

Abstract
Tri-methylation of histone H3 on lysine 4 (H3K4me3) localizes near the 5′ region of genes and is tightly associated with active loci. Several proteins, such as CHD1, BPTF, JMJD2A, and the ING tumor suppressor family, directly recognize this lysine methyl mark. However, how H3K4me3 recognition participates in active transcription remains poorly characterized. Here we identify specific CHD1-interacting proteins via H3K4me3 affinity purification, including numerous factors mediating post-initiation events. Conventional biochemical purification revealed a stable complex between CHD1 and components of the spliceosome. Depletion of CHD1 in extracts dramatically reduced splicing efficiency in vitro, indicating a functional link between CHD1 and the spliceosome. Knockdown of CHD1 and H3K4me3 levels by siRNA reduced association of U2 snRNP components with chromatin, and more importantly, altered the efficiency of pre-mRNA splicing on active genes in vivo. These findings suggest that methylated H3K4 serves to facilitate the competency of pre-mRNA maturation through the bridging of spliceosomal components to H3K4me3 via CHD1.
Introduction
Gene expression is greatly impacted by non-covalent and post-translational, covalent modification of chromatin. In particular, histone lysine methyltransferases (HKMTases) have received a great deal of attention, although how histone lysine methylation mechanistically contributes to gene expression remains poorly understood. Global changes in histone methylation and acetylation are associated with cancer, and may serve as a predictive indicator of clinical outcome (Seligson et al., 2005). Several human enzymes that catalyze methylation on histone H3 lysine 4 (H3K4me), including MLL and SMYD3, are altered in a variety of cancer types (Ayton and Cleary, 2001; Hamamoto et al., 2004). More recently, enzymes that counteract the effects of H3K4me through demethylation have been found associated with various disease states (Iwase et al., 2007; Tahiliani et al., 2007). To understand how these enzymes contribute to disease, it is essential to identify how H3K4me functionally relates to events pertinent to transcriptional activation.
H3K4 tri-methylation (H3K4me3) is tightly associated with the transcription start site at the 5′ end of active genes (Bernstein et al., 2005; Schneider et al., 2004). Histone methylation is thought to serve as a “mark” that can be recognized by specific proteins resulting in the recruitment of downstream effectors. For instance, the PHD finger of Yng1 was recently found to promote the histone acetyltransferase activity of NuA3 to facilitate H3K14 acetylation in yeast (Taverna et al., 2006). It was demonstrated using a functional transcription assay that the establishment of H3K4me3 requires the process of active transcription (Pavri et al., 2006); therefore, the functional implication of this modification must act downstream of transcription initiation, or perhaps during re-initiation. In vitro analyses also established that the H3K4 tri-methyl mark itself has no direct effect on transcription (Pavri et al., 2006), suggesting that H3K4me3 requires ancillary factors to carry out its effect on transcription. We and others previously demonstrated that the human CHD1 protein specifically interacts with methylated H3K4 via its tandem chromodomains (Flanagan et al., 2005; Sims et al., 2005). Studies in yeast, Drosophila, and mammalian systems implicate a role for CHD1 in transcript elongation and termination (Sims et al., 2004). However, whether CHD1 recruits downstream effectors in the context of H3K4me3 remains to be determined.
Transcript elongation is a highly regulated and dynamic stage of the transcription cycle (Sims et al., 2004). A current view of mRNA biogenesis incorporates multiple events simultaneously, including transcript elongation, CAP addition, pre-mRNA splicing and polyadenylation, in addition to mRNA surveillance and export (Hirose and Manley, 2000; Maniatis and Reed, 2002; Orphanides and Reinberg, 2002; Proudfoot et al., 2002). The involvement of chromatin dynamics in transcript elongation has been well documented, although how chromatin relates to pre-mRNA processing is far less understood. Chromatin immunoprecipitation experiments revealed that splicing factor recruitment in mammalian cells occurs co-transcriptionally, which differs from yeast (Listerman et al., 2006).
Our current findings demonstrate that human CHD1 recognition of H3K4me3 functions to recruit factors involved in the coordinated events of mRNA maturation, namely transcript elongation and pre-mRNA processing. CHD1 was found to bridge core spliceosomal components to H3K4me3 via specific interactions with the SF3a sub-complex of U2 snRNP. The recruitment of SF3a and the efficiency of pre-mRNA splicing were perturbed upon reduction of CHD1 and H3K4me3. Collectively, our results provide a mechanistic explanation for the H3K4me3 mark during active transcription: to facilitate the efficiency of post-initiation processes, such as pre-mRNA splicing.
Results
Identification of H3K4me3-associated polypeptides
To better understand the function of H3K4me3 during transcription, we sought to identify proteins that selectively recognize this methyl mark. We affinity-purified H3K4me3-binding factors from HeLa nuclear extracts using a histone H3 peptide (first eight residues) tri-methylated on lysine 4. The yeast protein Isw1p, and its human homologue SNF2h, selectively binds H3K4me2 and H3K4me3 indirectly through an unidentified protein (Santos-Rosa et al., 2003); thus, we scored initially for SNF2h binding. We observed clear specificity for SNF2h binding to H3K4me2/3 peptide columns (Supplementary Figure 1A and 1B). The ATP-dependent chromatin remodeling protein SNF2h is a component of numerous distinct complexes, including RSF and ACF among others (Eberharter and Becker, 2004). Interestingly, the Rsf-1 subunit of RSF did not bind to a column containing either the H3K4me0 or H3K4me3 peptides, while SNF2h bound only to H3K4me3 (Figure 1A). This result indicates that SNF2h binds to H3K4me3 within a distinct complex(es). Thus, our affinity-based purification method displays a clear specificity in its discrimination between the H3K4me0 and H3K4me3 peptides.
Figure 1.

CHD1 bridges post-initiation factors to H3K4me3. A, B, and C) Western blotting of input and eluate from H3K4me0 or H3K4me3 affinity columns. The antibodies used are indicated. D) Western blotting of the input and eluate from H3K4me0 or H3K4me3 affinity columns probed with the FACT subunit SSRP1. FT and Elu represent the flow through and elution fractions, respectively. E) Western blotting using anti-CHD1, anti-SNF2h, or anti-PAF1 antibodies of the eluate from the H3K4me3 affinity column fractionated by gel filtration (Superose 6). Sizes are indicated (kD). F) Co-immunoprecipitation from the H3K4me3 elution material using the indicated antibodies for precipitation (top) and detection by western blotting (bottom).
Next, we focused on identifying additional H3K4me3-bound proteins. Silver staining revealed a number of distinct polypeptides (Supplementary Figure 1C). A combination of mass spectrometry and western blot analyses of the H3K4me3 affinity purified material identified numerous proteins implicated in transcriptional post-initiation events, including transcript elongation, pre-mRNA splicing, mRNA surveillance, and mRNA export, among other activities (Supplementary Table 1). Our approach did not identify proteins involved in initiation of transcription by RNA polymerase (RNAP) II, or factors associated with RNAP I or III (data not shown). The ATP-dependent chromatin remodeling protein and histone chaperone CHD1 was observed to bind to the H3K4me3 affinity column (Supplementary Table 1, Figure 1B). The related chromodomain containing ATPase CHD3 did not associate with H3K4me3 peptides, a finding supported by previous studies (Figure 1B) (Nishioka et al., 2002; Zegerman et al., 2002). Mass spectroscopy of the H3K4me3 eluate identified components of PAF, a protein complex consisting of hPaf1, hCtr9, hLeo1, hCdc73, and hSki8 (Supplementary Table 1, Figure 1B) (Rozenblatt-Rosen et al., 2005; Zhu et al., 2005). PAF modulates covalent histone modifications involved in elongation such as methylation of H3 (K4 and K36) and monoubiquitination of H2B (Rozenblatt-Rosen et al., 2005; Sims et al., 2004; Zhu et al., 2005). PAF displayed specific binding to H3K4me3 when directly compared with unmodified peptides, a property also shared by CHD1 and SNF2h (Figure 1C). Aside from SNF2h and the PAF complex, the chromatin modulator FACT was present in the H3K4me3 elution (Figure 1B). FACT is a heterodimeric complex that allows RNAP II to traverse nucleosomes (Reinberg and Sims, 2006), and physically interacts with CHD1 (Kelley et al., 1999). Importantly, FACT demonstrated a selective specificity for H3K4me3, as compared to H3K4me0 (Figure 1D).
Insight into how SNF2h, PAF, and FACT were binding to H3K4me3 was provided by our partial conventional purification of SNF2h-containing complexes. We separated SNF2h derived from nuclear pellets into at least two different complexes, including one that co-eluted with CHD1. The SNF2h fraction containing CHD1 retained H3K4me3 binding, whereas the complex devoid of CHD1 did not (Supplementary Figure 2A). These results suggest that CHD1 functions to bridge SNF2h to H3K4me3. We further purified a native SNF2h/CHD1 complex by gel filtration, which revealed that SNF2h, CHD1, and PAF co-elute as a large (~2 MDa) complex (data not shown), perhaps signifying that these proteins directly interact with one another. We next analyzed whether CHD1, PAF, and SNF2h derived from the H3K4me3 affinity eluate co-fractionate as a large complex, similar to that seen during conventional purification. Analysis of the sizing fractions revealed that CHD1, PAF1, and SNF2h co-elute as a large molecular weight complex of nearly 2 MDa (Figure 1E). Co-immunoprecipitation experiments using the H3K4me3 eluate confirmed that CHD1 interacts with SNF2h and PAF (Figure 1F). Similarly, FACT co-precipitated CHD1 derived from the H3K4me3 eluate (Figure 1F). Gel filtration analysis of the H3K4me3 eluate revealed that CHD1 and FACT co-elute, corroborating that CHD1 and FACT interact when bound to H3K4me3 (data not shown). However, FACT did not co-elute together with SNF2h and PAF, indicating that the H3K4me3 eluate contains a mixture of protein complexes (data not shown). The latter results indicate that CHD1 functions to bridge FACT to H3K4me3 in addition to SNF2h and PAF.
Components of the spliceosome interact with H3K4me3 via CHD1
Consistent with the notion that H3K4me3 facilitates events operating downstream of transcription initiation, mass spectrometry of the H3K4me3 eluate identified the U2 snRNP subunit SF3A1 (Supplementary Table 1). Intron excision of most pre-mRNAs is carried out by the spliceosome, a large complex consisting of five small nuclear ribonucleoprotein (snRNP) particles (U1, U2, U4, U5, U6) and numerous non-snRNP proteins (Jurica and Moore, 2003). These include members of the SR (serine/arginine-rich) family of proteins that assist in early spliceosome assembly (Huang and Steitz, 2005). As the U2 snRNP is a component of the pre-spliceosome (Complex A, (Jurica and Moore, 2003)), we tested the H3K4me3 eluate for additional proteins involved in pre-spliceosome formation, including SR proteins, the U1 snRNP-specific protein U1-70K, and U2 snRNP-specific proteins. Neither U1-70K nor any of the canonical SR proteins were detected in the H3K4me3 elution (Figure 2A and 2B). In contrast, every U2 snRNP-specific protein analyzed was observed in the H3K4me3 eluate, as well as the splicing factor U2AF65 (Supplementary Table 1 and Figure 2A). Importantly, specificity of U2 snRNP binding to H3K4me3 compared to the unmodified H3 tail was observed (Figure 2C). Further analyses of the H3K4me3-bound proteins by mass spectrometry corroborate that H3K4me3 recruits factors associated with the pre-spliceosome (Supplementary Table 1). To further characterize the spliceosomal components associated with H3K4me3, we tested for the presence of snRNAs in the H3K4me3 elution using 32P-[pCp] labeling of RNAs (Michaud and Reed, 1991). We detected the 5 major spliceosomal snRNAs in the H3K4me3 eluate (Figure 2D). Importantly, a substantial increase in RNA purity was observed with the H3K4me3 elution material in the case of snRNAs compared to non-snRNAs, such as tRNA or 5S ribosomal RNAs. Closer inspection revealed that purification of snRNPs by H3K4me3 was most evident for U2 snRNA (11-fold), while U1 was only modestly enhanced (3 fold), consistent with the apparent absence of U1-70K. These collective findings demonstrate that H3K4me3 specifically enriches a subset of essential splicing factors that operate early in spliceosome assembly.
Figure 2.

Spliceosomal components associate with H3K4me3 via CHD1. A and B) Western blotting of nuclear extract or the H3K4me3 eluate. C) Total RNA labeling with [5′-32P]pCp from nuclear extract (NE) or the H3K4me3 eluate. The RNA species are indicated. For quantification, the signal from each snRNA band was calculated as a percentage of the total labeled RNA. The fold-increase in purity is shown graphically on the right (ratio of H3K4me3 eluate to NE). D) Co-immunoprecipitation experiments using the indicated antibodies for precipitation (top) and detection by western blotting (side). E) Western analysis of the eluate from H3K4me0 or H3K4me3 affinity columns.
A microarray approach that monitored alterations in mRNA and pre-mRNA levels in yeast was used to decipher functional connections between factors involved in the coordinated processes of mRNA biogenesis (Burckin et al., 2005). Interestingly, the splicing defects most closely related to mutations affecting spliceosome components corresponded to numerous factors identified in the H3K4me3 eluate, including PAF subunits and the Spt16 subunit of FACT. Given that CHD1 bridges FACT, SNF2h, and PAF to H3K4me3, we tested for physical interactions between the U2 snRNP and CHD1. The U2 snRNP includes U2-A′, U2-B″, p14, the SF3a sub-complex composed of SF3A1, SF3A2, and SF3A3, and the SF3b sub-complex that includes SF3B1, SF3B2, SF3B3, and SF3B4 (Jurica and Moore, 2003). Co-immunoprecipitation experiments indicate that CHD1 exhibits a robust interaction with SF3a subunits, as well as associations with U2-B″ and SF3b subunits (Figure 2E). A weak but specific interaction between CHD1 and a U5 snRNP subunit was also observed, and no associations were seen between CHD1 and U1 snRNP nor the canonical SR proteins (Figure 2E, data not shown). Therefore two independent methods, H3K4me3 affinity chromatography and anti-CHD1 co-precipitation, yielded comparable results suggesting that CHD1 specifically associates with distinct spliceosomal components, with a preference for the SF3a sub-complex of the U2 snRNP.
Components of the SF3a sub-complex are associated with CHD1
Based on the above results, we thought that CHD1 may bridge larger spliceosomal complexes via SF3a. To test this contention, we sought to characterize these interactions more rigorously. To further characterize the CHD1-U2 snRNP association, we performed conventional chromatography. CHD1 and U2 snRNP protein complexes were fractionated from HeLa nuclear extracts using a series of ion-exchange, size exclusion, and affinity chromatography as outlined in Figure 3A. CHD1 and subunits of the SF3a and SF3b sub-complexes bound tightly to phosphocellulose, eluting with 1M KCl (Figure 3A, and data not shown). Additional fractionation identified that CHD1, SF3a, and SF3b bound to DEAE-cellulose and co-eluted in several peaks when an HPLC DEAE-5PW column was eluted with a 0.1 to 0.8M KCl linear gradient (Figure 3A, and data not shown). Interestingly, when fractions from the DEAE-5PW column containing CHD1, SF3a, and SF3b were immunoaffinity purified using anti-CHD1 antibodies, SF3a but not to SF3b subunits were bound to the CHD1 affinity column (Figure 3B). When these fractions were analyzed by gel filtration (Superose 6), CHD1 appeared to peak at a molecular mass of approximately 500–600 kDa, while the SF3a and SF3b complexes co-eluted at a molecular mass of greater than 700 kDa (data not shown). These results indicate that the majority of SF3a and SF3b associate together in a larger complex lacking CHD1, perhaps representing U2 snRNP. However, subunits of the SF3a sub-complex did overlap with the peak fraction of CHD1 (data not shown).
Figure 3.

CHD1 is stably associated with the SF3a sub-complex of U2 snRNP. A) Purification scheme of CHD1/SF3a complexes. B) Immunoaffinity purification using the indicated antibodies for precipitation (top) and detection by western blotting (side). The input material was derived from the DEAE-5PW elution indicated as immunoaffinity in (A). C) Analyses of the gel filtration fractions from the Superose 6 PC 3.2/30 labeled in (A). Western blotting was performed using the indicated antibodies (side). Sizes are indicated (kD). D) Co-immunoprecipitation experiment using IgG or anti-CHD1 antibodies together with recombinant CHD1 and 35S-translated SF3A1, SF3A2, or SF3A3 protein. A Coomassie stain of recombinant CHD1 is indicated in the left panel.
Based on the tight association observed between CHD1 and SF3a subunits in this input fraction, we concluded that the SF3a that overlapped with CHD1 represents a smaller fraction of SF3a that directly interacts with CHD1. To test this, we pooled the CHD1 peak containing the overlapping SF3a sub-complex and further fractionated this material using a SMART Superose 6 gel filtration column. Analysis of these fractions confirmed that SF3a is indeed present in two complexes, a larger complex containing SF3b devoid of CHD1, and a smaller complex containing CHD1, but lacking SF3b subunits (Figure 3C). Experiments using recombinant CHD1 and in vitro translated SF3a subunits revealed that CHD1 interacts with SF3A1, but not SF3A2 or SF3A3, likely indicating a direct association between CHD1 and SF3A1 (Figure 3D). Consistently, DNase and RNase treatment had no effect on the interactions between CHD1 and SF3a, either from a partially purified source or from nuclear extracts (Supplementary Figure 2B).
CHD1 is functionally linked to splicing in vitro
Given that CHD1 physically associates with spliceosomal components, primarily through the SF3a sub-complex of U2 snRNP, we next assessed whether this association has functional relevance with respect to splicing. To test this, HeLa nuclear extracts depleted of CHD1 using specific anti-CHD1 antibodies were used in splicing assays in vitro. Western blotting indicated that specific U2 snRNP subunits were indeed depleted along with CHD1 (Figure 4A), but the U5 snRNP subunit PRPF8 or the SWI/SNF subunit BAF170 were not. Importantly, splicing reactions performed in vitro using the depleted CHD1 extracts displayed significantly reduced activity compared to the control antibody depleted extracts (Figure 4B, 4C). Generation of the mature mRNA (indicated by an asterisk in Figure 4B) was reduced by 70–80% when analyzing within a time course (Figure 4B, compare lanes 3 and 6) or when titrating the control or CHD1-depleted extracts (Figure 4C). Addition of 200 ng of purified U2 snRNP to the CHD1-depleted extracts resulted in a 20–30% restoration of splicing activity (Figure 4D); in comparison, a similar amount of purified U2 snRNP results in roughly 50% restoration of splicing activity in extracts depleted for the 17S U2 snRNP-associated protein hPrp5p (Will et al., 2002). While significant, the restoration of splicing activity observed in Figure 4D is noticeably not up to the levels observed for depleting the U2 snRNP itself. Therefore, it is apparent that additional factors are also diminished upon immunodepleting with anti-CHD1 antibodies. Nevertheless, the partial restoration of splicing activity by addition of purified U2 snRNP corroborates the link between CHD1 and U2 snRNP established above and demonstrates that the subset of spliceosomal proteins associated with CHD1 is functional for pre-mRNA splicing.
Figure 4.
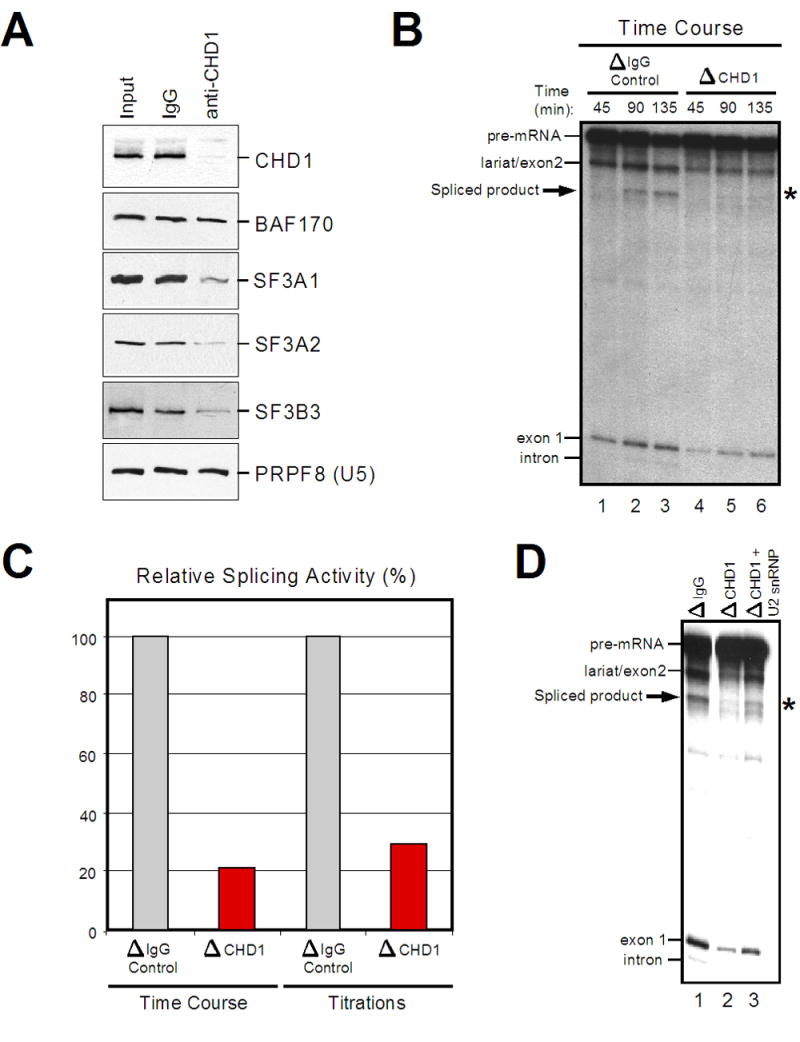
CHD1 is functionally linked to pre-mRNA splicing activity. A) Western analysis of control (IgG) or anti-CHD1 immunodepleted nuclear extracts. B) Time course in vitro splicing assays of β-globin using untreated or depleted extracts described in (A). The various splicing products are indicated. C) The relative splicing activity is shown as a percentage of control to CHD1 depleted extracts. D) In vitro splicing assays as performed in (B) using untreated, CHD1-depleted, or CHD1-depleted extracts supplemented with purified U2 snRNP.
Reduction of CHD1 or H3K4me3 levels alters the chromatin association of SF3a in vivo
Both H3K4me3-affinity purification and conventional chromatography have identified U2 snRNP components as CHD1/H3K4me3 associated proteins. We therefore next sought to determine if these interactions have significance in vivo. Toward this end, we monitored the chromatin-binding properties of SF3a subunits upon depletion of both CHD1 and H3K4me3 in HeLa cells by small interfering RNA-mediated (siRNA) knockdown (Figure 5A). H3K4me3 was specifically targeted using siRNA against ASH2, a subunit of H3K4 methyltransferase complexes that has been shown to specifically eliminate global levels of H3K4me3, but not H3K4me1 and H3K4me2 (Steward et al., 2006)(see Sup. Figure 3A). Sub-cellular fractionation of siRNA-treated HeLa cells revealed that depletion of H3K4me3 resulted in the partial loss of CHD1 from chromatin and a release of CHD1 into the soluble nuclear fraction (Figure 5B). These results are consistent with recent studies in Drosophila, which reported that changes of global H3K4me3 patterns induced altered localization of Chd1 (Eissenberg et al., 2007). A less dramatic effect was observed when examining the fractionation pattern of SF3a subunits upon H3K4me3 reduction, although depletion of CHD1 resulted in an increased release of SF3a components into the soluble nuclear fraction (Figure 5B).
Figure 5.
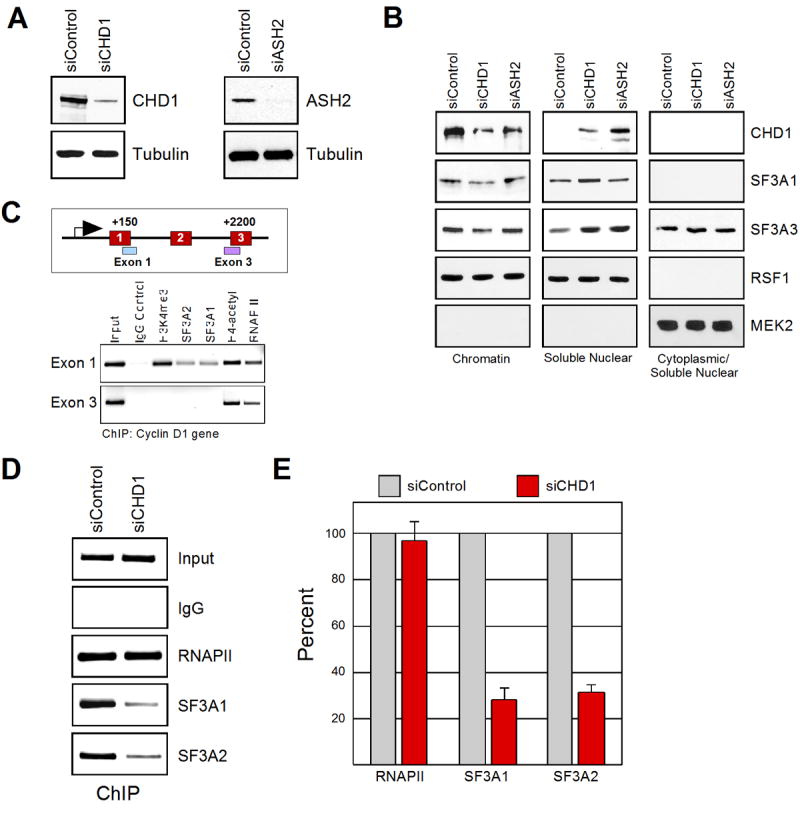
CHD1 and H3K4me3 modulate the association of SF3a on chromatin. A) Western blotting of whole cell extracts from control, CHD1, or ASH2 siRNA-treated cells (indicated on top). B) Sub-cellular fractionation from cells treated with the indicated siRNA (top). Western blotting was performed using the labeled antibodies (side). MEK2 is a cytoplasmic protein used as a control for the integrity of the fractionation. C) Chromatin immunoprecipitation on the cyclin D1 gene. A scheme of the cyclin D1 gene is indicated on top. The antibodies used for immunoprecipitation are labeled. D) Chromatin immunoprecipitations on the cyclin D1 gene derived from cells treated with control or CHD1 siRNAs. The antibodies used for immunoprecipitation are labeled (side). C) Graphical representation of three independent experiments as performed in (D). Results from three independent experiments are shown represented by the mean and standard error.
To examine if similar effects were evident on specific genes, we performed chromatin immunoprecipitation (ChIP) experiments following depletion of CHD1 by siRNA treatment. The SF3a subunits SF3A1 and SF3A2 were readily detectable at the 5′ region of the active cyclin D1 gene in HeLa cells (Figure 5C). Unfortunately, the two different antisera against CHD1 tested were unsuitable for ChIP analyses. H3K4me3 was undetectable using primers spanning the 3′ splice site of exon 3 (located 2200 bp downstream of the transcription initiation site), and consistent with the absence of H3K4me3, the SF3A1 or SF3A2 splicing factors were also absent, although RNAPII and acetyl-H4 were present at this region (Figure 5C). These results are consistent with the idea that components of U2 snRNP are enriched on H3K4me3. It is likely that U2 snRNP present downstream of H3K4me3 is either associated with the pre-mRNA at this stage, and/or our assay is not sensitive enough to allow its detection on chromatin. Next, we performed ChIP analyses for SF3a components on the cyclin D1 gene upon CHD1 depletion. Significantly, recruitment of SF3A1 and SF3A2 to the cyclin D1 gene was significantly impaired upon depletion of CHD1 in vivo (Figure 5D and 5E). Similar results were observed on the GAPDH gene (Supplementary Figure 3B). Taken together, these results provide evidence that CHD1 facilitates recruitment and/or placement of U2 snRNP components to active chromatin in vivo.
The rate of pre-mRNA splicing in vivo is compromised by perturbing CHD1 or H3K4me3
A reasonable prediction from the above results is that reducing the levels of the SF3a sub-complex on chromatin via CHD1 knockdown would alter the splicing efficiency of target transcripts. However, when we monitored the ratio of un-spliced to spliced mRNA transcripts by RT-PCR upon knockdown of CHD1, we did not detect any observable changes in splicing of cyclin D1 or GAPDH pre-mRNAs, or the total transcript levels (data not shown). Consistently, RNA immunoprecipitation experiments (Gilbert et al., 2004) revealed that comparable amounts of SF3A1 are associated with cyclin D1 and GAPDH transcripts derived from either control or HeLa cells targeted with CHD1 siRNA (Supplementary Figure 3C). Taking this into account, we hypothesized that CHD1/H3K4me3 is not necessary to determine the extent of splicing, which would be measured by analysis of steady-state RNA, but is required instead to enhance the rate of the reaction. To test this, we monitored pre-mRNA splicing of the rapidly inducible gene IRF1. Upon treatment of HeLa cells with IFN-γ, we observed induction of IRF1 transcription by real-time, quantitative PCR (qPCR). Primer sets were designed to detect the intron-containing pre-mRNA (un-spliced primer set), as well as the total mRNA (spliced primer set) that is produced upon IRF1 induction (Figure 6A). Both transcripts were detected when using the spliced primer set (Figure 6B). IRF1 mRNA levels were monitored at 0, 15, 30, and 60 minutes after the addition of IFN-γ (Supplementary Figure 4A). To monitor pre-mRNA splicing, we considered the ratio of un-spliced to total mRNA (splicing efficiency). Under normal conditions, the ratio of un-spliced to total mRNA was highest at the earliest time points after IRF1 induction (Supplementary Figure 4B), indicating that splicing is a slow step relative to transcription and increases over time during the generation of an mRNA. Next, we monitored the relative splicing efficiency of IRF1 in cells treated with CHD1 siRNA as compared to the control siRNA. Strikingly, at 15 minutes after the induction of IRF1, we observed an eight fold increase in the ratio of un-spliced to total mRNA in the cells treated with CHD1-targeted siRNA as compared to the control siRNA (Figure 6C). Transcription also appeared to be affected, as the total transcript level was reduced under these conditions (Figure 6B and data not shown). The effect on splicing was diminished by 30 minutes after induction of IRF1, and no effect was observed after 60 minutes of induction (Figure 6C).
Figure 6.
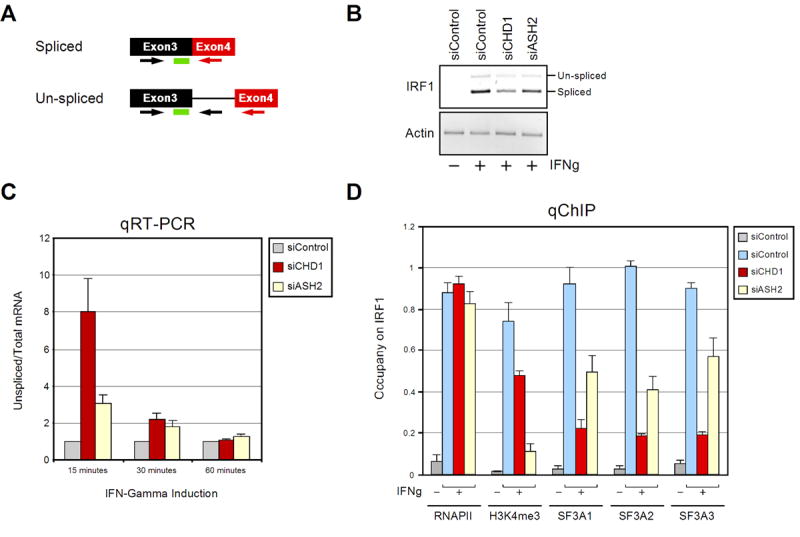
CHD1 and H3K4me3 modulate the efficiency of pre-mRNA splicing in vivo. A) Schematic representation of the probe and primer pairs used to detect the un-spliced pre-mRNA and total mRNA. Arrows indicate the primers and the green rectangle represents the Taqman probe used. B) Agarose gel of the IRF1 products generated from the total mRNA primer set 15–30 minutes after the induction of IFN-γ. The indicated siRNA treated HeLa cells are shown. C) Real-time RT-PCR analyses detecting the ratio of un-spliced to total mRNA of the IRF1 gene after the indicated time points following treatment with IFN-γ. The control siRNA treated cells were compared to cells treated with siRNA targeted against CHD1 and ASH2. The specific siRNA treated cells are indicated in the panel on the upper right side of the graph. Results from three independent experiments are shown represented as the mean and standard error. D) Real-time PCR analyses of the 5′ region of the IRF1 gene from ChIP-derived samples using the after 0 or 15 minutes of IFN-γ treatment (IFNg). The antibodies used for immunoprecipitation are shown on the bottom of the graph. The specific siRNA treated cells are indicated in the panel on the upper right side of the graph. Results from three independent experiments are shown represented as the mean and standard error.
To determine the contribution of H3K4me3 to pre-mRNA splicing, we performed similar experiments following depletion of ASH2 (Dou et al., 2006; Steward et al., 2006). A reduction in the efficiency of pre-mRNA splicing of IRF1 transcripts was also observed in the H3K4me3 knockdown cells when compared to the control siRNA treated cells (Figure 6C), although the decrease in splicing efficiency (~3-fold at 15 mins) was less when compared to cells that were targeted with CHD1 siRNA. As in the CHD1-depleted cells, the decrease in pre-mRNA splicing efficiency mediated by H3K4me3 appeared to diminish as the induction of IRF1 was allowed to proceed with time (Figure 6C). Importantly, similar effects mediated by CHD1 and ASH2 knockdown were observed on the IFN-γ inducible gene FRA1 (Supplementary Figure 5A).
To confirm that the in vivo alterations of splicing efficiency upon CHD1/H3K4me3 knockdown correlated with a reduction of SF3a subunits on the IRF1 gene, we monitored the occupancy of SF3a components early during IRF1 gene induction. The relative levels of SF3A1, SF3A2, and SF3A3 on the 5′ region of IRF1 were determined by quantitative ChIP upon knockdown of CHD1 and H3K4me3. As shown in Figure 6C, levels of the SF3a sub-complex were reduced on the IRF1 gene upon diminishing the levels of CHD1 and H3K4me3, as compared to the control siRNA-treated cells. Similar results were obtained when analyzing the FRA1 gene (Supplementary Figure 5B). Consistent with previously published reports, knockdown of ASH2 reduced the level of H3K4me3 present on the IRF1 and FRA1 genes (Figure 6C and Supplementary Figure 5B). Notably, no effect on RNAP II levels was observed. Thus, we observe a consistent correlation between reduced pre-mRNA splicing efficiency and a reduction of SF3a on active genes brought about by depletion of CHD1 or reduction of H3K4me3 levels.
Discussion
Despite considerable effort, our understanding of the mechanistic contribution of histone modifications during active transcription remains poor. Here, we provide biochemical and functional evidence that CHD1 functions to modulate the efficiency of pre-mRNA splicing in part through physical bridging of spliceosomal components to H3K4me3. Of the many steps involved in pre-mRNA processing, the recruitment of factors required for pre-mRNA splicing to nascent transcripts is perhaps the most poorly understood. In crude extracts, H3K4me3 has the ability to enrich components of the spliceosome, notably U2 snRNP and associated factors. Moreover, CHD1 can physically associate with multiple subunits of U2 snRNP in addition to the SF3a sub-complex. Considering that conventional chromatography identified a stable complex between CHD1 and SF3a, SF3a is likely required for U2 snRNP binding to H3K4me3. Currently, we are unaware what triggers the association of additional U2 snRNP components, besides the SF3a sub-complex, to CHD1. Perhaps CHD1 binding to the H3K4me3 mark itself facilitates the nucleation of larger spliceosomal complexes to the 5′ end of active genes. Overall, the observed effect on U2 snRNP chromatin association and the efficiency of pre-mRNA splicing is most dramatic when reducing the level of CHD1 protein, as compared to H3K4me3 levels. This could simply be due to the remaining H3K4me2 that is present in the absence of H3K4me3, as CHD1 binds both H3K4me2 and H3K4me3. CHD1 was also observed to affect the amount of H3K4me3 on active genes as well, perhaps indicating a combinatorial action of reducing both CHD1 and H3K4me3 levels at the same time. We are currently exploring these differing scenarios.
It is notable that the U1 snRNP and SR proteins, which act very early in spliceosome assembly, were largely absent from the H3K4me3 affinity and anti-CHD1 co-immunoprecipitation fractions. Thus alternative mechanisms for recruitment of spliceosomal components must exist. This is especially true given that pre-mRNA splicing in vivo fully recovers with time, even under reduced levels of CHD1 and H3K4me3 on active genes. As such, U1 snRNP and SR proteins associate, albeit perhaps indirectly, with the carboxy terminal domain of the largest subunit of RNAP II (for review see (Hirose and Manley, 2000)), indicating that splicing factors can be recruited in multiple fashions. It is also possible that the restored ratio of un-spliced to total transcripts observed in Figure 6C is a consequence of post-transcriptional splicing, or alternatively, via degradation of un-spliced pre-mRNAs by the mRNA surveillance pathway.
Mass spectrometry and western blot analyses of the H3K4me3 affinity purified material identified numerous proteins implicated in transcription post-initiation events, in addition to pre-mRNA splicing. Among the mixture of complexes affinity purified through H3K4me3, we identified a large protein complex consisting of CHD1, SNF2h, and the PAF complex. CHD1 was also demonstrated to bridge the FACT heterodimer to H3K4me3. The interactions between CHD1, SNF2h, PAF, and FACT described above are also supported by numerous previous findings, primarily in yeast (Alen et al., 2002; Gavin et al., 2002; Kelley et al., 1999; Krogan et al., 2002; Simic et al., 2003; Tsukiyama et al., 1999). Consistent with our results, human SNF2h precisely overlaps with H3K4me2/3 at active genes in vivo, as determined by quantitative chromatin immunoprecipitation analyses (Kouskouti and Talianidis, 2005). Knockdown of CHD1 by siRNA treatment resulted in the altered association of SNF2h, FACT and PAF on chromatin as determined by sub-cellular fractionation and ChIP (Supplementary Figure 3B and 3D). These collective results suggest that H3K4me3 may serve to position FACT, PAF, and SNF2h via their interactions with CHD1 near the 5′ region of active genes.
Of particular interest is FACT enrichment on H3K4me3. The functional role of FACT in transcript elongation on chromatin templates is well established (Sims et al., 2004). However, how FACT is properly placed near the first nucleosome after transcription initiation remained unclear. Our findings provide a possible mechanism for FACT recruitment and/or placement upon gene activation in human cells. The localization of H3K4me3 near the transcription start site is consistent with the required positioning of FACT to allow passage of the first nucleosomal barrier. Furthermore, ChIP studies in human and Drosophila cells identified the H3K4 tri-methyl mark co-localizing with FACT on active genes (Kouskouti and Talianidis, 2005; Saunders et al., 2003). Consistent with our findings, reduction of H3K4me3 on active genes via knock down of PAF subunits results in reduced recruitment of both FACT and the chromatin modulator Spt6, although RNAPII levels remained unaffected (Adelman et al., 2006). Interestingly, Spt6 was also identified by mass spectrometry in the H3K4me3 eluted material (Supplementary Table 1), although its relationship to CHD1 remains unknown at present.
In a fully reconstituted transcription system, the generation of H3K4me3 was observed to be coupled to transcription, but also dependent on H2B monoubiquitination, as previously described in yeast (Dover et al., 2002; Pavri et al., 2006; Sun and Allis, 2002). These results suggest that H3K4me3 must occur subsequent to transcription initiation and after FACT and the H2B monoubiquitination machinery has located the first nucleosome during the initial round of transcription. Thus, H3K4me3 may function to expedite FACT/PAF localization during subsequent rounds of transcription, similar to re-initiation, a process that is much faster than initial PIC formation (Orphanides and Reinberg, 2000). Given that H2B monoubiquitination is reversed during transcription (Henry et al., 2003), the more stable H3K4me3 mark would serve as a permanent recruitment platform for facilitating RNAPII passage through nucleosomes.
An interesting question is whether the function of histone methylation (and perhaps other histone modifications) during active transcription is to facilitate the timing, but not the absolute levels of gene expression. In this context, histone methylation on active genes would be critical during embryonic development of multi-cellular organisms as well as on genes that respond to stimuli. Perhaps this is why histone methyl “writers” and “readers” are so instrumental during mammalian development (Margueron et al., 2005). Moreover, tightly regulated gene regulatory networks may be particularly sensitive to the altered timing of gene expression, which may explain why histone methylation appears to play a prominent role in pleuripotency and oncogenesis.
H3K4me3, a histone tail modification associated with transcription activity, has now been shown to facilitate pre-mRNA maturation (Figure 7). This adds credence to the general belief that modifications within the histone tails are not passive, but function to recruit factors that regulate different steps of gene expression, some to maintain genes in a silent state, and others, like H3K4me3, to facilitate the production of a mature mRNA.
Figure 7.
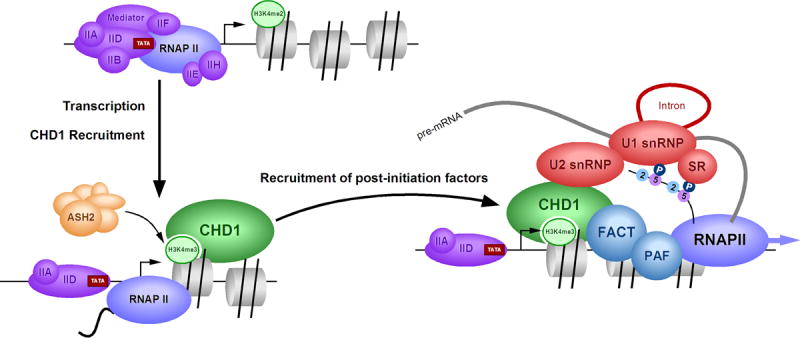
Model of H3K4me3 function during transcription. Upon transcriptional activation, H3K4me2 is converted to H3K4me3 by an ASH2 containing complex, which places CHD1 near the transcriptional start site. CHD1 then recruits U2 snRNP (via the SF3a sub-complex) and FACT/PAF near the first nucleosome in order to enhance the efficiency of pre-mRNA splicing and transcript elongation.
Experimental Procedures
Peptide affinity purification and protein identification
Peptide-affinity columns were generated using SulfoLink coupling gel (Pierce). The histone peptides were synthesized by Global Peptide Services. Bound proteins were washed extensively (60 column volumes of 25 mM Tris, pH 8, 150 mM NaCl, 2 mM EDTA, and 0.5% NP40) and eluted with either 0.5 mg/ml peptide or low pH buffer (100mM glycine, pH 3.0) from nuclear extracts or pellets. See Supplementary methods for details of protein identification by mass spectrometry and Supplementary Table 1 for specific details of the peptides identified.
Co-immunoprecipitation, western blotting, and antibodies
Co-immunoprecipitation assays were conducted using 0.5–2 ug IgG per reaction in RIPA buffer (150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 50 mM Tris, pH 8, 0.1% SDS) using nuclear extracts, the H3K4me3 elution material, or recombinant CHD1 mixed with in vitro translated SF3a subunits. Western blotting was performed using standard methods. Recombinant CHD1 was purified from insect cells using NTA resin (Qiagen). SF3A1, SF3A2, and SF3A3 were in vitro translated using the 35S-TNT Quick Coupled Transcription/Translation System from Promega. Antibodies used for western and ChIP analyses are listed in the Supplementary methods. Silver staining and autoradiography were performed using standard molecular biology techniques.
Conventional chromatography of CHD1/SF3a complexes
CHD1, SF3a, and SF3b complexes were purified by Western blotting from 3 grams of HeLa nuclear extracts, initially by fractionation over P11 resin (Whatman). The P11 resin was step eluted using 0.3M, 0.5M, and 1M KCl in BC buffer (20mM Tris pH 7.9, 20% glycerol, 0.5mM DTT, 0.2 mM EDTA). The 1M-P11 fraction was further purified using DEAE-52 resin (Whatman), which was step eluted using 0.5M KCl in BC buffer (BC500). The DE52 bound material (0.5M step) was then loaded onto an HPLC DEAE-5PW column (8 × 7.5 mm; Tosoh) equilibrated with BC100. CHD1, SF3a, and SF3b were eluted with a linear gradient of 0.1 to 0.8M KCl in BC buffer. Co-eluting fractions were pooled and run on a 24 ml Superose 6 10/300 gel filtration column (Amersham) equilibrated in 300 mM KCl in BC buffer. Overlapping fractions were pooled and run over a 2.4 ml Superose 6 PC 3.2/30 gel filtration column (Amerhsam). Calibration of all gel filtration chromatography was performed using known molecular weight standards (Sigma).
RNA analyses and in vitro splicing
Extracts (1–10 μl) were digested with proteinase K, and RNA was extracted with phenol/chloroform (1:1) and precipitated by adding 0.25 volumes 10M ammonium acetate, and 2.5 volumes ethanol. RNA was labeled with [5′-32P]pCp (Amersham Biosciences) using T4 RNA ligase (New England Biolabs) in reaction buffer supplemented with 10% DMSO. RNA was collected as before and separated on a 6% acrylamide 7M urea denaturing gel and visualized/quantified on a Molecular Dynamics phosphorimager. Pre-mRNA substrates were prepared as described previously (Tacke et al., 1998). In vitro splicing reactions were performed in 12.5-μl reaction mixtures, which contained 2– 6-μl nuclear extract and 1–2 ng labeled β-globin substrate. A more detailed protocol is provided in the Supplementary methods. ΔIgG, ΔCHD1, and untreated nuclear extracts contain near equal total protein concentration as determined by protein assays (BioRAD). Reactions supplemented with U2 snRNP contain 200 ng immunopurified 17S U2 snRNP (Will et al., 2002). Quantitation of gel images were performed using ImageJ software.
Immunodepletion, chromatin immunoprecipitation and siRNA treatment
Immunodepletions were performed essentially as described (Das et al., 2000), although the salt concentration was ~400 mM during the depletion. Chromatin immunoprecipitation assays were performed as described (Lewis et al., 2005). Three independent ChIP experiments were performed, imaged (Fotodyne), and quantitated using ImageJ software. Quantitative ChIPs were performed as above, although real-time PCR was performed at the final step (see below). siRNA transfections of HeLa cells were performed with the HiPerfect transfection reagent according to the manufactures instructions (Qiagen). Control, CHD1, or ASH2 siRNA’s were obtained from Ambion.
Quantitative RT-PCR and ChIP
Quantitative RT-PCR was carried out using an Mx3005P Real-Time PCR System (Stratagene). Taqman probes were ordered from Integrated DNA Technologies. The primers and Taqman probes for RT-PCR are the following. IRF1: 5′: CTGCCAGATATCGAGGAGGTGAAA; 3′-intron: GACCCAGAGTCCTTGGATACCTTT; 3′-exon: TCTTGGCCTTGCTCTTAGCAT-CTC; Probe: AGCTGTGCGAGTGTACCGGATGCTT. FRA1: 5′: TGGATGGTACAGCC-TCATTTCCTG; 3′ exon: TGTTCACAAGGCCTTCGACGTA; 3′ intron: CAATGCCTCTGT-GCCTTACCTGTT; Probe: AGGCCTCTGACCTACCCTCAGTACAGC. The primers and Taqman probes for ChIP are the following. IRF1: 5′: AGGGAATCGTGACCTACGGAGACT; 3′: CCTGAATCCATTCTACGCCTTCCT; Probe: AAGGGTTAGCGTCCTGGTCTTAGCG-TTGT. FRA1: 5′: AGTGGTTCAGCCCGAGAACTTT; 3′: CCGAAGTCTCGGAACATGCC; Probe: TGAGTCAGAACCCAGCAGCCGTGTA. For RT-PCR, the raw data were normalized to actin: 5′: CTCCTCCTGAGCGCAAGTACTC; 3′: TCGTCATACTCCTGCTTGCTGAT; Probe: CCATCCTGGCCTCGCTGTCCA. For ChIP, the raw data were normalized to input DNA.
Supplementary Material
Acknowledgments
We would like to thank Drs. C. Will and R. Luhrmann for U1 and U2 snRNPs and the anti-U5 116kD antibody, R.P. Perry and D. Stokes for CHD1 antibodies, A. Kramer for SF3a antibodies and SF3a plasmids for in vitro translation (Nesic and Kramer, 2001), S. C. Lee for SSRP1 antibodies, W. van Venrooij for U1-70K antibodies, R. Reed for the U2-B″ antibody, P. Trojer, T. Huang, A. Sims, B. Zhu and Y. Shi for helpful assistance, and R.P. Perry and L. Vales for critical reading of the manuscript. These studies were supported by the HHMI (to DR), and grants from NIH (GM-37120 to DR, GM-71166 to RJS, K01 DK60001 to BAL, and GM-37971 to JLM), and NCI Cancer Center (P30 CA08748 to PT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adelman K, Wei W, Ardehali MB, Werner J, Zhu B, Reinberg D, Lis JT. Drosophila paf1 modulates chromatin structure at actively transcribed genes. Mol Cell Biol. 2006;26:250–260. doi: 10.1128/MCB.26.1.250-260.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alen C, Kent NA, Jones HS, O’Sullivan J, Aranda A, Proudfoot NJ. A role for chromatin remodeling in transcriptional termination by RNA polymerase II. Mol Cell. 2002;10:1441–1452. doi: 10.1016/s1097-2765(02)00778-5. [DOI] [PubMed] [Google Scholar]
- Ayton PM, Cleary ML. Molecular mechanisms of leukemogenesis mediated by MLL fusion proteins. Oncogene. 2001;20:5695–5707. doi: 10.1038/sj.onc.1204639. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, 3rd, Gingeras TR, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Burckin T, Nagel R, Mandel-Gutfreund Y, Shiue L, Clark TA, Chong JL, Chang TH, Squazzo S, Hartzog G, Ares M. Exploring functional relationships between components of the gene expression machinery. Nat Struct Mol Biol. 2005;12:175–182. doi: 10.1038/nsmb891. [DOI] [PubMed] [Google Scholar]
- Das R, Zhou Z, Reed R. Functional association of U2 snRNP with the ATP-independent spliceosomal complex E. Mol Cell. 2000;5:779–787. doi: 10.1016/s1097-2765(00)80318-4. [DOI] [PubMed] [Google Scholar]
- Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- Dover J, Schneider J, Tawiah-Boateng MA, Wood A, Dean K, Johnston M, Shilatifard A. Methylation of histone H3 by COMPASS requires ubiquitination of histone H2B by Rad6. J Biol Chem. 2002;277:28368–28371. doi: 10.1074/jbc.C200348200. [DOI] [PubMed] [Google Scholar]
- Eberharter A, Becker PB. ATP-dependent nucleosome remodelling: factors and functions. J Cell Sci. 2004;117:3707–3711. doi: 10.1242/jcs.01175. [DOI] [PubMed] [Google Scholar]
- Eissenberg JC, Lee MG, Schneider J, Ilvarsonn A, Shiekhattar R, Shilatifard A. The trithorax-group gene in Drosophila little imaginal discs encodes a trimethylated histone H3 Lys4 demethylase. Nat Struct Mol Biol. 2007;14:344–346. doi: 10.1038/nsmb1217. [DOI] [PubMed] [Google Scholar]
- Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim Y, Minor W, Rastinejad F, Khorasanizadeh S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- Gavin AC, Bosche M, Krause R, Grandi P, Marzioch M, Bauer A, Schultz J, Rick JM, Michon AM, Cruciat CM, et al. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002;415:141–147. doi: 10.1038/415141a. [DOI] [PubMed] [Google Scholar]
- Gilbert C, Kristjuhan A, Winkler GS, Svejstrup JQ. Elongator interactions with nascent mRNA revealed by RNA immunoprecipitation. Mol Cell. 2004;14:457–464. doi: 10.1016/s1097-2765(04)00239-4. [DOI] [PubMed] [Google Scholar]
- Hamamoto R, Furukawa Y, Morita M, Iimura Y, Silva FP, Li M, Yagyu R, Nakamura Y. SMYD3 encodes a novel histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol. 2004 doi: 10.1038/ncb1151. [DOI] [PubMed] [Google Scholar]
- Henry KW, Wyce A, Lo WS, Duggan LJ, Emre NC, Kao CF, Pillus L, Shilatifard A, Osley MA, Berger SL. Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA-associated Ubp8. Genes Dev. 2003;17:2648–2663. doi: 10.1101/gad.1144003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirose Y, Manley JL. RNA polymerase II and the integration of nuclear events. Genes Dev. 2000;14:1415–1429. [PubMed] [Google Scholar]
- Huang Y, Steitz JA. SRprises along a messenger’s journey. Mol Cell. 2005;17:613–615. doi: 10.1016/j.molcel.2005.02.020. [DOI] [PubMed] [Google Scholar]
- Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell. 2007;128:1077–1088. doi: 10.1016/j.cell.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Jurica MS, Moore MJ. Pre-mRNA splicing: awash in a sea of proteins. Mol Cell. 2003;12:5–14. doi: 10.1016/s1097-2765(03)00270-3. [DOI] [PubMed] [Google Scholar]
- Kelley DE, Stokes DG, Perry RP. CHD1 interacts with SSRP1 and depends on both its chromodomain and its ATPase/helicase-like domain for proper association with chromatin. Chromosoma. 1999;108:10–25. doi: 10.1007/s004120050347. [DOI] [PubMed] [Google Scholar]
- Kouskouti A, Talianidis I. Histone modifications defining active genes persist after transcriptional and mitotic inactivation. Embo J. 2005;24:347–357. doi: 10.1038/sj.emboj.7600516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Kim M, Ahn SH, Zhong G, Kobor MS, Cagney G, Emili A, Shilatifard A, Buratowski S, Greenblatt JF. RNA polymerase II elongation factors of Saccharomyces cerevisiae: a targeted proteomics approach. Mol Cell Biol. 2002;22:6979–6992. doi: 10.1128/MCB.22.20.6979-6992.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BA, Sims RJ, 3rd, Lane WS, Reinberg D. Functional characterization of core promoter elements: DPE-specific transcription requires the protein kinase CK2 and the PC4 coactivator. Mol Cell. 2005;18:471–481. doi: 10.1016/j.molcel.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Listerman I, Sapra AK, Neugebauer KM. Cotranscriptional coupling of splicing factor recruitment and precursor messenger RNA splicing in mammalian cells. Nat Struct Mol Biol. 2006;13:815–822. doi: 10.1038/nsmb1135. [DOI] [PubMed] [Google Scholar]
- Maniatis T, Reed R. An extensive network of coupling among gene expression machines. Nature. 2002;416:499–506. doi: 10.1038/416499a. [DOI] [PubMed] [Google Scholar]
- Margueron R, Trojer P, Reinberg D. The key to development: interpreting the histone code? Curr Opin Genet Dev. 2005;15:163–176. doi: 10.1016/j.gde.2005.01.005. [DOI] [PubMed] [Google Scholar]
- Michaud S, Reed R. An ATP-independent complex commits pre-mRNA to the mammalian spliceosome assembly pathway. Genes Dev. 1991;5:2534–2546. doi: 10.1101/gad.5.12b.2534. [DOI] [PubMed] [Google Scholar]
- Nesic D, Kramer A. Domains in human splicing factors SF3a60 and SF3a66 required for binding to SF3a120, assembly of the 17S U2 snRNP, and prespliceosome formation. Mol Cell Biol. 2001;21:6406–6417. doi: 10.1128/MCB.21.19.6406-6417.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishioka K, Chuikov S, Sarma K, Erdjument-Bromage H, Allis CD, Tempst P, Reinberg D. Set9, a novel histone H3 methyltransferase that facilitates transcription by precluding histone tail modifications required for heterochromatin formation. Genes Dev. 2002;16:479–489. doi: 10.1101/gad.967202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanides G, Reinberg D. RNA polymerase II elongation through chromatin. Nature. 2000;407:471–475. doi: 10.1038/35035000. [DOI] [PubMed] [Google Scholar]
- Orphanides G, Reinberg D. A unified theory of gene expression. Cell. 2002;108:439–451. doi: 10.1016/s0092-8674(02)00655-4. [DOI] [PubMed] [Google Scholar]
- Pavri R, Zhu B, Li G, Trojer P, Mandal S, Shilatifard A, Reinberg D. Histone H2B Monoubiquitination Functions Cooperatively with FACT to Regulate Elongation by RNA Polymerase II. Cell. 2006;125:703–717. doi: 10.1016/j.cell.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Proudfoot NJ, Furger A, Dye MJ. Integrating mRNA processing with transcription. Cell. 2002;108:501–512. doi: 10.1016/s0092-8674(02)00617-7. [DOI] [PubMed] [Google Scholar]
- Reinberg D, Sims RJ., 3rd de FACTo nucleosome dynamics. J Biol Chem. 2006;281:23297–23301. doi: 10.1074/jbc.R600007200. [DOI] [PubMed] [Google Scholar]
- Rozenblatt-Rosen O, Hughes CM, Nannepaga SJ, Shanmugam KS, Copeland TD, Guszczynski T, Resau JH, Meyerson M. The parafibromin tumor suppressor protein is part of a human Paf1 complex. Mol Cell Biol. 2005;25:612–620. doi: 10.1128/MCB.25.2.612-620.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bernstein BE, Karabetsou N, Morillon A, Weise C, Schreiber SL, Mellor J, Kouzarides T. Methylation of histone H3 K4 mediates association of the Isw1p ATPase with chromatin. Mol Cell. 2003;12:1325–1332. doi: 10.1016/s1097-2765(03)00438-6. [DOI] [PubMed] [Google Scholar]
- Saunders A, Werner J, Andrulis ED, Nakayama T, Hirose S, Reinberg D, Lis JT. Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science. 2003;301:1094–1096. doi: 10.1126/science.1085712. [DOI] [PubMed] [Google Scholar]
- Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6:73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- Seligson DB, Horvath S, Shi T, Yu H, Tze S, Grunstein M, Kurdistani SK. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–1266. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- Simic R, Lindstrom DL, Tran HG, Roinick KL, Costa PJ, Johnson AD, Hartzog GA, Arndt KM. Chromatin remodeling protein Chd1 interacts with transcription elongation factors and localizes to transcribed genes. Embo J. 2003;22:1846–1856. doi: 10.1093/emboj/cdg179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims RJ, 3rd, Belotserkovskaya R, Reinberg D. Elongation by RNA polymerase II: the short and long of it. Genes Dev. 2004;18:2437–2468. doi: 10.1101/gad.1235904. [DOI] [PubMed] [Google Scholar]
- Sims RJ, 3rd, Chen CF, Santos-Rosa H, Kouzarides T, Patel SS, Reinberg D. Human but not yeast CHD1 binds directly and selectively to histone H3 methylated at lysine 4 via its tandem chromodomains. J Biol Chem. 2005 doi: 10.1074/jbc.C500395200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward MM, Lee JS, O’Donovan A, Wyatt M, Bernstein BE, Shilatifard A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol. 2006;13:852–854. doi: 10.1038/nsmb1131. [DOI] [PubMed] [Google Scholar]
- Sun ZW, Allis CD. Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature. 2002;418:104–108. doi: 10.1038/nature00883. [DOI] [PubMed] [Google Scholar]
- Tacke R, Tohyama M, Ogawa S, Manley JL. Human Tra2 proteins are sequence-specific activators of pre-mRNA splicing. Cell. 1998;93:139–148. doi: 10.1016/s0092-8674(00)81153-8. [DOI] [PubMed] [Google Scholar]
- Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F, Li J, Rao A, Shi Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447:601–605. doi: 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- Taverna SD, Ilin S, Rogers RS, Tanny JC, Lavender H, Li H, Baker L, Boyle J, Blair LP, Chait BT, et al. Yng1 PHD finger binding to H3 trimethylated at K4 promotes NuA3 HAT activity at K14 of H3 and transcription at a subset of targeted ORFs. Mol Cell. 2006;24:785–796. doi: 10.1016/j.molcel.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukiyama T, Palmer J, Landel CC, Shiloach J, Wu C. Characterization of the imitation switch subfamily of ATP-dependent chromatin-remodeling factors in Saccharomyces cerevisiae. Genes Dev. 1999;13:686–697. doi: 10.1101/gad.13.6.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will CL, Urlaub H, Achsel T, Gentzel M, Wilm M, Luhrmann R. Characterization of novel SF3b and 17S U2 snRNP proteins, including a human Prp5p homologue and an SF3b DEAD-box protein. Embo J. 2002;21:4978–4988. doi: 10.1093/emboj/cdf480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegerman P, Canas B, Pappin D, Kouzarides T. Histone H3 lysine 4 methylation disrupts binding of nucleosome remodeling and deacetylase (NuRD) repressor complex. J Biol Chem. 2002;277:11621–11624. doi: 10.1074/jbc.C200045200. [DOI] [PubMed] [Google Scholar]
- Zhu B, Mandal SS, Pham AD, Zheng Y, Erdjument-Bromage H, Batra SK, Tempst P, Reinberg D. The human PAF complex coordinates transcription with events downstream of RNA synthesis. Genes Dev. 2005;19:1668–1673. doi: 10.1101/gad.1292105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.