

Abstract
Background
Macrophage apoptosis is a critical process in the formation of necrotic cores in vulnerable atherosclerotic plaques. In-vitro and in-vivo data suggest that macrophage apoptosis in advanced atheromata may be triggered by a combination of endoplasmic reticulum (ER) stress and engagement of the type A scavenger receptor (SRA), which together induce death through a rise in cytosolic calcium and activation of toll-like receptor-4 (TLR4).
Methods and Results
Using both primary peritoneal macrophages and studies in advanced atheromata in vivo, we introduce Signal Transducer and Activator of Transcription-1 (STAT1) as a critical and necessary component of ER stress/SRA-induced macrophage apoptosis. We show that STAT1 is serine-phosphorylated in macrophages subjected to SRA ligands and ER stress in a manner requiring cytosolic calcium, calcium/calmodulin-dependent protein kinase II (CaMKII), and TLR4. Remarkably, apoptosis was inhibited by ~80–90% (p < 0.05) by STAT1 deficiency or CaMKII inhibition. In vivo, nuclear Ser-P-STAT1 was found in macrophage-rich regions of advanced murine and human atheromata. Most importantly, macrophage apoptosis was decreased by 61% (p = 0.034) and plaque necrosis by 34% (p = 0.02) in the plaques of fat-fed Ldlr−/− mice transplanted with Stat1−/− bone marrow.
Conclusions
STAT1 is critical for ER stress/SRA-induced apoptosis in primary tissue macrophages and in macrophage apoptosis in advanced atheromata. These findings suggest a potentially important role for STAT1-mediated macrophage apoptosis in atherosclerotic plaque progression.
Keywords: atherosclerosis, plaque necrosis, macrophage, apoptosis, STAT1
In advanced atherosclerosis, death of macrophages in the setting of defective phagocytic clearance of apoptotic cells contributes to the development of plaque necrosis.1,2 Plaque necrosis, in turn, is thought to promote plaque disruption and arterial thrombosis, which are the proximate causes of acute cardiovascular events.1–3 Our laboratory established an important principle of advanced lesional macrophage death, namely, involvement of the ER stress pathway known as the Unfolded Protein Response (UPR).4,5 The laboratories of Austin, Lusis, and others have important evidence that the UPR is activated in intimal cells, including macrophages, in advanced murine and human plaques.6–9 In particular, Myoishi et al.9 recently showed a dramatic rise in UPR markers, including CHOP, and intimal cell apoptosis in autopsy specimens from humans with vulnerable and ruptured plaques, but not stable lesions, and in atherectomy specimens from humans with unstable angina, but not stable angina. Although the UPR is primarily an ER repair pathway, a branch of the UPR involving the effector CHOP (GADD153) can trigger apoptosis when the cell senses that repair is no longer possible.4,10 In terms of causation, we have shown that advanced lesional macrophage death and plaque necrosis are decreased in atherosclerotic Apoe−/− mice in the setting of ER stress prevention5 or CHOP deficiency (E. Thorp and I. Tabas, unpublished data).
Our work on the UPR began with a model of advanced lesional macrophage death that is present in advanced plaques, namely, intracellular accumulation of lipoprotein-derived free cholesterol (FC).11 FC enrichment of macrophages, like many ER stressors, activates the UPR through depletion of ER lumenal calcium.12,13 Since then, mechanistic studies have led to a broader concept of advanced lesional macrophage death, beyond the FC model. These studies have shown that any combination of inducers of ER stress and ligands for the macrophage type A scavenger receptor (SRA), both of which are prominently expressed in advanced lesions, trigger macrophage apoptosis.14,15 Macrophage SRA recognizes a number of lesional molecules and atherogenic lipoproteins, including those used to enrich macrophages with cholesterol in the FC model.16 The SRA is also a pattern recognition receptor (PRR) of the innate immune system, and endotoxin-free SRA ligands also activate other PRRs, notably TLR4.15,17,18 In this context, our studies have shown that SRA ligands trigger two critical pro-apoptotic events in ER-stressed macrophages: (a) TLR4-mediated activation of a pro-apoptotic MyD88 pathway;15 and (b) SRA-mediated suppression of a pro-survival TLR4-TRIF-INFβ pathway.14,15
In this report, we show that apoptosis of ER-stressed macrophages also requires STAT1 and CaMKII in a process involving cytosolic calcium and TLR4. Most importantly, we provide evidence that activated STAT1 is present in atheromata and that lesional macrophage apoptosis is suppressed in the setting of STAT1 deficiency.
Methods (see Data Supplement for expanded methods)
Assay of Macrophage Apoptosis
Mid- and late-stage apoptosis in peritoneal macrophages was assayed by annexin V and propidium iodine (PI) staining, respectively, using the Vybrant Apoptosis Assay kit #2 (Molecular Probes). At the end of incubation, the macrophages were gently washed once with PBS and incubated for 15 min at room temperature with 120 μl of annexin-binding buffer (25 mM HEPES, 140 mM NaCl, 1 mM EDTA, pH 7.4, 0.1% bovine serum albumin) containing 10 μl of Alexa Fluor 488-conjugated annexin V and 1 μl of 100 μg/ml PI. The staining mixture was then removed and replaced with 120 μl of annexin-binding buffer. The cells were viewed immediately at room temperature using an Olympus IX-70 inverted fluorescent microscope equipped with filters appropriate for fluorescein and rhodamine, and images were obtained using a Cool Snap CCD camera (RS Photometrics) equipped with imaging software from Roper Scientific. Three fields of cells (~650 cells/field) were photographed for each condition, and the number of annexin V/PI–positive cells in each field were counted and expressed as a percent of the total number of cells.
Bone Marrow Transplantation
Ten-week-old female Ldlr−/− mice were lethally irradiated with 1000 rads from a cesium γ source 4–6 h before transplantation. Bone marrow cells were collected from the femurs and tibias of donor Stat1−/− or Stat1+/+ mice by flushing with sterile medium (RPMI 1640, 2% FBS, 10 U/ml heparin, 50 U/ml penicillin, 50 μg/ml streptomycin). The bone marrow cells were extensively washed and resuspended in RPMI medium containing 20 mM HEPES, 50 U/ml penicillin, and 50 μg/ml streptomycin. Each recipient mouse was injected with 5 × 106 bone marrow cells through the tail vein. The mice were given acidified water containing 100 mg/liter neomycin and 10 mg/liter polymyxin B sulfate 1 week before and 2 weeks after transplantation. Six weeks after transplantation, the mice were fed a “Western-type” diet (21% anhydrous milk fat and 0.15% cholesterol—TD88137 from Harlan-Teklad) for 10 or 12 weeks.
Atherosclerotic Lesion Analysis
On the day of the analysis, food was removed from the cages in the morning, and the mice were fasted for 8 h. The animals were then anesthetized using isoflurane, and blood was withdrawn by cardiac puncture. The heart was then perfused with PBS, and the heart and proximal aorta were harvested. The heart and aorta were perfused ex vivo with PBS and then transferred to 10% buffered formalin, processed, and embedded in paraffin. Starting from the atrial leaflets, serial sections (6-μm thick) were prepared such that every eighth section was stained with Harris hematoxylin and eosin. Atherosclerotic lesions in six sections were analyzed in a blinded fashion using a Nikon Labophot-2 microscope equipped with a Sony CCD-Iris/RGB color video camera attached to a computerized imaging system using IMAGE-PRO PLUS 3.0 software. Aortic lesion area was quantified by averaging the lesion areas of the six sections. Necrotic areas were defined as those regions of the lesions that lacked nuclei and cytoplasm.
In-situ TUNEL Assays
Apoptotic cells in the intima of atherosclerotic lesions were detected by the TUNEL (TdT-mediated dUTP nick-end labeling) technique using the TMR red in-situ cell death detection kit (Roche) and the stringent method of Kockx.19 Sections of proximal aorta were deparaffinized, rehydrated, and treated with 2 μg/ml Proteinase K (Roche) for 30 min at 37°C in a humidified chamber. The treated sections were incubated in TdT reaction mixture containing TMR red dUTP for 1 hr at 37°C in a humidified chamber. After washing, genomic DNA was stained with DAPI for 5 min at room temperature, and then the slides were mounted with coverslips. TUNEL staining was analyzed using an Olympus IX-70 inverted fluorescent microscope equipped with a Cool Snap CCD camera and imaging software (Roper Scientific). Fluorescent images were captured and analyzed using image analysis software Photoshop (Adobe).
Statistics
Data are presented as mean ± S.E.M.. Absent error bars in the bar graphs signify S.E.M. values smaller than the graphic symbols. Significance of paired data was determined by the Student’s t test. Data with >2 groups or ≥2 independent variables were analyzed with ANOVA followed by the Bonferroni post hoc test. Significance is indicated by an asterisk in the figures, with an explanation in the figure legends, while non-significance is indicated by ns in the figures.
Statement of Responsibility
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the mansucript as written.
Results
SRA-Induced Apoptosis in ER-Stressed Macrophages Requires STAT1 and is Preceded by Serine Phosphorylation of STAT1
During the course of another study investigating an ER stress response mediator called PKR, we conceived the hypothesis that STAT1, whose activity is modulated by PKR20, may play a role in ER stressed-induced macrophage apoptosis. To test this idea, we compared SRA/ER stress-induced apoptosis in peritoneal macrophages from wild-type vs. Stat1−/− mice. Confirming our previous work, both intracellular FC enrichment with an SRA-interacting lipoprotein and treatment with the SRA ligand fucoidan plus the UPR activator thapsigargin triggered apoptosis, as indicated by an increase in annexin V staining (Figure 1A–B, WT). In contrast, Stat1−/− macrophages were markedly protected from apoptosis by both inducers (80–90% inhibition of apoptosis, p < 0.05), indicating an essential role of STAT1 in this model of macrophage apoptosis (Figure 1A–B, Stat1−/−). The decrease in apoptosis in Stat1−/− macrophages could not be explained by a decrease in either SRA (not displayed) or CHOP induction (Figure 1C).
Figure 1.
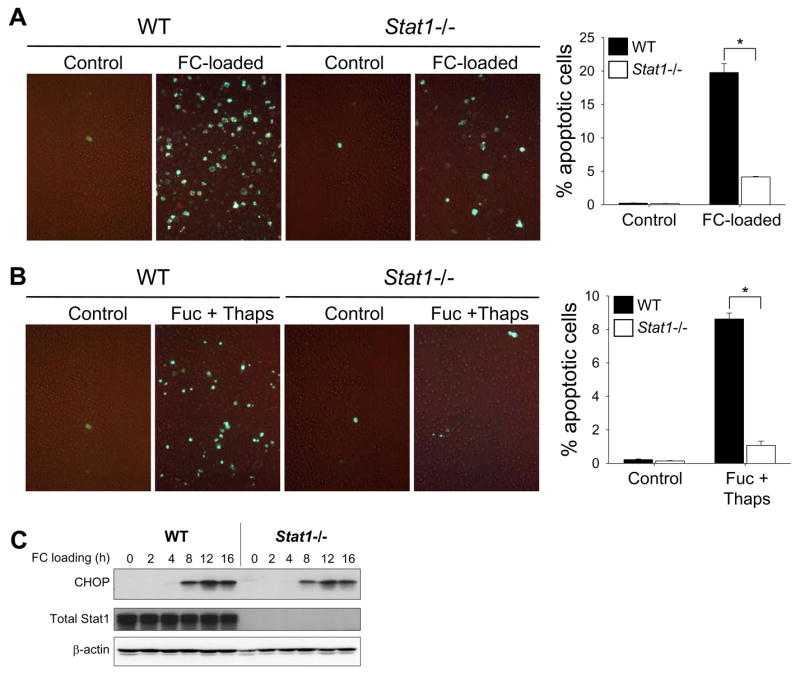
SRA/ER stress-induced macrophage apoptosis requires STAT1. ( A–B)Peritoneal macrophages from wild-type (WT) or Stat1−/− mice were incubated for 17 h with medium alone (Control) or medium containing acetyl-LDL plus the ACAT inhibitor 58035 (FC-loaded); or 21 h with medium alone (Control) of medium containing 50 μg/ml fucoidan and 0.5 μM thapsigargin (Fuc + Thaps). Mid- and late-stage apoptosis were assessed by staining with Alexa Fluor 488-conjugated annexin V (green) and PI (orange), respectively. Representative merged fluorescence and bright-field images and quantitative data from 3 fields of cells for each condition are shown. *, p = 0.001 by Bonferroni after ANOVA. (C) Lysates from wild-type and Stat1−/− macrophages were FC-loaded for the indicated times and subjected to immunoblot analysis to detect CHOP, total STAT1, and β-actin.
STAT1 is activated by phosphorylation of Y701 or S727.21 Y701 phosphorylation is essential for STAT1 dimerization, nuclear translocation, and DNA binding.21 S727 phosphorylation enhances the transcriptional activity of tyrosine-phosphorylated STAT1 or, in some cases, has been reported to participate in signaling in the absence of Y701 phosphorylation.21–23 As shown in Figure 2A, FC loading of macrophages induced serine, whereas tyrosine phosphorylation was not detected, and total STAT1 was not increased. In contrast, very little serine phosphorylation was seen in non-loaded or cholesteryl ester-loaded macrophages, which show no or very little evidence of ER stress.4 As expected, IFNγ induced highly detectable levels of tyrosine phosphorylation as well as serine phosphorylation of STAT1.21 Previous work has suggested that nuclear Ser-P-STAT1 can occur through serine phosphorylation of a constitutive pool of nuclear STAT1.23 We detected STAT1 in nuclear fractions isolated from untreated macrophages, and Ser-P-STAT1 was increased with FC loading. Although total nuclear STAT1 was modestly increased after FC loading, this increase was much less than that seen with IFNγ, which is known to induce STAT1 nuclear translocation21 (Figure 2B). These data suggest that at least a portion of FC-induced Ser-P-STAT1 occurs through phosphorylation of constitutively nuclear STAT1. It is also possible that at least a portion of the STAT1 was tyrosine phosphorylated but below the limits of detection of our immunoblot assay.
Figure 2.

FC loading induces serine- but not tyrosine-phosphorylation of STAT1. (A) Macrophages were incubated for the indicated times with medium alone (Control) or medium containing acetyl-LDL (CE-loaded), acetyl-LDL plus 58035 (FC-loaded), or 100 U/ml IFNγ. Whole-cell lysates were then prepared and subjected to immunoblot analysis to detect phospho-S727 STAT1 (Stat1 pS727), phospho-Y701 STAT1 (Stat1 pY701), and total STAT1. (B) Nuclear fractions from control, FC-loaded, and IFNγ-treated macrophages were subjected to immunoblot analysis to detect STAT1 pS727, total STAT1, and the nuclear marker nucleophosmin.
The ability of IFNγ to stimulate both serine and tyrosine phosphorylation of STAT1, the presence of IFNγ in atherosclerotic lesions, and recent evidence that IFNγ promotes advanced plaque progression24,25 led us to explore the effect of the combination of FC loading and IFNγ treatment on Ser-P-STAT1 and apoptosis. FC-loaded macrophages treated with IFNγ showed an increase in Ser-P-STAT1 that was greater than either condition alone (Figure 3A). Note that IFNγ alone did not induce CHOP, nor did it further increase CHOP in the setting of FC loading. Most importantly, under conditions in which IFNγ alone induced no apoptosis, IFNγ treatment led to a > 5-fold enhancement of FC-induced apoptosis (Figure 3B, WT). This effect of IFNγ required STAT1, because it was inhibited by 93% (p < 0.05) in Stat1−/− macrophages (Figure 3B, Stat1−/−). Thus, in atheromata, where macrophages are likely exposed to the combination of SRA ligands, ER stressors, and IFNγ, the role of STAT1 in macrophage apoptosis may be particularly important (below).
Figure 3.

IFNγ enhances FC-induced STAT1 serine phosphorylation and STAT1-dependent FC-induced apoptosis. (A) Macrophages were incubated for the indicated times with medium alone (Control) or medium containing acetyl-LDL plus 58035 (FC), 100U/ml IFNγ, or acetyl-LDL, 58035, and IFNγ (FC + IFNγ). Whole-cell lysates were then prepared and subjected to immunoblot analysis to detect STAT1 pS727, total STAT1, CHOP, and β-actin. In the CHOP blot, a non-specific band is indicated by the asterisk. (B) Macrophages from wild-type (WT) or Stat1−/− mice were incubated for 13 h with medium alone (Control) or medium containing 100U/ml IFNγ, acetyl-LDL plus 58035 (FC), or acetyl-LDL, 58035, and IFNγ (FC + IFNγ). Apoptosis was assayed and quantified as in Figure 1. *, p = 0.01 for FC and 0.001 for FC + INFγ by Bonferroni after ANOVA.
Cytosolic Calcium, TLR4, and CaMKII Activation are Required for Stat1 Serine Phosphorylation and Apoptosis in FC-Loaded Macrophages
Three kinases that are able to catalyze serine phosphorylation of STAT1 are p38, ERK, and protein kinase C-δ.23 However, using a combination of gene targeting and chemical inhibitors, we found that inhibition of these kinases did not abrogate FC-induced serine phosphorylation of STAT1 (data not shown). In the face of these neative data, we next asked whether two critical components of the multi-hit model, ER stress and TLR4 signaling, were necessary for STAT1 serine phosphorylation. The data in Figure 4A–B show that blocking FC-induced ER stress by the cholesterol trafficking inhibitor U18666A4 or omitting thapsigargin from the fucoidan-plus-thapsigargin model markedly suppressed Ser-P-STAT1. In addition, FC-induced serine phosphorylation of STAT1 was almost completely prevented in TLR4-deficient macrophages (Figure 4C). Note that all of these manipulations also block macrophage apoptosis.4,15
Figure 4.
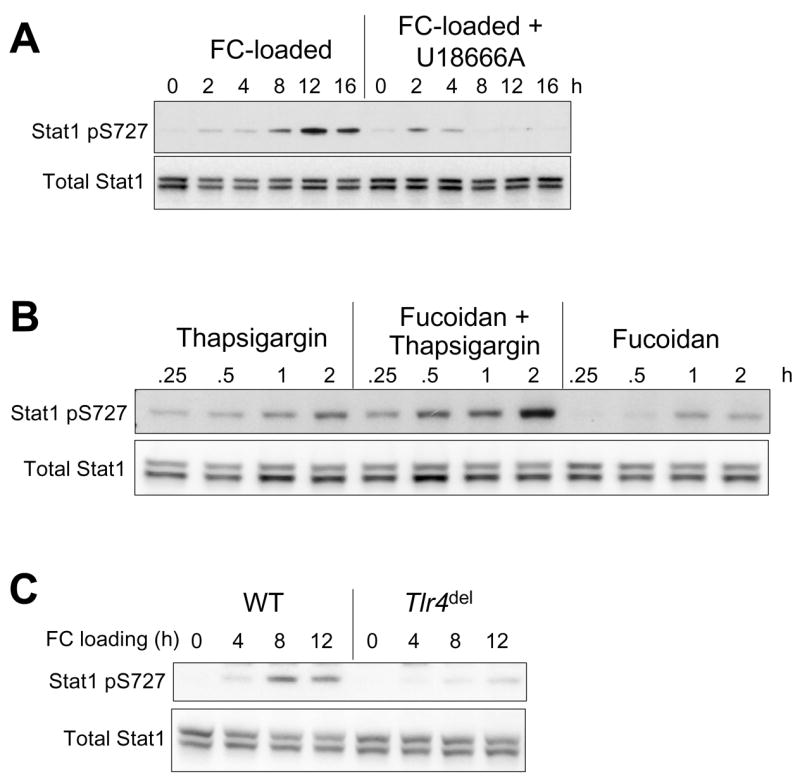
STAT1 serine phosphorylation in SRA-engaged, ER-stressed macrophages is amplified by ER stress and requires TLR4 activation. Whole-cell lysates were subjected to immunoblot analysis to detect STAT1 pS727 and total STAT1 under the following conditions: (A) Macrophages were incubated for the indicated times with acetyl-LDL and 58035 (FC-loaded) or acetyl-LDL, 58035, and 1μM U18666A (FC-loaded + U18666A). (B) Macrophages were incubated for the indicated times with 0.5 μM thapsigargin, 50 μg/ml fucoidan plus thapsigargin, or fucoidan alone. (C) Macrophages from wild-type (WT) or Tlr4del mice were incubated for the indicated times under FC-loading conditions.
Both ER stress and TLR4 signaling can affect cellular calcium metabolism (see Discussion).12,13,15,26,27 Moreover, we recently showed that buffering cytosolic calcium with BAPTA-AM markedly inhibited both FC-induced and thapsigargin/fucoidan-induced apoptosis.15 To test the role of cytosolic calcium in STAT1 serine phosphorylation, we incubated FC-loaded macrophages with increasing concentrations of BAPTA-AM or equivalent volumes of vehicle control. As shown in Figure 5A, BAPTA-AM suppressed FC-induced serine phosphorylation of STAT1 in a dose-dependent manner.
Figure 5.

Cytosolic calcium and CaMKII activation are required for FC-induced STAT1 serine phosphorylation, and inhibition of CaMKII blocks FC-induced apoptosis. (A–D), Whole-cell lysates were subjected to immunoblot analysis to detect STAT1 pS727, total STAT1, CaMKII pT286/287, β-actin, or CHOP, as indicated in the individual blots, under the following conditions: (A)Macrophages were incubated for 8 h in medium alone ( Control); medium containing acetyl-LDL and 58035 plus vehicle control (FC + Vehicle); or medium containing acetyl-LDL, 58035, and increasing concentrations of BAPTA (FC + BAPTA). The indicated microliters of vehicle or BAPTA-AM stock solution (1 mg/ml) were added per ml of medium. (B) Macrophages were incubated for the indicated times with medium alone (Control) or medium containing acetyl-LDL and 58035 (FC-loaded) or 2 μg/ml A23187. (C) Macrophages were incubated for the indicated times with acetyl-LDL and 58035 (FC) or acetyl-LDL and 58035 plus either 10 μM KN93 or 10 μM KN92 (FC + KN93 or FC + KN92). (D) Macrophages were incubated for 8 h with acetyl-LDL and 58035 (FC), 10 μM AIP, or acetyl-LDL, 58035, and AIP. (E) Macrophages were incubated for 24 h in medium alone (Control) or medium containing acetyl-LDL and 58035 (FC-loaded) or acetyl-LDL and 58035 plus either 10 μM KN93 or 10 μM KN92 (FC-loaded + KN93 or FC-loaded + KN92). Apoptosis was assayed and quantified as in Figure 1. For all experiments involving KN93, KN92, or AIP, the macrophages were pre-treated for 1 h with medium alone or medium containing these inhibitors prior to FC loading. *, p < 0.01 by Bonferroni after ANOVA.
One mechanism by which cytosolic calcium might participate in STAT1 serine phosphorylation is by activating CaMKII, which may directly phosphorylate STAT128 and/or lead to its phosphorylation through enhancing TLR4 signaling (see Discussion).15,29,30 As shown in Figure 5B, FC loading led to a rapid and marked enhancement of CaMKII threonine phosphorylation, which is a marker of its activation. At the 30- and 60-min timepoints, the degree of activation was similar to that of the calcium ionophore A23187, a known potent activator of CaMKII. Similar results were found with fucoidan plus thapsigargin (data not shown). Note that the time course of CaMKII activation by FC loading or by thapsigargin plus fucoidan precedes the onset of STAT1 serine phosphorylation in these cells. To show a functional role for CaMKII activation in both STAT1 serine phosphorylation and apoptosis in FC-loaded macrophages, we utilized two structurally diverse CaMKII inhibitors. The data in Figure 5C–D show that the chemical CaMKII inhibitor KN9331, but not the inactive homologue KN92, and the peptide CaMKII inhibitor AIP32 markedly suppressed FC-induced STAT1 serine phosphorylation. Most importantly, KN93, but not KN92, suppressed FC-induced apoptosis by 92% (p < 0.05) (Figure 5E). Note that neither KN93 nor AIP decreased the uptake or ER-trafficking of lipoprotein-derived FC or the induction of CHOP (data not shown and Figure 5D). In summary, these data indicate that cytosolic calcium and CaMKII are essential for STAT1 serine phosphorylation and apoptosis in the SRA-ER stress model. We also conducted experiments on two additional macrophage models, namely, mouse bone marrow-derived and human peripheral blood-derived macrophages. In both of these cell types, the SRA-ER stress model exclusively induced STAT1 serine phosphorylation via a pathway mediated by cytosolic calcium and CaMKII (see Figure in the online Data Supplement), suggesting the universality of this signaling pathway among macrophages.
STAT1 is Serine-Phosphorylated in Murine and Human Atherosclerotic Lesions, and STAT1 Plays a Role in Advanced Lesional Macrophage Apoptosis and Plaque Necrosis in Female Ldlr−/− Mice
To provide evidence for the relevance of Ser-P-STAT1 in atherosclerosis, we first used immunohistochemistry to assess expression of Ser-P-STAT1 in murine and human atheromata (Figure 6 and 7). In mouse lesions, Ser-P-STAT1 was present in numerous macrophage foam cells, as assessed by staining adjacent sections with anti-Mac-3 antibody (Figure 6A,B) and Oil-Red O (Figure 6D). As illustrated by these images, Ser-P-STAT1 staining was also observed in the endothelial cells lining the lumen, which was PECAM-1-positive (not shown), and in smooth muscle cells in the media, which were α-actin positive (Figure 6F). In human lesions, staining of Ser-P-STAT1 was found in the advanced stages termed pathological intimal thickening and fibroatheroma (Figure 7B,C), but not in the early stage of diffuse intimal thickening (Figure 7A). In the advanced lesions, most of the Ser-P-STAT1 co-localized with macrophages (Figure 7B,C). Note that Ser-P-STAT1 was found in the nuclei of these cells (Figure 7C, bottom middle image) and in areas that were TUNEL-positive, a marker of apoptosis (Figure 7C, bottom right image). Of interest, some of the Ser-P-STAT1 in the most advanced fibroatheroma was found in macrophages surrounding necrotic areas (Figure 7C, bottom left image, asterisk).
Figure 6.
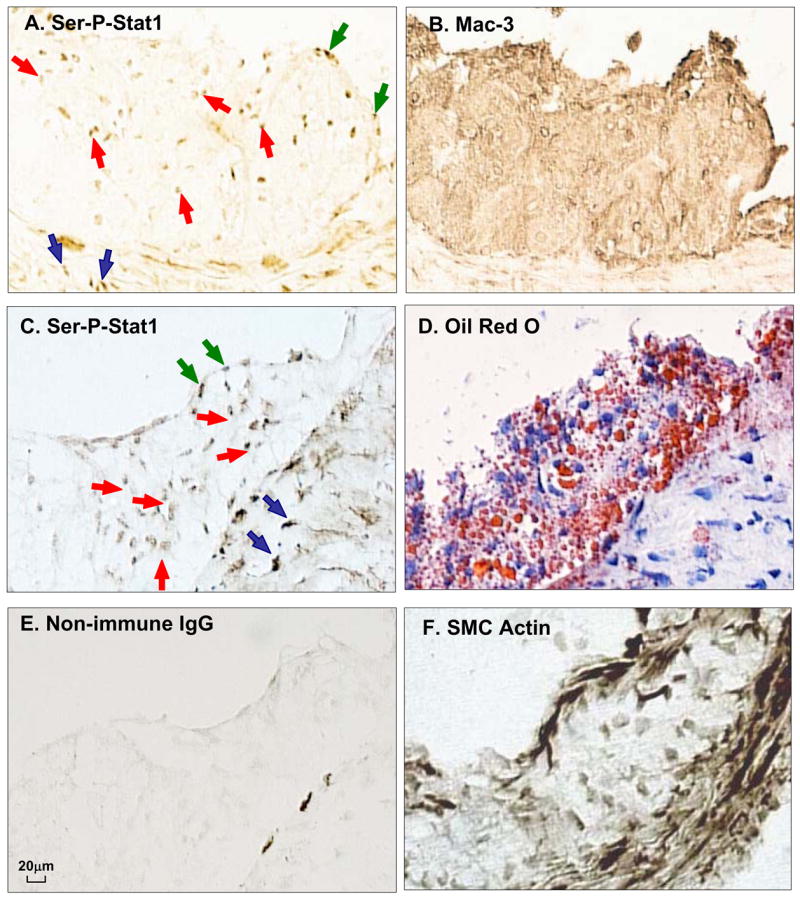
STAT1 is serine-phosphorylated in atherosclerotic lesions from Ldlr−/− mice. Adjacent frozen sections of an aortic root lesion from an Ldlr−/− mice fed a Western-type diet for 12 wks were immunostained with anti-Ser-P-STAT1 or anti-Mac3 (macrophages) (A,B) or anti-Ser-P-STAT1, Oil Red O, non-immune IgG, and α-actin (C–F). Note examples of brown stain in the nuclei of the intimal cells (red arrows), endothelial cells (blue arrows), and smooth muscle cells in the media (black arrows). The dark streaks at the intima-media interface in panel E represent non-specific staining.
Figure 7.

Ser-P- STAT1 is present in advanced human coronary atheromata but not in diffuse intimal thickening. The sections were stained with Movat pentachrome, anti-Ser-P-STAT1, anti-CD68, and non-immune IgG, as indicated in the images. (A) Diffuse intimal thickening. The CD68 and Ser-P-STAT1 images on the right are higher magnifications of the area indicated by the box in the low-magnification Ser-P-STAT1 image. As shown in the higher magnification images, only a few CD68-positive macrophages are present directly under the endothelium (arrowheads). Ser-P-STAT1 was not detected. Arrow indicates internal elastic lamina (IEL). (B) Pathological intimal thickening. Ser-P-STAT1 staining coincides with CD68-positive macrophages. (C) Fibroatheroma: Ser-P-STAT1 staining coincides with CD68-positive macrophages. Some of the Ser-P-STAT1 staining is in macrophages surrounding a necrotic area (asterisk). The lower middle and right images are higher magnifications of the area indicated by the box in the low-magnification CD68 image. These lower middle image shows the result of double immunostaining with anti-Ser-P-STAT1 (dark, punctate structures) and anti-CD68 (red), demonstrating Ser-P-STAT1 in the nuclei of macrophages. The lower right image shows the result of double immunostaining with TUNEL (dark, punctate structures) and anti-CD68 (red), demonstrating apoptotic macrophages. Nuclei of non-apoptotic cells are stained green. Note that exact alignment of the nuclei is not possible due to the fact that the sections are from separate tissue slices.
To further investigate a causal link between STAT1 and lesional macrophage apoptosis, we compared advanced plaques of Western diet-fed Ldlr−/− mice reconstituted with either wild-type or Stat1−/− bone marrow. The mice were fed the Western diet for 10 or 12 weeks. Plasma lipoprotein cholesterol and body weight were similar between the two groups of mice (Figure 8A for 10-wk protocol; data not shown for 12-wk protocol). In the 10-wk study, overall lesion area was similar (Figure 8B,C). However, the number of TUNEL-positive cells in macrophage-rich regions was deceased by 61% (p = 0.034) in the Stat1−/− → Ldlr −/− lesions, and there was a trend towards decreased plaque necrosis that did not quite reach statistical significance (p = 0.078) (Figure 8C). Note that total macrophage area was not affected by STAT1 deficiency (120.0 ± 11.8 ×103 μm2 and 111.5 ± 21.3 ×103 μm2 in wild-type and Stat1−/− bone marrow recipients respectively, p = 0.72; see Discussion).
Figure 8.

STAT1 plays a role in advanced lesional macrophage apoptosis and plaque necrosis in female Stat1−/− →Ldlr −/− mice. (A) The table shows plasma cholesterol and body weight of Ldlr−/− mice transplanted with Stat1+/+ or Stat1/bone marrow and then fed a Western-type diet for 10 wks starting at 6 wks after transplantation. The graph shows pooled plasma samples from three Stat1+/+ and three Stat1−/− recipient mice that were fractionated by FPLC gel-filtration chromatography and then assayed for cholesterol. None of the differences in cholesterol, lipoproteins, or body weight were statistically significant. (B) Hematoxylin and eosin staining of proximal aortas from 10-wk Western diet-fed Ldlr−/− mice transplanted with bone marrow from Stat1+/+ and Stat1−/− mice. Bar, 20 μm. Nec = necrotic areas. (C) TUNEL (red), DAPI (blue), and macrophage (brown) staining of lesions similar to those in (B). Bar, 20 μm. The graph in panel C shows quantification of lesion area, TUNEL-positive cells, and necrotic area in the lesions of Stat1+/+ →Ldlr −/− and Stat1−/− →Ldlr −/− mice. *, p = 0.034 by Student’s t test. (D) Hematoxylin and eosin staining of proximal aortas from 12-wk Western diet-fed Ldlr−/− mice transplanted with bone marrow from Stat1+/+ and Stat1−/− mice (n = 18 for both groups of mice). Bar, 20 μm. The graph in panel D shows quantification of TUNEL-positive cells and necrotic area in the lesions of Stat1+/+ →Ldlr −/− and Stat1−/− →Ldlr −/− mice. Total lesion area was 493.7 ± 40.5 and 380.6 ± 24.4 μm2 in Stat1+/+ →Ldlr −/− and Stat1−/− → Ldlr−/− mice, respectively. *, p = 0.02 by Student’s t test.
Plaque necrosis likely results from the eventual cellular necrosis of macrophages that become apoptotic but are not subsequently cleared by phagocytes.1,2 Therefore, we predicted that as the lesions in the two groups of mice progressed, the difference in necrotic core areas would become statistically significant, while apoptotic macrophages per se would become less numerous and less different between the two groups of mice. As shown by the data in Figure 8D, the necrotic cores were larger in the 12-wk-diet mice, and there was a statistically significant difference in the necrotic core area (34% decrease in the Stat1−/− → Ldlr −/− lesions, p = 0.02) but not the number of TUNEL-positive cells. In summary, STAT1 deficiency in bone marrow-derived cells in Ldlr−/− mice has a substantial protective effect on apoptosis in macrophage-rich lesions of advanced plaques and on plaque necrosis.
Discussion
There is increasing evidence from a number of laboratories that an ER stress-based model of macrophage apoptosis plays an important role in advanced lesional macrophage death and plaque necrosis.4–9 The work in this report adds critical new components to this model by demonstrating essential roles for STAT1 and CaMKII in macrophage apoptosis in vitro and for STAT1 in advanced lesional macrophage apoptosis and plaque necrosis in vivo.
Future studies will be required to define at a precise molecular level how the pro-apoptotic components elucidated in this study fit into the overall scheme of the multi-hit model of macrophage apoptosis. Our working hypothesis is depicted in Figure 9. We suggest that ER stress triggers two key pro-apoptotic processes: UPR/CHOP and another pathway in which ER stress-induced cytosolic calcium activates CaMKII, which in turn leads to serine phosphorylation of pro-apoptotic STAT1. Activation of the TLR4-MyD88 pathway by SRA ligands, which is critical for apoptosis15, also contributes to STAT1 serine phosphorylation. SRA ligands additionally promote apoptosis through SRA-dependent suppression of pro-survival IFNβ.14,15
Figure 9.

Integration of calcium, CaMKII, and STAT1 into the multi-hit pathway of macrophage apoptosis. According to this working hypothesis, ER stress-induced increase in cytosolic calcium triggers two pro-apoptotic hits: UPR/CHOP and a pathway involving CaMKII and Ser-P-STAT1. TLR4 activation also contributes to STAT1 serine phosphorylation. Ser-P-STAT1 is depicted as a separate pathway from CHOP, because studies with Chop−/− and Stat1−/− macrophages showed that CHOP is neither upstream nor downstream of Ser-P-STAT1 (data not shown). See Discussion for details and for a description of the areas of uncertainty in this model.
This scheme raises a number of critical issues that will require further investigation. Among these is whether STAT1 serine-phosphorylation per se is required for apoptosis, which is consistent with our data and with previous work showing a pro-apoptotic role of Ser-P-STAT1 in apoptosis in other systems.33 However, definitive proof will require comparing SRA/ER stress-induced apoptosis in macrophages containing S727- vs. Y701-mutated STAT1.22,33 Until then, we cannot definitively rule out the possibility that apoptosis requires Y701 phosphorylation and that Tyr-P-STAT1 in our SRA-ER stress model is below the limit of immunoblot detection. In pilot studies, we found that apoptosis induced by thapsigargin and fucoidan was markedly suppressed in peritoneal macrophages from S727A-STAT1 knock-in mice, but results with FC-induced apoptosis were difficult to interpret due to an as-yet-undefined compensatory pathway (J. Timmins, W. Lim, T. Decker, and I. Tabas, unpublished data). Assuming Ser-P-STAT1 is a key apoptosis mediator in this pathway, the next goal is to elucidate if and how serine-only phosphorylated STAT1 affects gene transcription in a manner that promotes apoptosis.33–35 A related issue is the precise mechanism by which STAT1 is serine-phosphorylated and how this process is linked to both CaMKII and TLR4 (Figure 9). One obvious possibility is that CaMKII directly phosphorylates STAT1, as has been described previously in other models.28 If this is the case in our model, a link to TLR4 could occur through IP3 receptor-mediated elevation of cytosolic calcium26, leading to a further increase in CaMKII activation. Alternatively, TLR4 signaling may more directly stimulate STAT1 serine phosphorylation, as has been reported in other models using the TLR4 ligand, LPS.29,30
The impetus for this study was to explore pathways that may be involved in promoting macrophage apoptosis in atherosclerosis. The ultimate significance of lesional macrophage apoptosis likely depends on lesion stage.1,2 In early lesions, rapid and efficient phagocytic clearance of apoptotic macrophages appears to limit lesion cellularity and progression. Of interest, STAT1 may have a separate role in these early lesions independent of macrophage death, because STAT1 deficiency in Apoe−/− mice blocks foam cell formation and early lesion development.36 In advanced lesions, however, there is evidence that clearance of apoptotic cells is defective, leading to post-apoptotic macrophage necrosis, inflammation, and, eventually, overall plaque necrosis.1,2 In this context, the multi-hit model of macrophage apoptosis is likely most relevant to advanced lesions. For example, immunoblots have shown that CHOP is expressed only in advanced lesions7, and manipulation of ER stress in vivo is positively associated with advanced lesional necrosis, not negatively associated with early lesion progression.5,37 In the case of STAT1 deficiency, there was a clear trend towards decreased plaque necrosis. However, the maximum effect on plaque necrosis may lag behind that of macrophage apoptosis, because plaque necrosis likely results from the progressive coalescence of apoptotic macrophages after they become secondarily necrotic.1,2 Another prediction from this idea and from the fact that the anti-macrophage antibody used in our study recognizes pre-necrotic apoptotic macrophages is that total macrophage area should be similar in Stat1+/+ → Ldlr−/− and Stat1−/− → Ldlr −/− lesions, exactly as we observed experimentally. More fundamentally, we clearly did not observe an increase in lesion area in the Stat1−/− group, which is what is found when early lesional macrophage apoptosis is blocked.38 In terms of other studies linking STAT1 to advanced plaque progression, in-vivo data suggest that interleukin-10, which suppresses STAT1 activity39, may protect advanced atheromata from macrophage apoptosis and plaque necrosis.40,41 Moreover, Koga et al.25 reported that blocking the function of the STAT1 activator IFNγ stabilized advanced plaques in Apoe−/− mice. Thus, pending further in-vivo studies, local inhibition of STAT1 activity may represent a potentially promising therapeutic strategy to prevent the progression of relatively benign lesions to those with increased macrophage apoptosis and plaque necrosis.
Supplementary Material
Acknowledgments
The authors thank Dr. Chris Schindler for helpful discussions and for supplying the Stat1−/− mice, Dr. Mary Reyland for providing the Pkcd−/− mice, and George Kuriakose assistance with the mouse atherosclerosis experiment.
Funding Sources
This work was supported by an American Heart Association-Heritage Affiliate Post-Doctoral Fellowship (to W.L.), a T32 training grant from the NIH (HL007343; to J.M.T.), an individual post-doctoral training grant from the the NIH (HL79801; to T.A.S.), NIH grants HL54591 and HL75662 (to I.T.), and US Army Medical Research and Materiel Command (USAMRMC) grant W81XWH-06-1-0212 (to I. T.).
Footnotes
Disclosures
None.
References
- 1.Tabas I. Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol. 2005;25:2255–64. doi: 10.1161/01.ATV.0000184783.04864.9f. [DOI] [PubMed] [Google Scholar]
- 2.Schrijvers DM, De Meyer GR, Herman AG, Martinet W. Phagocytosis in atherosclerosis: Molecular mechanisms and implications for plaque progression and stability. Cardiovasc Res. 2007;73:470–80. doi: 10.1016/j.cardiores.2006.09.005. [DOI] [PubMed] [Google Scholar]
- 3.Virmani R, Burke AP, Kolodgie FD, Farb A. Vulnerable plaque: the pathology of unstable coronary lesions. J Interv Cardiol. 2002;15:439–46. doi: 10.1111/j.1540-8183.2002.tb01087.x. [DOI] [PubMed] [Google Scholar]
- 4.Feng B, Yao PM, Li Y, Devlin CM, Zhang D, Harding HP, Sweeney M, Rong JX, Kuriakose G, Fisher EA, Marks AR, Ron D, Tabas I. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol. 2003;5:781–92. doi: 10.1038/ncb1035. [DOI] [PubMed] [Google Scholar]
- 5.Feng B, Zhang D, Kuriakose G, Devlin CM, Kockx M, Tabas I. Niemann-Pick C heterozygosity confers resistance to lesional necrosis and macrophage apoptosis in murine atherosclerosis. Proc Natl Acad Sci U S A. 2003;100:10423–8. doi: 10.1073/pnas.1732494100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hossain GS, van Thienen JV, Werstuck GH, Zhou J, Sood SK, Dickhout JG, de Koning AB, Tang D, Wu D, Falk E, Poddar R, Jacobsen DW, Zhang K, Kaufman RJ, Austin RC. TDAG51 is induced by homocysteine, promotes detachment-mediated programmed cell death, and contributes to the cevelopment of atherosclerosis in hyperhomocysteinemia. J Biol Chem. 2003;278:30317–27. doi: 10.1074/jbc.M212897200. [DOI] [PubMed] [Google Scholar]
- 7.Zhou J, Lhotak S, Hilditch BA, Austin RC. Activation of the unfolded protein response occurs at all stages of atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2005;111:1814–21. doi: 10.1161/01.CIR.0000160864.31351.C1. [DOI] [PubMed] [Google Scholar]
- 8.Gargalovic PS, Gharavi NM, Clark MJ, Pagnon J, Yang WP, He A, Truong A, Baruch-Oren T, Berliner JA, Kirchgessner TG, Lusis AJ. The unfolded protein response is an important regulator of inflammatory genes in endothelial cells. Arterioscler Thromb Vasc Biol. 2006;26:2490–6. doi: 10.1161/01.ATV.0000242903.41158.a1. [DOI] [PubMed] [Google Scholar]
- 9.Myoishi M, Hao H, Minamino T, Watanabe K, Nishihira K, Hatakeyama K, Asada Y, Okada K, Ishibashi-Ueda H, Gabbiani G, Bochaton-Piallat ML, Mochizuki N, Kitakaze M. Increased endoplasmic reticulum stress in atherosclerotic plaques associated with acute coronary syndrome. Circulation. 2007;116:1226–33. doi: 10.1161/CIRCULATIONAHA.106.682054. [DOI] [PubMed] [Google Scholar]
- 10.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–77. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tabas I. Consequences of cellular cholesterol accumulation: basic concepts and physiological implications. J Clin Invest. 2002;110:905–11. doi: 10.1172/JCI16452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li Y, Ge M, Ciani L, Kuriakose G, Westover E, Dura M, Covey D, Freed JH, Maxfield FR, Lytton J, Tabas I. Enrichment of endoplasmic reticulum with cholesterol inhibits SERCA2b activity in parallel with increased order of membrane lipids. Implications for depletion of ER calcium stores and apoptosis in cholesterol-loaded macrophages. J Biol Chem. 2004;279:37030–9. doi: 10.1074/jbc.M405195200. [DOI] [PubMed] [Google Scholar]
- 13.Chen LY, Chiang AS, Hung JJ, Hung HI, Lai YK. Thapsigargin-induced grp78 expression is mediated by the increase of cytosolic free calcium in 9L rat brain tumor cells. J Cell Biochem. 2000;78:404–16. doi: 10.1002/1097-4644(20000901)78:3<404::aid-jcb6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 14.DeVries-Seimon T, Li Y, Yao PM, Stone E, Wang Y, Davis RJ, Flavell R, Tabas I. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J Cell Biol. 2005;171:61–73. doi: 10.1083/jcb.200502078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seimon TA, Obstfeld A, Moore KJ, Golenbock DT, Tabas I. Combinatorial pattern recognition receptor signaling alters the balance of life and death in macrophages. Proc Natl Acad Sci U S A. 2006;103:19794–9. doi: 10.1073/pnas.0609671104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krieger M, Herz J. Structures and functions of multiligand lipoprotein receptors: macrophage scavenger receptors and LDL receptor-related protein (LRP) Annu Rev Biochem. 1994;63:601–37. doi: 10.1146/annurev.bi.63.070194.003125. [DOI] [PubMed] [Google Scholar]
- 17.Krieger M. The other side of scavenger receptors: pattern recognition for host defense. Curr Opin Lipidol. 1997;8:275–80. doi: 10.1097/00041433-199710000-00006. [DOI] [PubMed] [Google Scholar]
- 18.Dunzendorfer S, Lee HK, Soldau K, Tobias PS. TLR4 is the signaling but not the lipopolysaccharide uptake receptor. J Immunol. 2004;173:1166–70. doi: 10.4049/jimmunol.173.2.1166. [DOI] [PubMed] [Google Scholar]
- 19.Kockx MM. Apoptosis in the atherosclerotic plaque: quantitative and qualitative aspects. Arterioscler Thromb Vasc Biol. 1998;18:1519–22. doi: 10.1161/01.atv.18.10.1519. [DOI] [PubMed] [Google Scholar]
- 20.Wong AH, Tam NW, Yang YL, Cuddihy AR, Li S, Kirchhoff S, Hauser H, Decker T, Koromilas AE. Physical association between STAT1 and the interferon-inducible protein kinase PKR and implications for interferon and double-stranded RNA signaling pathways. EMBO J. 1997;16:1291–304. doi: 10.1093/emboj/16.6.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bromberg J, Darnell JE., Jr The role of STATs in transcriptional control and their impact on cellular function. Oncogene. 2000;19:2468–73. doi: 10.1038/sj.onc.1203476. [DOI] [PubMed] [Google Scholar]
- 22.Varinou L, Ramsauer K, Karaghiosoff M, Kolbe T, Pfeffer K, Muller M, Decker T. Phosphorylation of the Stat1 transactivation domain is required for full-fledged IFN-gamma-dependent innate immunity. Immunity. 2003;19:793–802. doi: 10.1016/s1074-7613(03)00322-4. [DOI] [PubMed] [Google Scholar]
- 23.Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19:2628–37. doi: 10.1038/sj.onc.1203481. [DOI] [PubMed] [Google Scholar]
- 24.Frostegard J, Ulfgren AK, Nyberg P, Hedin U, Swedenborg J, Andersson U, Hansson GK. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis. 1999;145:33–43. doi: 10.1016/s0021-9150(99)00011-8. [DOI] [PubMed] [Google Scholar]
- 25.Koga M, Kai H, Yasukawa H, Yamamoto T, Kawai Y, Kato S, Kusaba K, Kai M, Egashira K, Kataoka Y, Imaizumi T. Inhibition of Progression and Stabilization of Plaques by Postnatal Interferon-{gamma} Function Blocking in ApoE-Knockout Mice. Circ Res. 2007 doi: 10.1161/CIRCRESAHA.106.147256. [DOI] [PubMed] [Google Scholar]
- 26.Prpic V, Weiel JE, Somers SD, DiGuiseppi J, Gonias SL, Pizzo SV, Hamilton TA, Herman B, Adams DO. Effects of bacterial lipopolysaccharide on the hydrolysis of phosphatidylinositol-4,5-bisphosphate in murine peritoneal macrophages. J Immunol. 1987;139:526–33. [PubMed] [Google Scholar]
- 27.Cuschieri J, Bulger E, Garcia I, Jelacic S, Maier RV. Calcium/calmodulin-dependent kinase II is required for platelet-activating factor priming. Shock. 2005;23:99–106. doi: 10.1097/01.shk.0000148075.19190.db. [DOI] [PubMed] [Google Scholar]
- 28.Nair JS, DaFonseca CJ, Tjernberg A, Sun W, Darnell JE, Jr, Chait BT, Zhang JJ. Requirement of Ca2+ and CaMKII for Stat1 Ser-727 phosphorylation in response to IFN-gamma. Proc Natl Acad Sci U S A. 2002;99:5971–6. doi: 10.1073/pnas.052159099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rhee SH, Jones BW, Toshchakov V, Vogel SN, Fenton MJ. Toll-like receptors 2 and 4 activate STAT1 serine phosphorylation by distinct mechanisms in macrophages. J Biol Chem. 2003;278:22506–12. doi: 10.1074/jbc.M208633200. [DOI] [PubMed] [Google Scholar]
- 30.Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, Williams BR, Major J, Hamilton TA, Fenton MJ, Vogel SN. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol. 2002;3:392–8. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- 31.Sumi M, Kiuchi K, Ishikawa T, Ishii A, Hagiwara M, Nagatsu T, Hidaka H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem Biophys Res Commun. 1991;181:968–75. doi: 10.1016/0006-291x(91)92031-e. [DOI] [PubMed] [Google Scholar]
- 32.Ishida A, Shigeri Y, Tatsu Y, Uegaki K, Kameshita I, Okuno S, Kitani T, Yumoto N, Fujisawa H. Critical amino acid residues of AIP, a highly specific inhibitory peptide of calmodulin-dependent protein kinase II. FEBS Lett. 1998;427:115–8. doi: 10.1016/s0014-5793(98)00405-0. [DOI] [PubMed] [Google Scholar]
- 33.Kumar A, Commane M, Flickinger TW, Horvath CM, Stark GR. Defective TNF-alpha-induced apoptosis in STAT1-null cells due to low constitutive levels of caspases. Science. 1997;278:1630–2. doi: 10.1126/science.278.5343.1630. [DOI] [PubMed] [Google Scholar]
- 34.Stephanou A, Scarabelli TM, Brar BK, Nakanishi Y, Matsumura M, Knight RA, Latchman DS. Induction of apoptosis and Fas receptor/Fas ligand expression by ischemia/reperfusion in cardiac myocytes requires serine 727 of the STAT-1 transcription factor but not tyrosine 701. J Biol Chem. 2001;276:28340–7. doi: 10.1074/jbc.M101177200. [DOI] [PubMed] [Google Scholar]
- 35.Zhang JJ, Zhao Y, Chait BT, Lathem WW, Ritzi M, Knippers R, Darnell JE., Jr Ser727-dependent recruitment of MCM5 by Stat1alpha in IFN-gamma-induced transcriptional activation. EMBO J. 1998;17:6963–71. doi: 10.1093/emboj/17.23.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Agrawal S, Febbraio M, Podrez E, Cathcart MK, Stark GR, Chisolm GM. Signal transducer and activator of transcription 1 is required for optimal foam cell formation and atherosclerotic lesion development. Circulation. 2007;115:2939–47. doi: 10.1161/CIRCULATIONAHA.107.696922. [DOI] [PubMed] [Google Scholar]
- 37.Han S, Liang CP, DeVries-Seimon T, Ranalletta M, Welch CL, Collins-Fletcher K, Accili D, Tabas I, Tall AR. Macrophage insulin receptor deficiency increases ER stress-induced apoptosis and necrotic core formation in advanced atherosclerotic lesions. Cell Metab. 2006;3:257–66. doi: 10.1016/j.cmet.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 38.Liu J, Thewke DP, Su YR, Linton MF, Fazio S, Sinensky MS. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler Thromb Vasc Biol. 2005;25:174–9. doi: 10.1161/01.ATV.0000148548.47755.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ito S, Ansari P, Sakatsume M, Dickensheets H, Vazquez N, Donnelly RP, Larner AC, Finbloom DS. Interleukin-10 inhibits expression of both interferon alpha- and interferon gamma- induced genes by suppressing tyrosine phosphorylation of STAT1. Blood. 1999;93:1456–63. [PubMed] [Google Scholar]
- 40.Mallat Z, Heymes C, Ohan J, Faggin E, Leseche G, Tedgui A. Expression of interleukin-10 in advanced human atherosclerotic plaques: relation to inducible nitric oxide synthase expression and cell death. Arterioscler Thromb Vasc Biol. 1999;19:611–6. doi: 10.1161/01.atv.19.3.611. [DOI] [PubMed] [Google Scholar]
- 41.Pinderski LJ, Fischbein MP, Subbanagounder G, Fishbein MC, Kubo N, Cheroutre H, Curtiss LK, Berliner JA, Boisvert WA. Overexpression of interleukin-10 by activated T lymphocytes inhibits atherosclerosis in LDL receptor-deficient Mice by altering lymphocyte and macrophage phenotypes. Circ Res. 2002;90:1064–71. doi: 10.1161/01.res.0000018941.10726.fa. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.