

Abstract
A simple high performance liquid chromatography (HPLC) method was developed and validated for the determination of six phenolic compounds, five anthraquinones (rhein, aloe-emodin, emodin, chrysophanol and physcion) and a flavonoid (kaempferol), in root extracts from Cassia alata L. Solid-phase extraction, using C18 cartridges, was used to remove interfering substances from the root extracts. The extracts were analyzed on a C18 column using an isocratic mobile phase which consisted of acetonitrile, methanol, and 10 mM aqueous ammonium acetate (25: 55: 20, v/v). Identification of the analytes was performed by use of standards and on-line mass spectrometric detection using atmospheric pressure chemical ionization. The concentration of the phenolic compounds in the root extracts was determined using HPLC with ultraviolet detection at 260 nm. The limits of detection obtained for the anlytes were in the range of 0.23 – 4.61 ppm. The overall R.S.D. precision values (intra and inter-day) for the retention times and peak-areas were lower than 0.16% and 2.10%, respectively. In addition, the recovery of the developed method for the analysis of these phenolic compounds was determined, and ranged from 81.2 ± 4.3% to 106 ± 2%.
Keywords: Anthraquinones, Kaempferol, Phenolic compounds, LC-MS, Solid-phase extraction, Recovery
1. Introduction
Ringworm cassia or golden candle (Cassia alata Linn, Leguminosae), a wild-growing shrub often cultivated as an ornamental plant, is indigenous to South America, but now widely distributed in the tropics [1–5]. This worldwide important herbal medicine has been recognized for centuries in traditional medicine for its role as a laxative as well as in the treatment of a variety of skin and respiratory diseases [1,2, 5–7]. In Suriname, root extracts from C. alata are used for the treatment of uterus disorders [1]. Pharmacological investigations performed so far on C. alata have shown that this herb has several biological activities, such as antimicrobial [8,9], antifungal [2,10,11], purgative [4], anti-inflammatory [11,12], analgesic [1,11,13], anti-tumor [14], and hypoglycaemic [11] activities.
C. alata is one of the most important species of the genus Cassia which is rich in anthraquinones and polyphenols [13,15]. The leaves of C. alata have been qualitatively analyzed for the presence of primarily five pharmacologically active anthraquinones: rhein, aloe-emodin, chrysophanol, emodin, and physcion [16], as well as the flavonoid kaempferol [17]. Rhein and chrysophanol are known to be present in the roots [2], in addition to two other quinone pigments [18]. These anthraquinone derivatives are well known to exhibit a variety of biological activities [19], such as antimicrobial [2], antifungal [20], antitumor [21], antioxidant [22], cytotoxic [23], and hypoglycemic [24] activities. The flavonoid kaempferol has been reported to have anticancer properties [25,26]. It is clear that there is a correlation between the ethno-pharmacological properties ascribed to this herbal medicine and the biological activities of these phenolic compounds. Therefore, it is of great importance to determine the amount of these bioactive constituents in C. alata. However, to our knowledge there are no reports in the literature on the determination of these phenolic compounds in the roots of C. alata. This leads to the necessity of developing an analytical method for the simultaneous analysis and identification of these bioactive compounds in the roots of C. alata.
Previously, C. alata extracts were analyzed for the presence of kaempferol-3-O-gentiobioside in various parts of the plant (leaves, flowers, rachis, stem and seed) using high performance liquid chromatography (HPLC) [27]. Several analytical methods such as thin-layer chromatography [28], micellar electrokinetic capillary chromatography [29], capillary zone electrophoresis [30], capillary electrophoresis-mass spectrometry (CE-MS) [31], gas chromatography [32], and gas chromatography-mass spectrometry (GC-MS) [33] have been used for the determination of anthraquinones in various medicinal plants and natural products. In addition, analytical methods involving the use of HPLC [32–36] and liquid chromatography-mass spectrometry (LC-MS) [37–40] have also been used for anthraquinone analysis in several natural products.
Although CE, GC, and HPLC methods can afford sufficient sensitivity for the analysis of anthraquinones in natural products, they all lack accurate peak identification in comparison with CE-MS, GC-MS, and LC-MS methods. Generally, the use of LC-MS is advantageous over the use of CE-MS and GC-MS because it affords high separation efficiency and accurate identification of analytes over a wide range of molecular structures, chemical reactivities, polarities, and volatilities. Hence, separation of analytes as well as mass identification and confirmation of analytes can be achieved simultaneously in one experimental run using LC-MS. For example, LC-MS using electrospray ionization (ESI) [37,38] and atmospheric pressure chemical ionization (APCI) [39,40] has been used for accurate identification of anthraquinones in plant extracts. In comparison with APCI-MS detection, the use of ESI-MS detection for anthraquinone identification is less effective, because the latter requires mobile phase additives other than the buffer to enhance analyte ionization [37,38]. Moreover, aside from eluent additives, the efficiency of the mass detection of anthraquinones using an ESI interface depends on the pKa value of the analyte [38].
The aim of this study was to develop an HPLC-UV detection method for quantitative analysis of rhein (1), kaempferol (2), aloe-emodin (3), emodin (4), chrysophanol (5), and physcion (6) in root extracts of C. alata. The structures of these analytes are shown in Figure 1. The root extracts were purified by use of solid-phase extraction and separation of the phenolic compounds was performed by use of reversed-phase HPLC using isocratic elution. In addition, on-line mass spectrometric detection using APCI in the negative ionization mode was used to confirm the presence of these phenolics in the root extract. To the best of our knowledge, this study represents the first investigation of the simultaneous analysis and identification of these six phenolic compounds in root extracts of C. alata using LC-UV-APCI-MS.
Figure 1.
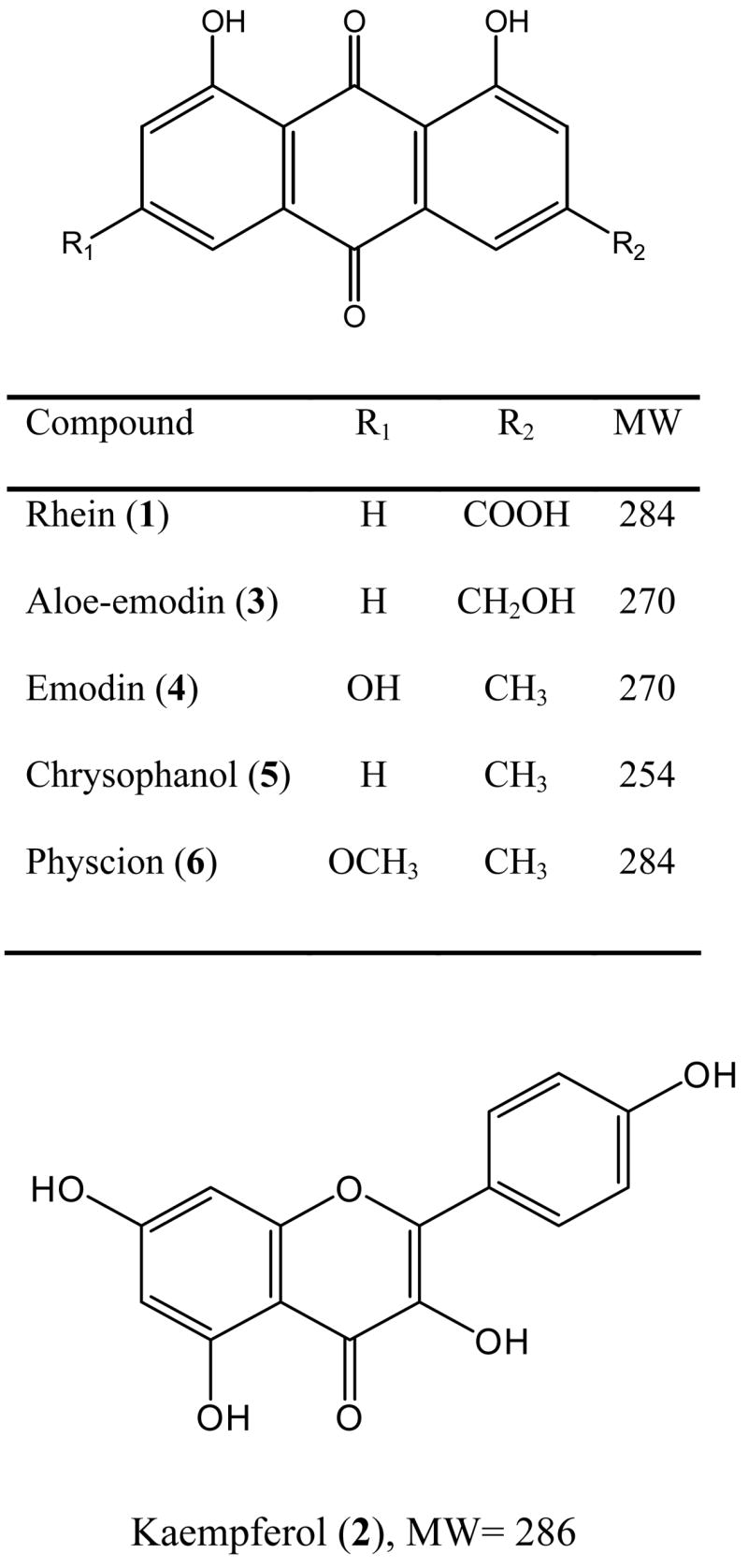
Chemical structures of six phenolic compounds in C. alata root extract.
2. Experimental
2.1. Chemicals and materials
Rhein, kaempferol, aloe-emodin, emodin, and chrysophanol standards were purchased from Sigma Aldrich (St. Louis, MO, USA), while the physcion standard was purchased from Micro Source Discovery System (Gaylordsville, CT, USA). Ammonium acetate (NH4Ac) buffer was purchased from Fluka (Amsterdam, the Netherlands). HPLC grade acetonitrile, ethanol, methanol, water, and Whatman no. 1 filter paper were obtained from Fisher Scientific (Houston, TX, USA). Doubly-distilled deionized water, used throughout the study, was obtained by use of an Elga Genetic Ultra-Pure water polishing system from US Filter (Lowell, MA, USA). Maxi-clean RP solid-phase extraction (SPE) C18 cartridges and 0.45 μm filters were purchased from Altech (Deerfield, IL, USA). C. alata root samples were collected by the Center of Agricultural Research, Suriname, and identified at the National Herbarium of the University of Suriname, Paramaribo, Suriname (Reference number: Jansen Jacobs 5027).
2.2. Preparation of standard solutions
Standard stock solutions of compounds 2–6 were prepared in ethanol at a concentration of 0.5 mg/mL, while rhein (1) was prepared at 0.25 mg/mL. Standard mixture solutions were prepared in ethanol at various concentration levels in the range of 5 – 250 ppm. All solutions were filtered prior to analysis through a 0.45 μm syringe filter and injected four times into the HPLC. The calibration curve for each compound was constructed by plotting the peak area as a function of the standard analyte concentration.
2.3. Sample preparation
The C. alata root samples were oven dried at 40 °C for five days. The dried roots were ground by use of a Wiley Mill grinder (Standard Model No. 3) to particle sizes of 6 mm or smaller. Ten grams of ground roots were extracted with 100 mL ethanol on an orbital shaker for 12 hours at room temperature. The extraction procedure was repeated two times, after which the two extracts were combined and filtered using Whatman no. 1 filter paper. The extraction solvent was removed by use of rotary evaporation and the residue was reconstituted in 10 mL ethanol and diluted with water (1:1 v/v).
Solid phase extraction (SPE) was used to remove unwanted interfering phytochemicals from the root extract. The SPE procedure was performed on an Altech extraction manifold system. SPE C18 cartridges (4 mL; 600 mg) were first conditioned with 4 mL methanol, followed with 4 mL water. Following the conditioning step, 4 mL of the diluted root extract was loaded onto the cartridge. After sample loading, the interfering compounds were removed with 2 mL of 10 % aqueous ethanol. Finally, the fraction containing compounds 1–6 were eluted with 2 mL of hot ethanol (60 °C). The vacuum pressure was kept at 10 mm Hg during the pre-conditioning step and was held constant at 2 mm Hg during the loading and eluting steps. Four replicate SPE extracts were collected. Each eluate was diluted to 5 mL with ethanol. The diluted SPE root extract, the eluate, was then filtered through a 0.45 μm syringe filter and injected into the HPLC. Each diluted SPE root extract was injected into the HPLC five times, and the average peak area was reported and used for analyte quantification.
2.4. HPLC analysis
Separation and quantitative analyses of compounds 1–6 were performed on a Shimadzu HPLC system (Kyoto, Japan) consisting of an SCL-10A system controller, two LC-10AD pumps, a DGU-14A degasser, an SIL-10AD auto injector and an SPD-10AV UV-VIS detector (λ = 260 nm). Separation of the analytes was performed at 40 °C on a Phenomenex Luna C18 (2) column, 100 Å pore size, 5 μm particle size, 250 × 4.6 mm ID column containing a guard column (Phenomenex, Torrance, CA, USA). The analytes were eluted isocratically at a flow rate of 0.4 mL/min using an acetonitrile/methanol/buffer (25: 55: 20, v/v), where the buffer is 10 mM ammonium acetate (NH4Ac) at pH 6.8. The injection volume was 10 μL.
2.5. LC-APCI-MS analysis
Analyte identification was performed by use of a Shimadzu LCMS-2010 system (Kyoto, Japan). Operating conditions for the HPLC were as described in the previous section. The mass spectrometer used for the identification of the analytes consists of a Q-array-octapole-quadrupole mass analyzer with an APCI interface used in the negative ionization mode and coupled to the Phenomenex Luna C18 (2) column described above. The APCI probe was operated at a temperature of 400 °C, while the CDL and block temperatures were operated at 200 °C. The detector voltage was 1.5 kV and the probe was operated in the negative ionization mode with a voltage of −4.0 kV. The nebulizing gas was nitrogen at a flow rate of 2.5 L/min. The optimum operating conditions for the LC-APCI-MS were determined for the separation and identification of compounds 1–6 in the scan mode with minimum fragmentation of the analytes. The scan rate of the mass analyzer was at 1s/scan within the mass range of m/z 100–1000.
2.6. Method validation
Precision (reproducibility) of the method was obtained by calculating the relative standard deviation (R.S.D.) from repeated injections of the standard mixture solutions at 15, 45, and 75 ppm for all analytes, except for kaempferol (2) that was determined at 30, 90, and 150 ppm. The intra-day precision was determined by six replicate injections, while the inter-day precision was determined by six injections for six days, for both the retention times and peak-areas [41].
A recovery study was performed to validate the accuracy of the developed method. Hence, root samples (10 grams) were spiked with standard stock solutions of the analytes in triplicate at two different concentrations. The spiked root samples were extracted with 100 mL ethanol following the procedure for sample preparation as described in section 2.3. Finally, the spiked samples were analyzed using the same experimental and instrumental conditions as previously described for the analysis of the un-spiked roots. The recovery was determined by comparing the amount of analyte added to the root sample and the amount of analyte detected during HPLC analysis [42].
3. Results and discussion
3.1. HPLC Optimization
Several preliminary studies were conducted to develop an HPLC method for the separation of compounds 1–6 in the C. alata root extract. The LC separation conditions of the analytes were optimized by systematically adjusting the methanol and acetonitrile content in the mobile phase with the addition of different buffers, such as trifluoroacetic acid, formic acid, and ammonium acetate to obtain better resolution of the phenolic compounds. The retention times of the analytes decreased with an increase in the amount of methanol in the eluent. This observation was in agreement with a previous report by Ding et al. [35]. An increase in the amount of acetonitrile in the eluent also resulted in a decrease in retention time of compounds 1–6. The addition of 10 mM NH4Ac buffer to the mobile phase resulted in the best peak resolution of compounds 1–6. Addition of NH4Ac buffer to the mobile phase not only improved the resolution, but also resulted in complete deprotonation of compounds 1–6 [M-H]−.
The pH of the mobile phase was also optimized to obtain better resolution of compounds 1–6. Separation at pH 4.8 using NH4Ac buffer resulted in co-elution of rhein (1) and kaempferol (2). Therefore, resolution of only compounds 3–6 could be obtained. At pH 8.8 (adjusted with NH4OH) compounds 1–3 co-eluted. Complete separation of compounds 1–6 were only achieved at pH 6.8 using NH4Ac buffer. The flow rate of the eluent was also optimized at 0.4 mL/min for best resolution and MS detection. The use of flow rates greater than 0.4 mL/min resulted in overloading of the mass spectrometer detector.
Optimal separation of the analytes was obtained within 45 minutes for standard mixtures as well as the C. alata root extract by use of an isocratic mobile phase of ACN/MeOH/NH4Ac buffer at pH 6.8 (25: 55: 20, v/v). We optimized the retention times of the analytes using a gradient elution system containing ACN/MeOH/NH4Ac buffer at pH 6.8 (25:55:20, v/v) for solvent A and ACN/MeOH/NH4Ac buffer at pH 6.8 (25:65:10, v/v) for solvent B, allowing successful separation of all analytes within 30.0 minutes. However, we did not use the gradient elution system for quantification of the analytes because it was not reproducible.
The phenolic compounds 1–6 were identified in the C. alata root extract by spiking the extracts with the respective standards. Prior to this procedure, all standards were run separately to determine the retention time of each analyte. The chromatographic separation of compounds 1–6 is shown in Figure 2A for the standard mixture at 30 ppm as an example, and in Figure 2B for the root extract (without spiking). In addition to the analyte peaks obtained in Figure 2B, an unidentified first eluting peak (at 6.83 min) was also observed. We have isolated this unknown using flash column chromatography, followed by purification using preparative HPLC. However, after performing spectroscopic studies (UV, IR, MS, and NMR), we concluded that this unknown peak is an impurity composed of a mixture of compounds. No further analysis of this peak was attempted.
Figure 2.
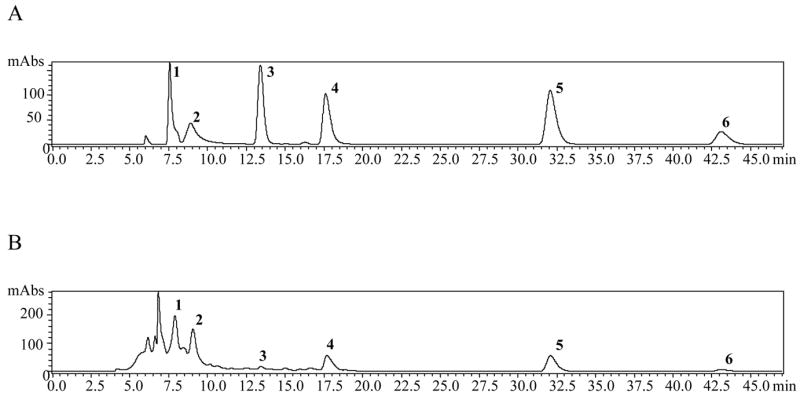
A) HPLC chromatogram of the phenolic standard mixture (30 ppm), λ = 260 nm. Rhein (1, 7.57 min); Kaempferol (2, 8.92 min); Aloe-emodin (3, 13.41 min); Emodin (4, 17.63 min); Chrysophanol (5, 32.06 min); Physcion (6, 43.07 min).
B) HPLC chromatogram of C. alata root extract, λ = 260 nm. Rhein (1, 7.91 min); Kaempferol (2, 9.05 min); Aloe-emodin (3, 13.43 min); Emodin (4, 17.69 min); Chrysophanol (5, 32.04 min); Physcion (6, 43.01 min).
3.2. LC-MS analysis
Simultaneous separation and identification of phenolic compounds 1–6 in the C. alata root extracts were performed by use of LC-APCI-MS detection. Identification of the peaks was achieved by comparison of the retention times, UV spectra, as well as MS data of the separated compounds with the respective standards. The total ion chromatograms (TIC) of analytes 1–6 in the standard mixture and root extract were recorded in the scan mode, and are shown in Figure 3A and B, respectively. As seen in Figure 3B, the peak intensities for aloe-emodin (3) and physcion (6) are very low, as a result of their low concentration (‘vide infra’) in the root extract as determined by HPLC in this study.
Figure 3.

Total ion chromatograms of A) Standard mixture (30 ppm); B) C. alata root extract
The mass spectra of the phenolic compounds 1–6 in the root extract are presented in Figure 4. The presence of each analyte in the root extract was confirmed by its respective [M-H]− m/z. In addition to the ions at [M-H]− of compounds 1–6, the ion at m/z 239 was registered in the mass spectrum of rhein (1) and aloe-emodin (3) due to fragmentation of molecular ions of the analyte resulting in [M-COOH]− and [M-CH2OH]−, respectively. The ions at m/z 253 and 271 were also recorded in the mass spectrum of rhein (1) which are assumed to be a fragment derived from the molecular ion resulting in [M-CH2OH]− and an adduct formation between the ion at m/z 239 and methanol ([M-COOH + MeOH]−), respectively. The ion at m/z 253 obtained in the mass spectrum of aloe-emodin (3) was due to the loss of a hydroxyl group, resulting in [M-OH]−. The ions at m/z 255 and 269 observed in the mass spectrum of kaempferol (2) are possibly due to fragmentation of molecular ion resulting in [M-CH2OH]− and [M-OH]−, respectively. Finally, the ion at m/z 269 recorded in the mass spectrum of physcion (6) is due to the loss of a methyl group leading to [M-CH3]−.
Figure 4.

Mass spectra of Rhein (1); Kaempferol (2); Aloe-emodin (3); Emodin (4); Chrysophanol (5); Physcion (6) from the C. alata root extract
3.3. Calibration curves
In this study, as well as in previous reports [38,39], the analysis of anthraquinones using on-line mass spectrometric detection was found to be less sensitive than on-line UV detection. Therefore, HPLC-UV was chosen for the determination of compounds 1–6. The investigated compounds in the root extract were quantified by integration of the peak areas at 260 nm using an external calibration method.
Calibration curves were constructed for each analyte by using a series of standard mixture solutions. Least-squares linear regression was used to determine the calibration parameters for each of the six standards. A summary of the calibration studies for the six analytes is presented in Table 1. The linearity of all calibration curves was determined by calculating the correlation coefficients, which varied from 0.9971 for kaempferol to 0.9999 for emodin and physcion.
Table 1.
Calibration parameters of HPLC analysis for phenolic compounds 1–6
| Compound | a | b | r | Linear range (ppm) | LOD (ppm) |
|---|---|---|---|---|---|
| Rhein (1) | 145321 | 69934 | 0.9985 | 13.1 – 115 | 4.61 |
| Kaempferol (2) | 60862 | −26171 | 0.9971 | 5.27 – 230 | 0.98 |
| Aloe-emodin (3) | 160496 | −43698 | 0.9998 | 3.06 – 145 | 0.54 |
| Emodin (4) | 98680 | −29660 | 0.9999 | 2.18 – 115 | 0.23 |
| Chrysophanol (5) | 170751 | 46224 | 0.9998 | 6.04 – 115 | 2.19 |
| Physcion (6) | 142579 | −43473 | 0.9999 | 3.26 – 105 | 0.55 |
Note: The relationship between peak area and analyte concentration is expressed as linear regression lines (y = ax + b), where y is the peak area measured by UV detector, x is the concentration (ppm) of the analytes, and a and b are the respective slope and intercept of the calibration curve. The correlation coefficient is r.
The limit of detection (LOD), defined as the lowest detectable concentration of an analyte, was calculated using the formula LOD = (b + 3 σb)/a, where a is the slope of the calibration curve; b is the intercept; and σb is the standard deviation associated with the intercept. The LOD for the analytes were in the range of 0.23 – 4.61 ppm. In addition, the limit of quantification (LOQ), defined as the lowest measurable analyte concentration was determined according to the formula LOQ = (b + 10 σb)/a, where all parameters are as defined for the LOD. The LOQ is reported in Table 1 as the lower limit from the linear range.
3.4. Method validation and Quantification
The method was validated by determining the intra- and inter-day precision. The relative standard deviation (R.S.D.) values for the retention times and peak-areas for the intra-day precision were 0.07–0.15% and 0.27–1.63% (n = 6), respectively. The R.S.D. values obtained for the inter-day precision study for the retention times and peak-areas were 0.07–0.16% and 0.39–2.10 (n = 6), respectively (Table 2).
Table 2.
Relative standard deviation values for intra- and inter-day precision for compounds 1–6
| Compound | Intra-day precision (n = 6) | Inter-day precision (n = 6) | ||
|---|---|---|---|---|
| Retention time (R.S.D., %) | Peak area (R.S.D., %) | Retention time (R.S.D., %) | Peak area (R.S.D., %) | |
| Rhein (1) | 0.08 | 1.25 | 0.07 | 1.33 |
| Kaempferol (2) | 0.07 | 1.63 | 0.07 | 2.10 |
| Aloe-emodin (3) | 0.08 | 0.32 | 0.08 | 0.45 |
| Emodin (4) | 0.09 | 0.28 | 0.11 | 0.46 |
| Chrysophanol (5) | 0.12 | 0.27 | 0.13 | 0.46 |
| Physcion (6) | 0.15 | 0.31 | 0.16 | 0.39 |
In order to determine the accuracy of the method, the mean recovery of the phenolic compounds 1–6 was calculated at two concentration levels as reported in Table 3 for each analyte. The overall mean recovery ranged between 81.2 ± 4.3% for aloe-emodin and 106 ± 2% for emodin (n = 3).
Table 3.
Concentrations and recoveries of phenolic compounds 1–6 in the root extract of C. alata
| Compound | Root concentration (ppm) | Spiked concentration (ppm) | Total concentration detected (ppm) | Recovery (%) (mean ± S.D.) |
|---|---|---|---|---|
| Rhein (1) | 68.4 ± 1.6 | 45.4
90.8 |
121 ± 1
151 ± 10 |
106 ± 1
90.4 ± 6.1 |
| Kaempferol (2) | 122 ± 7 | 65.0
130 |
192 ± 2
257 ± 1 |
103 ±1
102 ± 1 |
| Aloe-emodin(3) | 4.02 ± 0.37 | 3.30
6.61 |
6.43 ± 0.27
8.64 ± 0.46 |
87.7 ± 3.7
81.2 ± 4.3 |
| Emodin (4) | 26.5 ± 5.0 | 13.8
27.6 |
42.7 ± 0.9
52.5 ± 2.9 |
106 ± 2
97.0 ± 5.3 |
| Chrysophanol (5) | 21.6 ± 6.5 | 11.3
22.5 |
30.1 ± 0.6
37.1 ± 1.5 |
91.4 ± 1.8
84.1 ± 3.4 |
| Physcion (6) | 4.45 ± 0.71 | 2.62
5.24 |
7.37 ± 0.12
8.78 ± 0.20 |
104 ± 2
90.6 ± 2.0 |
The concentration of each investigated compound in the root extract was determined by substituting its peak area for y in the equation listed in Table 1. The concentrations of compounds 1–6 detected in the root extract are given in Table 3. Among the six analytes determined in the root extracts, the concentrations of rhein (68.4 ± 1.6 ppm) and kaempferol (122 ± 7 ppm) were found to be highest.
4. Conclusions
A simple and reliable LC-UV-APCI-MS method has been developed for the simultaneous analysis and identification of six pharmacologically active compounds in root extracts of C. alata. The extracts were purified by SPE and separated on a C18-HPLC column using isocratic elution. The investigated phenolic compounds in the root extract were identified by their UV spectra and MS data. The compounds were ionized using an APCI interface and their molecular masses as well as their fragmentation patterns were determined. Finally, HPLC-UV was used to determine the amount of these phenolic compounds in the root extract.
This valuable information concerning the concentration of these pharmacologically active compounds in the root extract of C. alata could aid in determining their active component(s), which can be of great importance for pharmaceutical industry. However, further studies involving complete characterization and bioassay analysis of these phenolic compounds in the root extract of C. alata are necessary. There is ongoing research in this direction in our laboratory which will be reported in a future manuscript.
Acknowledgments
We acknowledge the National Science Foundation (CHE0616824) and the National Institutes for Health (R1GM39844) for financial support. We also acknowledge the helpful discussions of Dr. Xiaodong Huang and Dr. Ginger Powe.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Heyde H. Medicijn Planten in Suriname, “Medicinal Plants in Suriname”. 3. Uitg. Stichting Gezondheidsplanten Informatie; Paramaribo: 1990. p. 120. [Google Scholar]
- 2.Co LL. Common Medicinal Plants of the Cordillera Region. Bustamante Press; Quezon City: 1989. p. 11. [Google Scholar]
- 3.Agarkar SV, Jadge DR. Asian J Chem. 1999;11:295. [Google Scholar]
- 4.Rai PP. Current Sci. 1978;47:271. [Google Scholar]
- 5.Irvine FR. Woody plants of Ghana with special reference to their uses. Oxford University Press; London: 1961. p. 178. [Google Scholar]
- 6.Kirtikar KR, Basu BD. Indian Medicinal Plants. Vol. 2. Periodical Experts; New Delhi: 1975. p. 870. [Google Scholar]
- 7.de Padua LS, Lugod GC, Pancho JV. Handbook on Phillippines Medicinal Plants. 2. Vol. 3. University of the Phillipines; Los Baños: 1980. p. 37. [Google Scholar]
- 8.Ibrahim D, Osman H. J Ethnopharmacol. 1995;45:151. doi: 10.1016/0378-8741(94)01200-j. [DOI] [PubMed] [Google Scholar]
- 9.Somchit MN, Reezal I, Elysha Nur I, Mutalib AR. J Ethnopharmacol. 2003;84:1. doi: 10.1016/s0378-8741(02)00146-0. [DOI] [PubMed] [Google Scholar]
- 10.Damodaran S, Venkataraman S. J Ethnopharmacol. 1994;42:19. doi: 10.1016/0378-8741(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 11.Villaseñor IM, Canlas AP, Pascua MP, Sabando MN, Soliven LAP. Phytother Res. 2002;16:S93. doi: 10.1002/ptr.768. [DOI] [PubMed] [Google Scholar]
- 12.Moriyama H, Iizuka T, Nagai M, Miyataka H, Satoh T. Yakugaku Zasshi. 2003;123:607. doi: 10.1248/yakushi.123.607. [DOI] [PubMed] [Google Scholar]
- 13.Palanichamy S, Nagarajan S. J Ethnopharmacol. 1990;29:73. doi: 10.1016/0378-8741(90)90099-f. [DOI] [PubMed] [Google Scholar]
- 14.Belkin M, Fitzgerald Dorothea B, Cogan W. J Natl Cancer Inst. 1952;13:139. [PubMed] [Google Scholar]
- 15.Yagi SM, El Tigani S, Adam SE. Phytother Res. 1998;12:324. [Google Scholar]
- 16.Smith R, Ali S. NZ J Sci. 1979;22:123. [Google Scholar]
- 17.Rao JVL, Sastry PSR, Rao RYK, Vimaladevi M. Curr Sci India. 1975;44:736. [Google Scholar]
- 18.Tiwari RD, Yadava OP. Planta Med. 1971;19:299. doi: 10.1055/s-0028-1099645. [DOI] [PubMed] [Google Scholar]
- 19.Thomson RH. Naturally Occurring Quinones III: Recent Advances. 3. Chapman and Hall Ltd, University Press; Cambridge: 1987. pp. 345–526. [Google Scholar]
- 20.Agarwal SK, Singh SS, Verma S, Kumar S. J Ethnopharmacol. 2000;72:43. doi: 10.1016/s0378-8741(00)00195-1. [DOI] [PubMed] [Google Scholar]
- 21.Koyama J, Morita I, Tagahara K, Ogata M, Mukainaka T, Tokuda H, Nishino H. Cancer Lett. 2001;170:15. doi: 10.1016/s0304-3835(01)00566-3. [DOI] [PubMed] [Google Scholar]
- 22.Yen G, Duh P, Chuang D. Food Chem. 2000;70:437. [Google Scholar]
- 23.Wang H, Chen T, Yang P, Ueng T. DMD. 2001;29:1229. [PubMed] [Google Scholar]
- 24.Choi SB, Ko BS, Park SK, Jang JS, Park S. Life Sci. 2006;78:934. doi: 10.1016/j.lfs.2005.05.101. [DOI] [PubMed] [Google Scholar]
- 25.Nakamura Y, Chang C, Mori T, Sato K, Ohtsuki K, Upham BL, Trosko JE. Carcinogenesis. 2005;26:665. doi: 10.1093/carcin/bgi003. [DOI] [PubMed] [Google Scholar]
- 26.Campbell JK, King JL, Harmston M, Lila M, Erdman JW., Jr J Food Sci. 2006;71:S358. [Google Scholar]
- 27.Moriyama H, Iizuka T, Nagai M, Murata Y. Fitoterapia. 2003;74:425. doi: 10.1016/s0367-326x(03)00058-3. [DOI] [PubMed] [Google Scholar]
- 28.Singh NP, Gupta AP, Sinha AK, Ahuja PS. J Chromatogr A. 2005;1077:202. doi: 10.1016/j.chroma.2005.03.130. [DOI] [PubMed] [Google Scholar]
- 29.Jiang TF, Lv ZH, Wang YH. J Sep Sci. 2005;28:2225. doi: 10.1002/jssc.200500144. [DOI] [PubMed] [Google Scholar]
- 30.Tian K, Wang Y, Chen Y, Chen X, Hu Z. Talanta. 2007;72:587. doi: 10.1016/j.talanta.2006.11.027. [DOI] [PubMed] [Google Scholar]
- 31.Puchalska M, Orli ska M, Ackacha MA, Pole-Pawlak K, Jarosz M. J Mass Spectrom. 2003;38:1252. doi: 10.1002/jms.549. [DOI] [PubMed] [Google Scholar]
- 32.Harruff LG, Vazquez MA. Tappi. 1981;64:109. [Google Scholar]
- 33.ElSohly MA, Gul W, Murphy TP. Int Immunopharmcol. 2004;4:1739. doi: 10.1016/j.intimp.2004.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Steinert J, Khalaf H, Rimpler M. J Chromatogr A. 1996;723:206. [Google Scholar]
- 35.Ding M, Ma S, Liu D. Anal Sci. 2003;19:1163. doi: 10.2116/analsci.19.1163. [DOI] [PubMed] [Google Scholar]
- 36.Derksen GCH, Lelyveld GP, van Beek TA, Capelle A, de Groot æ. Phytochem Anal. 2004;15:397. doi: 10.1002/pca.800. [DOI] [PubMed] [Google Scholar]
- 37.Nindi MM, Kgarebe BV, Wolfender JL, Abegaz BM. Phytochem Anal. 1999;10:69. [Google Scholar]
- 38.Derksen GCH, Niederländer HAG, van Beek TA. J Chromatogr A. 2002;978:119. doi: 10.1016/s0021-9673(02)01412-7. [DOI] [PubMed] [Google Scholar]
- 39.Mueller SO, Schmitt M, Dekant W, Stopper H, Schlatter J, Schreier P, Lutz WK. Food Chem Toxicol. 1999;37:481. doi: 10.1016/s0278-6915(99)00027-7. [DOI] [PubMed] [Google Scholar]
- 40.Li W, Chan C, Lueng H. J Pharm Pharmacol. 2000;52:723. doi: 10.1211/0022357001774408. [DOI] [PubMed] [Google Scholar]
- 41.Miller JM. Chromatography: Concepts and Contrasts. 2 . John Wiley & Sons, Inc.; 2005. pp. 24–29. [Google Scholar]
- 42.Skoog DA, Holler FJ, Nieman TA. Principles of Instrumental Analysis. 5. Thomson Learning; United States: 1998. pp. 11–15. [Google Scholar]