resulting from ROS contributes in the pathology of neurodegenerative diseases, cardiovascular dysfunctions, inflammation, cancer and promotes to the aging process [1]. DNA damage caused by oxidative stress may result in activation of growth arrest, DNA repair and/or cell death pathways. The tumor suppressor protein, p53, plays a critical role in regulating cell cycle checkpoints and apoptosis during genotoxic responses. The primary function of p53 is to transactivate gene targets involved in the damage response, since mice and cells expressing a transcriptionally inactive mutant of p53 lose the ability to undergo growth arrest and apoptosis during stress [2, 3].
P21Cip1/Waf1/Sdi1 (hereafter p21), is a transcriptional target of p53 which is responsible for initiating G1 arrest by binding and inhibiting cyclin-dependent kinases (cdk) and proliferating cell nuclear antigen (PCNA). Expression of p21 enhances survival during exposure to apoptotic stimuli such as ionizing radiation, doxorubicin, cisplatin, nitrogen mustard, Fas activation, PGA2 exposure, p53-overexpression and TNF activation [4–9]. While it is apparent that the extent of stress-induced damage is dependent on cell cycle phase of the cell, p21 possesses anti-apoptotic functions which are cell cycle-independent [10]. P21 has direct anti-apoptotic functions by blocking activation of procaspase-3, caspase-8 and ASK-1 [11, 12].
Apoptosis is tightly orchestrated through the interactions of the Bcl-2 protein family which includes several p53 gene targets, such as p53-upregulated modulator of apoptosis (PUMA), Bax and NOXA. Bcl-2 family proteins are classified by homology between Bcl-2 domains (BH1-4) and functional outcome during apoptotic activation. The anti-apoptotic proteins (Bcl-2, Bcl-XL, Mcl-1, Bcl-w, A1) interact with pro-apoptotic multidomain members (Bax, Bak) and the BH3-only proteins (PUMA, NOXA, tBid, Bad, Bim, Bmf, Bik, Hrk) to regulate the release of pro-apoptotic stimuli from the mitochondria. It is clear that BH3-only molecules activate the pro-apoptotic multidomain proteins and that anti-apoptotic members prevent this activation. It still remains unclear which Bcl-2 proteins are engaged during resting state and/or stress conditions [13].
Unlike most instantaneous genotoxic agents, exposure to hyperoxia results in the persistent production of reactive species which cause cellular damage resulting in DNA damage [14]. Even though mixed apoptotic and necrotic cell death occurs, abundant findings implicate the Bcl-2 family as mediators of the hyperoxic cell death response. Fibroblasts isolated from Bax−/− Bak−/− mice have increased resistance against hyperoxic death [15] and ROS activation of Bax causes cytochrome c release in MLE-12 cells which can be blocked by Bcl-XL overexpression [15, 16]. Caspase-8-dependent cleavage of Bid to tBid in hyperoxia can be blocked by FLIP [17, 18]. Hyperoxia stimulated expression of Bcl-XL, Bcl-2 and A1 in mouse lungs and deletion of A1 increased hyperoxic injury [19]. Overexpression of Bcl-XL in Rat1a cells protected against LDH release and cell death while siRNA knockdown of Bcl-XL in HCT116 wt cells increased cell death [15, 20]. Also, Bcl-2 overexpression in L929 cells was able to prevent mitochondrial release of apoptosis inducing factor (AIF) and cell death [21]. Together, these findings implicate members of the Bcl-2 family in hyperoxia-induced cell death.
Hyperoxia has been shown to activate p53 and p21 and loss of p21 sensitizes mice and cells to hyperoxic death [22–24]. Recent studies show that the p21 pro-survival function in hyperoxia is uncoupled from its growth suppressive activity and involves the regulation of Bcl-XL [20, 25]. Also, it is clear that p21 disruption can promote p53-dependent cell death which is thought to be mediated through increased PUMA transcription [26–28]. The current studies manipulate PUMA, Bcl-XL, Bax, Bak and p21 in HCT116 colon cancer cells to investigate if PUMA regulates p53 activation of p21 utilizing hyperoxia as a model of persistent oxidative stress. Since p21 regulates Bcl-XL, the pathway of Bcl-XL inhibition of cell death was also investigated.
MATERIALS AND METHODS
Cell lines and exposures
The human colon carcinoma HCT116 wild-type (wt) and isogenic cell lines lacking PUMA (PUMA−), P21/PUMA (p21−/PUMA−) or BAX (Bax−) were obtained from Dr. Bert Vogelstein (Johns Hopkins Oncology Center and the Program in Human Genetics and Molecular Biology) and genetic deletions have previously been verified by our laboratory [20]. Cells were cultured in McCoy’s medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 µg/mL streptomycin (Gibco). Cells were counted with a hemocytometer and plated at 5 × 105 cells per 100-mm dish and allowed to adhere overnight. The following day plates were exposed to normoxia (room air with 5% CO2) or hyperoxia (95% O2/5% CO2) by being placed into a Plexiglas box (Belco Glass) that was sealed and flooded with gas at a flow rate of 5 L/min for 10 min and then maintained at a flow rate of 0.2 L/min [20]. Oxygen concentrations were monitored with a miniOXI for the duration of exposure (Catalyst Research Corporation).
Cell death and cell cycle measurements
Following treatment, cells were trypsinized, resuspended in medium and centrifuged before overnight fixation in 75% ethanol. Cells were treated with 1 mg/mL RNase for 30 min and resuspended in phosphate buffered saline containing 10 µg/mL propidium iodide (Sigma-Aldrich). Samples were analyzed using a BD FACSCalibur flow cytometer set to collect 10,000 events. The percentage of cells with subG1 DNA content was determined by using CellQuest v3.3 software (BD Biosciences). The percentage of cells in G1, S and G2/M was determined using ModFit LT software (Verity Software House).
Western blot analysis
Cells were lysed in 50 mM Tris (pH 7.4), 150 mM NaCl, 2 mM EDTA, 25 mM sodium fluoride, 25 mM sodium β-glycerophosphate, 0.1 mM sodium vanadate, 0.1 mM phenylmethylsulfonyl fluoride, 0.2% Triton X-100, 0.3% IGEPAL CA-630, 0.1 µg/ml pepstatin A, 1.9 µg/ml aprotinin and 2 µg/ml leupeptin. Protein concentrations were determined by the Lowry method (BioRad). Cell lysates were diluted in 3X Laemmli Buffer and boiled for 5 minutes. Laemmli at 1X contains 50 mM Tris (pH 6.8), 1% β-mercaptoethanol, 2% SDS, 0.1% bromophenol blue and 10% glycerol. The extracted protein was separated by SDS-PAGE and transferred to polyvinylidene difluoride membranes (Pall Life Sciences). The membranes were then incubated with anti-p53 clone DO-1 (1:1000, Oncogene Research Products), anti-p53 ser-15 (1:1000, Cell Signaling), anti-p21 clone SX118 (1:500, BD Pharmingen), anti-Bax clone N-20 (1:500, Santa Cruz Biotechnology), anti-Bcl-XL clone 2H12 (1:500, Sigma-Aldrich) and anti-Bak clone Ab-1 (1:500, Calbiochem) with actin (1:1000, Sigma-Aldrich) as a loading control. Membranes were then incubated in horseradish peroxidase conjugated secondary anti-mouse (1:4000, Southern Biotechnology) or anti-rabbit (1:5000, Jackson Immunoresearch) antibodies and visualized by chemiluminescence (Amersham Biosciences). Densitometric analysis was performed by quantifying band intensities and normalizing to actin using a Fluorochem8900 (Alpha Innotech).
RNAi treatment
Cells were plated overnight in 12-well plates at 7 × 104 cells per well in medium lacking antibiotics. Cells were transfected with annealed luciferase (Dharmacon), Bcl-X (5’-AAGAGAATCACTAACCAGAGA-3’, Invitrogen) or Bak (SMARTpool, Dharmacon) oligonucleotides using Lipofectamine 2000 [20, 29]. After 12 hrs cells were washed and exposed to room air or hyperoxia for 4 days.
Statistical analysis
Group means were compared by ANOVA with Fisher’s PLSD using Statview software (Abacus Concepts). Values represent means ± standard deviation for three to four separate exposures to hyperoxia with p ≤ 0.05 considered to be significant.
RESULTS
Loss of PUMA Activates p21-Dependent Growth Arrest in Hyperoxia
Since p21 is able to affect p53 activity during the DNA damage response, we studied the ability of PUMA to regulate p53 and p21 during oxidative stress caused by treatment in hyperoxia. HCT116 cells exposed to hyperoxia accumulated total p53, increased phosphorylation of p53 at serine-15 and increased levels of p21 (Fig. 1A). Disruption of the PUMA gene altered the response to hyperoxia characterized by increased p53 total and phosphorylated levels along with increased p21 levels. Cell cycle analysis was performed on PUMA− cells due to altered p21 expression in response to hyperoxia after 4 days. HCT116 wt and PUMA− cells had similar cell cycle profiles in room air (Fig 1C), but a greater number of PUMA− cells were found in G1 in hyperoxia (Fig. 1B, C). The number of HCT116 wt cells in G1 following hyperoxia was 24.3±1.6% compared to 38±2.83% of the PUMA− cells which had increased levels of p21 (p = 0.03). This finding is consistent with the ability of p21 to elicit growth checkpoints in G1 during environmental stress. Since PUMA− cells expressed more p21 and accumulated in G1, p21−/PUMA− were used to test for p21-dependency. Fewer p21−/PUMA− cells were in G1 following hyperoxic exposure (12.7±3.13%), suggesting that these cell cycle changes are p21-dependent. These data suggest lack of PUMA leads to increased p53 and p21 levels which enhanced G1 growth arrest during oxidative stress.
Figure 1. Loss of PUMA activates cell cycle checkpoints through p21.
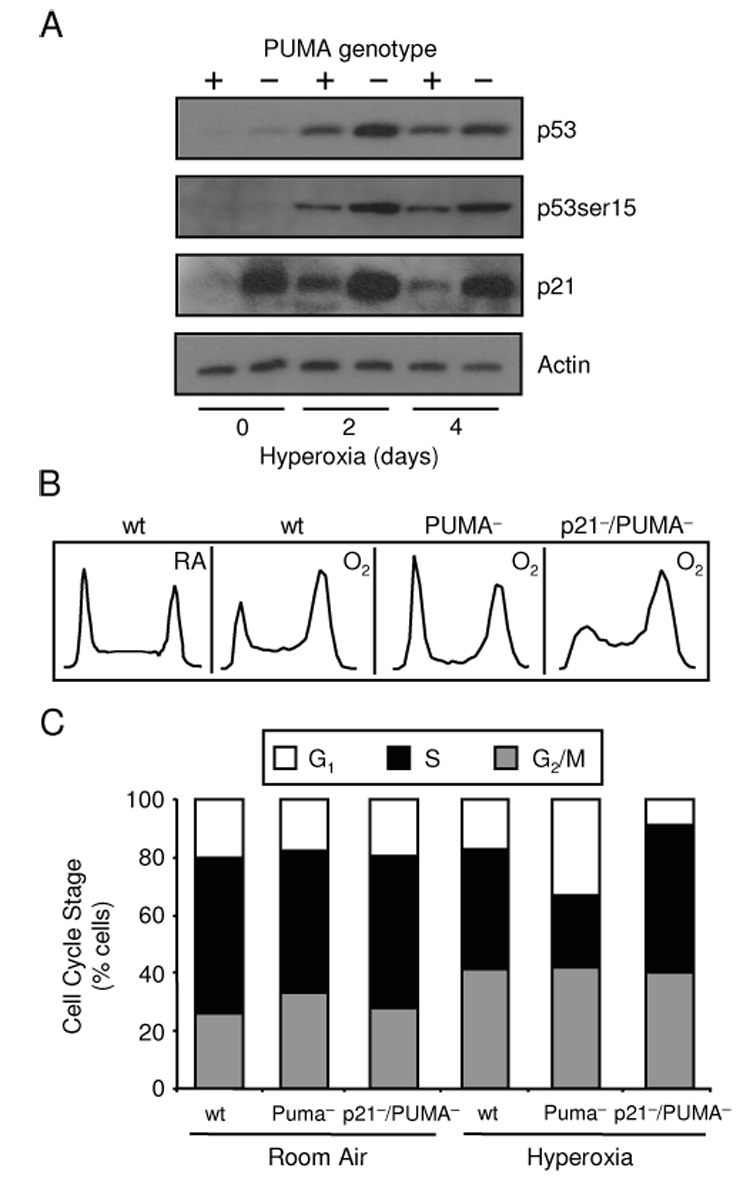
(A) Expression of p53, p53 ser-15 and p21 in HCT116 wt and PUMA− cells exposed to a hyperoxic time course with actin used as a loading control. Immunoblots were representative of three separate experiments. (B) Representative DNA histograms of HCT116 wt, PUMA− and p21−/PUMA− cells exposed to 4 days of room air (RA) or hyperoxia (O2). (C) Mean average of HCT116 wt, PUMA− and p21−/PUMA− cells in G1, S and G2/M exposed to 4 days of room air or hyperoxia (n=3).
Cells Lacking PUMA are Protected Against Hyperoxic Death via p21/Bcl-XL
P21 is able to protect cells against hyperoxia by regulating Bcl-XL levels [20, 25]. Since PUMA− cells have higher levels of p21, changes in Bcl-XL and survival were examined. PUMA− cells had increased Bcl-XL protein levels compared to HCT116 wt in room air and during the hyperoxic timecourse (Fig. 2A). To determine if changes in Bcl-XL were p21-dependent, expression of Bcl-XL was investigated in HCT116 wt, PUMA− and p21−/PUMA− cells exposed to room air or hyperoxia. While hyperoxic treatment caused loss of Bcl-XL, it remained higher in PUMA–cells. While Bcl-XL remained elevated in p21−/PUMA− cells, it was not as high as PUMA− cells, suggesting Bcl-XL expression in PUMA− cells was regulated by both p21-dependent and independent mechanisms (Fig 2B,C). Since Bcl-XL inhibits cell death under numerous stress stimuli including hyperoxia, cell survival studies were performed to test if PUMA− were protected against hyperoxia and Bcl-XL knockdown was perfomed to see how PUMA− cells responsd to loss of Bcl-XL. Successful RNAi knockdown of Bcl-XL in HCT116 cells has been performed previously [20] and was performed in both HCT116 wt and PUMA− cells prior to 4 days of hyperoxia exposure. Efficient knockdown of Bcl-XL levels was achieved with no effect of the transfection reagent alone (mock) or siRNAs targeting luciferase (Fig. 2D). PUMA− cells had reduced death after 4 days hyperoxia by measuring subG1 DNA content (HCT116 mock siRNA, 32.54±6.38% death; PUMA− mock siRNA, 21.68±3.82% death; Fig. 2E). RNAi knockdown of Bcl-XL increased death of both HCT116 wt and PUMA− cells, implicating that p21 regulation of Bcl-XL affects survival of PUMA− cells exposed to hyperoxia. These data also indicate that PUMA is not required to cause cell during Bcl-XL knockdown.
Figure 2. PUMA inactivation protects cells against hyperoxic cell death via p21/Bcl-XL.
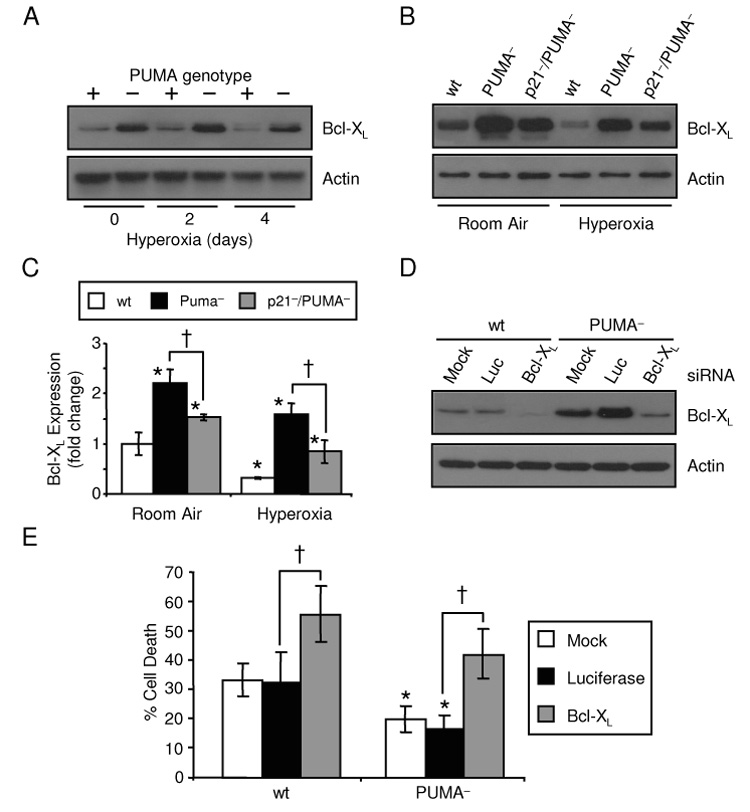
(A) Bcl-XL protein expression in HCT116 wt and PUMA− cells exposed to a hyperoxic time course with actin used as a loading control. (B) Representative immunoblots of Bcl-XL and actin expression in HCT116 wt, PUMA− and p21−/PUMA− cells exposed to 4 days of room air or hyperoxia. (C) Bcl-XL levels represented in (B) were quantified by densitometry and normalized to actin. (D) Bcl-XL and actin levels in HCT116 wt and PUMA− cells either mock transfected or treated with siRNAs targeting luciferase (Luc) or Bcl-XL prior to hyperoxic exposure for 4 days. (E) Percentage of death in HCT116 wt and PUMA− cells transfected with Bcl-XL siRNAs and treated with hyperoxia for 4 days (*p=0.04 compared between cell lines; †p=0.006 compared between siRNA treatment; n=3).
Bax and Bak promote hyperoxic cell death
Bax and Bak are Bcl-2 pro-apoptotic multidomain proteins which are required for cell death in response to a variety of stress stimuli [30, 31]. Bax activation in response to hyperoxia has been identified and hyperoxic cell death was reduced in murine embryonic fibroblasts from Bax−/−Bak−/− mice, but it is not clear whether it is Bax, Bak or both proteins which are required to promote cell death [15, 16]. Less cell death was seen in Bax− cells after 2 and 4 days hyperoxia (Fig. 3A). Only 16.6±4.35% cell death occurred in Bax− cells after 4 days hyperoxia compared to 28.92±5.47% cell death of HCT116 wt cells. After 4 days hyperoxic treatment, Bax− cells had increased cell death compared to untreated controls indicating that cell death was not completely inhibited by genetic deletion of Bax. To test for Bak-dependent cell death in hyperoxia, RNAi was used to knockdown Bak in Bax− cells. Bak protein levels increased in response to hyperoxia, but Bak siRNA treatment was sufficient to knockdown protein levels 87% in Bax− cells (Fig. 3B). Cell death was reduced with Bak siRNA treatment indicating that Bak also promotes hyperoxic cell death (Fig. 3C). SiRNA knockdown of Bak also increased death of HCT116 wt cells exposed to hyperoxia (data not shown). Taken together these data show that Bax and Bak both activate cell death pathways in response to hyperoxia.
Figure 3. Bax and Bak promote hyperoxic cell death.
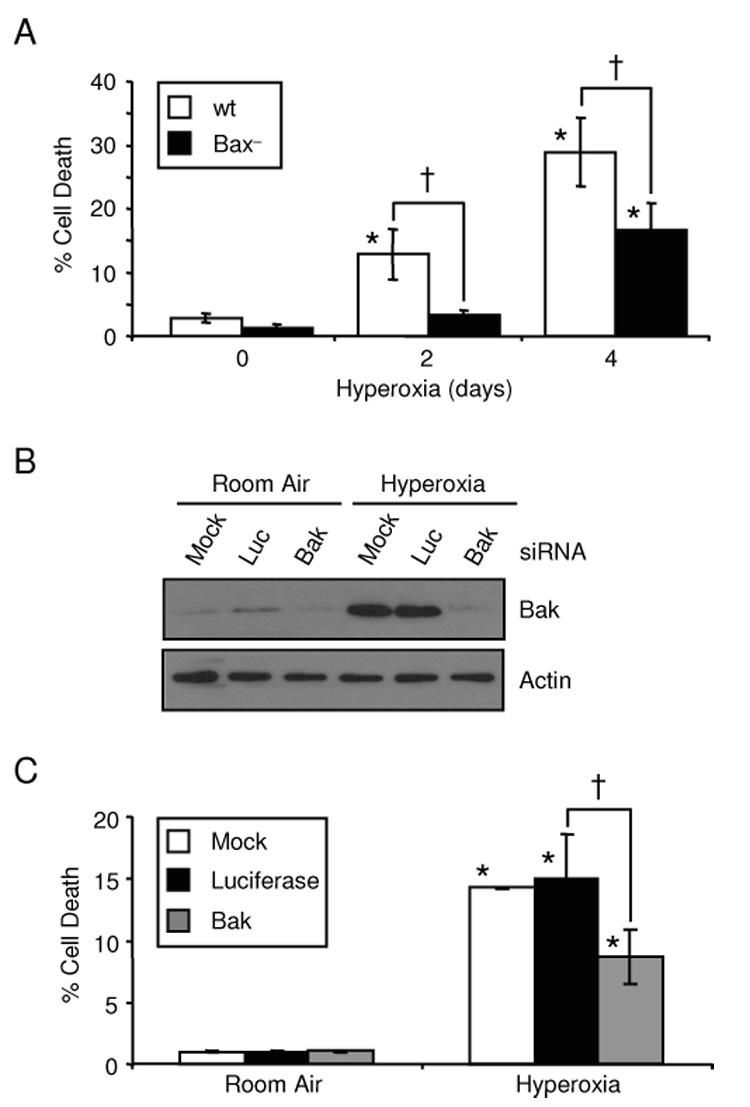
(A) Percentage of cell death in HCT116 wt and Bax− cells exposed to a hyperoxic time course. (B) Bak expression in HCT116 Bax− cells either mock transfected or treated with siRNAs targeting luciferase (Luc) or Bak prior to hyperoxic exposure for 4 days. Actin expression was used as a loading control (C) Percentage of death in HCT116 Bax− cells transfected with Bak siRNAs and cultured for 4 days in room air or hyperoxia (*p=0.0002 compared to untreated controls; †p=0.0013 compared between cell lines or siRNA treatment; n=3).
Bcl-XL blocks Bax-dependent hyperoxic cell death
Bcl-XL can block death by binding and inhibiting Bax and Bak or by inactivating specific BH3-only proteins [13]. To test if Bcl-XL protected against Bax-induced hyperoxic cell death, RNAi targeting Bcl-XL was performed in Bax− cells. Bcl-XL knockdown significantly increased hyperoxic death of HCT116 wt cells (luciferase siRNA, 23.39±6.34% death; Bcl-XL siRNA, 48.11±8.57% death), while Bcl-XL siRNA treatment had no change in the survival of Bax− cells exposed to hyperoxia (luciferase siRNA, 16.93±1.55% death; Bcl-XL siRNA, 17.72±1.27% death; Fig. 4A, B). An interesting observation was that siRNA knockdown of Bcl-XL significantly increased the number of HCT116 wt cells in the G1 phase (luciferase siRNA, 18.76±1.6% G1; Bcl-XL siRNA, 31.41±3.36% G1) of the cell cycle and decreased the number of cells in the G2/M phase (luciferase siRNA, 32.81±2.17% G2/M; Bcl-XL siRNA 13.79±0.02% G2/M) after 4 days of hyperoxic treatment (Fig. 4C, D). Bax− cells responded similarly to the Bcl-XL siRNA treatment, justifying that Bcl-XL knockdown was sufficient to elicit a cell cycle response even though there were no changes in survival (Fig. 4B, D). Cycling effects were not secondary to changes in viability, since Bax− cells had similar changes in cell cycle with Bcl-XL knockdown even though survival was unaffected. P21 expression was investigated due to its role in the G1 checkpoint, but there were no changes in p21 expression during Bcl-XL knockdown (data not shown). To test if Bcl-XL was involved in G1 progression or a G2/M block, HCT116 wt cells were treated with colcemid to prevent progression through G2/M. Cells treated with Bcl-XL siRNA still had more cell in G1 after colcemid treatment in hyperoxia suggesting that Bcl-XL plays a role in promoting cell cycle progression through G1 phase (data not shown). These results suggest that Bcl-XL plays a role in cell cycle regulation independent of p21 and is able to inhibit Bax-dependent cell death during hyperoxia.
Figure 4. Bcl-XL blocks Bax-dependent hyperoxic cell death.
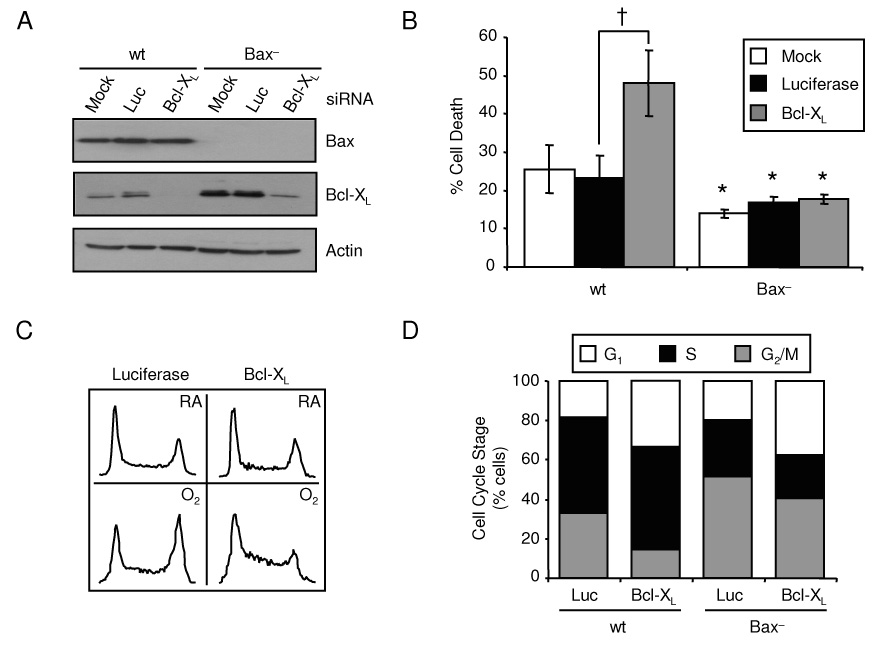
(A) Expression of Bax and Bcl-XL in HCT116 wt and Bax− cells mock transfected or treated with siRNAs targeting luciferase (Luc) or Bcl-XL prior to hyperoxic exposure for 4 days with actin used as a loading control. Immunoblots were representative of three separate experiments. (B) Percentage of cell death in HCT116 wt and Bax− cells transfected with Bcl-XL siRNAs then exposed to hyperoxia for 4 days. (C) Representative DNA histograms HCT116 wt cells transfected with Bcl-XL siRNAs then exposed to room air or hyperoxia for 4 days. (D) Mean average of HCT116 wt and Bax− cells in G1, S and G2/M after Bcl-XL siRNA treatment cultured in room air or hyperoxia for 4 days (*p=0.02 compared between cell lines; †p=0.0004 compared between siRNA treatments; n=4)‥
DISCUSSION
P53 plays a pivotal role in regulating the damage response, inducing growth arrest which prevents the replication of damaged DNA or programmed cell death which is important for eliminating defective cells [32]. It was previously established that p21 exerted negative feedback on p53 transactivation of PUMA [26–28]. As illustrated in Figure 5, we report that PUMA regulates p53 and p21 levels during oxidative stress and that this affects p21-dependent growth arrest and survival pathways. Loss of PUMA led to elevated expression of p21 and growth arrest in G1 and decreased cell death was associated with higher Bcl-XL levels in hyperoxia. Bax and Bak activation both contributed to hyperoxic cell death, but only Bax-dependent cell death was blocked by Bcl-XL. Also, increased numbers of cells were seen in G1 after Bcl-XL knockdown without any changes in p21 expression.
Figure 5. Model of cell survival and death pathways activated during hyperoxia.
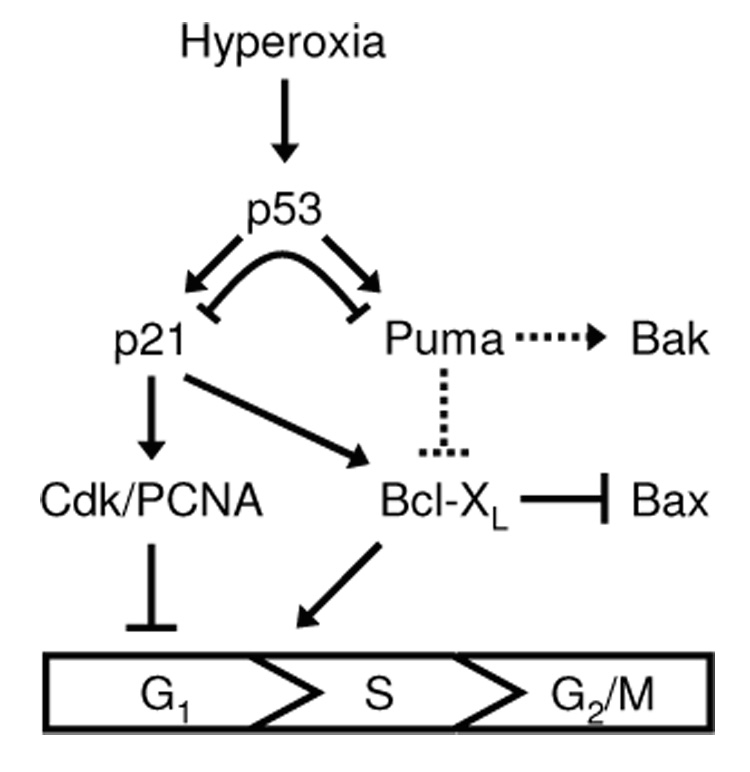
Hyperoxia-induced cell survival and death pathways downstream of p53. Solid lines represent interactions described in this study or previous studies and dotted arrows represent putative relationships. Arrows indicate pathway activation while perpendicular lines indicate pathway inhibition.
The novel finding of PUMA feedback inhibition of p53 and p21 is consistent with the idea that pro-apoptotic factors exist to antagonize the protective effects of p21. Polyak and colleagues [28] showed that one colorectal cell line, DLD1, undergoes apoptosis in response to transfected p53 while HCT116 wt cells preferentially growth arrest. Tetraploid fusions between each cell type resulted in an apoptotic response to p53 that was described to be dominant because of a cell specific pro-apoptotic factor that inhibited p21-dependent survival. Another key piece of evidence suggesting that survival factors are important regulators of the p53 response is from data that loss of P53 in mice or HCT116 cells does not alter the rate of cell death during hyperoxia [33, 34]. The chronic nature of hyperoxia leads to a balanced increase in both p53-dependent survival (p21) and death (PUMA) pathways. These observations question if p53 itself can contribute in choosing the response to oxidative stress [35]. P53 may activate default pathways following stress and there are accessory signals which help coordinate the downstream response such as activation of caspase-8 via Fas signaling [17, 35]. Alternatively, specific post-translational modifications, such as phosphorylation, may direct p53 activation of preferential response pathways. This is evidenced by different substrate specificity of the phosphatidylinositol 3-kinase-like kinases ATM and ATR in response to hyperoxia, ionizing radiation and UV light. Cofactors, such as Slug, p300 and p21, have been shown to preferentially direct p53 transactivation of gene targets [26, 36–38]. There is also evidence that negative feedback p53 loops are integrated in the p53 response. It is known that p53 gene targets, cyclin G and HDM2, function together to promote degradation of p53 [39]. Also, p63 and p73 have overlapping functions to p53 and can regulate p53 gene transcription [37]. P21 and PUMA, respectively, regulate pro-survival and pro-death pathways and these studies demonstrate that PUMA has the ability to moderate p21/Bcl-XL pro-survival signaling. It is suggested that p21 regulates p53 activity through p14ARF and it is possible that PUMA also targets a protein which regulates p53 stability.
Our studies also clearly show that both Bax and Bak are able to activate cell death in response to hyperoxia. This is consistent with multiple studies implicating Bcl-2 family proteins in the regulation of hyperoxic cell death [15–21]. Bax or Bak oligomerization must take place in order to initiate mitochondrial release of cytochrome c and AIF. Studies suggest that Bcl-XL can inhibit Bax and Bak to block cell death, yet our data support the idea that Bcl-XL works only via Bax inhibition under oxidative stress, since deletion of BAX completely blocked the death response to Bcl-XL knockdown. Support for Bcl-XL inhibition of Bak comes from experiments showing that Bcl-XL binds Bak in healthy cells but UV light treatment causes Bak-dependent cell death via NOXA. While a direct genetic link between NOXA and Bcl-XL was established, Bcl-XL binding to Bak could be an artifact of detergent conditions during protein co-immunoprecipitation [40]. However, our studies investigate cell death pathways using a genetic approach with a physiological outcome measure, thereby, reducing the likelihood of experimental artifacts. Cell death was not abolished in Bax− cells treated with Bak siRNA indicating that additional pro-death pathways including Bid cleavage may remain intact. Another important finding was that hyperoxia upregulated Bak protein levels. Bak may be regulated transcriptionally by WT1 or Sp3, or it may be post-translationally modified by interacting proteins [41, 42]. Current studies are aimed at investigating the pathway of Bak upregulation by hyperoxia.
Currently, there are two working models for activation of Bax and Bak [13, 43]. In the direct model of activation a genotoxic signal activates BH3 only proteins which then block anti-apoptotic protein inhibition of Bax and Bak causing cell death [44]. While it is clear that anti-apoptotic proteins block the activation of Bax and Bak, it is thought that the biochemical inhibition is due to direct binding and sequestration of Bax and Bak in this model. Recent evidence argues for a hierarchical model of activation where a higher class of BH3 only proteins disrupts anti-apoptotic protein inhibition of more potent BH3 only proteins. The free potent BH3 only proteins then stimulate Bax and Bak activation [45]. Our findings argue that PUMA is not directly downstream of Bcl-XL during hyperoxic death, since PUMA was not necessary to promote death in cells lacking Bcl-XL. Studies by Ming et al. show that adriamycin stimulates Bcl-XL dissociation from Bax in a PUMA-dependent manner [46]. During hyperoxia, it is possible that either PUMA is targeting a different anti-apoptotic protein upstream of Bax or that it activates Bak directly in this model. P21-independent regulation of Bcl-XL was seen in PUMA− cells and one explanation is that PUMA binding to Bcl-XL promotes degradation of Bcl-XL, since NOXA binding to Mcl-1 initiates proteasome-dependent degradation of Mcl-1 [40]. The ability to visualize changes in Bcl-XL degradation may be unique to hyperoxia due to the chronic nature of the stress given the relatively long protein half-life of Bcl-XL [47]. Cytoplasmic p53 may promote cell death by engaging Bcl-XL, Bcl-2 and Bax [48, 49], but there is currently no evidence supporting that this pathway is functional during hyperoxia as p53 was not required to promote cell death with knockdown of Bcl-XL (data not shown).
Similar to the dual-functions of p21, specific Bcl-2 family proteins integrate regulation of apoptotic and cell cycle pathways. It has been reported that Bcl-2 and Bcl-XL are able to delay G1 and S phase progression by driving cells into G0 [50]. Also, many of the anti-apoptotic proteins have cell cycle effects which can be mediated through binding of Bcl-2 and Bcl-XL. The precise mechanism for these growth effects is not known and appears to be cell specific. Interestingly, our studies found no cell cycle effects due to Bcl-XL knockdown in room air, but increased cells in G1 with hyperoxia. Since the effect is damage-dependent, one explanation for the unexpected Bcl-XL changes in cell cycle is compounded due to hyperoxia-induced growth changes [51].
This study utilizes hyperoxia as a model of persistent oxidative stress to identify pathways which may be associated with chronic oxidative diseases such as atherosclerosis, neurodegenerative diseases, ischemia-reperfusion injuries and Crohn’s disease. In summary, this study demonstrates that PUMA regulates p53 and p21 levels during oxidative stress resulting in cell cycle and survival effects. Our data suggest that survival was enhanced in PUMA− cells due to increased levels of Bcl-XL which were partly dependent on p21. Hyperoxic exposure resulted in activation of Bax and Bak but Bcl-XL inhibition of Bax activation prevented cell death. Since many disease processes and aging are associated with ROS accumulation and DNA damage, knowledge of which specific cell survival and death pathways are activated in response to oxidative stress could provide new opportunities for effective disease treatment.
ACKNOWLEDGEMENTS
This work was supported in part by NIH grant HL-067392. NIH training grant ES-07026 supported P. Vitiello. The flow cytometry core was supported in part by ES-01247.
LIST OF ABBREVIATIONS
- ROS
reactive oxygen species
- PCNA
proliferating cell nuclear antigen
- wt
wild-type
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Maccarrone M, Ullrich V. Redox regulation in disease and ageing. Cell Death Differ. 2004;11(8):949–951. doi: 10.1038/sj.cdd.4401458. [DOI] [PubMed] [Google Scholar]
- 2.Chao C, et al. p53 transcriptional activity is essential for p53-dependent apoptosis following DNA damage. Embo J. 2000;19(18):4967–4975. doi: 10.1093/emboj/19.18.4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jimenez GS, et al. A transactivation-deficient mouse model provides insights into Trp53 regulation and function. Nat Genet. 2000;26(1):37–43. doi: 10.1038/79152. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Blandino G, Givol D. Induced p21waf expression in H1299 cell line promotes cell senescence and protects against cytotoxic effect of radiation and doxorubicin. Oncogene. 1999;18(16):2643–2649. doi: 10.1038/sj.onc.1202632. [DOI] [PubMed] [Google Scholar]
- 5.Gorospe M, et al. Protective role of p21(Waf1/Cip1) against prostaglandin A2-mediated apoptosis of human colorectal carcinoma cells. Mol Cell Biol. 1996;16(12):6654–6660. doi: 10.1128/mcb.16.12.6654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fan S, et al. Cells lacking CIP1/WAF1 genes exhibit preferential sensitivity to cisplatin and nitrogen mustard. Oncogene. 1997;14(18):2127–2136. doi: 10.1038/sj.onc.1201052. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki A, et al. Resistance to Fas-mediated apoptosis: activation of caspase 3 is regulated by cell cycle regulator p21WAF1 and IAP gene family ILP. Oncogene. 1998;17(8):931–939. doi: 10.1038/sj.onc.1202021. [DOI] [PubMed] [Google Scholar]
- 8.Gorospe M, et al. p21(Waf1/Cip1) protects against p53-mediated apoptosis of human melanoma cells. Oncogene. 1997;14(8):929–935. doi: 10.1038/sj.onc.1200897. [DOI] [PubMed] [Google Scholar]
- 9.Xu SQ, El-Deiry WS. p21(WAF1/CIP1) inhibits initiator caspase cleavage by TRAIL death receptor DR4. Biochem Biophys Res Commun. 2000;269(1):179–190. doi: 10.1006/bbrc.2000.2247. [DOI] [PubMed] [Google Scholar]
- 10.Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1(8):639–649. [PubMed] [Google Scholar]
- 11.Asada M, et al. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. Embo J. 1999;18(5):1223–1234. doi: 10.1093/emboj/18.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Suzuki A, et al. Procaspase 3/p21 complex formation to resist fas-mediated cell death is initiated as a result of the phosphorylation of p21 by protein kinase A. Cell Death Differ. 2000;7(8):721–728. doi: 10.1038/sj.cdd.4400706. [DOI] [PubMed] [Google Scholar]
- 13.Galonek HL, Hardwick JM. Upgrading the BCL-2 network. Nat Cell Biol. 2006;8(12):1317–1319. doi: 10.1038/ncb1206-1317. [DOI] [PubMed] [Google Scholar]
- 14.Joenje H. Genetic toxicology of oxygen. Mutat Res. 1989;219(4):193–208. doi: 10.1016/0921-8734(89)90001-5. [DOI] [PubMed] [Google Scholar]
- 15.Budinger GR, et al. Hyperoxia-induced apoptosis does not require mitochondrial reactive oxygen species and is regulated by Bcl-2 proteins. J Biol Chem. 2002;277(18):15654–15660. doi: 10.1074/jbc.M109317200. [DOI] [PubMed] [Google Scholar]
- 16.Buccellato LJ, et al. Reactive oxygen species are required for hyperoxia-induced Bax activation and cell death in alveolar epithelial cells. J Biol Chem. 2004;279(8):6753–6760. doi: 10.1074/jbc.M310145200. [DOI] [PubMed] [Google Scholar]
- 17.Wang X, et al. Necrotic cell death in response to oxidant stress involves the activation of the apoptogenic caspase-8/bid pathway. J Biol Chem. 2003;278(31):29184–29191. doi: 10.1074/jbc.M301624200. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, et al. FLIP inhibits endothelial cell apoptosis during hyperoxia by suppressing Bax. Free Radic Biol Med. 2007;42(10):1599–1609. doi: 10.1016/j.freeradbiomed.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 19.He CH, et al. Bcl-2-related protein A1 is an endogenous and cytokine-stimulated mediator of cytoprotection in hyperoxic acute lung injury. J Clin Invest. 2005;115(4):1039–1048. doi: 10.1172/JCI23004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Staversky RJ, et al. p21(Cip1/Waf1/Sdi1) protects against hyperoxia by maintaining expression of Bcl-X(L) Free Radic Biol Med. 2006;41(4):601–609. doi: 10.1016/j.freeradbiomed.2006.04.029. [DOI] [PubMed] [Google Scholar]
- 21.Metrailler-Ruchonnet I, et al. Bcl-2 protects against hyperoxia-induced apoptosis through inhibition of the mitochondria-dependent pathway. Free Radic Biol Med. 2007;42(7):1062–1074. doi: 10.1016/j.freeradbiomed.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 22.O'Reilly MA, et al. The cyclin-dependent kinase inhibitor p21 protects the lung from oxidative stress. Am J Respir Cell Mol Biol. 2001;24(6):703–710. doi: 10.1165/ajrcmb.24.6.4355. [DOI] [PubMed] [Google Scholar]
- 23.Helt CE, et al. p53-dependent induction of p21(Cip1/WAF1/Sdi1) protects against oxygen-induced toxicity. Toxicol Sci. 2001;63(2):214–222. doi: 10.1093/toxsci/63.2.214. [DOI] [PubMed] [Google Scholar]
- 24.Helt CE, et al. Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage. J Biol Chem. 2005;280(2):1186–1192. doi: 10.1074/jbc.M410873200. [DOI] [PubMed] [Google Scholar]
- 25.Vitiello PF, et al. p21Cip1 protection against hyperoxia requires Bcl-XL and is uncoupled from its ability to suppress growth. Am J Pathol. 2006;168(6):1838–1847. doi: 10.2353/ajpath.2006.051162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iyer NG, et al. p300 regulates p53-dependent apoptosis after DNA damage in colorectal cancer cells by modulation of PUMA/p21 levels. Proc Natl Acad Sci U S A. 2004;101(19):7386–7391. doi: 10.1073/pnas.0401002101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Javelaud D, Besancon F. Inactivation of p21WAF1 sensitizes cells to apoptosis via an increase of both p14ARF and p53 levels and an alteration of the Bax/Bcl-2 ratio. J Biol Chem. 2002;277(40):37949–37954. doi: 10.1074/jbc.M204497200. [DOI] [PubMed] [Google Scholar]
- 28.Polyak K, et al. Genetic determinants of p53-induced apoptosis and growth arrest. Genes Dev. 1996;10(15):1945–1952. doi: 10.1101/gad.10.15.1945. [DOI] [PubMed] [Google Scholar]
- 29.Chandra D, et al. Bax-dependent regulation of Bak by voltage-dependent anion channel 2. J Biol Chem. 2005;280(19):19051–19061. doi: 10.1074/jbc.M501391200. [DOI] [PubMed] [Google Scholar]
- 30.Hetz C, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312(5773):572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- 31.Wei MC, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science. 2001;292(5517):727–730. doi: 10.1126/science.1059108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer. 2002;2(8):594–604. doi: 10.1038/nrc864. [DOI] [PubMed] [Google Scholar]
- 33.Barazzone C, et al. Oxygen toxicity in mouse lung: pathways to cell death. Am J Respir Cell Mol Biol. 1998;19(4):573–581. doi: 10.1165/ajrcmb.19.4.3173. [DOI] [PubMed] [Google Scholar]
- 34.O'Reilly MA, et al. p53-independent induction of GADD45 and GADD153 in mouse lungs exposed to hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2000;278(3):L552–L559. doi: 10.1152/ajplung.2000.278.3.L552. [DOI] [PubMed] [Google Scholar]
- 35.Vousden KH. p53: death star. Cell. 2000;103(5):691–694. doi: 10.1016/s0092-8674(00)00171-9. [DOI] [PubMed] [Google Scholar]
- 36.Wu WS, et al. Slug antagonizes p53-mediated apoptosis of hematopoietic progenitors by repressing puma. Cell. 2005;123(4):641–653. doi: 10.1016/j.cell.2005.09.029. [DOI] [PubMed] [Google Scholar]
- 37.Flores ER, et al. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416(6880):560–564. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- 38.Lohr K, et al. p21/CDKN1A mediates negative regulation of transcription by p53. J Biol Chem. 2003;278(35):32507–32516. doi: 10.1074/jbc.M212517200. [DOI] [PubMed] [Google Scholar]
- 39.Okamoto K, et al. Cyclin G recruits PP2A to dephosphorylate Mdm2. Mol Cell. 2002;9(4):761–771. doi: 10.1016/s1097-2765(02)00504-x. [DOI] [PubMed] [Google Scholar]
- 40.Willis SN, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19(11):1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Morrison DJ, English MA, Licht JD. WT1 induces apoptosis through transcriptional regulation of the proapoptotic Bcl-2 family member Bak. Cancer Res. 2005;65(18):8174–8182. doi: 10.1158/0008-5472.CAN-04-3657. [DOI] [PubMed] [Google Scholar]
- 42.Chirakkal H, et al. Upregulation of BAK by butyrate in the colon is associated with increased Sp3 binding. Oncogene. 2006;25(54):7192–7200. doi: 10.1038/sj.onc.1209702. [DOI] [PubMed] [Google Scholar]
- 43.Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17(6):617–625. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Willis SN, et al. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315(5813):856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 45.Kim H, et al. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8(12):1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- 46.Ming L, et al. PUMA Dissociates Bax and Bcl-X(L) to induce apoptosis in colon cancer cells. J Biol Chem. 2006;281(23):16034–16042. doi: 10.1074/jbc.M513587200. [DOI] [PubMed] [Google Scholar]
- 47.Pardo OE, et al. Fibroblast growth factor-2 induces translational regulation of Bcl-XL and Bcl-2 via a MEK-dependent pathway: correlation with resistance to etoposide-induced apoptosis. J Biol Chem. 2002;277(14):12040–12046. doi: 10.1074/jbc.M109006200. [DOI] [PubMed] [Google Scholar]
- 48.Chipuk JE, et al. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309(5741):1732–1735. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- 49.Chipuk JE, et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303(5660):1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- 50.Janumyan YM, et al. Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. Embo J. 2003;22(20):5459–5470. doi: 10.1093/emboj/cdg533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O'Reilly MA. DNA damage and cell cycle checkpoints in hyperoxic lung injury: braking to facilitate repair. Am J Physiol Lung Cell Mol Physiol. 2001;281(2):L291–L305. doi: 10.1152/ajplung.2001.281.2.L291. [DOI] [PubMed] [Google Scholar]