

Abstract
The herpes simplex virus type 1 (HSV-1) latency associated transcript (LAT) gene’s anti-apoptosis activity plays a central, but not fully elucidated, role in enhancing the virus’s reactivation phenotype. In transient transfection experiments, LAT increases cell survival following an apoptotic insult in the absence of other HSV-1 genes. However, the high background of untransfected cells has made it difficult to demonstrate that LAT inhibits specific apoptotic factors such as caspases. Here we report that in mouse neuroblastoma cell lines (C1300) stably expressing high levels of LAT, cold shock induced apoptosis was blocked as judged by increased survival, protection against DNA fragmentation (by DNA ladder assay), and inhibition of caspase 3 cleavage and activation (Western blots). To our knowledge this is the first report providing direct evidence that LAT blocks two biochemical hallmarks of apoptosis, caspase 3 cleavage and DNA laddering, in the absence of other HSV-1 gene products.
Keywords: HSV-1, LAT, latency, apoptosis, latency associated transcript
In the US, herpes simplex virus type 1 (HSV-1) is the leading cause of corneal blindness due to an infectious agent (Nesburn, 1983). HSV-1 induced corneal blindness results from corneal scarring, most of which is due to recurrent rather than primary infection. HSV-1 also causes cold sores in and around the mouth, genital herpes, and encephalitis. Estimates are that 60-90% of the adult US population harbors latent HSV-1 or HSV-2 (Liesegang et al., 1989; Whitley and Roizman, 2001). Most severe herpetic disease is due to recurrent rather than primary disease. Understanding the molecular mechanisms by which the HSV-1 latency--reactivation cycle is regulated is therefore important for the eventual control and elimination of herpetic disease. During primary infection at a peripheral mucosal site the virus enters sensory nerves, travels up the nerves to the ganglionic nerve bodies, and establishes a latent infection. The virus can reactivate sporadically throughout the life of the infected individual, return to and be shed at the original peripheral site, and cause recurrent disease.
During neuronal latency a single HSV-1 gene, the latency associated transcript, or LAT gene, is abundantly transcribed (Rock et al., 1987; Stevens et al., 1987). LAT null mutants have a significantly reduced reactivation phenotype in small animal models indicating that LAT plays an important role in enhancing the virus’s reactivation phenotype (Block et al., 1993; Bloom et al., 1994; Devi-Rao et al., 1994; Leib et al., 1989; Perng et al., 1994; Perng et al., 2001; Trousdale et al., 1991). Although it is not yet clear exactly how LAT enhances the reactivation phenotype, recent work indicates that LAT has anti-apoptosis activity and that this anti-apoptosis activity is important (Ahmed et al., 2002; Branco and Fraser, 2005; Gupta et al., 2006; Henderson et al., 2002; Inman et al., 2001; Jin et al., 2003; Jin et al., 2005; Mott et al., 2003; Peng et al., 2004; Perng et al., 2000; Perng et al., 2002). The first 1.5 kb of LAT which is approximately the first 18% of the primary LAT transcript, is sufficient for supporting a wild type virus reactivation phenotype (Perng et al., 1996a). The same LAT region can interfere with apoptosis as efficiently as much longer stretches of LAT (Inman et al., 2001; Peng et al., 2004). Thus LAT’s anti-apoptosis activity and LAT’s ability to enhance the reactivation phenotype co-map, suggesting a functional relationship. In addition, viruses in which the functional region of LAT has been replaced by an alternative anti-apoptosis gene have a LAT(+) wild type reactivation phenotype (Jin et al., 2005; Perng et al., 2002). These findings indicate that LAT’s anti-apoptosis function is involved in how LAT enhances the HSV-1 reactivation phenotype.
Studying the mechanism, or even the steps in the apoptosis pathway that are affected by LAT has been limited to either studies using LAT(+) versus LAT(-) virus infected animals or cells, or transient transfection assays in tissue culture. The former is complicated by the fact that several HSV-1 genes other than LAT can affect apoptosis (Aubert and Blaho, 1999; Galvan, Brandimarti, and Roizman, 1999; Galvan and Roizman, 1998; Leopardi, Van Sant, and Roizman, 1997; Zhou et al., 2000). The latter is complicated by the high background of untransfected cells present in cell cultures following transient transfection. One obvious solution would be to study LAT’s anti-apoptosis activity in a cell line in which LAT is stably expressed, thus eliminating background problems due to cells not expressing LAT, while also isolating LAT from other viral gene products. We have taken this approach here.
We report here the construction and the preliminary characterization of stable C1300 derived cell lines that express high levels of LAT. In addition, we show that compared to similar LAT(-) cells that contain the same plasmid and the same LAT coding region, but without a promoter, the LAT(+) cells are refractory to death following cold shock. Furthermore, cell death following cold shock was due to apoptosis as judged by DNA fragmentation assays (DNA laddering) and by caspase 3 activation (caspase 3 cleavage by Western analysis), both of which were inhibited in the high expression LAT(+) cells.
RESULTS
Establishment of C1300 cells stably expressing high levels of LAT
pLNotI contains a NotI-NotI restriction fragment corresponding to LAT nts -361 to +3,225 and thus contains 361 nts of the LAT promoter followed by the first 3,225 nts of the primary LAT transcript (+1 to +3225) (Fig. 1). It can therefore efficiently transcribe the first 3,225 nts of the primary LAT transcript (indicated by the transcript under the plasmid in Fig. 1). pLNotIΔPstI, was derived from pLNotI by removing a PstI-PstI restriction fragment corresponding to LAT nts -130 to +64. This removed a critical region of the LAT promoter, including the LAT TATA box, and the start site of RNA transcription, rendering the plasmid extremely restricted for transcribing LAT RNA (Fig 1; dashed lines). Three stable cell lines containing pLNotI (DC-LAT2, DC-LAT6, DC-LAT11) and two cell lines containing pLNotIΔPstI (DC-ΔLAT35 and DC-ΔLAT311) were generated using mouse neuroblastoma C1300 cells.
Figure 1.
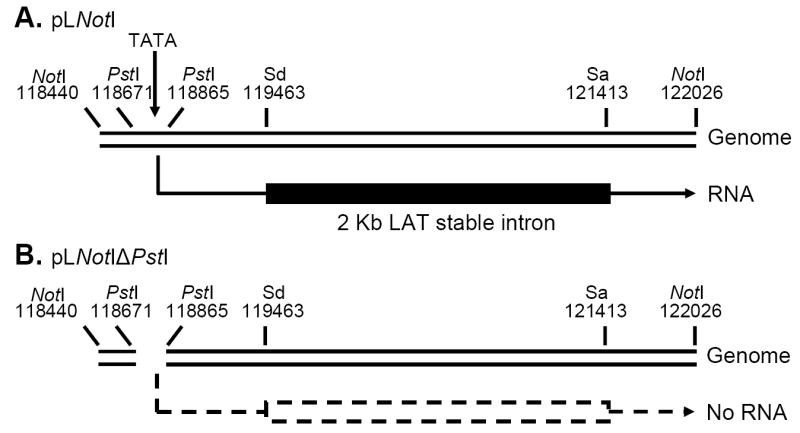
Schematic diagram of the cloned LAT region expressed in stable C1300 cell lines. Panel A: pLNotI, a 3,586bp NotI-NotI DNA restriction fragment containing LAT promoter nts -361 to +1 followed by the first 3225 nts of the primary LAT transcript (+1 to +3225). This restriction fragment was used to generate the cell lines DC-LAT2, DC-LAT6, and DC-LAT11. The LAT RNA expressed is shown under the plasmid. Panel B: pLNotIΔPstI is identical to pLNotI but with a PstI-PstI deletion corresponding to LAT nts -130 to +64. This deletion removes essential LAT promoter elements including the LAT TATA box and no detectable LAT RNA is made (dashed lines). This plasmid was used to generate DC-ΔLAT35 and DC-ΔLAT311 cells. Sd=Splice donor; Sa=Spice acceptor.
Northern blots using a LAT derived BspMI-HpaI probe specific for the 2 kb LAT, detected a band from DC-LAT2, DC-LAT6, and DC-LAT11 cells, indicating that these cell lines each expressed LAT (Fig. 2). The band detected with this 2 kb LAT specific probe co-migrated with 18S ribosomal RNA (detected by ethidium bromide staining) which migrates with an apparent size of approximately 2 kb. Also as expected, no LAT RNA was detected in parental C1300 cells, in DC-ΔLAT311 (Fig. 2) or DC-ΔLAT35 (not shown). DC-LAT6, which had the highest LAT RNA levels, was chosen as the main LAT positive cell line for further studies. No primary LAT transcript corresponding to an unspliced 3.2 kb primary transcript was detected using the above 2 kb LAT specific probe even upon overexposure. The primary transcript was also not detected using a StyI-StyI probe (corresponding to LAT nts 76-447) specific for LAT exon 1. The ability to detect the 2 kb LAT intron but not the primary transcript encoded by the plasmid is not surprising because during both latent and acute infection the 2 kb LAT is stable and easily detected by Northern blots, while the much less stable primary 8.3 kb LAT transcript is not.
Figure 2.

Northern blot of LAT-RNA in stable C1300 derived cell lines. Total RNA was isolated from cells and analyzed by Northern blot using a probe specific for the stable 2 kb LAT intron as described in Materials and Methods. The cell lines are indicated at the top. The arrow indicates the location of 18S ribosomal RNA (with an apparent mobility of 2 kb) based on ethidium bromide staining.
The amount of 2 kb LAT present in DC-LAT2, DC-LAT6, and DC-ΔLAT311 cells was estimated by real time RT-PCR and compared to the amount of 2 kb LAT we detected 7 hours after acute infection with 3 PFU/cell of wild type (strain 17syn+) HSV-1. Acutely infected cells and the high LAT expressing DC-LAT6 cells had an average of 564 and 597 copies of LAT RNA/cell. In contrast, the DC-LAT2 cells which appeared to have much less LAT by Northern blot (see Fig. 2) had 131 copies/cell. As expected, C1300 cells and the LAT negative DC-ΔLAT311 cells had no detectable LAT.
Cold shock
It was recently reported that transient transfection of SY5Y cells with a LAT expressing plasmid protected the cells from cold stress (Atanasiu et al., 2006). To determine how DC-LAT6 cells reacted to cold, we cold shocked DC-LAT6, DC-ΔLAT311, and parental C1300 cells on ice for 2 hours and returned them to 37 °C for 4 hours. Many more DC-LAT6 compared to DC-ΔLAT311 cells and C1300 cells appeared to survive the cold shock/recovery period as judged by phase contrast microscopy (not shown). To quantitate the number of cells that remained adherent, the monolayers were rinsed, trypsinized, and the number of cells that were attached prior to trypsinization were counted (Fig. 3). Following recovery from cold shock, the number of attached LAT expressing DC-LAT6 cells was significantly higher (~2.5 fold) compared to the DC-ΔLAT311 cells and C1300 cells (P<0.001, ANOVA). Other LAT(+) and LAT(-) expressing cells (DC-LAT11 and DC-ΔLAT35; not shown) behaved similarly to DC-LAT6 and DC-ΔLAT311 respectively. Thus, expression of LAT appeared to provide significant protection against death of C1300 cells following recovery from cold shock. Similar cell loss was seen following UV induction of apoptosis (not shown). In addition, we have previously shown that in Neuro2A cells, a cell line derived from the same neuroblastoma as C1300 cells and which in our hands are very similar to C1300 cells, apoptosis can be induced by numerous standard methods (Inman et al., 2001; Jin et al., 2003; Peng et al., 2005; Peng et al., 2004; Perng et al., 2000). Note that compared to no cold shock, cold shock appeared to have slightly reduced the number of DC-LAT6 cells. This may have been the result of a small amount of cell death during recovery from cold shock.
Figure 3.

Cell survival following cold shock. Each cell line was plated into 6 T25 flasks and allowed to recover overnight in a 37 °C CO2 incubator. The next day the flasks were tightly capped. Three flasks of each cell line were incubated for 2 hrs on ice (cold shocked) and 3 flasks of each cell line were incubated at 37 °C without CO2 (not cold shocked). After 2 hrs the caps were loosened and all flasks were returned to a 37 °C CO2 incubator to recover for 4 hours. After treatment the cells were washed once, trypsinized, and the number of cells that had remained attached was determined using a hemacytometer. The bars represent the average of 3 repeats +/- SD. The numbers above each bar indicate the percent of cells after cold shock [(cold shock/no cold shock) × 100%] for each cell line. The results for each cell line are representative of at least 3 independent experiments.
LAT decreased DNA laddering in C1300 cells following recovery from cold shock
C1300, DC-LAT6, DC-LAT11 and DC-ΔLAT311 cells were maintained at 37 °C, cold shocked for 2 hours and immediately harvested, or cold shocked for 2 hours and then recovered at 37 °C for 2 hours. Fragmented DNA was isolated from the monolayers as described in Materials and Methods, run on an agarose gel, and visualized by ethidium bromide staining (Fig. 4). Note that in this assay the harvested cells are incubated in hypotonic buffer for 2 hrs at 4°C. This lyses the cells without disrupting the nuclei and allows fragmented DNA to be eluted from the nuclei while leaving the large unfragmented chromosomal DNA trapped inside the nucleus. The nuclei (and unfragmented chromosomal DNA) are removed by centrifugation and equal aliquots are subjected to electrophoresis. This results in both more “laddering” and more total (fragmented) DNA in samples from apoptotic cultures, with little or no intact chromosomal DNA at the top of the gel. A typical apoptotic DNA laddering pattern was seen in the C1300 parental cells and in DC-ΔLAT311 cells. Much less DNA laddering was seen in DC-LAT6 or DC-LAT11 cells. Little or no DNA laddering was seen in any cell line maintained at 37 °C or cold shocked with no 37 °C recovery. Thus, apoptosis induced by recovery from cold shock in C1300 cells appeared greatly inhibited by LAT.
Figure 4.

Decreased DNA laddering in LAT(+) cell lines following cold shock. Cells were maintained at 37 °C, cold shocked for 2 hours but not recovered, or cold shocked for 2 hours and recovered at 37 °C for 2hours as indicated. Fragmented chromosomal DNA was isolated using a hypotonic buffer that releases fragmented but not intact chromosomal DNA from the nucleus and equal aliquots were run on a gel as described in Materials and Methods.
LAT decreased cold shock induced caspase 3 cleavage in C1300 cells
LAT(+) DC-LAT2, DC-LAT6, and DC-LAT11 cells, and LAT(-) DC-ΔLAT311 cells were cold shocked as above and either harvested immediately or allowed to recover for 2 hours at 37 °C. Attached and unattached cells were pooled, total cell extracts prepared and Western blots were performed using an antibody specific for cleaved (activated) caspase 3 as described in Materials and Methods. At the exposure time shown, little or no cleaved caspase 3 was detected in any of the cells maintained at 37 °C or in any of the cells immediately after 2 hrs on ice. However, after 2 hrs of recovery from cold shock at 37 °C, cleaved caspase 3 appeared abundant in the LAT(-) DC-ΔLAT311 cells compared to the high LAT expressing DC-LAT6 and DC-LAT11 cells. As expected, DC-LAT2 cells, which express less LAT than do the DC-LAT6 and DC-LAT11 cells (see Fig. 2) did not block caspase 3 cleavage as efficiently as the DC-LAT6 and DC-LAT11 cells. Also as expected, the DC-LAT2 cells had less cleaved caspase 3 than the LAT(-) DC-ΔLAT311 cells. This confirmed that recovery from cold shock induces apoptosis in C1300 cells not expressing LAT and that high levels of LAT expression efficiently decreased cold shock induced apoptosis. The apoptosis occurred during recovery from cold shock rather than while cells were on ice, since maintaining cells on ice for 4 hours instead of 2 hours did not alter caspase 3 cleavage (not shown). These results suggest that there is a minimum level of LAT expression required for efficient blocking of caspase 3 cleavage. Thus, the ability of LAT to block cold shock related apoptosis appeared to be dose dependent, providing further evidence that LAT protects against cold shock related apoptosis.
DISCUSSION
The HSV-1 LAT gene is the only viral gene abundantly expressed during neuronal latency (Rock et al., 1987; Stevens et al., 1987). LAT plays an important role in the reactivation phenotype since LAT(-) mutants have a significantly reduced reactivation phenotype in small animal models (Block et al., 1993; Bloom et al., 1994; Devi-Rao et al., 1994; Leib et al., 1989; Perng et al., 1994; Perng et al., 2001; Trousdale et al., 1991). LAT has anti-apoptosis activity (Ahmed et al., 2002; Branco and Fraser, 2005; Gupta et al., 2006; Henderson et al., 2002; Inman et al., 2001; Jin et al., 2003; Jin et al., 2005; Mott et al., 2003; Peng et al., 2004; Perng et al., 2000; Perng et al., 2002). The first 1.5 kb of the 8.3-8.5 kb primary LAT transcript is sufficient and necessary for both enhancing the reactivation phenotype and for efficiently interfering with apoptosis (Inman et al., 2001; Perng et al., 1996a). In addition, mutants in which the functional region of LAT was replaced by an alternative anti-apoptosis gene have reactivation phenotypes at least equivalent to that of LAT(+) (wild type) virus (Jin et al., 2005; Perng et al., 2002). Thus, at least one of the mechanisms by which LAT enhances the reactivation phenotype in small animal models is through its anti-apoptosis activity.
Although we and others have reported that LAT can block apoptosis, the studies that suggest that LAT can block activation of caspases have been limited to comparisons using LAT(-) versus LAT(+) viruses (Henderson et al., 2002; Henderson et al., 2004). These experiments by necessity were done in the presence of all 80+ herpes gene products, several of which have powerful anti-apoptosis activity (Aubert and Blaho, 1999; Galvan, Brandimarti, and Roizman, 1999; Galvan and Roizman, 1998; Leopardi, Van Sant, and Roizman, 1997; Zhou et al., 2000), thus potentially hindering interpretation of the results. Previous studies of LAT in the absence of other viral proteins have relied on indirect anti-apoptosis assays following transient transfection of cells with LAT expressing plasmids. Although these studies clearly showed that LAT plasmids can inhibit cell death following various apoptotic insults (Branco and Fraser, 2005; Henderson et al., 2002; Inman et al., 2001; Jin et al., 2003; Peng et al., 2004; Perng et al., 2000), the high background from untransfected cells has not allowed analysis of LAT’s effect on apoptosis events such as DNA laddering or caspase 3 cleavage. In contrast, by using stable LAT expressing cells and thus eliminating high background problems, here we were able to show that LAT in the absence of other HSV-1 genes can significantly decrease death, apoptotic DNA fragmentation, and caspase 3 cleavage, thus directly demonstrating for the first time that LAT expression in the absence of other HSV-1 gene expression can block specific apoptotic events.
Further standardization was achieved by using cold shock to induce apoptosis. This is an extremely simple, highly reproducible method in which a flask of cells is placed on ice for 2 hrs and then returned to 37 °C. Variations between chemical lots, solution preparations, and UV light sources that are routinely used to induce apoptosis are therefore eliminated. It should be noted that the use of C1300 cells was important here because cold shock does not induce apoptosis in all cell lines.
It has recently been reported that LAT encodes a microRNA (miRNA) from within LAT exon 1 that is capable of interfering with apoptosis (Gupta et al., 2006). The region encoding this miRNA is completely contained within the LAT expressed in the stable cell lines in this report. However we think this miRNA is unlikely to be fully responsible for LAT’s anti-apoptosis activity since a plasmid expressing the full LAT exon 1 [LAT(1-661)], which completely contains this miRNA, has little or no anti-apoptosis activity in transient transfection assays. In addition, a mutant virus (LAT2.5A) expressing only this region of LAT has a low, LAT(-)-like reactivation phenotype (Inman et al., 2001). Finally, dLAT371, a mutant deleted from LAT nts 76-447 which includes a critical portion of the miRNA, has a wild type reactivation phenotype (Perng et al., 1996b). Interestingly, and in contrast, a mutant deleted for the same region but expressing only the first 1.5 kb of LAT has a low LAT(-)-like reactivation phenotype (Perng et al., 1999) and the corresponding LAT plasmid does not block apoptosis in transient transfection assays (Jin et al., 2003) suggesting that the region containing the miRNA is more important in this context. These previous findings strongly suggest that LAT has multiple functional domains and that the LAT miRNA described by Gupta et al contributes to, but is not sufficient for, or in the context of the entire LAT transcript, essential for, LAT’s function.
In conclusion, these stable cell lines expressing LAT, especially when used in conjunction with cold shock, should be a useful model for further detailed investigation of LAT’s effects on apoptosis.
MATERIALS AND METHODS
Plasmids
A 3,586 bp NotI-NotI DNA restriction fragment from HSV-1 strain 17 syn+ (HSV-1 DNA nts 118,440 to122,026) (McGeoch et al., 1988; Perry and McGeoch, 1988) was cloned into the NotI site of the plasmid pSL301 (Invitrogen) using standard methods. This plasmid was designated pLNotI. A second plasmid, designated pLNotIΔ PstI, was derived from pLNotI by digestion with PstI to remove a 194 bp PstI-PstI restriction fragment corresponding to LAT nts -130 to +64. For selection of stably transfected cells a SalI-EcoRI DNA fragment containing a G418 resistance cassette from the plasmid pBAG (Price, Turner, and Cepko, 1987) was cloned into SalI-EcoRI sites of the pSL301 vector with transcription occurring in the direction opposite to that of LAT.
Cell lines
Mouse neuroblastoma C1300 cells were grown as monolayers in MEM with 10% FCS in a 37 °C incubator with 5% CO2. C1300 cells were transfected with either 15ug of pLNotI or 15ug of pLNotIΔPstI by lipofectin (invitrogen) following the manufacturer’s standard protocol. Clonal cell lines were selected with 0.5 mg/ml Geneticin/G418 Sulfate (Gibco BRL) and grown for 2 weeks before isolation by ring cloning. Cell numbers were quantitated using a hemacytometer.
Northern Blot
Total RNA was extracted from cell lines using TRIzol Reagent (Invitrogen) following the manufacturer’s recommendations. The RNA (10 ug per sample) was separated by electrophoresis on a 1% agarose formaldehyde denaturing gel and transferred to Hybond-N+ membrane (Amersham). The blot was hybridized with a 32P-radiolabelled 1805 bp BspMI-HpaI DNA restriction fragment corresponding to LAT nts +675 to +1499 and visualized on X-ray film.
Cold shock
Cell culture flasks were sealed with Parafilm and placed on ice for 2 hours. The Paraflim was then removed and the cells returned to a 37 °C incubator with 5% CO2.
DNA Laddering assay
Fragmented chromosomal DNA was isolated from cells using a hypotonic buffer that lyses the cell without disrupting the nucleus and allows fragmented but not intact chromosomal DNA to leave the nucleus, as we previously described (Jin et al., 2004). DNA laddering was visualized as we previously described (Jin et al., 2004).
Western Blot
Proteins were extracted in protein extraction buffer (50mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP-40, 0.25% sodium deoxycholate, 1× protease inhibitor cocktail [P8340, Sigma-Aldrich, St. Louis, MO], 10 mM NaF and 1 mM sodium orthovanadate). Proteins (30ug per lane) were separated on 4-20% precast gels (Invitrogen) by SDS-PAGE and then transferred to polyvinylidene fluoride (PVDF) membrane (Millipore Corporation, Bedford, MA). Anti-cleaved caspase-3 (Asp175) and anti-GAPDH (14C10) antibodies were purchased from Cell Signaling Technology (Beverly, MA). All primary antibodies were used at a dilution of 1:1,000 in 5% BSA. Primary antibodies were detected with goat anti-rabbit HRP-conjugated secondary antibody (dilution 1:1,000) and visualized using SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford, IL) and X-ray film.
Figure 5.
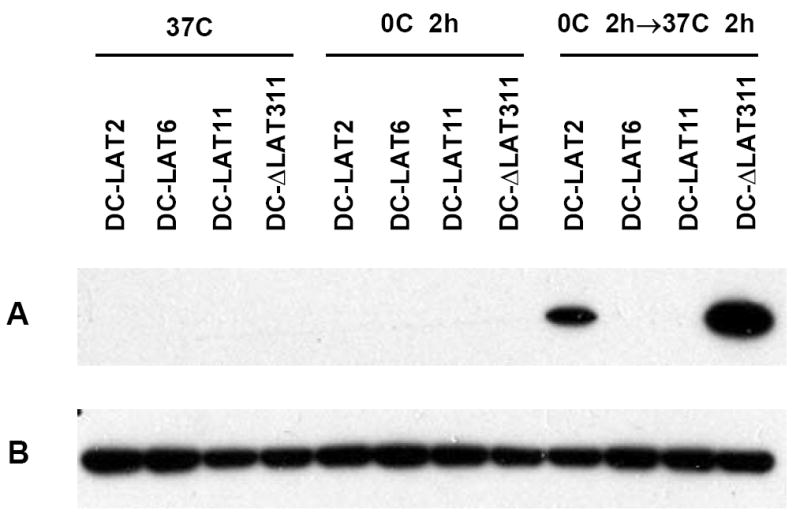
LAT(+) cell lines are protected against cold shock induced caspase 3 cleavage. Cells were maintained at 37 °C (left panel) cold shocked and harvested immediately (middle panel) or allowed to recover for 2 hours at 37 °C (right panel) as described in Materials and Methods. Western blotting was done using an antibody specific for activated (cleaved) caspase 3 as described in Materials and Methods. A. Cleaved caspase-3. B. GAPDH loading controls.
Acknowledgments
This work was supported by Public Health Service grants EY13191, EY12823, EY14900; P20RR15635, USDA grants 2005-01554, 2006-01627, The Discovery Eye Foundation, The Henry L. Guenther Foundation, and Research to Prevent Blindness. Dr. Wechsler is an RPB Senior Scientific Investigator. Dr. BenMohamed is an RPB Special Award Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Dale Carpenter, Email: DCarpent@uci.edu.
Chinhui Hsiang, Email: CHsiang@uci.edu.
Ling Jin, Email: Ling.Jin@oregonstate.edu.
Nelson Osorio, Email: NOsorio@uci.edu.
Lbachir BenMohamed, Email: Lbenmoha@uci.edu.
Clinton Jones, Email: cjones@unlnotes.unl.edu.
Steven L. Wechsler, Email: Wechsler@uci.edu.
References
- Ahmed M, Lock M, Miller CG, Fraser NW. Regions of the herpes simplex virus type 1 latency-associated transcript that protect cells from apoptosis in vitro and protect neuronal cells in vivo. J Virol. 2002;76(2):717–29. doi: 10.1128/JVI.76.2.717-729.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasiu D, Kent JR, Gartner JJ, Fraser NW. The stable 2-kb LAT intron of herpes simplex stimulates the expression of heat shock proteins and protects cells from stress. Virology. 2006;350(1):26–33. doi: 10.1016/j.virol.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Aubert M, Blaho JA. The herpes simplex virus type 1 regulatory protein ICP27 is required for the prevention of apoptosis in infected human cells. J Virol. 1999;73(4):2803–13. doi: 10.1128/jvi.73.4.2803-2813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block TM, Deshmane S, Masonis J, Maggioncalda J, Valyi-Nagi T, Fraser NW. An HSV LAT null mutant reactivates slowly from latent infection and makes small plaques on CV-1 monolayers. Virology. 1993;192(2):618–630. doi: 10.1006/viro.1993.1078. [DOI] [PubMed] [Google Scholar]
- Bloom DC, Devi-Rao GB, Hill JM, Stevens JG, Wagner EK. Molecular analysis of herpes simplex virus type 1 during epinephrine- induced reactivation of latently infected rabbits in vivo. J Virol. 1994;68(3):1283–1292. doi: 10.1128/jvi.68.3.1283-1292.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco FJ, Fraser NW. Herpes simplex virus type 1 latency-associated transcript expression protects trigeminal ganglion neurons from apoptosis. J Virol. 2005;79(14):9019–25. doi: 10.1128/JVI.79.14.9019-9025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi-Rao GB, Bloom DC, Stevens JG, Wagner EK. Herpes simplex virus type 1 DNA replication and gene expression during explant-induced reactivation of latently infected murine sensory ganglia. J Virol. 1994;68(3):1271–1282. doi: 10.1128/jvi.68.3.1271-1282.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan V, Brandimarti R, Roizman B. Herpes simplex virus 1 blocks caspase-3-independent and caspase- dependent pathways to cell death. J Virol. 1999;73(4):3219–26. doi: 10.1128/jvi.73.4.3219-3226.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan V, Roizman B. Herpes simplex virus 1 induces and blocks apoptosis at multiple steps during infection and protects cells from exogenous inducers in a cell- type-dependent manner. Proc Natl Acad Sci U S A. 1998;95(7):3931–6. doi: 10.1073/pnas.95.7.3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Gartner JJ, Sethupathy P, Hatzigeorgiou AG, Fraser NW. Anti-apoptotic function of a microRNA encoded by the HSV-1 latency-associated transcript. Nature. 2006;442(7098):82–5. doi: 10.1038/nature04836. [DOI] [PubMed] [Google Scholar]
- Henderson G, Peng W, Jin L, Perng GC, Nesburn AB, Wechsler SL, Jones C. Regulation of caspase 8- and caspase 9-induced apoptosis by the herpes simplex virus type 1 latency-associated transcript. J Neurovirol. 2002;8(Suppl 2):103–11. doi: 10.1080/13550280290101085. [DOI] [PubMed] [Google Scholar]
- Henderson G, Perng GC, Nesburn AB, Wechsler SL, Jones C. The latency-related gene encoded by bovine herpesvirus 1 can suppress caspase 3 and caspase 9 cleavage during productive infection. J Neurovirol. 2004;10(1):64–70. doi: 10.1080/13550280490261716. [DOI] [PubMed] [Google Scholar]
- Inman M, Perng G, Henderson G, Ghiasi H, Nesburn A, Wechsler S, Jones C. Region of Herpes Simplex Virus type 1 latency-associated transcript sufficient for wild type spontaneous reactivation promotes cell survival in tissue culture. J Virol. 2001;75:3636–3646. doi: 10.1128/JVI.75.8.3636-3646.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Peng W, Perng GC, Brick DJ, Nesburn AB, Jones C, Wechsler SL. Identification of Herpes Simplex Virus Type 1 Latency-Associated Transcript Sequences That both Inhibit Apoptosis and Enhance the Spontaneous Reactivation Phenotype. J Virol. 2003;77(11):6556–61. doi: 10.1128/JVI.77.11.6556-6561.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Perng GC, Brick DJ, Naito J, Nesburn AB, Jones C, Wechsler SL. Methods for detecting the HSV-1 LAT anti-apoptosis activity in virus infected tissue culture cells. J Virol Methods. 2004;118(1):9–13. doi: 10.1016/j.jviromet.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Jin L, Perng GC, Mott KR, Osorio N, Naito J, Brick DJ, Carpenter D, Jones C, Wechsler SL. A herpes simplex virus type 1 mutant expressing a baculovirus inhibitor of apoptosis gene in place of latency-associated transcript has a wild-type reactivation phenotype in the mouse. J Virol. 2005;79(19):12286–95. doi: 10.1128/JVI.79.19.12286-12295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol. 1989;63(7):2893–2900. doi: 10.1128/jvi.63.7.2893-2900.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopardi R, Van Sant C, Roizman B. The herpes simplex virus 1 protein kinase US3 is required for protection from apoptosis induced by the virus. Proc Natl Acad Sci U S A. 1997;94(15):7891–6. doi: 10.1073/pnas.94.15.7891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liesegang TJ, Melton LJ, 3rd, Daly PJ, Ilstrup DM. Epidemiology of ocular herpes simplex. Incidence in Rochester, Minn, 1950 through 1982. Arch Ophthalmol. 1989;107(8):1155–9. doi: 10.1001/archopht.1989.01070020221029. [DOI] [PubMed] [Google Scholar]
- McGeoch DJ, Dalrymple MA, Davison AJ, Dolan A, Frame MC, McNab D, Perry LJ, Scott JE, Taylor P. The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol. 1988;69(Pt 7):1531–1574. doi: 10.1099/0022-1317-69-7-1531. [DOI] [PubMed] [Google Scholar]
- Mott KR, Osorio N, Jin L, Brick DJ, Naito J, Cooper J, Henderson G, Inman M, Jones C, Wechsler SL, Perng GC. The bovine herpesvirus-1 LR ORF2 is critical for this gene’s ability to restore the high wild-type reactivation phenotype to a herpes simplex virus-1 LAT null mutant. J Gen Virol. 2003;84(Pt 11):2975–85. doi: 10.1099/vir.0.19421-0. [DOI] [PubMed] [Google Scholar]
- Nesburn AB, editor. Report of the corneal disease panel: Vision research: A national plan 1983-1987. part III. II. St Louis: C.V Mosby Co; 1983. [Google Scholar]
- Peng W, Henderson G, Inman M, BenMohamed L, Perng GC, Wechsler SL, Jones C. The locus encompassing the latency-associated transcript of herpes simplex virus type 1 interferes with and delays interferon expression in productively infected neuroblastoma cells and trigeminal Ganglia of acutely infected mice. J Virol. 2005;79(10):6162–71. doi: 10.1128/JVI.79.10.6162-6171.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng W, Jin L, Henderson G, Perng GC, Brick DJ, Nesburn AB, Wechsler SL, Jones C. Mapping herpes simplex virus type 1 latency-associated transcript sequences that protect from apoptosis mediated by a plasmid expressing caspase-8. J Neurovirol. 2004;10(4):260–5. doi: 10.1080/13550280490468690. [DOI] [PubMed] [Google Scholar]
- Perng G, Jones C, Ciacci-Zanella H, Henderson G, Yukht A, Slanina S, Hofman F, Ghiasi H, Nesburn A, Wechsler S. Virus induced neuronal apoptosis blocked by the herpes simplex virus latency associated transcript (LAT) Science. 2000;287:1500–1503. doi: 10.1126/science.287.5457.1500. [DOI] [PubMed] [Google Scholar]
- Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol. 1994;68(12):8045–8055. doi: 10.1128/jvi.68.12.8045-8055.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng GC, Ghiasi H, Slanina SM, Nesburn AB, Wechsler SL. The spontaneous reactivation function of the herpes simplex virus type 1 LAT gene resides completely within the first 1.5 kilobases of the 8.3- kilobase primary transcript. J Virol. 1996a;70(2):976–984. doi: 10.1128/jvi.70.2.976-984.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng GC, Maguen B, Jin L, Mott KR, Osorio N, Slanina SM, Yukht A, Ghiasi H, Nesburn AB, Inman M, Henderson G, Jones C, Wechsler SL. A gene capable of blocking apoptosis can substitute for the herpes simplex virus type 1 latency-associated transcript gene and restore wild-type reactivation levels. J Virol. 2002;76(3):1224–35. doi: 10.1128/JVI.76.3.1224-1235.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng GC, Slanina SM, Ghiasi H, Nesburn AB, Wechsler SL. A 371-nucleotide region between the herpes simplex virus type 1 (HSV-1) LAT promoter and the 2-kilobase LAT is not essential for efficient spontaneous reactivation of latent HSV-1. J Virol. 1996b;70(3):2014–2018. doi: 10.1128/jvi.70.3.2014-2018.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng GC, Slanina SM, Ghiasi H, Nesburn AB, Wechsler SL. The effect of latency-associated transcript on the herpes simplex virus type 1 latency-reactivation phenotype is mouse strain-dependent. J Gen Virol. 2001;82(Pt 5):1117–22. doi: 10.1099/0022-1317-82-5-1117. [DOI] [PubMed] [Google Scholar]
- Perng GC, Slanina SM, Yuhkt A, Drolet BS, Keleher WJ, Ghiasi H, Nesburn AB, Wechsler SL. A herpes simplex virus type 1 latency associated transcript (LAT) mutant with increased virulence and reduced spontaneous reactivation. J Virol. 1999;73:920–929. doi: 10.1128/jvi.73.2.920-929.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry LJ, McGeoch DJ. The DNA sequences of the long repeat region and adjoining parts of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol. 1988;69(Pt 11):2831–2846. doi: 10.1099/0022-1317-69-11-2831. [DOI] [PubMed] [Google Scholar]
- Price J, Turner D, Cepko C. Lineage analysis in the vertebrate nervous system by retrovirus-mediated gene transfer. Proc Natl Acad Sci U S A. 1987;84(1):156–60. doi: 10.1073/pnas.84.1.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock DL, Nesburn AB, Ghiasi H, Ong J, Lewis TL, Lokensgard JR, Wechsler SL. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J Virol. 1987;61(12):3820–3826. doi: 10.1128/jvi.61.12.3820-3826.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science. 1987;235(4792):1056–1059. doi: 10.1126/science.2434993. [DOI] [PubMed] [Google Scholar]
- Trousdale MD, Steiner I, Spivack JG, Deshmane SL, Brown SM, MacLean AR, Subak-Sharpe JH, Fraser NW. In vivo and in vitro reactivation impairment of a herpes simplex virus type 1 latency-associated transcript variant in a rabbit eye model. J Virol. 1991;65(12):6989–6993. doi: 10.1128/jvi.65.12.6989-6993.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitley RJ, Roizman B. Herpes simplex virus infections. Lancet. 2001;357(9267):1513–8. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- Zhou G, Galvan V, Campadelli-Fiume G, Roizman B. Glycoprotein D or J delivered in trans blocks apoptosis in SK-N-SH cells induced by a herpes simplex virus 1 mutant lacking intact genes expressing both glycoproteins. J Virol. 2000;74(24):11782–91. doi: 10.1128/jvi.74.24.11782-11791.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]