

Abstract
A hyperadrenergic state is a seminal aspect of chronic heart failure. Also, “Takotsubo stress cardiomyopathy,” is associated with increased plasma catecholamine levels. The mechanisms of myocyte damage secondary to excess catecholamine exposure as well as the consequence of this neurohumoral burst on cardiac stem cells (CSCs) are unknown. Cardiomyocytes and CSCs were exposed to high doses of isoproterenol (ISO), in vivo and in vitro. Male Wistar rats received a single injection of ISO (5 mg kg-1) and were sacrificed 1, 3, and 6 days later. In comparison with controls, LV function was impaired in rats 1 day after ISO and started to improve at 3 days. The fraction of dead myocytes peaked 1 day after ISO and decreased thereafter. ISO administration resulted in significant ryanodine receptor 2 (RyR2) hyperphosphorylation and RyR2-calstabin dissociation. JTV519, a RyR2 stabilizer, prevented the ISO-induced death of adult myocytes in vitro. In contrast, CSCs were resistant to the acute neurohumoral overload. Indeed, CSCs expressed a decreased and inverted complement of β1/β2-adrenoreceptors and absence of RyR2, which may explain their survival to ISO insult. Thus, a single injection of ISO causes diffuse myocyte death through Ca2+ leakage secondary to the acutely dysfunctional RyR2. CSCs are resistant to the noxious effects of an acute hyperadrenergic state and through their activation participate in the response to the ISO-induced myocardial injury. The latter could contribute to the ability of the myocardium to rapidly recover from acute hyperadrenergic damage.
Heart failure (HF),3 the complex clinical syndrome characterized by impaired ability of the ventricle to fill with or eject blood, is the leading cause of death in the United States (1). Left ventricular (LV) dysfunction is usually a progressive process, and the main morphological manifestation of such progression is a poorly understood process known as cardiac remodeling. The latter is a rather complex phenomenon secondary to, as well as responsible for, anatomical, cellular, molecular, and humoral changes (2, 3).
Recent data have added new pieces to the puzzle of cardiac remodeling. We and others (see Ref. 4) have identified a population of cells with stem and progenitor cell characteristics in the hearts of adult mammals, including humans. These cardiac stem/progenitor cells, hereafter identified together as CSCs, are essential for maintaining normal cardiac cellular homeostasis throughout life by regenerating myocardial cells lost by wear and tear (5). CSCs, when injected into an infarcted myocardium, give rise to new functionally competent myocytes and vascular structures (6). In addition, CSCs participate in the pathologic remodeling of hearts from aortic stenosis, myocardial infarction, and end stage cardiomyopathy (7-9). However, the role, if any, of CSCs during cardiac adaptation to stress remains unclear. Also unknown is whether cardiac insults that severely damage myocytes also affect the CSCs.
In HF, cardiac contractility is impaired by abnormalities in the structure and/or function of molecules responsible for calcium (Ca2+) handling within the myocytes (3, 10). Chronically elevated concentrations of catecholamines are a hallmark of chronic HF and contribute to the alterations in intracellular Ca2+ handling (11). Indeed, chronic hyperactivity of the β-adrenoreceptor (AR) signaling pathway in HF leads to PKA-mediated hyperphosphorylation of the ryanodine receptor 2 (RyR2) of the sarcoplamic reticulum, which in turn results in continuous intracellular Ca2+ leakage (12). Moreover, acute increased levels of catecholamines are cardio-toxic (13-16) and cause significant myocyte death and hypertrophy in the long term (14-16). In human HF, this increased myocyte death is accompanied by increased new myocyte formation (4, 7-9) produced by the differentiation of the CSCs. Interestingly, spikes of excessive circulating catecholamines correlate with acute episodes of heart failure (i.e.“Takotsubo stress” cardiomyopathy) (17).
Despite its biological and clinical significance, the mechanism(s) by which acute cathecolaminergic overload causes LV impairment remains unknown. Furthermore, regardless of the mechanism mediating myocyte damage and dysfunction, the effects of adrenergic overload on CSCs remain to be elucidated.
Therefore, in the present study, a single injection of isoproterenol (ISO) was employed to induce acute diffuse myocyte damage and RyR2 expression and function in adult rat ventricular myocytes (ARVMs) were assessed in vivo and in vitro. We show here that acute β-cathecolaminergic overload causes significant myocyte apoptosis and necrosis, leading to acute but reversible cardiac failure, resembling “stress cardiomyopathy.” In addition, our data suggest that the acute ISO-induced myocyte damage is mediated by Ca2+ leakage through transiently dysfunctional RyR2 protein complex. The differentiation stage-specific pattern of expression of β1- and β2-ARs and the absence of RyR2 in the CSCs partially accounts for the resistance of these cells to ISO damage. Thus, CSCs are available to potentially contribute to cardiac recovery following acute neurohumoral stress damage.
EXPERIMENTAL PROCEDURES
Animals
Male Wistar adult rats (336 ± 18 g) received a single injection (subcutaneously) of 5 mg kg-1 ISO or saline (CTRL), and they were killed (n = 7 per group) 1, 3, and 6 days later.
Cardiac Hemodynamics and Echocardiography
Rats were anesthetized with 30% chloral hydrate (400 mg kg-1, intraperitoneally). A Millar microtip pressure transducer (Houston, TX) was advanced into the LV cavity through the right carotid artery, and LV end-diastolic and end-systolic pressures, LV developed pressure, and dP/dtmax and dP/dtmin were measured (6, 18). EKG tracing was obtained in anesthetized rats by PowerLab/16e (ADInstruments, Australia). Also, parasternal long and short axis views were obtained with both M-mode and two-dimensional echocardiography. LV dimensions (LVEDD and LVESD) were measured perpendicular to the long axis of the ventricle at the midchordal level. Fractional shortening and LV ejection fraction were calculated (6).
Myocyte Death and Hypertrophy
At the time of sacrifice, the rat heart was excised, and LV and RV were separated and weighed. In separate experiments, after obtaining LV wet weights, the LV dried weights were obtained by drying the cardiac tissue at 70 °C for 72 h. Heart tissue was fixed and embedded in paraffin before 5-μm LV cross-sections were prepared using a cryotome (Sakura). Total myocyte mass was extrapolated through morphometric analysis (18). Myocyte-specific necrotic damage was detected using a mouse anti-myosin monoclonal antibody injected in vivo (13). All animals received 1 mg kg-1 of the anti-myosin (intraperitoneally) 1 h before ISO injection and 3 h prior to euthanasia at 1, 3, and 6 days. To detect cellular apoptosis, LV sections were stained with rabbit anti-caspase-3 primary antibody (R&D Systems) as well as using the Terminal deoxynucleotidyltransferase (TdT) assay (Roche Applied Science). Dead myocytes were detected and quantified using fluorescent and confocal microscopy. Myocyte diameter was measured across the nucleus in transverse hematoxylin and eosin sections of the subendocardium layer. Myocyte volume was calculated assuming a spherical cross-section.
Blood samples were obtained from animals sacrificed 1 day after ISO injection. The measurement of cardiac troponin I (cTnI) plasma level was performed using the Access 2 immunochemiluminometric assay (Beckman Coulter).
Myocyte and CSC Isolation, Immunocytochemistry, and Histochemistry
ARVMs and CSCs were isolated from hearts of male Wistar rats (268 ± 30 g) by enzymatic dissociation (6, 18). CSCs were identified, in vivo or in vitro, using an antibody against c-Kit (6). More specifically, c-Kitpos CSCs were obtained by magnetic immunobead sorting (6), and the purity of the preparation was assessed by flow cytometry. CSCs were identified as lineage-negative (Linneg), by staining negative for markers of hematopoietic, neural, and skeletal muscle lineages (6). β1-ARs and β2-ARs on CSCs were detected with rabbit polyclonal antibodies (Santa Cruz Biotechnology, Inc., Santa Cruz, CA).
In Vitro Response of CSCs And ARVMs to ISO
ARVMs were maintained in α-MEM medium for 16 h (19) and then were supplemented with fresh ascorbic acid (0.1 mmol/liter) before being treated with 10 nm, 100 nm,1 μm,or 10 μm ISO. The 1,4-benzothiazepine derivative, JTV519 (100 nm or 1.0 μm; a kind gift of Dr. Andrew Marks, Center for Molecular Cardiology, Columbia University, New York) was added 45 min before ISO. In separate dishes, the L-type Ca2+ channel blockers, verapamil (20 μm) or diltiazem (20 μm), were added 30 min before ISO. ARVMs were fixed with 3.7% paraformaldehyde, and TdT staining was performed. The percentage of apoptotic TdT-positive (TdTpos) myocytes was determined and quantified using confocal microscopy. The same experiments were repeated using freshly isolated ARVMs.
CSCs were plated on 35-mm dishes in modified F12K medium supplemented with 3% fetal calf serum (Invitrogen) and were treated with ISO at the same concentrations as ARVMs (see above). All dishes were supplemented with ascorbic acid (0.1 mmol/liter). The β1- and β2-AR blockers, CGP20172A (0.3 μm) and ICI 118551 (0.1 μm), were added 30 min before ISO, respectively. Eight hours after ISO treatment, cells were fixed and stained, and the percentage of apoptotic TdTpos CSCs was determined (18).
Quantitative Real Time RT-PCR Analysis
Total RNA was extracted from ARVMs and CSCs using the FastTrack® MAG kit (Invitrogen). Real time RT-PCR was performed using the Taqman detection protocol in an ABI Prism 7700 thermocycler (Applied Biosystems, Foster City, CA). The results for β1-AR, β2-AR, and RyR2 real time RT-PCR assays were averaged and normalized to product produced by the housekeeping gene, GAPDH, providing a relative quantitation value. Primers were designed using the Primer Express™ program (Applied Biosystems), and the specific sequences were as follows: AAGTCCTCCCTCAAGCTCCTAAGT (forward) and TTGCTTTGCCTTTGCCC (reverse) for β-MHC; 5′-CCCGCCTCGCTGCTGCCTCC-3′ (forward) and 5′-AGCCAGCAGAGCGTGAAC-3′ (reverse) for β1-AR; 5′-ACGAGCTCAGTGTGCAGGACGCGCC-3′ (forward) and 5′-TCAAATCCCTGCCTACAACACTCCA-3′ (reverse) for β2-AR; 5′-GCAAACTTGGAGTTCTTGTCAGGCAT-3′ (forward) and 5′-TCGTTGACGTCAACAGAACTT-3′ (reverse) for RyR2; 5′-ATTGCTCTCAATGACAACTT-3′ (forward) and 5′-GAACTTTATTGATGGTATTCG-3′ (reverse) for GAPDH.
Western Blot Analysis
Immunoblots and immunoprecipitations were carried out using protein lysates obtained from freshly isolated CSCs or ARVMs (18). Generally, the equivalent of ∼50 μg of proteins were separated on gradient (6-15%) SDS-polyacrylamide gels. After electrophoresis, proteins were transferred onto nitrocellulose filters, blocked with either 5% dry milk or 5% bovine serum albumin, and incubated with rabbit polyclonal Abs against β1-AR (Santa Cruz Biotechnology), β2-AR (Santa Cruz Biotechnology), calstabin, RyR2-5029, phospho-Ser2808-RyR2 (PKA site of RyR2-phosphorylation), phospho-Ser2814-RyR2 (CaMKII site of RyR2-phosphorylation) (a kind gift of Dr. A. R. Marks), CaMKII, phospho-CaMKII, cleaved caspase-3, cleaved poly(ADP-ribose) polymerase (PARP) (Cell Signaling), PKA, phospho-PKAα (Abcam) at dilutions suggested by the manufacturers. Proteins were detected by chemiluminescence using horseradish peroxidase-conjugated anti-rabbit secondary antibodies and the Chemidoc XRS system (Bio-Rad).
Statistical Analysis
Data are reported as mean ± S.D. Significance between two groups was determined by Student’s t test and in multiple comparisons by the analysis of variance. The Bonferroni post hoc method was used to locate the differences. Significance was set at p < 0.05.
RESULTS
ISO Produces Acute but Transient Cardiac Failure
Subcutaneous administration of 5 mg kg-1 ISO resulted in ∼20% mortality of the treated animals. In these ISO-treated rats, the EKG showed ST ischemic changes followed by rapid sustained ventricular tachycardia, which developed into ventricular fibrillation and death of the animal (Fig. 1, A-F). In the majority of the rats treated with ISO (n = 21), ST changes were evident, and periods of short bursts of ventricular tachycardia were registered during the EKG monitoring immediately after ISO administration (Fig. 1, G and H). Blood pressure of ISO-treated animals was lower than in CTRL at 1 day but returned to normal values at 3-6 days after ISO injection (Table 1). Heart rates in ISO-treated rats were significantly higher at 6-24 h but returned to normal at 3 days after ISO injection (Table 1). Since the plasma half-time for ISO is typically in the range of 4-5 min, the presence of tachycardia at 1 day after ISO is mainly the result of the pathophysiological adaptation of LV mechanics to the acute and temporary decompensated functional state secondary to the ISO injection (see below).
FIGURE 1. Representative EKG tracings monitoring the acute effects of subcutaneous administration of ISO (5 mg kg-1).
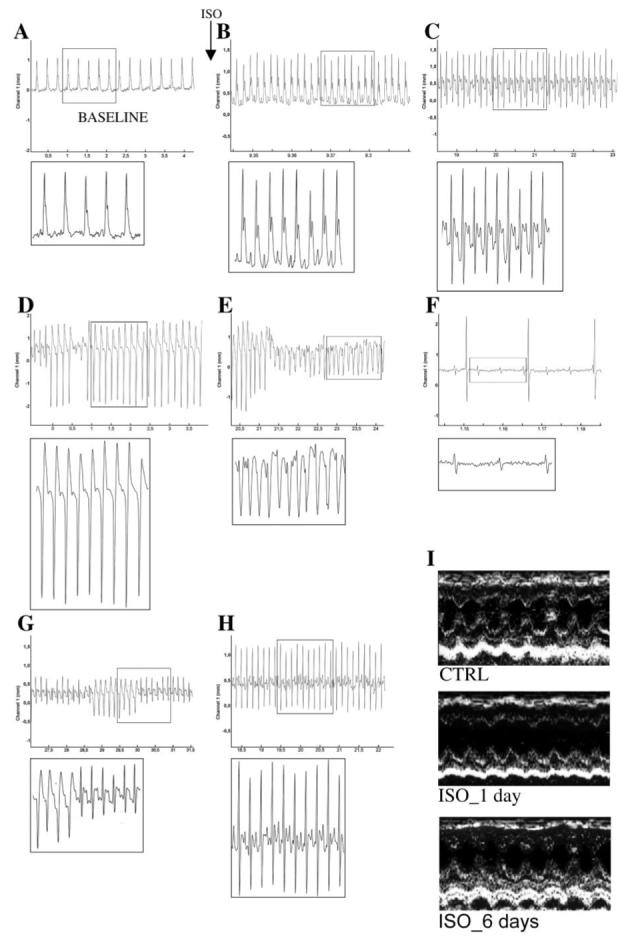
EKG monitoring was continued for up to 6 h after ISO or placebo administration. After this time period, no deaths have been observed. From every representative tracing, an insert was created to highlight the specific morphologic EKG modifications (A-H). A, EKG output of CTRL animals (BASELINE). B-F, in ∼20% of the treated rats, the EKG showed ST ischemic changes (B and C) followed by rapid sustained ventricular tachycardia, which turned into ventricular fibrillation and death of the animal (D-F). G and H, in the majority of the rats treated with ISO, the same acute ST changes were evident, and periods of short bursts of ventricular tachycardia were registered during the EKG monitoring after ISO administration. I, the representative two-dimensional echocardiogram of a surviving animal shows a dramatic dilatation of the LV at day 1, as compared with the recording prior to ISO administration (CTRL). The chamber dimensions have returned to normal at day 6.
TABLE 1.
Hemodynamic, echocardiographic, and anatomical measurements
| CTRL | ISO 1 day | ISO 3 days | ISO 6 days | |
|---|---|---|---|---|
| SBPa (mm Hg) | 93 ± 4 | 81 ± 5b | 94 ± 4 | 95 ± 4 |
| DBPc (mm Hg) | 70 ± 1 | 59 ± 5b | 69 ± 9 | 72 ± 8 |
| HRd (beats/min) | 402 ± 20 | 446 ± 20b | 408 ± 23 | 405 ± 15 |
| LVDDe (mm) | 4.7 ± 0.4 | 5.8 ± 0.1b | NDf | 5.1 ± 0.3g |
| LVSDh (mm) | 2.6 ± 0.4 | 4.3 ± 0.3b | ND | 3.1 ± 0.4g |
| Fractional shortening (%) | 45 ± 4 | 26 ± 3b | ND | 40 ± 4g |
| Ejection fraction (%) | 78 ± 6 | 57 ± 6b | ND | 72 ± 5g |
| LVEDPi (mm Hg) | 3.8 ± 1.5 | 21.0 ± 4.2b | 15.4 ± 7.4b | 10.4 ± 2.1b,g |
| LVDevPj (mm Hg) | 88.5 ± 8.2 | 64.3 ± 12.7b | 81.7 ± 9.1g | 86.7 ± 7.9g |
| dP/dtmax (mm Hg/s) | 8124 ± 1027 | 6546 ± 888b | 7576 ± 365 | 8116 ± 722g |
| dP/dtmin (mm Hg/s) | 7889 ± 1240 | 5730 ± 625b | 6739 ± 964 | 7906 ± 743g |
| LV wet weight (mg) | 767 ± 32 | 830 ± 58 | 873 ± 43b | 825 ± 26 |
| LV dry weight (mg) | 165 ± 7 | 153 ± 9 | 192 ± 7b,g | 181 ± 6b,g |
| Myocyte volume (mm3) | 12,827 ± 1612 | 13,360 ± 2600 | 20,213 ± 3664b,g | 17,132 ± 2248b |
Systolic blood pressure.
p < 0.05 versus CTRL.
Diastolic blood pressure.
Heart rate.
Left ventricular diastolic diameter.
ND, not determined.
p < 0.05 versus ISO 1 day.
Left ventricular systolic diameter.
LV end-diastolic pressure.
LV developed pressure.
Indeed, ventricular function parameters document that the single ISO injection caused marked changes in LV performance (Table 1). At 1 day after injection, when compared with CTRL, ISO-treated animals exhibited a significantly decreased (p < 0.05) LV developed pressure, dP/dtmax and dP/dtmin and increased LV end-diastolic pressure, uncovering acute and severe LV failure in all treated animals. These parameters started to improve 3 days after treatment and had returned to control values by day 6 except for end-systolic pressure, which still was lower than at 1 day after ISO (Table 1). Echo data confirmed that ISO caused acute LV failure at 1 day and that LV function spontaneously recovered at 6 days (Fig. 1I and Table 1).
ISO Causes Myocyte Death through both Necrosis and Apoptosis
In the rat heart with patent coronary circulation, ISO exposure caused diffuse necrotic and apoptotic myocyte death throughout the myocardium (Figs. 2 and 3). Hematoxylin and eosin cross-sections from ISO-treated hearts displayed myocytes with disrupted sarcolemmal membranes and pale cytoplasmic staining, implicating focal necrosis. This was most evident in the subendocardium (Fig. 2A). Furthermore, 3 days after ISO-induced damage, an increased inflammatory reaction was evident (Fig. 2A). Myocyte-specific necrosis was detected by injecting in vivo a mouse anti-myosin monoclonal antibody, which exclusively tags necrotic myocytes (13) (Fig. 2B). The fraction of necrotic myocytes was 8 ± 2% at 1 day in the subendocardial layer (p < 0.001 versus CTRL; 0 ± 0%; Fig. 2C). The other two layers were less affected by ISO (data not shown).
FIGURE 2. Isoproterenol caused extensive acute myocardial necrosis in the LV.
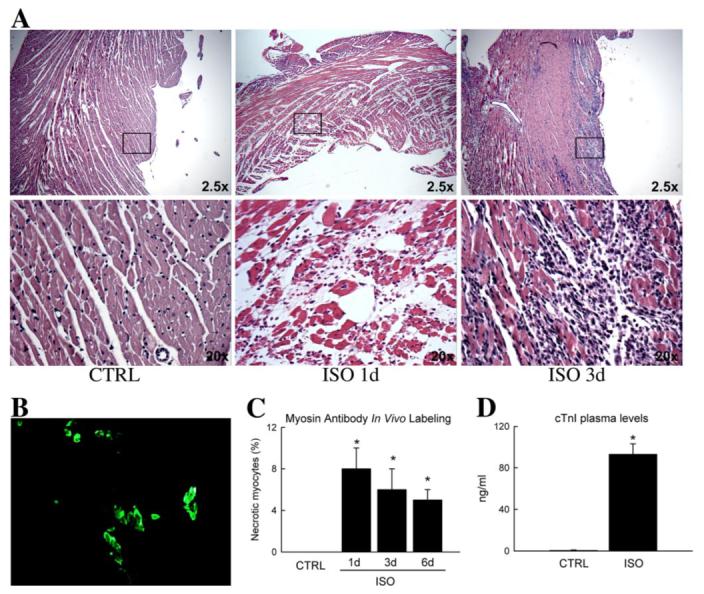
A, low magnification (×2.5) representative hematoxylin and eosin cross-sections of the apical LV wall, epi- to endocardial, from CTRL and ISO-treated hearts. The higher magnification (×20) inset at 1 and 3 days after ISO shows myocytes with disrupted sarcolemmal membranes and pale cytoplasmic staining, implicating focal necrosis. B, representative picture of specific necrotic myocytes labeled in vivo with anti-myosin antibody (green). C, the fraction of necrotic myocytes were significantly increased at 1 day in the LV and decreased over time. *, p < 0.05 versus CTRL. D, plasma cTnI was significantly elevated at 1 day after ISO. *, p < 0.001 versus CTRL.
FIGURE 3. Isoproterenol caused acute myocyte apoptosis along with RyR2 dysfunction and reactive myocyte hypertrophy in the LV.
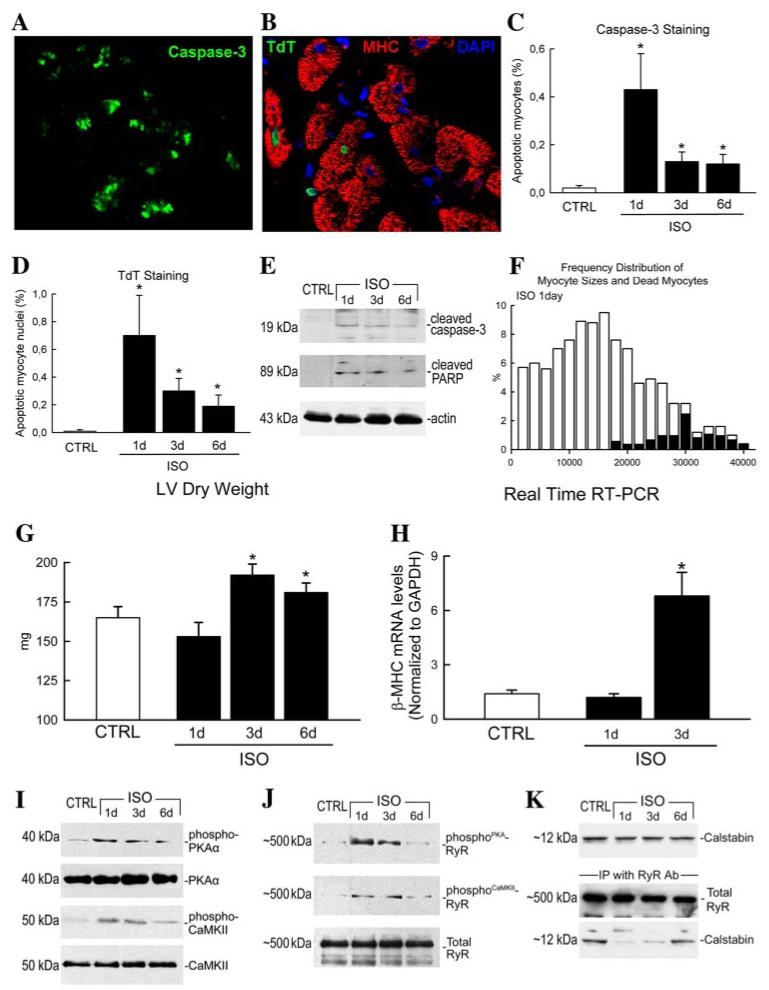
A-D, representative picture of apoptotic myocytes stained by activated caspase-3 (A)(green) and TdT (B) (TdT (green), MHC (red), and DAPI (blue)). C and D, apoptotic (caspase-3- and TdT-positive) myocytes were significantly increased at 1 day in the LV and progressively decreased over time. *, p < 0.05 versus CTRL. E, representative Western blots of cleaved caspase-3 and PARP in isolated myocytes after ISO injection. Changes to caspase-3 and PARP progressively decreased over 6 days. F, distribution of myocyte sizes (open bars) at 1 day after ISO showing the fraction of dead myocytes (necrotic and apoptotic; solid bars) only in the largest myocytes. G, LV dry weight was slightly decreased at 1 day but significantly increased at 3 days, indicating net loss of myocytes followed by reactive cardiac hypertrophy at days 1-3 after ISO injection. *, p < 0.05 versus CTRL. H, quantitative real-time RT-PCR showed increased mRNA transcripts of β-MHC in ARVMs isolated from ISO-treated hearts at 3 days when compared with CTRL and 1 day. Data are presented as the ratio between numbers of β-MHC and GAPDH mRNA molecules per μg of RNA. *, p < 0.01. I and J, representative Western blots of phospho-PKA, PKAα, phospho-CaMKII, CaMKII, total RyR2, and PKA- and CaMKII-mediated RyR2 phosphorylation levels in ARVMs isolated from hearts of rats at 1, 3, and 6 days after ISO or saline vehicle (CTRL). K, the levels of calstabin were not affected by ISO injection, whereas calstabin was displaced from the RyR2 complex at 1 day as shown by the immunoprecipitation with RyR2 and the Western blot for calstabin.
Necrotic myocytes progressively decreased from day 3 to 6 but were still higher than CTRL (p < 0.05; Fig. 2C). All evidence indicates that most, if not all, of the cardiac damage occurred during the first 24 h, but the scavenger system was overloaded to efficiently remove the cell debris.
To confirm myocyte necrosis after ISO injection, we measured cTnI in blood samples taken from the animals sacrificed 1 day after ISO. Increased plasma levels of cTnI or cardiac troponin T are highly specific for and correlate with the size of the damaged myocardium (20). ISO treatment resulted in a significant increase (p < 0.001) in blood cTnI (Fig. 2D), reaching 93 ± 10 ng/ml, which in humans corresponds to large infarct sizes (20). These values are in agreement with the loss of 17 ± 4% of myocyte mass 1 day after ISO administration (see below).
ISO-induced myocyte apoptosis was identified by labeling for caspase-3 and was confirmed using the TdT (terminal dUTP nick-end labeling) assay with dUTP (Fig. 3, A-D). The number of apoptotic myocytes in CTRL animals was minimal (caspase-3-labeled myocytes, 0.02 ± 0.01%; TdT-labeled myocyte nuclei, 0.01 ± 0.01%). In contrast, the percentage of apoptotic myocytes in the LV of ISO-treated animals was significantly (p < 0.05) increased at 1 day (caspase-3, 0.4 ± 0.16%; TdT, 0.7 ± 0.29%) in the severely damaged subendocardial layer (Fig. 3, C and D), being less in the midwall and subepicardial layers (data not shown), compared with CTRL. Like necrotic myocytes, the myocyte apoptosis rate had decreased by 3-6 days after ISO but was still significantly greater than CTRL (Fig. 3, C and D). As was the case for necrotic cell death, most death by apoptosis also occurred at the early time points. The cell bodies evident at late time points could most likely represent a delay in their removal due to saturation of the scavenger system, as indicated above, and not continuous cell loss.
To obtain further evidence of programmed myocyte death, ARVMs were isolated from ISO-treated rats at 1, 3, and 6 days and CTRL rats. Caspase-3 activation was assessed by detecting both its active cleavage product and its activity by evaluating cleavage of PARP into its 89-kDa form, which is considered a hallmark of apoptosis (21). Immunoblot analysis of protein extracts from isolated ARVMs showed an absence of cleavage of caspase-3 in CTRL rats (Fig. 3E). In contrast, ISO injection produced a remarkable caspase-3 cleavage in ARVMs 1 day after ISO (Fig. 3E). Consistent with these findings, PARP cleavage was significantly increased in ARVMs from ISO-treated compared with CTRL rats (Fig. 3E). In agreement with the quantitative immunohistochemistry data, the molecular changes to caspase-3 and PARP progressively decreased 3-6 days after ISO (Fig. 3E).
Not all myocytes were equally vulnerable to ISO damage and death by necrosis and/or apoptosis. In all cases examined, the myocyte death affected almost exclusively the larger myocytes while sparing those of normal and smaller size (Fig. 3F). As a consequence of the selective death of the larger myocytes, the loss of myocardial mass in response to ISO, calculated as a fraction of myocardial volume corresponding to myocytes (18), is disproportionate to the number of myocytes lost. The discrepancy between this value with the loss of only ∼8% of myocytes by necrosis and ∼0.5% dying by apoptosis translates into a loss of 17 ± 4% of myocyte mass 1 day after ISO administration.
Reactive Myocyte Hypertrophy Follows Diffuse Myocyte Death Produced by Acute Catecholaminergic Overload
LV wet weight was significantly increased (p < 0.05) at 1-3 days, when compared with CTRL. This was probably due to myocardial edema (Table 1). Indeed, LV dry weight slightly decreased 1 day after ISO injection, strongly suggesting the presence of myocardial edema. The fact that LV dry weight decreased but did not reach statistical significance at 1 day after ISO despite the calculated myocardial mass loss of ∼17% could be partially explained by the persistence of the necrotic myocytes that have not yet been removed by the scavenger system. Then LV dry mass increased at 3 days (Fig. 3G and Table 1), indicative of cardiac hypertrophy after ISO-induced myocyte loss. Accordingly, a significant reactive myocyte hypertrophic response was observed at 3 days after ISO. The average myocyte volume was markedly increased (p < 0.05) after 3 days, and it decreased slightly at day 6 but remained consistently higher (p < 0.05) than CTRL (Table 1). These values were consistent with LV dry weight (Table 1 and Fig. 3G). A myocyte hypertrophic response was further suggested by quantitative real time RT-PCR, which showed increased transcription for β-MHC in myocytes isolated from hearts 3 days after ISO injection (Fig. 3H), since it occurs during hypertrophy.
Acute ISO-induced Myocyte Damage Is Associated with Hyperphosphorylation of RyR2
RyR2 is hyperphosphorylated in end stage heart failure (3, 10). PKA- and CaMKII-dependent hyperphosphorylation of RyR2 induces myocyte death secondary to Ca2+ cell leakage (12, 22, 23). Specifically, PKA phosphorylation dissociates the regulatory protein calstabin (also known as FKBP 12.6) from RyR2, altering the gating of the channel, which results in Ca2+ leakage during diastole in failing hearts (10). Increased systemic catecholamine levels produced by isoproterenol infusion can activate hyperphosphorylation of RyR2 in vivo (14). Therefore, we investigated whether the myocyte death produced by a single dose of ISO employed in the present study might be the consequence of RyR2 protein complex dysfunction.
PKA and CaMKII activity was significantly elevated as shown by the increased phosphorylation of the two kinases in ARVMs isolated 1 day after ISO administration (Fig. 3I). This resulted in significant PKA- and CaMKII-dependent RyR2 phosphorylation, compared with CTRL (Fig. 3J). Importantly, PKA and CaMKII activation progressively decreased and RyR2 returned from the hyperphosphorylated calcium-leaking form present at 1 day to the normal calcium-handling form at days 3-6 after ISO (Fig. 3, I and J). Immunoprecipitation of RyR2 from myocytes isolated from ISO-treated hearts showed displacement of calstabin from the RyR2 complex at 1 day (Fig. 3K). The return of the normal calcium-handling form of RyR2 at 3-6 days after ISO (Fig. 3, I and J) was associated with reestablishment of a normal calstabin-RyR2 complex (Fig. 3K).
Hyperphosphorylation of RyR2 Mediates Myocyte Death in Vitro
To assess whether hyperphosphorylation of RyR2 directly mediates the high myocyte death measured after ISO, we exposed ARVMs isolated from CTRL rats to ISO in the absence or the presence of JTV519 (1 μm) in vitro (Fig. 4, A-F). This molecule is a member of a class known as calcium channel stabilizers, shown to increase calstabin binding to RyR2 (24) and therefore to stabilize the RyR2. Increasing doses (10 nm to 10 μm) of ISO led to increased myocyte apoptosis (Fig. 4C). ARVMs at 5, 10, and 20 min after ISO (10 μm) exposure exhibited an increased phosphorylation of PKA and CaMKII and significant PKA- and CaMKII-dependent RyR2 phosphorylation compared with CTRL (Fig. 4G). This hyperphosphorylation of the RyR2 by excess ISO exposure was associated with dissociation of calstabin from the RyR complex (Fig. 4G). Interestingly, JTV519 (1 μm) significantly prevented myocyte apoptosis induced by ISO (Fig. 4D). Furthermore, JTV519 prevented displacement of calstabin from RyR2 complex, indicating that JTV519 acts downstream of PKA and CaMKII. This is so because activation of the two kinases was not affected by JTV519 treatment (Fig. 4G). Thus, despite the kinase activation, JTV-induced calstabin-RyR2 protein complex protection was associated with a significant reduction of both PKA and CaMKII-dependent RyR phosphorylation (Fig. 4G). Also at a concentration of 100 nm, JTV519 reduced ISO-induced ARVM apoptosis (data not shown).
FIGURE 4. Isoproterenol induces myocyte death through RyR-2 hyperphosphorylation and calstabin complex dissociation.
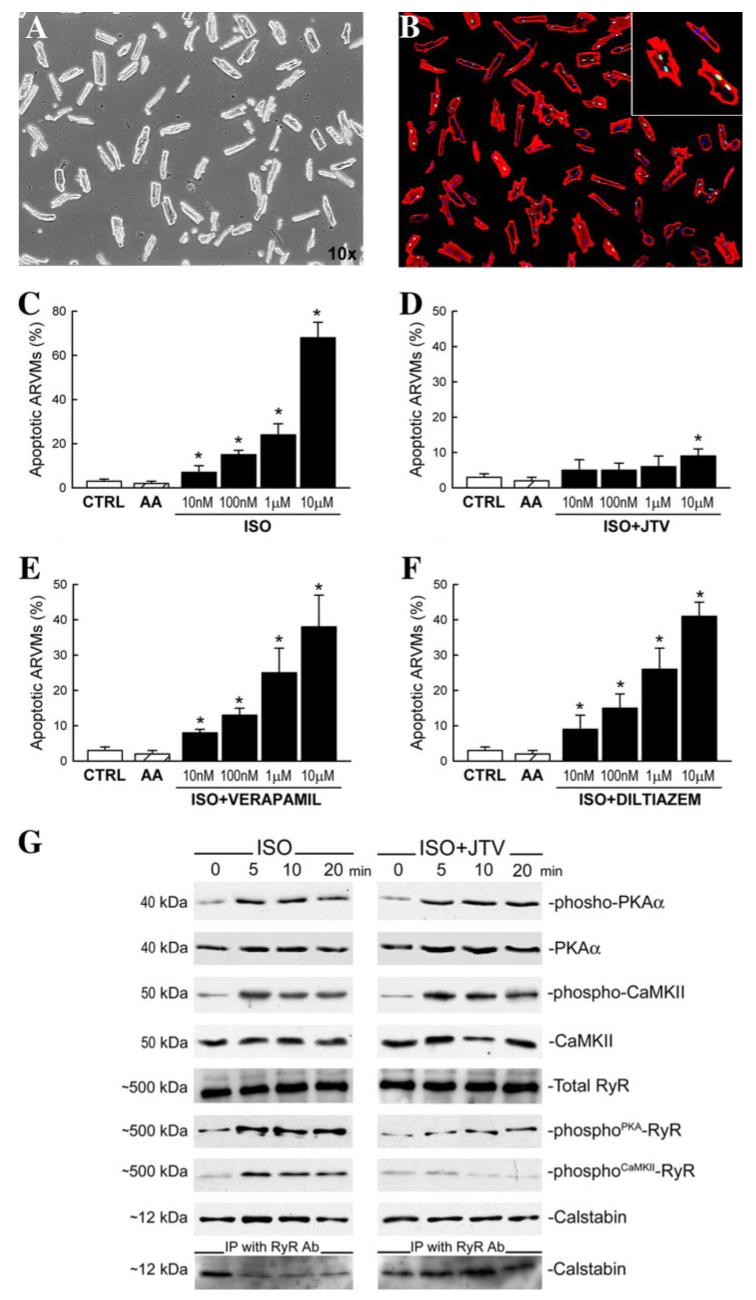
A, ARVMs were isolated and plated in culture dishes before ISO was administered in vitro, and cell apoptosis was measured by TdT labeling (B, TdT (green), MHC (red), and DAPI (blue)). C, ISO caused increased apoptosis of ARVMs in a dose-dependent manner (all dishes were supplemented with 0.1 mm ascorbic acid (AA); hatched bars). *, p < 0.001 versus CTRL and ascorbic acid alone. D, the presence of JTV519 (1.0 μm) prevented ISO-induced myocyte apoptosis. E and F, on the other hand, verapamil (20 μm) and diltiazem (20 μm) had no effect on attenuating ISO-induced apoptosis. *, p < 0.001 versus CTRL and ascorbic acid alone. G, representative Western blots of phospho-PKA, PKAα, phospho-CaMKII, CaMKII, total RyR2, and PKA- and CaMKII-mediated RyR2 phosphorylation levels and calstabin in in vitro ARVMs cultured with ISO for 0, 5, 10, and 20 min in the absence or presence of JTV. Also, representative immunoprecipitation with RyR2 antibody and the Western blot for calstabin is shown.
On the other hand, both specific L-type calcium channel blockers verapamil and diltiazem had no significant effect on ISO-induced ARVM death (Fig. 4, E and F). Thus, acute catecholaminergic excess causes hyperphosphorylation of RyR2, which, in turn, destabilizes the channel with consequent Ca2+ leakage from the SR followed by myocyte death by either apoptosis or necrosis.
Cardiac Stem Cells Are Resistant to the Damaging Effects of ISO because of Low Levels of β-ARs and the Absence of RyR2
Apoptotic c-Kitpos CSCs were a very rare, if not negligible, finding in the CTRL rat heart (Fig. 5A). Following 5 mg kg-1 ISO injection, the fraction of apoptotic CSCs was not significantly different from CTRL rats (Fig. 5A). Considering that ISO cardiotoxicity is mediated via β1-ARs (13, 16), we investigated whether the low level expression of β1/β2-ARs in CSCs could explain their resistance to the deleterious effects of ISO. To this aim, c-Kitpos CSCs were freshly isolated by magnetic bead immunosorting from CTRL rats (Fig. 5B) and analyzed for β1- and β2-AR mRNA and protein expression using quantitative real time RT-PCR, Western blot, and immunocytochemistry. Data analysis showed that c-Kitpos CSCs expressed both β1- and β2-ARs, but in considerably lower, if not negligible, amounts when compared with ARVMs (Fig. 5, C-E; p < 0.01). Moreover, although β1 is the most abundant AR in myocytes, the β1/β2-AR ratio was inverted in CSCs (Fig. 5, C-E; p < 0.01).
FIGURE 5. Adult c-Kitpos CSCs exhibit decreased and inverted β1- and β2-AR expression.
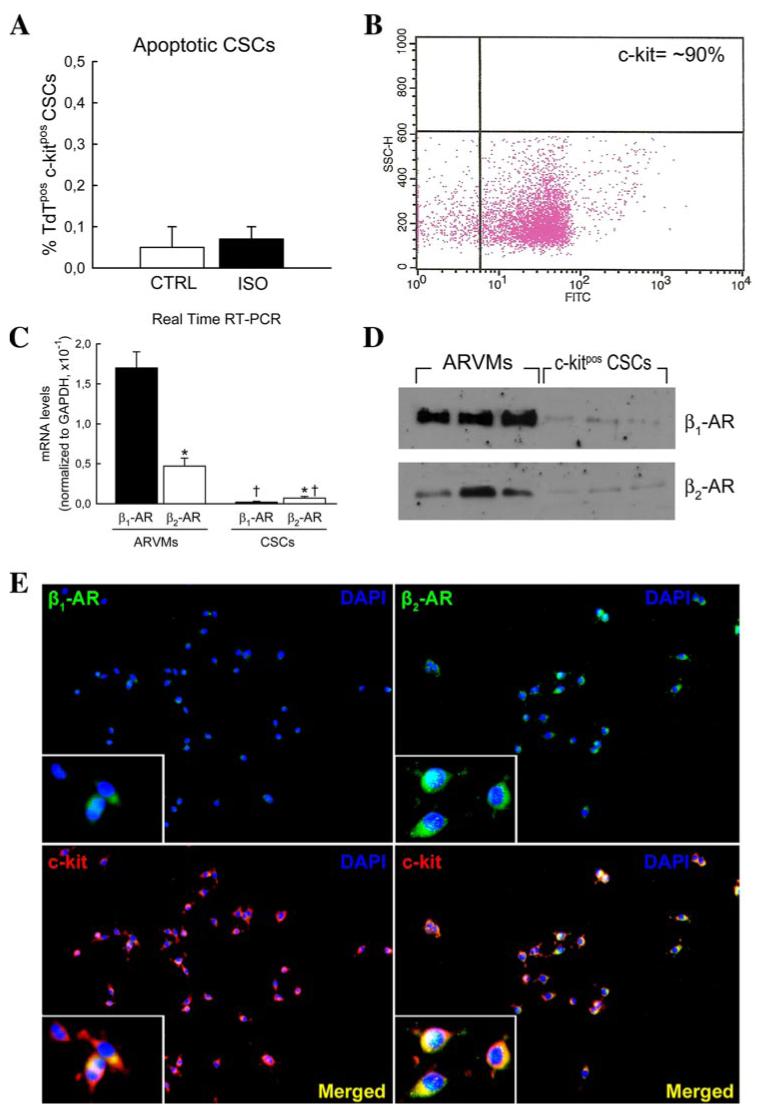
A, fraction of apoptotic c-Kitpos CSCs in the hearts of CTRL and ISO-treated (1 day) rats (p = not significant). B, c-Kitpos CSCs sorted by immunomagnetic beads showed a purity of ∼90% when analyzed by flow cytometry. C and D, representative real time RT-PCR and Western blot results showing decreased mRNA transcripts and protein expression of β1- and β2-ARs in isolated c-Kitpos CSCs compared with ARVMs. c-Kitpos CSCs exhibited an inverted expression of β1- and β2-ARs. RT-PCR data are presented as the ratio between numbers of β1- or β2-ARs and GAPDH mRNA molecules per μg of RNA. *, p < 0.01 versus β1-AR; ‡, p < 0.01 versus ARVMs. E, β1- and β2-ARs were also detected on isolated c-Kitpos CSCs using immunocytochemistry. Left, β1-ARs (green), c-Kit (red), and DAPI (blue). Right, β2-ARs (green), c-Kit (red), and DAPI (blue). Merged images (yellow) are shown at the bottom. Insets, magnified images.
To further ascertain whether these phenotypic differences were in fact responsible for the differential response to ISO in vivo, we directly tested in vitro the effects of the drug on survival of CSCs (Fig. 6, A-E), as compared with ARVMs. Although ARVMs showed a progressive increase of apoptosis with increasing ISO concentrations (Fig. 4C), this was in fact 3-fold higher than CSC apoptosis (Fig. 6, A-C). Importantly, the rate of apoptotic death in CSCs at the highest dose of 10 μm was similar to that of ARVMs at 10 nm ISO concentration (Figs. 4C and 6C). We then investigated separately the role of β1- and β2-AR signaling in CSC apoptosis. Interestingly, we found that β1- and β2-ARs differentially trigger apoptosis in CSCs. In the presence of ISO, a specific β1 blocker (CGP20172A; 0.3 μm) prevented the CSC apoptotic cell death (Fig. 6D). No such protection was apparent in the presence of the β2 blocker, ICI 118551 (0.1 μm) (Fig. 6E). Similar results were obtained with ARVMs (data not shown). Thus, the deleterious effects of β-adrenergic overstimulation in both myocytes and CSCs signals preferentially through the β1-AR. However, whether β-AR signaling is functionally intact in CSCs remains to be elucidated.
FIGURE 6. Adult c-Kitpos CSCs are resistant to the damaging effects of ISO.
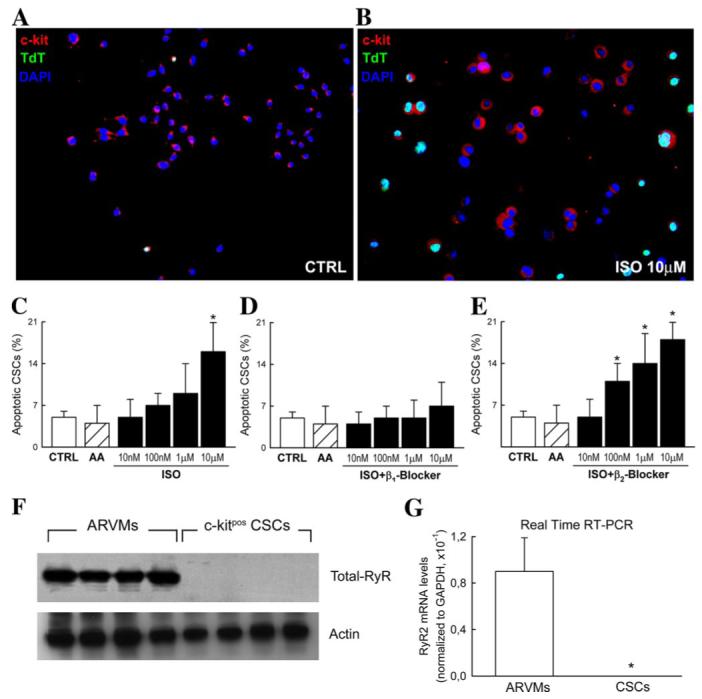
A and B, representative immunostaining showing apoptotic CSCs (c-Kit (red), TdT (green), and DAPI (blue)) in CTRL (A) and ISO-treated (B) CSCs in vitro. C, isolated c-Kitpos CSCs exposed to the same doses of ISO as ARVMs (Fig. 4C) were 2 orders of magnitude more resistant. Subsequently, only the highest dose of ISO (10 μm) caused significant CSC apoptosis. *, p < 0.01 versus CTRL and AA alone. D, the specific β1-AR blocker, CGP20172A (0.3 μm), attenuated CSC apoptosis. E, in contrast, in the presence of a specific β2-AR blocker (ICI 118551; 0.1 μm) CSC apoptosis increased with ISO dose. F and G, representative Western blot and quantitative real time RT-PCR results showing the absence of RyR2 in c-Kitpos CSCs when compared with ARVMs. Data are presented as the ratio between numbers of RyR2 and GAPDH mRNA molecules per μg of RNA. *, p < 0.01 versus ARVMs.
ISO induced myocyte death by Ca2+ leakage through acutely dysfunctional RyR2. Consequently, we evaluated the presence of RyR2 mRNA and protein in isolated CSCs. As shown in Fig. 6, F and G, CSCs do not express RyR2, reflecting their undifferentiated state, despite their potential to acquire the cardiac myocyte phenotype. Taken together, the difference in expression of β1-ARs and RyR2 between CSCs and myocytes provides a satisfactory explanation for the resistance of the former and sensitivity of the latter to the β-adrenergic overload in vivo.
Cardiac Stem Cells Are Activated as a Consequence of ISO-induced Myocyte Damage
Considering the survival of CSCs after ISO exposure, we investigated their exact response to the ISO challenge. A significant (p < 0.05) increase (∼8-fold) in c-Kitpos CSC number after ISO injection in vivo was observed. This peaked at 3 days and remained elevated at 6 days, compared with CTRL (Fig. 7A). When analyzed by quantitative immunohistochemistry, the percentage of proliferating CSCs (c-Kitpos/Ki67pos) was 3% in CTRL rat hearts, reaching ∼16% (p < 0.05), 1 day after ISO injection (Fig. 7, B and C). Of note, CSC activation was not the result of increased ISO per se, since excess ISO exposure in vitro had no effect on CSC proliferation (Fig. 7D). These findings document the responsive activation and reentry of the cell cycle of CSCs after ISO, which is mediated by activators other than ISO exposure per se.
FIGURE 7. Adult c-Kitpos CSCs are activated in response to ISO-induced damage.
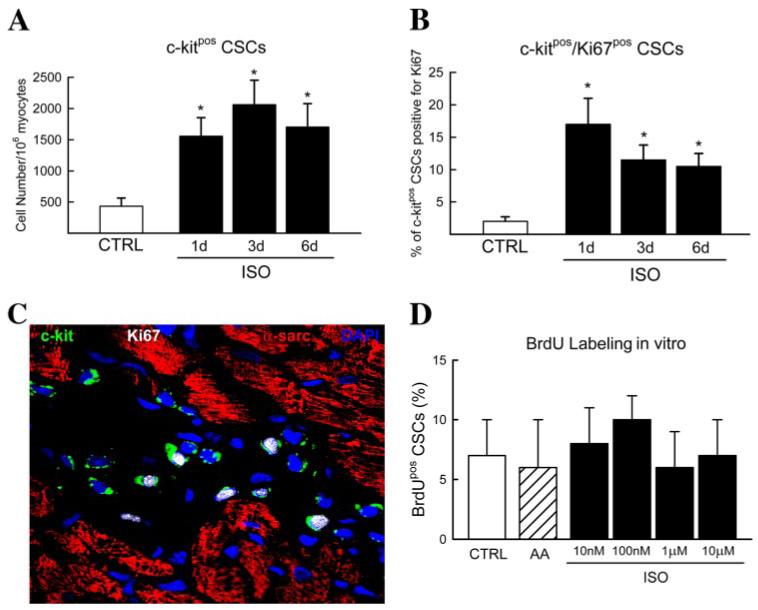
A, c-Kitpos CSC number was elevated in the myocardium after ISO administration. *, p < 0.05 versus CTRL (B). The percentage of c-Kitpos CSCs positive for the cell cycle marker, Ki67, increased after ISO injection, peaking at 1 day. *, p < 0.05 versus CTRL. C, representative immunohistochemistry staining of c-Kitpos/Ki67pos CSCs in ISO-treated rat myocardium (c-Kit (green), Ki67 (white), α-sarcomeric actin (red), and DAPI (blue)). D, ISO exposure had no effect on CSC proliferation, measured by bromodeoxyuridine incorporation, in vitro (p = not significant).
DISCUSSION
Five major findings emanate from the present study: (i) acute isoproterenol overload causes extensive myocyte necrosis and apoptosis; (ii) this diffuse myocyte loss is followed by acute but transient LV function impairment; (iii) the ISO-induced myocyte damage is due to increased sarcolemmal Ca2+ in the myocytes as a consequence of its leakage from the SR through a dysfunctional RyR2; (iv) the CSCs are spared from the ISO-induced myocardial damage because of a very low level of expression and inverse ratio of β1/β2-ARs and the absence of RyR2; (v) the resistance of CSCs to acute adrenergic overload and their subsequent activation leave these primitive cells readily available to regenerate the myocyte cell loss modulated by ISO.
Acute β-Adrenergic Overload Causes Myocyte Death through Hyperphosphorylation of RyR2 and Calstabin Displacement
A variety of pathological states are associated with increased myocyte necrosis and apoptosis, which can result in a “myocyte deficit” and impaired performance and/or failure (5, 25, 26). It has been suggested that increased sympathetic activity that accompanies low cardiac output states, through stimulation of β-ARs, plays an important role in the pathophysiology of myocardial failure (11). Chronic elevated levels of catecholamines can generate continuous dropout of myocytes from the heart by apoptosis and necrosis, contributing to progressive worsening of LV function (13-16).
As shown here, acute ISO injection produced extensive focal cell death by necrosis and apoptosis, which in the first day affected 8 ± 4% of the LV myocytes. Because the larger myocytes are the most susceptible to stress and respond by triggering apoptosis and necrosis, the total number of cells lost represented 17 ± 4% of the myocyte mass. The greatest amount of myocyte death occurred in the apical region and the subendocardial layer, which is in agreement with our previous findings (13) and the regions that are also most susceptible to ischemic injury (27). It is well documented that similar levels of myocyte loss are significantly more deleterious to ventricular function when they occur dispersed throughout the myocardium, as in certain myocardiopathies, than when they are restricted to a specific segment, as occurs in a myocardial infarction (28). It is not surprising therefore that the extensive cell death produced by the ISO overdose resulted in acute LV dysfunction.
Increased levels of circulating catecholamines activate β-ARs, elevate intracellular cAMP, and activate PKA (14). PKA as well as CaMKII modulates RyR2 and phospholamban function both in vitro and in vivo, acutely increasing short term myocardial contractility (14). In the long term, maladaptation of this pathway plays an important role in the pathophysiology of heart failure (14). Accordingly, Zhang et al. (29) have recently shown that CaMKII is dispensable for physiological “fight or flight” myocardial response, but CaMKII inhibition protects against myocardial infarction and damage produced by excessive β-AR stimulation. RyR2 is PKA- and CaMKII-hyperphosphorylated in end stage heart failure, leading to dissociation of calstabin, Ca2+ leakage from the SR, and impaired contractility (3, 10). The results of Zhang et al. (29), together with the data presented here, suggest that phosphorylation of RyR2 by CaMKII is as significant as the phosphorylation by PKA in producing the functional impairment of the RyR2 complex.
Defective Ca2+ handling in the excitation-contraction cycle has been implicated not only in heart failure but also in certain arrhythmias (30, 31), supporting the conclusion that proper regulation of the RyR2 channels are of key importance in determining myocyte cell fate (3, 10). Similar mechanisms induce cardiomyocyte death after acute high dose ISO administration. Indeed, myocytes from ISO-treated animals show hyperphosphorylation of the RyR2 and displacement of calstabin from the RyR2 protein complex at 1 day, in correlation with the peak of myocyte death. The hyperphosphorylated Ca2+-leaking form of RyR2 progressively declined over the time period of examination and was replaced by the normofunctioning phosphorylated form. The return of the normophosphorylated form of RyR2 at 3-7 days after ISO was coupled with reestablishment of normal calstabin-RyR2 complex.
It should be pointed out that the antibodies used in this study only monitor two possible phosphorylation sites on RyR2. Therefore, we cannot exclude that other unknown phosphorylation sites on RyR2 can be activated by isoproterenol. However, it should be also noted that PKA specifically phosphorylates Ser2808 on RyR2. Indeed, when this serine phosphorylation site was genetically ablated, RyR2 could not be PKA-phosphorylated during activation of the β-AR signaling pathway through isoproterenol infusion in vivo (32).
The dysfunctional RyR2 was associated with acute LV impairment, and the progressive normalization of both RyR2 phosphorylation and RyR2-calstabin complex tracked with restoration of LV function. This interpretation is supported by the finding that when the RyR2-calstabin complex displacement was antagonized with JTV519, the rate of ISO-induced ARVM death was decreased by ∼7-fold. In addition to enhancing calstabin binding to RyR, JTV519 has been shown to inhibit annexin-V-dependent Ca2+ influx and to block voltage-gated Na+, K+, and Ca2+ channels. However, because none of these other known targets of JTV519 have been associated with therapeutic benefit in the treatment of heart failure, it is unlikely that these off target activities of JTV519 are responsible for the beneficial effects of the drug in preventing the detrimental consequences of excess catecolaminergic drive. Also, it is important to highlight that JTV519 did not improve cardiac function in calstabin-2-/- mice, strongly suggesting that the main therapeutic effect of JTV519 depends on calstabin-2 binding to RyR2 (24).
Together, these findings support the view that catecholamine cardiotoxicity to myocytes results from increased Ca2+ secondary to the dysfunction of the homotetrameric RyR2-calstabin complex and consequent leakiness of this channel (3, 10, 24).
CSCs Are Resistant to Acute Hyperadrenergic Stimulation
Recent data tend to present a new view of a more plastic cardiac cellular homeostasis (4, 5, 33). Although the vast majority of adult myocytes are unquestionably terminally differentiated and incapable of reentering the cell cycle (34), evidence of actual new myocyte formation in the adult heart has been obtained (5). These cells are the result of the differentiation of CSCs, which represent a “cardiac cellular reserve” involved in maintaining cardiac cell homeostasis throughout life and potentially could be a valuable resource for myocardial cell replacement protocols (4, 5, 33).
The understanding of CSC biology and their roles under different physiological and pathological conditions is still in its infancy and is likely to take significant experimental work to be fully elucidated (33). One of the key questions is whether the mature myocytes and their progenitors, the CSCs, are similarly or differentially affected by specific insults. In the first case, it would be expected that damage to the working myocardium would, at the same time, hamper the prospects for recovery because of the concomitant loss of the cells with regenerative potential, the CSCs. In contrast, another alternative would be to expect that under proper conditions the myocardial damage should be reversible by the spared CSCs. The latter appears to be the case.
CSCs displayed a differential and apparently opposite response to acute β-adrenergic overstimulation compared with myocytes in vivo. Indeed, whereas myocytes were severely damaged, CSC survival was not affected by ISO injection. Interestingly, when CSCs were exposed to serial and increasing concentrations of ISO in vitro, CSC death was apparent only at the highest dose (10 μm), compared with myocytes.
The low level of expression of β1- and β2-AR together with the absence of RyR2 in CSCs explains their resistance to ISO-induced death and their preferential survival. Additionally, it is speculated that CSC resistance to ISO-induced apoptosis could also be improved by the relatively higher abundance of β2-ARs and an inverted β1/β2-AR ratio compared with myocytes, probably reflecting the presence of immature CSC-derived myogenic progenitors/precursors in the CSC pool. This could produce a β2-AR-mediated antiapoptotic effect through the Gi-stimulatory pathway (19, 35, 36). Since the β-adrenergic receptor system is developmentally less defined in CSCs, whether the β-AR signaling system is functionally intact in CSCs warrants further investigation.
The fact that CSCs are resistant to ISO overload leaves these cells viable and functional for their physiologic/pathophysiologic regenerative potential. Indeed, CSC number increased, and the percentage of proliferating CSCs rapidly elevated after ISO-induced myocardial damage. Thus, one intriguing possibility, still under investigation, is that following ISO injury, LV functional progressive recovery is also due to a phenomenon of myocyte replacement by CSC activation along with adaptative myocyte hypertrophy.4 Nevertheless, it could also be speculated that LV cardiac function amelioration after the single high dose of ISO is the product of progressive recovery from catecholamine-induced myocardial stunning.
ISO-induced Damage, a Model for Takotsubo Cardiomyopathy?
The pattern of damage (mainly apical and endomyocardial) and rapid restoration of LV function after ISO shock has many similarities to the stress (Takotsubo) cardiomyopathy, which occurs predominantly in women after an episode of acute emotional distress (37). The mechanism responsible for this phenomenon is still under investigation.
Wittstein et al. (17) reported that exaggerated sympathetic stimulation in stress cardiomyopathy patients is central to the syndrome. Acutely, these patients exhibited elevated cTnI levels and supraphysiologic plasma catecholamine levels. Endomyocardial biopsies were consistent with cell death and an elevated catecholamine state. Furthermore, all had severe LV dysfunction, which resolved in ∼21 days (17). Thus, if the stress cardiomyopathy is due to direct myocyte injury from increased catecholamines, it should be determined whether CSC activation is an important contributor to the rapid clinical recovery.
ISO-induced Damage: An in Vivo Assay to Study Myocardial Regeneration
Despite a burgeoning set of data on CSCs, the role of CSCs in the cardiac response to injury is still questioned and challenged (see Refs. 4 and 33). This skepticism is mainly due to the absence of myocyte regeneration sufficient to restore myocardial integrity and function after coronary occlusion. Unfortunately, most animal models of myocardial regeneration have consisted of acute myocardial infarction produced by permanent ligation of a major coronary artery, which minimally resembles the human situation. That leaves without coronary flow the area to be regenerated and sets a very high and probably unrealistic hurdle to be overcome by the regenerative process. Indeed, in the myocardial area supplied by the infarct-related artery, practically all of the myocytes as well as CSCs rapidly die following the permanent artery ligation (38). Therefore, this also represents an extreme and difficult experimental setting to study CSC function and regenerative potential in vivo.
In the present study, we have shown that an acute high dose of isoproterenol causes extensive myocyte loss without affecting coronary patency and CSC survival. After a transient period of LV dysfunction, the heart compensates for the damage, recovering to a normal function. Although it can be argued that myocardial damage by ISO overdose is not a physiological situation, we propose that diffuse damage throughout the myocardium in the presence of a patent coronary circulation could represent a more helpful experimental method by which to study the endogenous myogenic potential of CSCs. However, it is not our aim to downplay the value of established experimental myocardial infarction techniques, which remain of paramount importance. The two experimental approaches are likely to provide complementary information about the regenerative potential of CSCs in ischemic cardiomyopathy and chronic heart failure.
Acknowledgments
We thank Drs. Eugenia Pasceri and Stefania Zinzi (Magna Graecia University, Catanzaro, Italy) for valuable collaboration in performing the echocardiography. Confocal laser-scanning microscopy was performed at the Mount Sinai School of Medicine-Microscopy Shared Resource Facility.
Footnotes
This work was supported by British Heart Foundation Ph.D. Studentship FS/2001028/12895 (to G. M. E.), the Louis B. Meyer Foundation, a fellowship award from the American Heart Association (to G. M. E.), and Italian Health Minister Grant PRIN2005 2005060509-003. The Mount Sinai School of Medicine-Microscopy Shared Resource Facility was supported with funding from NCI, National Institutes of Health (NIH), shared resources Grant 5R24 CA095823-04, National Science Foundation Major Research Instrumentation Grant (DBI-9724504), and NIH shared instrumentation grant 1 S10 RR0 9145-01. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
- HF
- heart failure
- LV
- left ventricle
- AR
- adrenoreceptor
- PKA
- protein kinase A
- RyR2
- ryanodine receptor 2
- ISO
- isoproterenol
- ARVM
- adult rat ventricular myocyte
- EKG
- electrocardiogram
- TdT
- terminal deoxynucleotidyltransferase
- RT
- reverse transcription
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- PARP
- poly(ADP-ribose) polymerase
- CTRL
- control
- MHC
- myosin heavy chain
- CaMKII
- calcium/calmodulin-dependent protein kinase II
- DAPI
- 4′,6-diamidino-2-phenylindole
- cTnI
- cardiac troponin I
REFERENCES
- 1.Rosamond W, Flegal K, Friday G, Furie K, Go A, Greenlund K, Haase N, Ho M, Howard V, Kissela B, Kittner S, Lloyd-Jones D, McDermott M, Meigs J, Moy C, Nichol G, O’Donnell CJ, Roger V, Rumsfeld J, Sorlie P, Steinberger J, Thom T, Wasserthiel-Smoller S, Hong Y. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2007;115:e69–e171. doi: 10.1161/CIRCULATIONAHA.106.179918. [DOI] [PubMed] [Google Scholar]
- 2.Anversa P, Nadal-Ginard B. Nature. 2002;415:240–243. doi: 10.1038/415240a. [DOI] [PubMed] [Google Scholar]
- 3.Wehrens XHT, Marks AR. Nat. Rev. Drug Discov. 2004;3:565–573. doi: 10.1038/nrd1440. [DOI] [PubMed] [Google Scholar]
- 4.Torella D, Ellison GM, Karakikes I, Nadal-Ginard B. Cell. Mol. Life Sci. 2007;64:661–673. doi: 10.1007/s00018-007-6519-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Circ. Res. 2003;92:139–150. doi: 10.1161/01.res.0000053618.86362.df. [DOI] [PubMed] [Google Scholar]
- 6.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, Kasahara H, Rota M, Musso E, Urbanek K, Leri A, Kajstura J, Nadal-Ginard B, Anversa P. Cell. 2003;114:763–776. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 7.Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R, Nadal-Ginard B, Silvestri F, Leri A, Beltrami CA, Anversa P. N. Engl. J. Med. 2001;344:1750–1757. doi: 10.1056/NEJM200106073442303. [DOI] [PubMed] [Google Scholar]
- 8.Urbanek K, Quaini F, Tasca G, Torella D, Castaldo C, Nadal-Ginard B, Leri A, Kajstura J, Quaini E, Anversa P. Proc. Natl. Acad. Sci. U. S. A. 2003;100:10440–10445. doi: 10.1073/pnas.1832855100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Torella D, Ellison GM, Mendez-Ferrer S, Ibanez B, Nadal-Ginard B. Nat. Clin. Pract. Cardiovasc. Med. 2006;3:S8–S13. doi: 10.1038/ncpcardio0409. [DOI] [PubMed] [Google Scholar]
- 10.Wehrens XH, Lehnart SE, Marks AR. Annu. Rev. Physiol. 2005;67:69–98. doi: 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- 11.Cohn JN, Levine TB, Olivari MT, Garberg V, Lura D, Francis GS, Simon AB, Rector T. N. Engl. J. Med. 1984;311:819–823. doi: 10.1056/NEJM198409273111303. [DOI] [PubMed] [Google Scholar]
- 12.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 13.Goldspink DF, Burniston JG, Ellison GM, Clark WA, Tan LB. Exp. Physiol. 2004;89:407–416. doi: 10.1113/expphysiol.2004.027482. [DOI] [PubMed] [Google Scholar]
- 14.Reiken S, Gaburjakova M, Guatimosim S, Gomez AM, D’Armiento J, Burkhoff D, Wang J, Vassort G, Lederer WJ, Marks AR. J. Biol. Chem. 2003;278:444–453. doi: 10.1074/jbc.M207028200. [DOI] [PubMed] [Google Scholar]
- 15.Teerlink JR, Pfeffer JM, Pfeffer MA. Circ. Res. 1994;75:105–113. doi: 10.1161/01.res.75.1.105. [DOI] [PubMed] [Google Scholar]
- 16.Shizukuda Y, Buttrick PM, Geenen DL, Borczuk AC, Kitsis RN, Sonnenblick EH. Am. J. Physiol. 1998;275:H961–H968. doi: 10.1152/ajpheart.1998.275.3.H961. [DOI] [PubMed] [Google Scholar]
- 17.Wittstein IS, Thiemann DR, Lima JA, Baughman KL, Schulman SP, Gerstenblith G, Wu KC, Rade JJ, Bivalacqua TJ, Champion HC. N. Engl. J. Med. 2005;10:539–548. doi: 10.1056/NEJMoa043046. [DOI] [PubMed] [Google Scholar]
- 18.Torella D, Rota M, Nurzynska D, Musso E, Monsen A, Shiraishi I, Zias E, Walsh K, Rosenzweig A, Sussman MA, Urbanek K, Nadal-Ginard B, Kajstura J, Anversa P, Leri A. Circ. Res. 2004;94:514–524. doi: 10.1161/01.RES.0000117306.10142.50. [DOI] [PubMed] [Google Scholar]
- 19.Communal C, Singh K, Sawyer DB, Colucci WS. Circulation. 1999;100:2210–2212. doi: 10.1161/01.cir.100.22.2210. [DOI] [PubMed] [Google Scholar]
- 20.Jaffe AS, Ravkilde J, Roberts R, Naslund U, Apple FS, Galvani M, Katus H. Circulation. 2000;102:1216–1220. doi: 10.1161/01.cir.102.11.1216. [DOI] [PubMed] [Google Scholar]
- 21.Torella D, Leosco D, Indolfi C, Curcio A, Coppola C, Ellison GM, Russo VG, Torella M, Li Volti G, Rengo F, Chiariello M. Am. J. Physiol. 2004;287:H2850–H2860. doi: 10.1152/ajpheart.01119.2003. [DOI] [PubMed] [Google Scholar]
- 22.Maruyama R, Takemura G, Tohse N, Ohkusa T, Ikeda Y, Tsuchiya K, Minatoguchi S, Matsuzaki M, Fujiwara T, Fujiwara H. Am. J. Physiol. 2006;290:H1493–H1502. doi: 10.1152/ajpheart.00669.2005. [DOI] [PubMed] [Google Scholar]
- 23.Wehrens XH, Lehnart SE, Reiken SR, Marks AR. Circ. Res. 2004;94:e61–e70. doi: 10.1161/01.RES.0000125626.33738.E2. [DOI] [PubMed] [Google Scholar]
- 24.Wehrens XH, Lehnart SE, Reiken S, van der Nagel R, Morales R, Sun J, Cheng Z, Deng SX, de Windt LJ, Landry DW, Marks AR. Proc. Natl. Acad. Sci. U. S. A. 2005;102:9607–9612. doi: 10.1073/pnas.0500353102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nadal-Ginard B, Kajstura J, Anversa P, Leri A. J. Clin. Invest. 2003;111:1457–1459. doi: 10.1172/JCI18611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foo RS, Mani K, Kitsis RN. J. Clin. Invest. 2005;115:565–571. doi: 10.1172/JCI24569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shannon RP, Komamura K, Shen YT, Bishop SP, Vatner SF. Am. J. Physiol. 1993;265:H801–809. doi: 10.1152/ajpheart.1993.265.3.H801. [DOI] [PubMed] [Google Scholar]
- 28.Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB. Circulation. 2006;113:1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 29.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr., Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Nat. Med. 2005;11:409–417. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 30.Lehnart SE, Terrenoire C, Reiken S, Wehrens XH, Song LS, Tillman EJ, Mancarella S, Coromilas J, Lederer WJ, Kass RS, Marks AR. Proc. Natl. Acad. Sci. U. S. A. 2006;103:7906–10. doi: 10.1073/pnas.0602133103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Circ. Res. 2006;99:292–298. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- 32.Wehrens XH, Lehnart SE, Reiken S, Vest JA, Wronska A, Marks AR. Proc. Natl. Acad. Sci. U. S. A. 2006;103:511–518. doi: 10.1073/pnas.0510113103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Torella D, Ellison GM, Nadal-Ginard B, Indolfi C. Trends Cardiovasc. Med. 2005;15:229–236. doi: 10.1016/j.tcm.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 34.Tam SK, Gu W, Mahdavi V, Nadal-Ginard B. Ann. N. Y. Acad. Sci. 1995;752:72–79. doi: 10.1111/j.1749-6632.1995.tb17407.x. [DOI] [PubMed] [Google Scholar]
- 35.Chesley A, Lundberg MS, Asai T, Xiao RP, Ohtani S, Lakatta EG, Crow MT. Circ. Res. 2000;87:1172–1179. doi: 10.1161/01.res.87.12.1172. [DOI] [PubMed] [Google Scholar]
- 36.Zaugg M, Xu W, Lucchinetti E, Shafiq SA, Jamali NZ, Siddiqui MA. Circulation. 2000;102:344–350. doi: 10.1161/01.cir.102.3.344. [DOI] [PubMed] [Google Scholar]
- 37.Bybee KA, Kara T, Prasad A, Lerman A, Barsness GW, Wright RS, Rihal CS. Ann. Intern. Med. 2004;14:858–865. doi: 10.7326/0003-4819-141-11-200412070-00010. [DOI] [PubMed] [Google Scholar]
- 38.Mouquet F, Pfister O, Jain M, Oikonomopoulos A, Ngoy S, Summer R, Fine A, Liao R. Circ. Res. 2005;97:1090–1092. doi: 10.1161/01.RES.0000194330.66545.f5. [DOI] [PubMed] [Google Scholar]