

Abstract
Macrophages serve as a major reservoir for HIV-1 because a large number of macrophages in the brain and lung are infected with HIV-1 during late stage disease. Recent evidence suggests that those HIV-1-infected macrophages play a key role in contributing to tissue damage in AIDS pathogenesis. Macrophages undergo apoptosis upon HIV-1 infection; however, the mechanisms of this process are not well-defined. Previously, we demonstrated that HIV-1 infection inhibits Akt-1, a critical protein for cell survival of macrophages. In the present study, we investigated the involvement of transcription factor FOXO3a in the regulation of HIV-1-mediated apoptosis in macrophages. HIV-1 infection significantly decreased phosphorylation of FOXO3a and promoted FOXO3a translocation to the nucleus in human monocyte-derived macrophages (MDM). Overexpression of a constitutively active FOXO3a increased DNA fragmentation with decreased cell viability in MDM, whereas a dominant-negative mutant of FOXO3a or siRNA for FOXO3a to knockdown the function of FOXO3a in HIV1-infected MDM decreased DNA fragmentation and protected macrophages from death in HIV-1-infected MDM. Overexpression of constitutively active Akt-1 increased FOXO3a phosphorylation, suggesting that FOXO3a phosphorylation in human MDM is dependent on Akt-1. We therefore conclude that FOXO3a plays an important role in HIV-1-induced cell death of human macrophage. Understanding the PI3K/Akt-1/FOXO3a pathway and its associated death mechanism in macrophages during HIV-1 infection would lead to identification of potential therapeutic avenues for the treatment of HIV-1 infection.
Macrophages reside in a variety of tissues including the liver, spleen, lymph nodes, lung, and brain, and link the innate immune system and the adaptive immune system. Macrophages are among the first cells infected by HIV (HIV-1) and act as a viral reservoir. Macrophage-tropic (CCR5-tropic) strains of HIV-1 are the predominant strains for viral transmission in vivo, but later in disease T cell-tropic (X4-tropic) strains evolve (1, 2). Macrophages are also important sources of proinflammatory cytokines that affect cell function and viral replication. Macrophages have been demonstrated to be critical to HIV-1 disease progression (3-5).
Pathogen-infected or -activated macrophages are targeted by the immune system for elimination by apoptosis. Macrophage apoptosis has been observed in various diseases and contribute to pathogenesis in pathological conditions. For example, in advanced atherosclerotic lesions, type A scavenger receptor activation triggered macrophage apoptosis and led to plaque necrosis. Bacterial and viral infections also induce macrophage cell death (6-9). It has been reported that macrophage cell death contributes to atherosclerotic plaque formation and stability in atherogenesis (10, 11). Ebola virus infection causing massive macrophage and lymphocyte death also contributes to Ebola pathogenesis (12). HIV-1-infected macrophages are found in tissues including the brain and lung (13-15). Various host factors promote cell death of infected cells during the viral infection process (16, 17). The removal and elimination of activated macrophages is considered to be important to the prevention of chronic inflammation and promoting tissue repair. Further elimination of infected host cells particularly may control viral dissemination. Understanding the mechanism of HIV-1-induced macrophage cell death is crucial to the treatment of AIDS- and HIV-1-associated diseases.
FOXO3a, a member of the forkhead family of transcription factors, is the predominant FOXO member expressed in peripheral lymphoid organs and plays a pivotal role in regulating cellular differentiation, proliferation, and apoptosis in the immune system (18). Gene knockout of FOXO3a revealed spontaneous lymphoproliferation associated with inflammation of several organs in mice, suggesting FOXO3a regulates homeostasis of immune cells (19, 20). However, the detailed regulation and function of FOXO3a in peripheral immune cells is still not clear. In mammals, FOXO3a contains a highly conserved phosphorylation motif that is posttranslationally modified by phosphorylation, acetylation, and the nuclear transport machinery (21, 22). The most common upstream influence of FOXO3a is the PI3K/Akt-1 pathway. In the absence of insulin or growth factors, FOXO3a is activated and localized to the nucleus as a consequence of Akt-1 deactivation. This activation and nuclear localization of FOXO3a regulates a series of key target genes, causing cell cycle arrest, stress resistance, or apoptosis (23).
Our recent work demonstrated HIV-1 infection significantly down-regulates the Akt-1 activity in human macrophages (17). In this report, we investigated FOXO3a, a potential downstream factor of Akt-1, in HIV-1-mediated macrophage death. Our data found HIV-1 infection decreased phosphorylation of FOXO3a and promoted nuclear translocation in infected monocyte-derived macrophages (MDM).5 Azidothymidine (AZT), an inhibitor for HIV-1 reverse transcriptase, blocked these effects. Overexpression of constitutively active FOXO3a decreased cell viability and increased DNA fragmentation in MDM. Moreover, both a dominant-negative (DN) mutant of FOXO3a and FOXO3a siRNA knockdown partially reduced cell death in HIV-infected MDM. These studies provide insight into the apoptotic signaling events of HIV-1-induced cell death and identify FOXO3a as an important apoptotic factor in HIV-1-infected macrophages.
Materials and Methods
Isolation and culturing of primary human monocytes
Human monocytes were recovered from PBMC of HIV-1, HIV-2, and hepatitis B virus-seronegative donors after leukophoresis and purified by counter-current centrifugal elutriation (24). Monocytes were cultured in DMEM (Sigma-Aldrich) supplemented with 10% heat-inactivated human serum, gentamicin (50 μg/ml), ciprofloxacin (10 μg/ml), and M-CSF (1000 U/ml, a gift from Genetics Institute). Monocytes were allowed to differentiate for 7 days at which time they were considered MDM. All human subject studies were performed in full compliance with the University of Nebraska Medical Center and National Institutes of Health ethical guidelines.
HIV-1 infection of MDM
Seven days after plating, MDM were infected with HIV-1 virus at a multiplicity of infection (MOI) of 0.1 virus/target cell (24). Briefly, viral stocks were diluted 1/15 in medium for overnight incubation with MDM. On the second day, medium was removed and substituted with MDM culture medium that was half-exchanged every 2 days (24). Stock virus was screened for mycoplasma and endotoxin using hybridization and Limulus amebocyte lysate assays, respectively.
Measurements of reverse transcriptase (RT) activity
RT activity was determined in triplicate samples of cell culture fluids. For this assay, 10 μl of supernatant was incubated in a reaction mixture of 0.05% Nonidet P-40, 10 μg of poly(A)/ml, 0.25 μg of oligo(dT)/ml, 5 mM DTT, 150 mM KCl, 15 mM MgCl2, and [3H]TTP in Tris-HCl buffer (pH 7.9) for 24 h at 37°C. Radiolabeled nucleotides were precipitated with cold 10% trichloroacetic acid on paper filters in an automatic cell harvester and washed with 95% ethanol. Radioactivity was determined by liquid scintillation spectroscopy (13).
Immunofluorescence and confocal microscopy
Cells on coverslips were cultured 3 days following HIV-1ADA infection, fixed for 15 min in 4% paraformaldehyde in PBS, dried in methanol for 5 min, permeabilized, and blocked 30-60 min with 0.1% Triton X-100 with 2% BSA in PBS. Cells were incubated overnight with mouse anti-P24 mAb (DakoCytomation) and rabbit anti-FOXO3a Ab (Cell Signaling Technology), then conjugated with anti-mouse Alexa Fluor 488 nm (green; Invitrogen Life Technologies) and anti-rabbit Alexa Fluor 647 nm (red; Invitrogen Life Technologies) secondary Abs. Hoechst 33342 (Sigma-Aldrich) was used for nuclear staining. Triple immunostaining was examined by a Bio-Rad MRC1024ES laser scanning confocal microscope, using a triple laser line and simultaneous triple display mode of the Bio-Rad LaserSharp imaging program.
Western blot analysis
Cell lysates from MDM were prepared with M-PER Mammalian Protein Extraction Buffer (Pierce). Nuclear and cytoplasmic fractions were prepared from MDM with the Nucleus Protein Extraction kit according to the manufacturer’s instruction (Pierce). Protein concentration was determined by a BCA Protein Assay kit (Pierce). Protein (30 μg) was electrophoresed on 10% SDS-PAGE (Bio-Rad) and transferred to an Immune-Blot polyvinylidene difluoride membrane (Bio-Rad). Total FOXO3a, phospho-FOXO3a, Puma, Akt-1, β-actin, and TATA box binding protein (TBP) proteins were detected using anti-FOXO3a (Cell Signaling Technology), anti-phospho-FOXO3a at Ser318/321 (Cell Signaling Technology), anti-Puma (Cell Signaling Technology), anti-Akt-1 (Cell Signaling Technology), anti-β-actin (Sigma-Aldrich), and anti-TBP (Abcam) Abs. Membranes were treated overnight with primary Ab at 4°C followed by HRP-ligand anti-rabbit (Cell Signaling Technology) or anti-mouse (Cell Signaling Technology) secondary Ab for 1 hat room temperature. Ag-Ab complexes were visualized by ECL (Amersham Biosciences) and captured with CL-XPosure Film (Pierce). For data quantification, the films were scanned with a CanoScan 9950F scanner; the acquired images were then analyzed on a Macintosh computer using the public domain NIH image program (developed at the U.S. National Institutes of Health and available on the internet at http://rsb.info.nih.gov/nih-image/).
Adenoviral infection
Replication-defective adenoviral vectors expressing DN FOXO3a (Ad-DN-O3a) and constitutively active FOXO3a (Ad-AAA-O3a) were purchased from Vector Biolabs. The constitutively active FOXO3a was prepared by substituting alanine for amino acids threonine 32, serine 253, and serine 315. Both Ad-DN-O3a and Ad-AAA-O3a have a hemagglutinin tag at the N terminus and express GFP. Constitutively active Akt-1 (Ad-myrAkt-1) was generated as described (25) and provided by K. Walsh (Boston University School of Medicine, Boston, MA). The constitutively active Akt-1 construct has the c-src myristoylation sequence fused in-frame to the N terminus of the HA-Akt-1 (wild-type) coding sequence. Adenoviral constructs were amplified in HEK 293 cells and purified by ultracentrifugation through a CsCl gradient. MDM were infected with recombinant adenovirus at ∼90% confluency in serum-free DMEM for 2 h and then incubated for 48 h in a growth medium. Infection efficiency was close to 70% as determined by the GFP expression. For adenovirus and HIV-1 double infection, MDM were infected with HIV-1 for 24 h followed by infection with adenovirus. Apoptosis assays were performed 3 days after adenovirus infection.
siRNA transfection
Predesigned siRNA duplexes targeting FOXO3a (ID: 144672) and a control siRNA (catalog no. D-001206-01-05) were synthesized by Ambion and Dharmacon, respectively. Control siRNA is the number one siCONTROL nontargeting siRNA from Dharmacon that has been tested to have minimal off-target effects. At 24 h after HIV-1 infection, cells were transfected with 100 nM siRNA duplexes for 4 h in DMEM culture medium without serum, in the presence of siIMPORTER reagent (Upstate Cell Signaling Solutions) according to the manufacturer’s instructions. To evaluate transfection efficiency, cells were transfected with siGLO (red fluorescence tagged nonspecific siRNA; Qiagen). At 48 h posttransfection, cells were incubated with Hoechst 33342 (Sigma-Aldrich) for nuclear staining. Transfection efficiency was measured by counting siGLO-positive cells and total cells in fluorescence microscopy.
Assessment of cell viability and apoptosis
To assess apoptosis, MDM were plated in 24-well plates with 1.1 × 106 cells/well. After 48 h of adenoviral infection with or without HIV-1 infection, cells were lysed and assayed by Cell Death Detection ELISA (Roche), as previously described (26). After 72 h of adenoviral infection, a colorimetric MTT assay was performed to measure the cell viability as previously described (26).
FACS analysis
After 3 days of adenoviral infection, MDM were dissociated with Accutase (Chemicon International) for 15 min. Cells were then washed with 3% FBS/PBS and incubated with 1 μg/ml propidium iodide (PI; catalog no. 537059; Calbiochem) for 15 min at 4°C. Cells were washed twice with 3% FBS/PBS and resuspended in PBS containing 4% paraformaldehyde. Cell death was determined by flow cytometry and analyzed with CellQuest software. At least 10,000 cells were analyzed per sample.
Statistical tests
Data were analyzed as means ± SD of the mean (SD) unless specified. The data were evaluated statistically by the ANOVA, followed by the Tukey test for paired observations. Significance was considered to be <0.05 unless specified. To account for any donor-specific differences, all experiments were performed with at least three donors. All assays were performed at least three times with triplicate or quadruplicate determinations for each time.
Results
HIV-1 infection significantly decreased phosphorylation of FOXO3a in human macrophages
Phosphorylation regulates the function of FOXO3a. When in an unphosphorylated form, FOXO3a resides inside the nucleus. Phosphorylation of Foxo3a in the forkhead domain leads to disruption of DNA binding, cytosol exportation, abrogating its transcriptional activity (27-29). To assess the regulation of FOXO3a in macrophages during HIV-1 infection, we used three macrophage-tropic HIV-1 strains (HIV-1ADA, HIV-1JRFL, and HIV-1BAL) to infect cultured human MDM. The phosphorylated FOXO3a, total FOXO3a, and β-actin were determined through Western blotting (Fig. 1, A and B). After densimetric quantification of the Western blotting, we found HIV-1ADA, HIV-1JRFL, and HIV-1BAL infection decreased phosphorylation of FOXO3a to 28.7, 34, and 45.7% of control MDM, respectively (Fig. 1), when normalized with total FOXO3a. HIV-1 infection did not change total FOXO3a and β-actin levels. AZT, a reverse transcriptase analog, was used to inhibit HIV-1 replication. AZT treatment blocked the observed decrease of FOXO3a phosphorylation (Fig. 1). Infection levels of MDM by HIV-1 were monitored by RT activity assay to confirm infection of MDM by all the viral strains tested (data not shown). To assess whether the inhibition of FOXO3a phosphorylation is in transcription level, we examined the mRNA expression of FOXO3a after HIV-1 infection. There were no significant changes in the mRNA levels of FOXO3a following HIV-1 infection (data not shown), suggesting the decrease of FOXO3a phosphorylation occurred posttranscriptionally. Among all HIV-1 strains tested, HIV-1ADA represented typical HIV-1 infection and was used thereafter.
FIGURE 1.

Phosphorylation of FOXO3a decreases in different HIV-1 strains infection of human MDM. A, Human MDM were infected by HIV-1ADA, HIV-1JRFL, HIV-1BAL strains. Cell lysates were collected 5 days after infection and subjected to Western blotting for p-FOXO3a (phosphorylated FOXO3a) and FOXO3a (total FOXO3a). Control denotes uninfected cells. β-actin was used as a loading control. B, Viral strain infections were treated with AZT (5 μM). C and D, Results of A and B were densimetrically quantified as a ratio of p-FOXO3a to FOXO3a and presented as percentage of control in C and D, respectively. Average ± SD of three independent experiments from three donors were shown. **, p < 0.01 compared with control.
Next, we examined phosphorylation of FOXO3a over time during HIV-1 infection. Phosphorylated FOXO3a decreased in a time-dependent manner following HIV-1 infection (Fig. 2A). Densimetric quantification determined that phosphorylation of FOXO3a was reduced to 60% of control MDM 3 days after infection and to 23% 5 days after infection (Fig. 2B). Furthermore, we infected MDM at different MOIs of HIV-1, and quantitated FOXO3a phosphorylation by Western blotting (Fig. 2C). The infection levels by different MOIs of HIV-1 were monitored by RT activity assay, which showed a dose-dependent increase of RT activities as the MOI increased (Fig. 2E). Densimetric quantification showed a dose-dependent decrease of FOXO3a phosphorylation as HIV-1 infection levels increased (Fig. 2D). We observed a significant negative correlation between RT value and the ratio of p-FOXO3a to FOXO3a in HIV-1-infected MDM (Fig. 2F, r2 = 0.7171), suggesting the inhibition of FOXO3a phosphorylation is dependent on the levels of HIV-1 infection. Together, these results indicate HIV infection decreases the phosphorylation of FOXO3a, an effect that is dependent on HIV-1 replication.
FIGURE 2.

HIV-1 infection decreases the phosphorylation of FOXO3a in a time and dose-dependent manner. A, MDM were infected with HIV-1 and cell lysates were collected 1, 3, and 5 days after infection. p-FOXO3a and total FOXO3a were detected by Western blotting. B, Results of A were normalized as a ratio of p-FOXO3a to FOXO3a and shown as percentage of 1 dpi control. C, MDM were infected with different HIV-1 dilutions. Control denotes uninfected control cells and the HIV-1 dilutions used ranged from 0.001 to 1 (MOI). Cell lysates were collected 3 days after infection and then subjected to Western blotting for p-FOXO3a and total FOXO3a. D, Results of C were normalized as a ratio of p-FOXO3a to FOXO3a and then compared with control. Results were shown as average ± SD in representative experiments performed with three different donors. *, p < 0.05; **, p < 0.01 compared with control. E, The infection levels of different HIV-1 dilutions were monitored by RT activity assays. F, The GAPDH illustrates the linear relationships between p-FOXO3a/FOXO3a and the HIV-1 infection levels (RT activity). r2, Coefficient determination, were determined as 0.7171 through scatterplot and linear regression (p = 0.008).
HIV-1 infection causes nuclear translocation of FOXO3a
Phosphorylation of amino acids tyrosine 32, serine 253, and serine 315 of FOXO3a retains FOXO3a in the cytosol; loss of phosphorylation at those sites causes nuclear translocation of FOXO3a (28). To confirm decreased FOXO3a phosphorylation promotes nuclear translocation of FOXO3a, we immunostained MDM with or without HIV-1 infection and determined the subcellular localization of FOXO3a by confocal microscopy. p24 Ab against HIV-1 viral Ag was used to differentiate infected cells from noninfected cells. In control and AZT-treated cells, HIV-1 was not present or unable to replicate thus p24 staining was absent (Fig. 3, A, I, and M). In these cells, the majority of FOXO3a staining was localized in the cytoplasm (Fig. 3, B, J, and N). In HIV-1-infected MDM, there was staining of FOXO3a in the nucleus with minimal FOXO3a detected in the cytoplasm (Fig. 3, E-H). These results indicate HIV infection increases FOXO3a nuclear translocation. Furthermore, when treated with AZT, the nuclear translocation was blocked, confirming the translocation of FOXO3a was reliant upon HIV-1 replication (Fig. 3, M-P).
FIGURE 3.
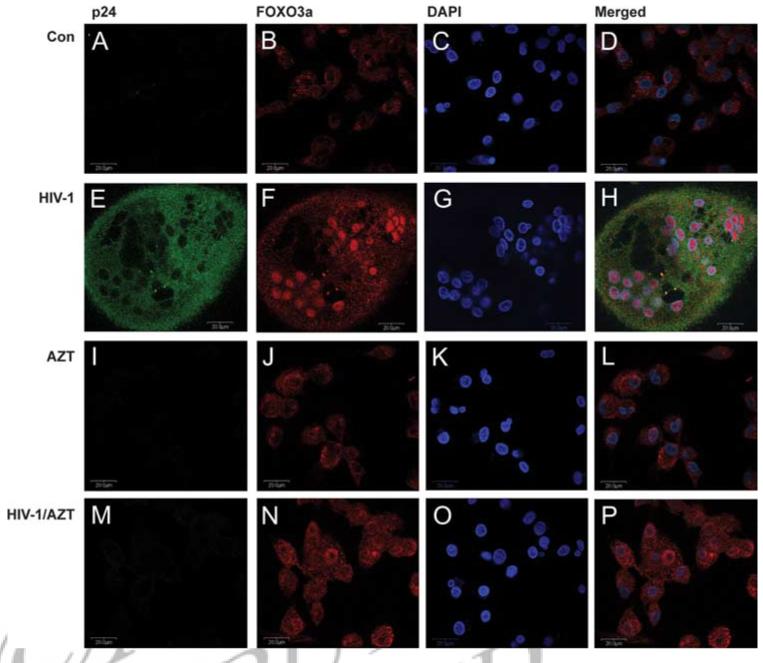
HIV-1 infection induces FOXO3a translocation from the cytoplasm to the nucleus (immunofluorescence staining). Human MDM were infected with HIV-1 for 3 days with or without AZT treatment and then stained with Abs to p24 (HIV-1 infection marker, green) and FOXO3a (red), conjugated with anti-mouse Alexa Fluor 488 nm and anti-rabbit Alexa Fluor 647 nm secondary Abs, respectively. 4′,6′-Di amidino-2-phenylindole (DAPI; blue) was used for DNA staining. A-D, Control uninfected MDM. E-H, HIV-1-infected MDM. I-L, AZT-treated control MDM. M-P, AZT-treated HIV-1-infected MDM. D, H, L, and P are merged pictures of A-C, E-G, I-K, and M-O, respectively. Images were acquired from a Bio-Rad MRC1024ES laser scanning confocal microscope. Magnifications: A-F, ×600. Panels are representative of four separate donors.
We also used subcellular fractionation as another approach to study the nuclear translocation of FOXO3a. Whole cells, cytoplasm, and nuclear protein lysates were prepared 72 h postinfection. Levels of phospho-FOXO3a and total FOXO3a, along with their corresponding loading control, were determined by Western blotting. The phosphorylation of FOXO3a decreased in a manner consistent with Fig. 1 in HIV-1-infected MDM in the whole cell lysates. In the subcellular fractions, the phosphorylation of FOXO3a decreased in the cytoplasm, whereas nuclear FOXO3a increased in HIV-infected MDM (Fig. 4A). After densimetric quantification of the Western blotting, we found HIV-1 infection significantly decreased phosphorylation of FOXO3a in the cytosolic fractions, while it increased phosphorylation of FOXO3a in nuclear fractions (Fig. 4, C and D). Decreased phosphorylation of FOXO3a in the cytosol and increased phosphorylation of FOXO3a in the nucleus was blocked by AZT treatment, suggesting this effect is reliant on HIV-1 replication. (Fig. 4B). Together, these data further confirmed HIV-infection inhibited phosphorylation of FOXO3a, leading to FOXO3a translocation to the nucleus.
FIGURE 4.

HIV-1 infection induces FOXO3a translocation from the cytoplasm to the nucleus (immunoblotting). A, After 3 days of infection by HIV-1, with or without AZT treatment (5 μM), proteins from whole cell lysates, cytoplasmic fractions, and nuclear fractions were examined for phosphorylated FOXO3a (p-FOXO3a) and total FOXO3a (FOXO3a) by Western blotting. β-actin was used as loading control for the whole cell lysates and cytoplasmic proteins; TBP was used as nuclear protein loading control. B-D, Results were densimetrically quantified as a ratio of p-FOXO3a to FOXO3a and then shown as percentage of control in different fractions. Error bars denote average ± SD and results are representative of experiments performed with three different donors. **, p < 0.01 compared with control; ##, p < 0.01 compared with AZT-treated MDM.
Constitutively active FOXO3a induces cell death in MDM
To investigate the function of FOXO3a in MDM, we used an adenoviral gene-delivery system to manipulate the activity of FOXO3a in MDM. Constitutively active FOXO3a (AAA-FOXO3a) delivered by adenovirus (Ad-AAA-FOXO3a) was used to increase activity of FOXO3a. The constitutively active FOXO3a was modified from native FOXO3a by mutating all three phosphorylation sites to alanine. Ad-DN-Foxo3a (DN-FOXO3a) was constructed by deletion of the transactivation domain from the C terminus of FOXO3a. Thus, DN-FOXO3a can competitively inhibit the regulatory function of the endogenous FOXO3a. GFP adenovirus was used as a vector control in all adenoviral infections. After 48h of adenoviral infection, the infection efficiency was monitored by fluorescence microscopy (Fig. 5A). At 2000 MOI, the infection efficiency was ∼70% for each adenovirus. FOXO3a overexpression in Ad-AAA-O3a and Ad-DN-O3a was confirmed by Western blotting (Fig. 5B), which also showed the endogenous FOXO3a of Ad-GFP group, 98 kDa of AAA-O3a, and 40 kDa of DN-O3a. We also examined Puma, one of possible downstream factors of FOXO3a. After infection with constitutively active FOXO3a for 72 h, the expression of Puma is increased as compared with the GFP vector control, whereas there are no significant changes in MDM infected with Ad-DN-Foxo3a (Fig. 5B). This result indicated an induction of downstream factor Puma by FOXO3a overexpression in human MDM.
FIGURE 5.
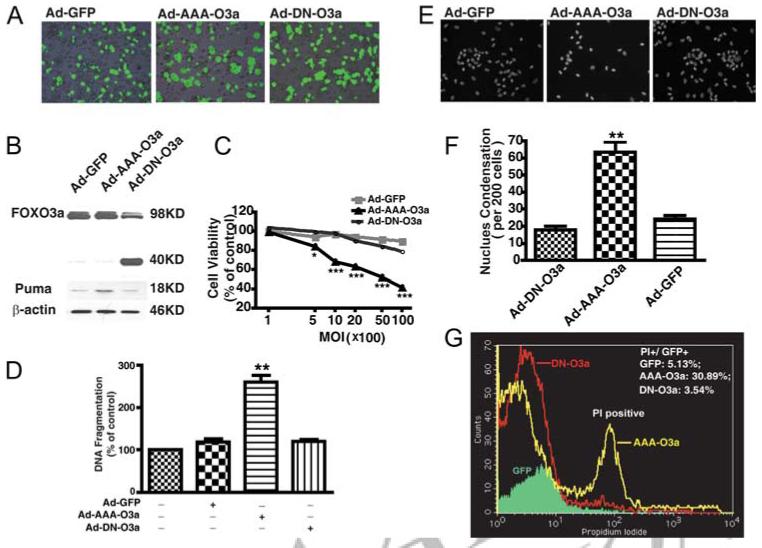
Overexpression of constitutively active FOXO3a induces macrophage cell death. A, Human MDM were infected with adenovirus vector containing a GFP reporter system. Infection efficiencies were monitored by taking fluorescence images 48 h after adenovirus infection. Ad-GFP is GFP control vector adenovirus; Ad-AAA-O3a is adenovirus expressing constitutively active FOXO3a; Ad-DN-FOXO3a is adenovirus expressing DN FOXO3a. B, After 72 h of adenoviral infection at 2000 MOI, FOXO3a overexpression was determined by immunoblotting for total FOXO3a. Constitutively active FOXO3a and endogenous FOXO3a both appeared at 98 kDa, while DN FOXO3a appeared at 40 kDa. In these samples, induction of Puma by FOXO3a overexpression was also determined by Western blotting. β-actin was used as a loading control. C, MDM were infected with various MOI of viral particles for Ad-GFP, Ad-AAA-O3a, and Ad-DN-FOXO3a. Seventy-two hours after infection, cell viability was determined by MTT assay; results shown were performed in triplicate and normalized as a percentage of GFP vector control. *, p < 0.05, ***, p < 0.001 compared with GFP vector control. D, At 2000 MOI of adenovirus, after 48 h of infection, apoptosis was determined by DNA fragmentation apoptosis ELISA. Results were representative of experiments performed in triplicate and normalized as percentage of control. **, p < 0.01 compared with GFP control vector. NS denotes no statistical difference from GFP control. E, Hoechst staining was performed to detect nuclear condensation after delivery of FOXO3a to MDM. F, Positive cells with nuclear condensation were counted every 200 cells per well with triplicate. **, p < 0.01 compared with GFP vector control. NS denotes no statistical difference from GFP control. G, After 72 h of adenoviral infection, human MDM were harvested and apoptosis was determined by PI staining and subsequent flow cytometry analysis. Numbers appearing in the upper right corner are percentage of PI-positive cells in GFP-positive cells. Results are representative of three independent experiments.
Next, cell viability and apoptosis were assessed by MTT, DNA fragmentation ELISA, nuclear condensation, and flow cytometry followed by adenoviral infection. Cell viability in AAA-FOXO3a cells was significantly decreased in a dose-dependent manner as compared with Ad-GFP and Ad-DN-FOXO3a (Fig. 5C). DNA fragmentation in Ad-AAA-FOXO3a dramatically increased after 48 h (Fig. 5D) and, consequently, nuclear condensation and nuclear fragmentation also increased (Fig. 5, E and F). Flow cytometric analysis showed PI-positive cells also dramatically increased following Ad-AAA-FOXO3a infection (Fig. 5G). These results demonstrated constitutively active FOXO3a induced significant cell death in MDM.
DN-FOXO3a and FOXO3a knockdown protected MDM from HIV-induced cell death
To explore the role of FOXO3a in HIV-1-induced cell death, DN-FOXO3a was delivered to MDM by adenovirus after 1 day of HIV-1 infection. We tested DNA fragmentation and cell viability 3 days after adenoviral infection. As compared with non-HIV-infected cells, HIV infection elevated DNA fragmentation and decreased cell viability (Fig. 6, A and B). More importantly, in HIV-infected MDM, we observed DN-FOXO3a partially prevented the increase of DNA fragmentation and the decrease of cell viability induced by HIV-1 infection (Fig. 6, A and B).
FIGURE 6.

DN FOXO3a or FOXO3a siRNA reduces apoptosis in HIV-1-infected MDM. A, Apoptosis was detected by DNA fragmentation apoptosis ELISA after 48 h of DN FOXO3a delivery to HIV-1-infected MDM. B, Cell viability was detected by MTT assay after 3 day of DN FOXO3a delivery to HIV-1-infected MDM. Results shown in A and B are the average ± SEM of triplicates performed with three different donors. **, p < 0.01 compared with Ad-GFP control; ##, p < 0.01 HIV-1/Ad-DN-FOXO3a vs HIV-1/Ad-GFP. C, FOXO3a knockdown was determined by Western blotting. β-actin was used as a loading control. D, Densimetrically quantified data of C are shown. **, p < 0.01 compared with nonspecific siRNA control; ##, p < 0.01 compared with nonspecific siRNA control in HIV-1-infected MDM. E, Apoptosis was detected by DNA fragmentation apoptosis ELISA in MDM after 48 h of siRNA transfection. F, Cell viability was detected by MTT assay after 72 h of siRNA transfection. Results shown in E and F are the average ± SEM of experiments performed in triplicate (n = 3 donors). **, p < 0.01 compared with nonspecific siRNA control; ##, p < 0.01 compared with nonspecific siRNA control in HIV-1-infected MDM.
We also used siRNA to knockdown the expression of FOXO3a. First, we assessed FOXO3a expression 72 h after siRNA transfection, and found the FOXO3a protein was reduced ∼70% by FOXO3a siRNA. Compared with nonspecific siRNA (Fig. 6, C and D), knockdown of FOXO3a by siRNA decreased DNA fragmentation by 50% in HIV-infected MDM (Fig. 6E). FOXO3a knockdown partially prevented the decreased cell viability in HIV-1-infected MDM (Fig. 6F). These results demonstrated that DN FOXO3a and FOXO3a siRNA knockdown prevented HIV-induced macrophage death, suggesting HIV-1-induced macrophage death is at least in part via FOXO3a transcription factor.
Reduced phosphorylation of FOXO3a in HIV-1-infected macrophages is mediated by inhibition of Akt-1
FOXO3a has been reported to be an important transcription factor downstream of PI3K/Akt-1 in various cell types (27). To test whether FOXO3a is a downstream factor of PI3K/Akt-1 pathway in MDM, PI3K inhibitor LY294002 was used at 0.5 and 5 μM to treat MDM (Fig. 7A). Phosphorylation of FOXO3a dramatically decreased after 2 h of both 0.5 or 5 μM LY294002 treatment. These results demonstrated that the PI3K pathway is necessary for FOXO3a phosphorylation in human macrophages.
FIGURE 7.

Reduced phosphorylation of FOXO3a in HIV-1-infected macrophages is mediated through inhibition of Akt-1. A, Human MDM were treated with 0.5 or 5 μM of PI3K inhibitor LY294002, and cell lysates were collected 2 h later. DMSO was used as a vehicle control without LY294002 treatment. Phosphorylated FOXO3a, total FOXO3a, and β-actin were determined by Western blotting. B, After 2 days of infection by HIV-1, MDM were infected with adenovirus expressing constitutively active Akt-1 (ad-myr-Akt) for 72 h at 10,000 MOI, p-FOXO3a, total FOXO3a, Akt-1, and β-actin were determined by Western blotting. C, Ratio of p-FOXO3a to FOXO3a was generated from the densimetrical quantification of B with four different donors. p < 0.05, compared with GFP vector control; #, p < 0.05, HIV-1/Ad-myr-Akt vs HIV-1/Ad-GFP.
To further confirm FOXO3a regulation by the PI3K pathway, we delivered constitutively active Akt-1 with an adenoviral vector (Ad-myr-Akt) to control and HIV-1-infected MDM. First, we measured Akt-1 expression through Western blotting after 72 h of infection with Ad-myr-Akt and confirmed Akt-1 was overexpressed (Fig. 7B). Then, we tested the phosphorylation of FOXO3a with or without Ad-myr-Akt infection. Densimetrical quantification of FOXO3a phosphorylation were shown in Fig. 7C. Compared with Ad-GFP vehicle control, HIV-1 infection reduced the phosphorylation of FOXO3a by 30%. Ad-myr-Akt infection increases the phosphorylation of FOXO3a in both the control and HIV-1-infected MDM (p < 0.05) (Fig. 7C). More importantly, there was no significant decrease of phosphorylated FOXO3a in HIV-1-infeced MDM after Ad-myr-Akt infection. These results further demonstrated that HIV-1 reduced phosphorylation of FOXO3a via down-regulating the PI3K/Akt-1 pathway and FOXO3a is a downstream factor of Akt-1 in human MDM.
Discussion
Eliminating HIV reservoirs and combating long-term viral persistence is critical to the treatment of AIDS (30-32). In the present study, the regulation and function of FOXO3a in HIV-induced macrophage cell death was investigated. First, we demonstrated that phosphorylation of FOXO3a was decreased in HIV-1-infected macrophages facilitating FOXO3a translocation to the nucleus (Figs. 1-4). Second, we applied a gene delivery system implementing adenoviral delivery of constitutively active FOXO3a. This rFOXO3a contains three mutated phosphorylation sites maintaining a transcriptionally active FOXO3 and was found to induce apoptosis in macrophages (Fig. 5). Third, both DN FOXO3a and FOXO3a knockdown with siRNA prevented macrophages from HIV-induced cell death (Fig. 6). Fourth, the PI3K inhibitor LY294002 inhibited FOXO3a phosphorylation, whereas constitutively active Akt-1 increased FOXO3a phosphorylation (Fig. 7). Together, these results suggest HIV infection increases the activity of transcription factor FOXO3a, and elevated FOXO3a activity promotes HIV-1-infected macrophage cell death. Although FOXO3a has been shown to influence cell death signaling, to our knowledge, we are the first to report that FOXO3a is a pivotal transcription factor involved in HIV-1-induced human macrophage death.
The physiological relevance of FOXO3a-mediated macrophage death is significant because a large number of macrophages in the brain and lung are infected with HIV-1 during late stage disease (13-15). Although macrophage are usually resistant to HIV-1-mediated cell death, targeted cell death of infected macrophage in tissue is likely a host immune response to prevent viral dissemination (17). Nonetheless, excessive cell death may also promote chronic inflammation and thus contribute to AIDS pathogenesis. Clearance of activated or HIV-1-infected macrophages from inflamed tissue is important to the host-mediated immune response. Understanding the signaling pathways regulating these processes, such as Akt-1 inhibition and FOXO3a activation, provides important information about macrophage survival and is relevant to HIV-1 pathogenesis, and could potentially identify therapeutic avenues for the treatment of HIV-1 infection.
Forkhead transcription factors play key roles in immunoregulation. Members of the FOXO subfamily have been implicated in the regulation of the cell cycle and/or apoptosis. However, the function of FOXO3a in peripheral immune cells, particularly in macrophages, is not clear. Gene knockout of FOXO3a led to spontaneous lymphoproliferation associated with inflammation of several organs in mice, suggesting FOXO3a regulates homeostasis of immune cells (19, 20). Indeed, FOXO3a appears to be a critical factor regulating apoptosis in lymphocytes (33-35). Our results support a similar role for FOXO3a in human macrophages. Exogenously expressed FOXO3a induced apoptosis in human macrophages and was found to significantly reduce viability of macrophages in vitro (Fig. 5).
In mammals, FOXO3a contains a highly conserved motif that is posttranslationally modified by phosphorylation. PI3K/Akt activation is the major kinase for phosphorylation of FOXO3a on T32, S253, and S315 or on homologous sites in other family members (29, 36, 37). Akt-1-mediated phosphorylation of FOXO proteins leads to cytoplasmic retention and impairment of nuclear transcription (38-40). Our data demonstrated an increase of nuclear translocation of FOXO3a by HIV-1 infection (Figs. 3 and 4). We demonstrated that inhibition of the PI3K/Akt pathway caused a decrease of FOXO3a phosphorylation in human macrophages and overexpression of constitutive Akt-1 blocked the HIV-1-mediated decrease of FOXO3a phosphorylation (Fig. 7). Notably, phosphorylated Akt-1 is also critical for macrophage survival (41). We recently demonstrated that Akt-1 is inhibited by HIV-1 infection and this inhibition contributes significantly to TRAIL-induced apoptosis (17). Together, these observations suggest the decrease of FOXO3a phosphorylation is due to inhibition of Akt-1 after HIV-1 infection. Although downstream factors of Akt-1 inhibition in our model are not clear, our study suggests the Akt-1-related signaling pathway plays a critical role in macrophage survival and is relevant to HIV-1 pathogenesis.
Akt-1-associated pathways and signaling cross-talk between Akt-1 and different apoptotic molecules remain a complex story. Recent studies have identified interactions between molecules downstream of Akt-1 and apoptosis pathways (see review in Ref. 42). For example, phosphorylation of BAD at Ser136 by phospho-Akt-1 prevents mitochondrial translocation of BAD (43, 44); phospho-Akt-1 phosphorylates procaspase-9 at Ser196 (45); phosphorylation of the Forkhead family of transcription factors by phospho Akt-1 leads to decreased transcriptional activity for proapoptotic genes including Fas, TRAIL, Bim,or Puma (46-48). Furthermore, Akt-1 phosphorylation activates NF-κB, a transcription factor promoting the transcription of many essential survival genes (49). In our study, we demonstrate the inhibition of FOXO3a phosphorylation during HIV-1 infection of human macrophage is at least in part responsible for HIV-1-mediated cell death. Thus, FOXO3a appears to be one downstream factor of Akt-1 mediating apoptosis in HIV-1infected macrophages.
HIV-gp120 has been reported to up-regulate the PI3K pathway and, thus, enhanced viral replication (50). We observed phosphorylation of Akt-1 decreased following 3 days of HIV-1 infection in human macrophages (17). Activation of FOXO3a by productive HIV-1 infection further supports this earlier observation. Variation in Akt-1 phosphorylation status is likely due to the state of HIV-1 infection. In addition, although we found HIV-1-infection decreased Akt-1 phosphorylation, and concurrently inhibited FOXO3a phosphorylation, there are other kinases that regulate the phosphorylation and translocation of FOXO3a, such as SGK and DYRK1A. Among those kinases, only phosphorylation of the Akt site in the forkhead domain on FOXO3a protein is indispensable for FOXO3a translocation (38). The exact roles of other kinases involved in the FOXO3a regulation require further investigation.
How downstream factors of FOXO3a are involved in HIV-1-mediated macrophage death is also an interesting and important topic. FOXO3a has been reported to induce proapoptotic signaling by regulating the expression of TRAIL, FasL, Bim, and Puma (23, 48, 51), and regulates cell cycle arrest by activation of p27, p130, and p21, repression of cyclin D expression (G1/S arrest) (29, 52), or activation of cyclin G2 (G0/G1 arrest) (53). Intriguingly, recent reports suggest FOXO3a also promotes cell type-specific antioxidative stress of MnSOD, catalase, and GADD45 in addition to promoting apoptosis (29, 54). Our study supports the role of FOXO3a in the induction of Puma in human MDM (Fig. 5B), and this would likely shift the cellular equilibrium of survival-apoptosis, and promote an apoptotic phenotype in macrophage. The specific mechanism by which Puma participates in FOXO3a- or HIV-1-mediated macrophage apoptosis is certainly interesting and remains further investigation.
In summary, the evidence presented in this study demonstrates that HIV-1 infection inhibits the phosphorylation and increases the activity of transcription factor FOXO3a activation, resulting in cell death of human macrophages. Our study may help unveil the mechanism of macrophage cell death in HIV-1 pathology, which facilitates the elimination of HIV-1 reservoirs during disease, making this pathway a potential therapeutic target during HIV-1 infection.
Acknowledgments
We acknowledge Dr. You Zhou, Dr. Hui Peng, Angelique Walstrom, and Yu Zhen who provided technical support for this work. Dr. Howard E. Gendelman, Dr. Terry Hexum, Nathan Erdmann, Nick Whitney, and Matthew Beaver provided valuable comments and suggestions about the manuscript. Dr. Charles Kuszynski, Linda Wilkie, and Victoria Smith performed the FACS analyses. Julie Ditter, Robin Taylor, Johna Belling, Na Ly, Myhanh Che, and Emilie Scoggins provided outstanding administrative support.
Footnotes
This work was supported in part by National Institutes of Health Research Grants R01 NS 41858, P20 RR15635, and P01 NS043985 (to J.Z.).
- MDM
- monocyte-derived macrophage
- AZT
- azidothymidine
- DN
- dominant negative
- MOI
- multiplicity of infection
- RT
- reverse transcription
- TBP
- TATA box binding protein
- PI
- propidium iodide
Disclosures
The authors have no financial conflict of interest.
References
- 1.Schuitemaker H, Koot M, Kootstra NA, Dercksen MW, de Goede RE, van Steenwijk RP, Lange JM, Schattenkerk JK, Miedema F, Tersmette M. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J. Virol. 1992;66:1354–1360. doi: 10.1128/jvi.66.3.1354-1360.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorry PR, Bristol G, Zack JA, Ritola K, Swanstrom R, Birch CJ, Bell JE, Bannert N, Crawford K, Wang H, et al. Macrophage tropism of human immunodeficiency virus type 1 isolates from brain and lymphoid tissues predicts neurotropism independent of coreceptor specificity. J. Virol. 2001;75:10073–10089. doi: 10.1128/JVI.75.21.10073-10089.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meltzer MS, Skillman DR, Gomatos PJ, Kalter DC, Gendelman HE. Role of mononuclear phagocytes in the pathogenesis of human immunodeficiency virus infection. Annu. Rev. Immunol. 1990;8:169–194. doi: 10.1146/annurev.iy.08.040190.001125. [DOI] [PubMed] [Google Scholar]
- 4.He J, Chen Y, Farzan M, Choe H, Ohagen A, Gartner S, Busciglio J, Yang X, Hofmann W, Newman W, et al. CCR3 and CCR5 are coreceptors for HIV-1 infection of microglia. Nature. 1997;385:645–649. doi: 10.1038/385645a0. [DOI] [PubMed] [Google Scholar]
- 5.Aquaro S, Balestra E, Cenci A, Francesconi M, Calio R, Perno CF. HIV infection in macrophage: role of long-lived cells and related therapeutical strategies. J. Biol. Regul. Homeost. Agents. 1997;11:69–73. [PubMed] [Google Scholar]
- 6.Keane J, Balcewicz-Sablinska MK, Remold HG, Chupp GL, Meek BB, Fenton MJ, Kornfeld H. Infection by Mycobacterium tuberculosis promotes human alveolar macrophage apoptosis. Infect. Immun. 1997;65:298–304. doi: 10.1128/iai.65.1.298-304.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Manickan E, Smith JS, Tian J, Eggerman TL, Lozier JN, Muller J, Byrnes AP. Rapid Kupffer cell death after intravenous injection of adenovirus vectors. Mol. Ther. 2006;13:108–117. doi: 10.1016/j.ymthe.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 8.Dalton JE, Pearson J, Scott P, Carding SR. The interaction of γδ T cells with activated macrophages is a property of the Vγ1 subset. J. Immunol. 2003;171:6488–6494. doi: 10.4049/jimmunol.171.12.6488. [DOI] [PubMed] [Google Scholar]
- 9.Egan PJ, Carding SR. Downmodulation of the inflammatory response to bacterial infection by γδ T cells cytotoxic for activated macrophages. J. Exp. Med. 2000;191:2145–2158. doi: 10.1084/jem.191.12.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stoneman V, Braganza D, Figg N, Mercer J, Lang R, Goddard M, Bennett M. Monocyte/macrophage suppression in CD11b diphtheria toxin receptor transgenic mice differentially affects atherogenesis and established plaques. Circ. Res. 2007;100:884–893. doi: 10.1161/01.RES.0000260802.75766.00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Verheye S, Martinet W, Kockx MM, Knaapen MW, Salu K, Timmermans JP, Ellis JT, Kilpatrick DL, De Meyer GR. Selective clearance of macrophages in atherosclerotic plaques by autophagy. J. Am. Coll. Cardiol. 2007;49:706–715. doi: 10.1016/j.jacc.2006.09.047. [DOI] [PubMed] [Google Scholar]
- 12.Gupta M, Spiropoulou C, Rollin PE. Ebola virus infection of human PBMCs causes massive death of macrophages, CD4 and CD8 T cell sub-populations in vitro. Virology. 2007;364:45–54. doi: 10.1016/j.virol.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 13.Koenig S, Gendelman HE, Orenstein JM, Canto MCD, Pezeshkpour GH, Yungbluth M, Janotta F, Aksamit A, Martin MA, Fauci AS. Detection of AIDS virus in macrophages in brain tissue from AIDS patients with encephalopathy. Science. 1986;233:1089–1093. doi: 10.1126/science.3016903. [DOI] [PubMed] [Google Scholar]
- 14.Gartner S, Markovits P, Markovits DM, Kaplan MH, Gallo RC, Popovic M. The role of mononuclear phagocytes in HTLV-III LAV infection. Science. 1986;233:214–218. doi: 10.1126/science.3014648. [DOI] [PubMed] [Google Scholar]
- 15.Gabuzda DH, Ho DD, Monte MSDL, Rota TR, Sobel RA. Immunohistochemical identification of HTLV-III antigen in brains of patients with AIDS. Ann. Neurol. 1986;20:289–295. doi: 10.1002/ana.410200304. [DOI] [PubMed] [Google Scholar]
- 16.Lum JJ, Pilon AA, Sanchez-Dardon J, Phenix BN, Kim JE, Mihowich J, Jamison K, Hawley-Foss N, Lynch DH, Badley AD. induction of cell death in human immunodeficiency virus-infected macrophages and resting memory CD4 T cells by TRAIL/Apo2L. J. Virol. 2001;75:11128–11136. doi: 10.1128/JVI.75.22.11128-11136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang Y, Erdmann N, Peng H, Herek S, Davis JS, Luo X, Ikezu T, Zheng J. TRAIL-mediated apoptosis in HIV-1-infected macrophages is dependent on the inhibition of Akt-1 phosphorylation. J. Immunol. 2006;177:2304–2313. doi: 10.4049/jimmunol.177.4.2304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin L, Hron JD, Peng SL. Regulation of NF-κB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity. 2004;21:203–213. doi: 10.1016/j.immuni.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 19.Hosaka T, Biggs WH, 3rd, Tieu D, Boyer AD, Varki NM, Cavenee WK, Arden KC. Disruption of forkhead transcription factor (FOXO) family members in mice reveals their functional diversification. Proc. Natl. Acad. Sci. USA. 2004;101:2975–2980. doi: 10.1073/pnas.0400093101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003;301:215–218. doi: 10.1126/science.1086336. [DOI] [PubMed] [Google Scholar]
- 21.Ghaffari S, Jagani Z, Kitidis C, Lodish HF, Khosravi-Far R. Cytokines and BCR-ABL mediate suppression of TRAIL-induced apoptosis through inhibition of forkhead FOXO3a transcription factor. Proc. Natl. Acad. Sci. USA. 2003;100:6523–6528. doi: 10.1073/pnas.0731871100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang H, Regan KM, Lou Z, Chen J, Tindall DJ. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science. 2006;314:294–297. doi: 10.1126/science.1130512. [DOI] [PubMed] [Google Scholar]
- 23.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 24.Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, Phipps T, Wahl LA, Lane HC, Fauci AS, Burke DS. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J. Exp. Med. 1988;167:1428–1441. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shiojima I, Yefremashvili M, Luo Z, Kureishi Y, Takahashi A, Tao J, Rosenzweig A, Kahn CR, Abel ED, Walsh K. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J. Biol. Chem. 2002;277:37670–37677. doi: 10.1074/jbc.M204572200. [DOI] [PubMed] [Google Scholar]
- 26.Zheng J, Thylin M, Ghorpade A, Xiong H, Persidsky Y, Cotter R, Niemann D, Che M, Zeng Y, Gelbard H, et al. Intracellular CXCR4 signaling, neuronal apoptosis and neuropathogenic mechanisms of HIV-1-associated dementia. J. Neuroimmunol. 1999;98:185–200. doi: 10.1016/s0165-5728(99)00049-1. [DOI] [PubMed] [Google Scholar]
- 27.Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ, Jr., DiStefano PS, Chiang LW, Greenberg ME. DNA repair pathway stimulated by the forkhead transcription factor FOXO3a through the Gadd45 protein. Science. 2002;296:530–534. doi: 10.1126/science.1068712. [DOI] [PubMed] [Google Scholar]
- 28.Moller C, Alfredsson J, Engstrom M, Wootz H, Xiang Z, Lennartsson J, Jonsson JI, Nilsson G. Stem cell factor promotes mast cell survival via inactivation of FOXO3a-mediated transcriptional induction and MEK-regulated phosphorylation of the proapoptotic protein Bim. Blood. 2005;106:1330–1336. doi: 10.1182/blood-2004-12-4792. [DOI] [PubMed] [Google Scholar]
- 29.Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- 30.McArthur JC, Sacktor N, Selnes O. Human immunodeficiency virus-associated dementia. Semin. Neurol. 1999;19:129–150. doi: 10.1055/s-2008-1040831. [DOI] [PubMed] [Google Scholar]
- 31.Carpenter CC, Cooper DA, Fischl MA, Gatell JM, Gazzard BG, Hammer SM, Hirsch MS, Jacobsen DM, Katzenstein DA, Montaner JS, et al. Antiretroviral therapy in adults: updated recommendations of the International AIDS Society-USA Panel. J. Am. Med. Assoc. 2000;283:381–390. doi: 10.1001/jama.283.3.381. [DOI] [PubMed] [Google Scholar]
- 32.Krebs FC, Ross H, McAllister J, Wigdahl B. HIV-1-associated central nervous system dysfunction. Adv. Pharmacol. 2000;49:315–385. doi: 10.1016/s1054-3589(00)49031-9. [DOI] [PubMed] [Google Scholar]
- 33.Asselin-Labat ML, David M, Biola-Vidamment A, Lecoeuche D, Zennaro MC, Bertoglio J, Pallardy M. GILZ, a new target for the transcription factor FoxO3, protects T lymphocytes from interleukin-2 withdrawal-induced apoptosis. Blood. 2004;104:215–223. doi: 10.1182/blood-2003-12-4295. [DOI] [PubMed] [Google Scholar]
- 34.Yusuf I, Zhu X, Kharas MG, Chen J, Fruman DA. Optimal B-cell proliferation requires phosphoinositide 3-kinase-dependent inactivation of FOXO transcription factors. Blood. 2004;104:784–787. doi: 10.1182/blood-2003-09-3071. [DOI] [PubMed] [Google Scholar]
- 35.Riou C, Yassine-Diab B, Van Grevenynghe J, Somogyi R, Greller LD, Gagnon D, Gimmig S, Wilkinson P, Shi Y, Cameron MJ, et al. Convergence of TCR and cytokine signaling leads to FOXO3a phosphorylation and drives the survival of CD4 + central memory T cells. J. Exp. Med. 2007;204:79–91. doi: 10.1084/jem.20061681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin L, Huang L, Kufe D. MUC1 oncoprotein activates the FOXO3a transcription factor in a survival response to oxidative stress. J. Biol. Chem. 2004;279:45721–45727. doi: 10.1074/jbc.M408027200. [DOI] [PubMed] [Google Scholar]
- 37.Trotman LC, Alimonti A, Scaglioni PP, Koutcher JA, Cordon-Cardo C, Pandolfi PP. Identification of a tumour suppressor network opposing nuclear Akt function. Nature. 2006;441:523–527. doi: 10.1038/nature04809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Der Heide LP, Hoekman MF, Smidt MP. The ins and outs of FoxO shuttling: mechanisms of FoxO translocation and transcriptional regulation. Biochem. J. 2004;380:297–309. doi: 10.1042/BJ20040167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Furukawa-Hibi Y, Kobayashi Y, Chen C, Motoyama N. FOXO transcription factors in cell-cycle regulation and the response to oxidative stress. Antioxid. Redox. Signal. 2005;7:752–760. doi: 10.1089/ars.2005.7.752. [DOI] [PubMed] [Google Scholar]
- 40.Skurk C, Izumiya Y, Maatz H, Razeghi P, Shiojima I, Sandri M, Sato K, Zeng L, Schiekofer S, Pimentel D, et al. The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signaling. J. Biol. Chem. 2005;280:20814–20823. doi: 10.1074/jbc.M500528200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu H, Perlman H, Pagliari LJ, Pope RM. Constitutively activated Akt-1 is vital for the survival of human monocyte-differentiated macrophages: role of Mcl-1, independent of nuclear factor (NF)-κB, Bad, or caspase activation. J. Exp. Med. 2001;194:113–126. doi: 10.1084/jem.194.2.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hanada M, Feng J, Hemmings BA. Structure, regulation and function of PKB/AKT-a major therapeutic target. Biochim. Biophys. Acta. 2004;1697:3–16. doi: 10.1016/j.bbapap.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 43.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 44.del Peso L, Gonzalez-Garcia M, Page C, Herrera R, Nunez G. Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science. 1997;278:687–689. doi: 10.1126/science.278.5338.687. [DOI] [PubMed] [Google Scholar]
- 45.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC. Regulation of cell death protease caspase-9 by phosphorylation. Science. 1998;282:1318–1321. doi: 10.1126/science.282.5392.1318. [DOI] [PubMed] [Google Scholar]
- 46.Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem. 1999;274:17179–17183. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 47.Burgering BM, Medema RH. Decisions on life and death: FOXO forkhead transcription factors are in command when PKB/Akt is off duty. J. Leukocyte Biol. 2003;73:689–701. doi: 10.1189/jlb.1202629. [DOI] [PubMed] [Google Scholar]
- 48.You H, Pellegrini M, Tsuchihara K, Yamamoto K, Hacker G, Erlacher M, Villunger A, Mak TW. FOXO3a-dependent regulation of Puma in response to cytokine/growth factor withdrawal. J. Exp. Med. 2006;203:1657–1663. doi: 10.1084/jem.20060353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 50.Francois F, Klotman ME. Phosphatidylinositol 3-kinase regulates human immunodeficiency virus type 1 replication following viral entry in primary CD4 + T lymphocytes and macrophages. J. Virol. 2003;77:2539–2549. doi: 10.1128/JVI.77.4.2539-2549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dijkers PF, Medema RH, Lammers JW, Koenderman L, Coffer PJ. Expression of the pro-apoptotic Bcl-2 family member Bim is regulated by the forkhead transcription factor FKHR-L1. Curr. Biol. 2000;10:1201–1204. doi: 10.1016/s0960-9822(00)00728-4. [DOI] [PubMed] [Google Scholar]
- 52.Medema R, Kops G, Bos J, Burgering B. AFX-like forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- 53.Martinez-Gac L, Marques M, Garcia Z, Campanero MR, Carrera AC. Control of cyclin G2 mRNA expression by forkhead transcription factors: novel mechanism for cell cycle control by phosphoinositide 3-kinase and forkhead. Mol. Cell. Biol. 2004;24:2181–2189. doi: 10.1128/MCB.24.5.2181-2189.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295:2450–2452. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]