

Summary
Protein kinase B, also known as Akt, is a serine/threonine kinase and plays a critical role in the modulation of cell development, growth, and survival. Interestingly, Akt is ubiquitously expressed throughout the body, but its expression in the nervous system is substantially up-regulated during cellular stress, suggesting a more expansive role for Akt in the nervous system that may involve cellular protection. In this regard, a body of recent work has identified a robust capacity for Akt and its downstream substrates to foster both neuronal and vascular survival during apoptotic injury. Cell survival by Akt is driven by the modulation of both intrinsic cellular pathways that oversee genomic DNA integrity and extrinsic mechanisms that control inflammatory microglial activation. A series of distinct pathways are regulated by Akt that include the Forkhead family of transcription factors, GSK-3ß, ß-catenin, c-Jun, CREB, Bad, IKK, and p53. Culminating below these substrates of Akt are the control of caspase mediated pathways that promote genomic integrity as well as prevent inflammatory cell demise. With further levels of progress in defining the cellular role of Akt, the attractiveness of Akt as a vital and broad cytoprotectant for both neuronal and vascular cell populations should continue to escalate.
Keywords: Bad, ß-catenin, caspases, CREB, FOXO, GSK-3ß, Iκß kinase, microglia, p53
Protein kinase B (Akt) as a molecular therapeutic target
Protein kinase B (PKB), also known as Akt, is ubiquitously expressed in mammals, but is initially present at low levels in the adult brain. However, expression of Akt in both neurons and vascular cells increases dramatically during cellular stress or injury, suggesting a prominent role for Akt during the maintenance of cell integrity and survival. Activation of Akt occurs through its phosphorylation that is dependent upon the signaling pathways of phosphoinositide 3 kinase (PI 3-K). Subsequently, the biological activity of Akt can be controlled through mechanisms that involve lipid phosphatases, proteins such as carboxyl-terminal modulator protein (CTMP), and heat shock proteins. Cytoprotection by Akt is considered to be broad in nature and applicable to a host of central nervous system (CNS) injuries that range form cerebral ischemia to Alzheimer's disease. Preservation of cell survival by Akt is driven by the modulation of both intrinsic cellular pathways that maintain genomic DNA integrity as well as extrinsic mechanisms that pertain directly to inflammatory cell activation. In this review, we will focus on the role of Akt in primarily neuronal and vascular cells in the CNS as a potential therapeutic molecular agent directed against neurodegenerative disorders.
Phosphorylation and activity of Akt originates with phosphoinositide 3 kinase
In mammals, three family members of the serine/threonine kinase protein kinase B (PKB), or Akt named after the oncogene v-Akt, have been identified. They are PKBα or Akt1, PKBß or Akt2, and PKBγ or Akt3. Akt belongs to the AGC (cAMP-dependent kinase/protein kinase G/protein kinase C) superfamily of protein kinases and consists of three functional domains (Fig. 1). The N-terminal pleckstrin homology (PH) domain provides binding sites for membrane phospholipids, which is involved in the recruitment of Akt to the plasma membrane. The catalytic domain of Akt has specificity for serine or threonine residues of proteins that are substrates for Akt. The C-terminal hydrophobic motif (HM) functions to provide a docking site for the activation of kinases.
Fig. 1.
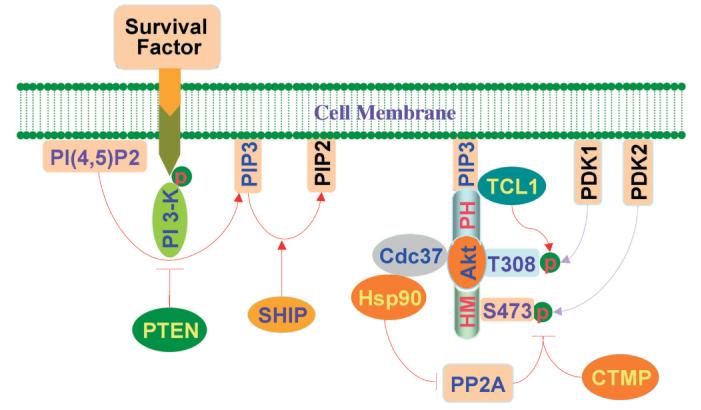
Controlling the activity of Akt. Following the activation of phosphoinositol-3- kinase (PI 3-K), PI 3-K phosphorylates membrane glycerophospholipid phosphatidylinositol 4,5-bisphosphate (PI(4,5)P2) with production of phosphatidylinositol 3,4, 5 trisphosphate (PIP3) and phosphatidylinositol 3,4 bisphosphate (PIP2), which is required for Akt recruitment to the cell membrane through its PH domain. The enzyme 3-phosphoinositide-dependent kinase-1 (PDK1) is responsible for phosphorylation of Thr308 (T308) of Akt. The residue Ser473 (S473) cannot directly be phosphorylated by PDK1, but the phosphorylation may be dependent on the postulated kinase PDK2. Akt activity can be negatively regulated by the phosphatase and tensin homolog deleted from chromosome 10 (PTEN), SH2 domain-containing inositol phosphatase (SHIP), and carboxyl-terminal modulator protein (CTMP). In contrast, the T cell leukemia/lymphoma 1 (TCL1) protein binds to Akt to enhance its activity. A 90 kDa heat shock protein (Hsp90) complexed with Cdc37 also can enhance Akt activity by inhibition of the protein phosphatase 2A (PP2A).
Activation of Akt is dependent upon PI 3-K (Fig. 1). The activation of the receptor tyrosine kinase (RTK) and the G protein-coupled receptor (CPCR) are required to activate PI 3-K. PI 3-K is composed of a catalytic p110 subunit and a regulatory p85 subunit. Survival factors, such as trophic factors or cytokines, can stimulate the recruitment of PI 3-K to the plasma membrane. PI 3-K preferentially phosphorylates the D3 position of the inositol ring of phosphoinositides. Following activation, PI 3-K phosphorylates membrane glycerophospholipid phosphatidylinositol 4,5-bisphosphate [PI (4,5)P2] resulting in the production of phosphatidylinositol 3,4, 5 trisphosphate (PIP3) and phosphatidylinositol 3,4 bisphosphate (PIP2). Both PIP2 and PIP3 are required for Akt activation. The critical step for activation of Akt is its transition from the cytosol to the plasma membrane, which is accomplished by the binding of Akt to PIP2 and PIP3 through its PH domain (Stephens et al., 1998). As a result of this sequence of events, Akt becomes available for phosphorylation by several upstream kinases.
The phosphorylation of two major residues, Thr308 and Ser473, are considered necessary for the activation of Akt. The site of Thr308 is located within the activation T-loop of Akt1. For Akt2 and Akt3, the equivalent residues are Thr309 and Thr305 respectively (Walker et al., 1998). These phosphorylation sites are believed to be critical for the activation of Akt. Yet, the phosphorylation of Ser473 at the C-terminal HM domain also is necessary for the complete activation of Akt (Bellacosa et al., 1998).
The phosphorylation of Thr308 is dependent upon its upstream kinase, 3-phosphoinositide-dependent kinase-1 (PDK1) (Wick et al., 2000). PDK1 contains a C-terminal PH domain by which PDK1 is recruited by PIP3 to the cell membrane, and to a lesser extend by PIP2. Similar to other AGC kinases, the C-terminal HM domain of Akt provides a docking site for PDK1, which is essential for the activation of the T-loop (Frodin et al., 2002). Moreover, the HM domain also can stabilize the N-lobe of PDK1 by association with the protein kinase C-related kinase-2 interacting fragment (PIF) and thus increase the activity of PDK1 by 10 fold (Balendran et al., 1999). Phosphorylation of Thr308 results in conformation change in PIF to allow it to become more suitable for HM binding. PDK1 cannot directly phosphorylate Ser473, but a distinct phosphoinositide-dependent kinase PDK2 (Ser473 kinase) has been postulated to promote Akt phosphorylation on Ser473. The existence of PDK2 is pending further confirmation.
The “brakes and gas” for Akt activity
A number of pathways can function to either block Akt activity or enhance the biological activity of Akt. Some lipid phosphatases have been shown to negatively modulate the activity of Akt. The phosphatase and tensin homolog deleted from chromosome 10 (PTEN) appears to be a critical regulator of PI 3-K signaling (Fig.1). The PTEN gene was originally identified as a tumor suppressor gene and is a common mutant gene in various human cancers. The PTEN protein has a C2 domain which functions to regulate the membrane affinity of the protein to membrane phospholipids. A phosphatase domain also exists that can interact with either lipids or proteins (Lee et al., 1999). PTEN can dephosphorylate tyrosine-, serine-, and threonine-phosphorylated peptides (Lee et al., 1999). PTEN negatively regulates PI 3-K pathways by specifically dephosphorylating PIP2 and PIP3 at the D3 position (Maehama and Dixon, 1998). As a result, a reduction in the membrane phospholipid pool that is necessary for the recruitment of Akt can ensue during PTEN activity.
Other lipid phosphatases, such as SHIP (SH2 domain- containing inositol phosphatase), can regulate Akt activity (Fig. 1). SHIP is an inositol 5' phosphatase that dephosphorylates inositides and phosphoinositides on the 5'-position resulting in the transformation of PIP3 into PIP2. Interestingly, the SHIP2 gene appears to modulate insulin signaling, since targeted disruption of this gene leads to increased insulin sensitivity that occurs as a result of enhanced phosphorylation of Akt2 at the plasma membrane (Sasaoka et al., 2004). In other cell systems that involve hematopoietic proliferation, SHIP also functions to block activation of Akt (Carver et al., 2000). The Src homology domain 2 (SH2)-containing tyrosine phosphatases (SHP) also have been implicated in the control of the Akt pathway. In regards to SHP1 and SHP2, SHP1 is predominantly expressed in hematopoietic cells, but SHP2 is more ubiquitously expressed and occurs in the nervous system (Chong et al., 2003e). Through the activation of Akt, SHP1 can selectively bind and dephosphorylate PTEN to reduce the stability of this protein (Lu et al., 2003). SHP2 also appears to be required for the activation of Akt (Ivins Zito et al., 2004) and prevent cellular death from apoptosis through inhibition of either caspase 1 or 3-like activities (Chong et al., 2003e; Ivins Zito et al., 2004).
A third protein, CTMP, also can negatively regulate the activity of Akt. CTMP is a 27 kDa protein that binds specifically to the carboxyl-terminal regulatory domain of Akt1 at the plasma membrane (Maira et al., 2001). The binding of CTMP to Akt1 decreases the activity of Akt1 by inhibiting the phosphorylation of Akt1 on Ser473 and Thr308 (Maira et al., 2001).
Alternate cellular systems are responsible for the enhancement of Akt activity. The T cell leukemia/lymphoma 1 (TCL1) protein functions as a co-activator of Akt (Fig. 1). The TCL1 gene at chromosome 14q32.1 is associated with the development of human mature T cell leukemia. In addition, TCL1 can stabilize mitochondrial membrane potential and promote cell proliferation and survival (Laine et al., 2000). TCL1 binds to Akt1 and increases Akt1 kinase activity to promote its nuclear translocation (Pekarsky et al., 2000). Further studies indicate that TCL1 binds to the PH domain of Akt and the formation of TCL1 trimers facilitates the formation of the Akt/TCL1 complex. Within this complex, Akt is phosphorylated and activated in vivo (Laine et al., 2000).
Akt activity also can be facilitated by a 90 kDa heat shock protein (Hsp90). Hsps are characterized by their mass in kilodaltons, are induced in response to heat in essentially all organisms, and are highly conserved between different species. Hsps, such as Hsp90 can be cytoprotective, such as preventing cell injury against heat thermal stress (Latchman, 2004). Akt binds to Hsp90 through its 229-309 residues resulting in stabilization of the phosphorylated Akt (Fig. 1). Interestingly, inhibition of Akt binding to Hsp90 leads to dephosphorylation of Akt by protein phosphatase 2A (PP2A) and induction of apoptosis (Sato et al., 2000). More recent investigations indicate that intracellular Akt can become complexed with Hsp90 and Cdc37. As a result of this association, increased Akt activity is present, but is closely dependent upon the presence of Hsp90 in the complex (Basso et al., 2002).
Akt is expressed in a variety of tissues and is intimately bound to cellular injury
Akt is expressed in mammals, but can vary in the level of expression in a variety of tissues and cells. Akt1 is the most highly expressed isoform. Although Akt2 is expressed at a lower level than Akt1, significant expression of Akt2 occurs in insulin-responsive tissues, such as skeletal muscle, liver, heart, kidney, and adipose tissue (Altomare et al., 1995). This distribution for Akt2 expression may suggest a critical role for this protein in insulin signaling pathways. In the central nervous system, the expression of Akt1 and Akt2 can be observed at increased levels during development, but is gradually decreased during postnatal development (Owada et al., 1997). Yet, in the adult brain, expression of Akt1 and Akt2 is initially weak with a dramatic increase in the expression of Akt1 mRNA and Akt1 protein in cells that are subjected to injury (Owada et al., 1997; Kang et al., 2003; Chong et al., 2005), suggesting that Akt may play an important role during paradigms that involve cell injury. Different from Akt1 and Akt2, Akt3 is only expressed in a limited number of tissues, such as in the brain and testes, with lower expression evident in skeletal muscle, pancreas, heart, and kidney (Nakatani et al., 1999).
Akt is a critical survival factor that can modulate cellular pathways involved in programmed cell death (PCD), also known as apoptosis. Early studies have demonstrated that over-expression of Akt in cerebellar granule neurons prevents apoptosis during growth factor withdrawal (Dudek et al., 1997). In contrast, the expression of a dominant-negative Akt or inhibition of PI 3-K attenuates cell survival normally supported by growth factors (Chong et al., 2002b; Maiese et al., 2004). Subsequent work has illustrated an important role for Akt for the survival of various cell types during injury paradigms. Akt promotes cell survival during free radical exposure in primary hippocampal neurons (Matsuzaki et al., 1999; Chong et al., 2003b), neuronal cell lines (Kang et al., 2003a,b), and cerebral vascular endothelial cells (ECs) (Chong, 2005). Enhanced Akt activity can foster cell survival against several other toxic insults that include matrix detachment (Rytomaa et al., 2000), DNA damage (Chong, 2002, 2005; Kang, 2003; Henry, 2001), anti-Fas antibody administration (Suhara et al., 2001), oxidative stress (Chong, 2005; Kang, 2003), and transforming growth factor-ß (TGF-ß) application (Conery et al., 2004).
As a result of the broad protective nature of Akt, many agents or growth factors appear to prevent apoptotic cellular injury through Akt activation. In the vascular system, angiopoietin-1 is an endothelium-specific ligand essential for embryonic vascular stabilization, branching, morphogenesis, and post-natal angiogenesis. Angiopoietin-1 also supports endothelial cell survival and prevents apoptosis through the activation of Akt that requires a PI 3-K dependent pathway (Papapetropoulos et al., 2000). Furthermore, in the cardiovascular system, myocardial protection by insulin during myocardial ischemia/reperfusion is abolished by PI 3-K inhibition, suggesting that cardioprotection of insulin is mediated by Akt activation (Jonassen et al., 2001). The involvement of the PI 3-K/Akt pathway also has been demonstrated during the protection of retinal ganglion cells from axotomy (Kermer et al., 2000).
Interestingly, a number of trophic factors and cytokines, such as erythropoietin (EPO), may depend upon Akt to offer cellular protection (Maiese et al., 2003). For example, EPO can phosphorylate Akt and is dependent upon the activation of PI 3-K and Janus Kinase 2 (Jak2) (Witthuhn et al., 1993; Chong et al., 2002b). Activation of Jak2 promotes the phosphorylation of tyrosine residues in the intracellular portion of the EPO receptor (Witthuhn et al., 1993). Phosphorylation of the last tyrosine of the EPO receptor initiates binding of the 85 kDa regulatory subunit of PI 3-K, a heterodimer consisting of a 110 kDa catalytic subunit and an 85 kDa regulatory subunit. As a result of the binding of the 85 kDa regulatory subunit, the 110 kDa catalytic subunit becomes active and leads to the phosphorylation of Akt.
Central to the ability of EPO to prevent cellular apoptosis is the activation of Akt by EPO. During anoxia or free radical exposure, expression of the active form of Akt (phospho-Akt) is increased (Kang et al., 2003b; Kang et al., 2003a). EPO can significantly enhance the activity of Akt during oxidative stress and prevent inflammatory activation of microglia (Chong et al., 2003a,b,d). This up-regulation of Akt activity during injury paradigms appears to be vital for EPO protection, since prevention of Akt phosphorylation blocks cellular protection by EPO (Chong et al., 2003a,b,d). Through the regulation of the PI 3-K/Akt dependent pathway, EPO can prevent cellular apoptosis following N-methyl-D-aspartate toxicity, neuronal axotomy, hypoxia, and oxidative stress (Maiese et al., 2003, 2004).
Akt can play a decisive role during several disease states that involve apoptosis
Given the intimate association between Akt and cytoprotective agents, Akt may be viewed as an essential target for therapeutic strategies against a number of diseases that involve apoptotic cell death. Although necessary for tissue re-modeling during development, apoptosis is recognized as a central pathway that can lead to a cell's demise in a variety of tissues and has recently been identified in organisms as diverse as plants (Hatsugai et al., 2004). Apoptotic injury is believed to contribute significantly to a variety of disease states that especially involve the nervous system such as ischemic stroke, Alzheimer's disease, Parkinson's disease, and spinal cord injury (Maiese, 2004; Chong, 2005). Outside of the nervous system, such as during cardiovascular injury, PCD also may be a significant precipitant of cell death. For example, ischemic-reperfusion injury can lead to apoptosis in cardiomyocytes (Cai et al., 2003).
Apoptotic cellular injury consists of two independent processes that involve membrane phosphatidylserine (PS) exposure and DNA fragmentation. The biological role of membrane PS externalization can vary in different cell populations. In many cell systems, membrane PS externalization can become a signal for the phagocytosis of cells (Li, 2004; Chong, 2005 ). In the nervous system, cells expressing externalized PS may be removed by microglia. An additional role for membrane PS externalization in the vascular cell system is the activation of coagulation cascades. The externalization of membrane PS residues in ECs can promote the formation of a procoagulant surface (Chong, 2005).
In contrast to the early externalization of membrane PS residues, the cleavage of genomic DNA into fragments is a delayed event that occurs late during apoptosis (Maiese and Vincent, 2000; Jessel et al., 2002). Several enzymes responsible for DNA degradation have been differentiated based on their ionic sensitivities to zinc and magnesium (Li, 2004; Chong, 2005). DNA degradation can proceed through mechanisms that involve calcium/magnesium - dependent endonucleases such as DNase I, the acidic, cation independent endonuclease (DNase II), cyclophilins, and the 97 kDa magnesium - dependent endonuclease. Modulation of endonuclease activity can influence cell survival in both vascular systems and the nervous system. For example, three separate endonuclease activities are present in neurons. They are a constitutive acidic cation-independent endonuclease, a constitutive calcium/magnesium-dependent endonuclease, and an inducible magnesium dependent endonuclease (Vincent and Maiese, 1999). The inducible magnesium-dependent endonuclease may be unique for the nervous system (Vincent and Maiese, 1999). The physiologic characteristics of the magnesium dependent endonuclease, such as a pH range of 7.4-8.0, a dependence on magnesium, and a molecular weight of 95-108 kDa, are consistent with a constitutive 97 kDa endonuclease in non-neuronal tissues, but the endonuclease in the nervous system is believed to be inducible rather than constitutive in nature.
Akt possesses the ability to offer a broad level of cytoprotection in cells through both intrinsic cell mechanisms that involve the maintenance of genomic DNA and the exposure of membrane PS residues. Through the over-expression of a myristoylated (active) forms of Akt and a kinase-deficient dominant-negative Akt, recent work has shown that Akt is both necessary and sufficient to protect cells, such as neurons and ECs from injury associated with oxidative stress (Kang, 2003; Chong, 2005). Over-expression of myr-Akt significantly protects cells from free radical injury and prevents degradation of genomic DNA. Yet, cells with a dominant-negative over-expression that lack kinase activity suffer a significant loss in cell survival during oxidative stress. Interestingly, through the inhibition of PI 3-K phosphorylation of Akt or through the over-expression of a kinase-deficient dominant-negative Akt, endogenous cellular reserves of Akt also can provide an additional level of protection during cell injury that can function in concert with the exogenous activation of Akt to achieve increased cellular protection (Kang, 2003; Chong, 2005). It is important to note that activation of Akt is not always desirable under certain conditions, such as that involve the control of neoplasms. Recent work has identified Akt as a potential target to block during the treatment of nonsmall cell lung cancers that contain mutations in the epidermal growth factor (Sordella et al., 2004).
Akt is a critical modulator of inflammatory cell injury
In disease states in areas of the body such as the CNS, modulation of extrinsic cell homeostasis through microglial activation is as vital to cellular survival as the maintenance of cellular DNA integrity. The analysis of brain tissue in patients has supported the premise that microglia may lead to the progression of some neurodegenerative disorders. In Huntington's disease and amyotrophic lateral sclerosis, significant microglial activation has been reported in regions of the nervous system that are specific for these disease entities (Singhrao et al., 1999; Obal et al., 2001). During cerebral ischemia, activation of microglia can parallel the induction of cellular apoptosis and correlate well with the severity of the ischemic insult (Chong, 2005). In patients with Alzheimer's disease, microglial cells co-localize with the perivascular deposits of ß-amyloid (Aß). In addition, microglial activation has been observed to occur in concert with the evolution of amyloid plaques (Maiese and Chong, 2004). The generation of oxidative stress by microglia during Aß deposition further suggests that microglia may play an important role in the pathogenesis of Alzheimer's disease.
As a result, oxidative stress generated by microglia can be responsible for cellular injury. Activated microglia up-regulate a variety of surface receptors and yield significant pro-inflammatory and toxic factors. These can include nitric oxide, superoxide, and fatty acid metabolites such as eicosanoids that can precipitate cell death (Chong et al., 2004b; Li et al., 2004; Maiese and Chong, 2004). The secretion of cytokines by microglia also may represent another source of cytotoxicity for microglia. Microglia produce a variety of cytokines in response to toxic stimulation, such as interleukins and tumor necrosis factor (TNF). TNF-α production by microglia may be linked to neurodegeneration by increasing the sensitivity of neurons to free radical exposure (Chong et al., 2004b; Maiese and Chong, 2004).
During microglia activation, the phagocytic removal of apoptotic cells within the CNS play an important role during development, tissue homeostasis, and host defense. Several potential mechanisms regulate the phagocytosis of cells that have become injured and may be destined to die. Secreted factors by either apoptotic or phagocytic cells, such as milk fat globule-EGF-factor 8, fractalkine, and lipid lysophosphosphatidylcholine also have been shown to assist with the phagocytic removal of injured cells (Chong et al., 2004; Li et al., 2004). Yet, a common denominator that appears to be critical for the removal of apoptotic cells is the translocation of membrane PS residues from the inner cellular membrane to the outer surface. During normal cellular function, the phospholipids of the plasma membrane are asymmetrical with the outer leaflet of the plasma membrane consisting primarily of choline-containing lipids, such as phosphatidylcholine and sphingomyelin, and the inner leaflets consisting of aminophospholipids that include phosphatidylethanolamine and PS. The disruption of membrane phospholipid asymmetry leads to the externalization of membrane PS residues and serves to identify cells for phagocytosis (Chong et al., 2004b; Li et al., 2004; Maiese et al., 2004).
Expression of the phosphatidylserine receptor (PSR) on microglia works in concert with cellular membrane PS externalization. Cells exposed to oxidative stress can lead to the induction of both microglial activation and microglial PSR expression. Treatment with an anti-PS receptor neutralizing antibody in microglia prevents this microglial activation (Chong et al., 2003b; Kang et al., 2003a). In addition, application of PS can directly result in microglial activation that is blocked by a PSR neutralizing antibody (Chong et al., 2003b; Kang et al., 2003b), suggesting that both PS exposure in target cells and PSR expression in microglia are necessary for microglial recognition of apoptotic cells. Recognition of cellular membrane PS by the PS-specific receptors on microglia may require cofactors, such as Gas6. In addition, microglia recognition of injured cells through membrane PS mediated mechanisms also may involve other agents, such as integrin and lectin (Chong et al., 2004b; Li et al., 2004; Maiese et al., 2004).
Akt prevents inflammatory cell demise through extrinsic cellular mechanisms that involve membrane PS exposure and the subsequent activation of microglia. Enhanced Akt activity can prevent cellular membrane PS externalization in both neurons and ECs during a variety of insults that involve anoxia, free radical exposure, and oxygen-glucose deprivation (Chong et al., 2004b; Li et al., 2004; Maiese et al., 2004). In addition, Akt appears to employ the modulation of membrane PS externalization to prevent microglial activation (Kang et al., 2003b). Activation of Akt can prevent membrane PS exposure on injured cells and block the activation of microglia that are exposed to media taken from cells that over-express active, phosphorylated Akt during cellular injury (Kang et al., 2003a,b). Cytoprotective agents, such as nicotinamide and EPO, also employ mechanisms that involve Akt to regulate microglial activation and proliferation (Maiese and Chong, 2003; Li et al., 2004; Maiese et al., 2004). These protective agents block membrane PS exposure on cells and possibly prevent the shedding of membrane PS residues that is known to occur during apoptosis (Simak et al., 2002). In addition to targeting the activity of membrane PS exposure and microglial activation, Akt also may directly address cellular inflammation by inhibiting several pro-inflammatory cytokines, such as TNF-α (Fontaine et al., 2002).
Akt is dependent upon the intimate modulation of its downstream cellular pathways
Investigations into the cellular pathways of Akt have begun to identify pathways that provide us with a clearer understanding of the significant role that Akt plays during cellular development, function, and survival. In the following sections, we discuss novel cellular pathways that originate from substrates of Akt that include the Forkhead family of transcription factors and the glycogen synthase kinase-3ß (GSK-3ß) and depend upon downstream mechanisms that involve ß-catenin, the translation initiation factor 2B (eIF2B), c-Jun, the cAMP-response element-binding protein (CREB), Bad, IkB kinase (IKK), p53, and c-Jun N-terminal kinase (JNK) interacting proteins (JIPs) (Fig. 2).
Fig. 2.
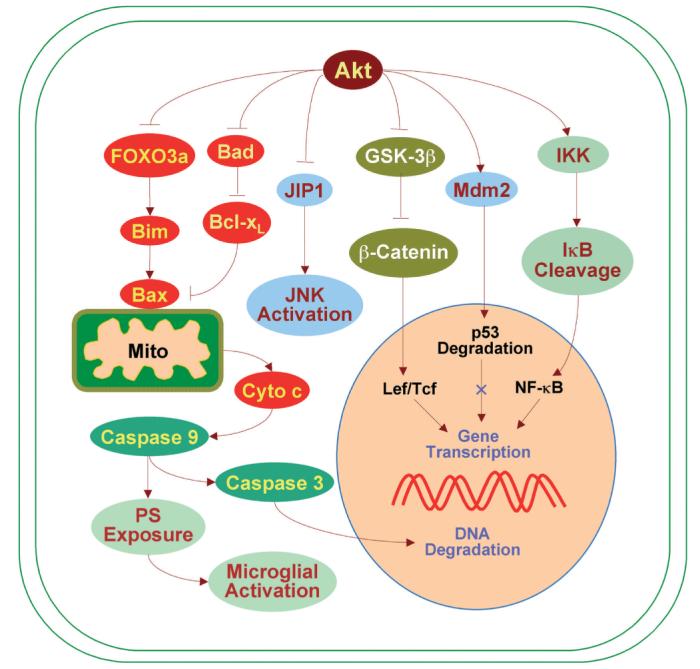
Akt provides cellular protection against apoptosis through a series of cellular pathways. Akt phosphorylates the Forkhead family member FOXO3a to block the induction of the pro-apoptotic member Bim resulting in inhibition of mitochondrial (Mito) depolarization and Bax-mediated cytochrome c release. The inactivation of glycogen synthase kinase-3ß (GSK-3ß) by Akt sends survival signals to block ß-catenin degradation and the subsequent activation of lymphocyte enhancer factor (Lef) and T cell factor (Tcf) in the nucleus. Akt prevents the association of the c-Jun N-terminal kinase (JNK) interacting protein 1 (JIP1) with JNK to prevent cellular demise. Following activation by Akt, IkB kinase (IKK) phosphorylates IkB allowing NF-kB to activate a host of genes that can block apoptosis. In addition, Akt phosphorylates murine double minute (Mdm2) to inactivate p53 and prevent cellular injury. Akt also inactivates Bad, leading to the inhibition of Bax through Bcl-xL and the prevention of cytochrome c (Cyto c) release. Without cytochrome c release, caspase activity is subsequently attenuated to prevent both DNA degradation and inflammatory microglial activation.
Akt and the Forkhead transcription factors
Three members of the mammalian Forkhead family have been identified and consist of the Forkhead in rhabdomyosarcoma (FKHR, also named as FOXO1), the FKHRL-like 1 (FKHRL1, FOXO3a), and the acute-lymphocytic-leukemia-1 fused gene from chromosome X (AFX, also named as FOXO 4). The Forkhead family is characterized by the forkhead box or winged helix that contains a 110 amino acid binding domain. Upon activation such as during oxidative stress, the Forkhead family members are translocated to the nucleus (Fig. 3). They function as transcription factors by preferentially binding to the core consensus DNA sequence 5'-TTGTTTAG-3', the Forkhead response element. The cellular function of Forkhead family is broad in nature and is involved in cell processes such as apoptotic injury (Gilley et al., 2003), cell cycle progression (Schmidt et al., 2002), oxidative stress (Kops et al., 2002), and the longevity of an organism (Taub et al., 1999).
Fig. 3.

FOXO3a translocation in cerebral microvascular endothelial cells (ECs) following oxygen-glucose deprivation (OGD). Immunohistochemical staining for FOXO3a was obtained in ECs 6 hours following an eight hour period of OGD by using a primary rabbit anti-FOXO3a antibody with Texas-red avidin. OGD in ECs was performed by replacing media with glucose-free HBSS containing 116 mM NaCl, 5.4 mM KCl, 0.8 mM MgSO4, 1mM NaH2PO4, 0.9 mM CaCl2, and 10 mg/L phenol red (pH 7.4) and cultures were maintained in an anoxic environment (95% N2 and 5% CO2) at 37 °C for 8 hours. Cell nuclei were stained with DAPI (4′-6-diamidino-2-phenylinodole). Representative images illustrate the translocation of FOXO3a from the cytoplasm (yellow arrows) to the nucleus (white arrows) during OGD, but both cytoplasmic and nuclear expression of FOXO3a is present without cellular translocation in control (untreated) ECs.
Modulation of activity of the Forkhead family has been closely associated with Akt. Following activation, Akt can phosphorylate all three members of the Forkhead family resulting in their retention in the cytosol of cells and the subsequent blockade of their transcriptional activities. Three phosphorylation sites for Akt have been identified in all three members of Forkhead family. In FOXO1, the phosphorylation sites reside at Thr24, Ser256, and Ser319. In FOXO4, the sites include Thr28, Ser128, and Ser258. Among the three phosphorylation sites for FOXO3a that include Thr32, Ser253, and Ser315, Akt preferentially phosphorylates Ser253 (Brunet et al., 1999). Mutation of the Akt phosphoacceptor sites to alanine residues on FOXO can enhance the transcriptional activity of FOXO and render it resistant to Akt inhibition, suggesting that Akt requires these sites of phosphorylation to modulate the transcriptional activity of FOXO (Tang et al., 1999).
The phosphorylation of the FOXO proteins results in their inactivation through cytoplasmic retention. In the absence of Akt activity or following the mutation of the FOXO phosphorylation sites, FOXO is exclusively localized to the nucleus. Translocation of FOXO also can occur during a cellular insult, such as in the presence of oxygen-glucose deprivation (Fig. 3). Following Akt activation, Akt translocates to the nucleus in which Akt phosphorylates the FOXO transcription factor resulting in the export of FOXO into the cytosol. This translocation of FOXO by Akt is associated with protein 14-3-3. The 14-3-3 family of proteins function through binding to their protein ligands in a phosphorylation-dependent manner (Rena et al., 2001). Two binding motifs of 14-3-3 proteins have been identified, namely RSXpSXP and RXY/FXpSXP (Yaffe et al., 1997), which are present in nearly all known 14-3-3 proteins. The phosphorylation of FOXO transcription factors by Akt leads to the generation of consensus binding sites for the 14-3-3 protein and the subsequent export of FOXO from the nucleus. For example, the phosphorylation of FOXO3a promotes the interaction of FOXO3a with the 14-3-3 protein and results in retention of the phosphorylated form of FOXO3a in the cytoplasm (Brunet et al., 1999).
The transcriptional activity of the FOXO family is closely related to the induction of apoptosis. In the nucleus, FOXO can bind to specific DNA sequences and mediate the activation of transcription that plays essential roles in the regulation of cell survival. Expression of an Akt-resistant mutant of FOXO that promotes the activity of FOXO can lead to apoptosis (Tang et al., 1999). On the other hand, activation of Akt can lead to the inhibition of FOXO-dependent transcription and block of apoptosis (Brunet et al., 1999). FOXO3a can result in PCD in cerebral granule neurons, sympathetic neurons, and a variety of other cell types in a transcription-dependent manner following its translocation to the nucleus (Brunet et al., 1999; Dijkers et al., 2002; Gilley et al., 2003). Further work has demonstrated that activation of FOXO3a can disrupt mitochondrial membrane potential and result in cytochrome c release (Dijkers et al., 2002).
Several transcriptional targets of FOXO in mammalian cells that are necessary for the induction of apoptosis have been identified. The death receptor Fas ligand appears to be an important target of FOXO. For example, a Forkhead binding sequence is present in the regulatory region of the Fas ligand gene, suggesting that FOXO may control the expression of the endogenous Fas ligand gene. In addition, FOXO3a can activate a Fas ligand promoter-driven reporter and result in apoptosis in cerebellar neurons, fibroblasts, and Jurkat cells (Brunet et al., 1999). Yet, induction of apoptosis by a FOXO3a mutant that cannot be phosphorylated by Akt and would normally lead to cell injury can be blocked when expressed in Fas mutant or FADD (Fas-associated protein with death domain) mutant cells (Brunet et al., 1999), further suggesting that FOXO3a is dependent upon the Fas ligand for the generation of apoptosis.
FOXO also may promote apoptotic injury through the regulation of Bim. Bim, a BH3-only family member, functions upstream of Bax-mediated cytochrome c release from the mitochondria. Over-expression of FOXO3a has been reported to enhance Bim expression during cell stress such as trophic factor withdrawal and lead to PCD that requires Bim activity (Gilley et al., 2003). In addition, FOXO3a can activate the Bim promoter through two conserved FOXO binding sites. Mutation of these sites blocks Bim promoter activation (Gilley et al., 2003).
Closely linked to cell survival and longevity is the control of the cell cycle by FOXO. FOXO controls the cell cycle through the increased expression of p27KIP1 (Medema et al., 2000) which can ultimately lead to PCD. p27KIP1 has been suggested to be critical in the regulation of cell survival, since even ectopic expression of p27KIP1 is sufficient to result in cellular apoptosis (Dijkers et al., 2000). p27KIP1 triggers apoptosis in several different human cancer cell lines. In human prostate carcinoma cells, a recombinant adenovirus expression of p27KIP1 is associated with the induction of apoptosis (Katner et al., 2002). In cholangiocarcinoma cell lines, over-expression of p27KIP1 leads to apoptosis that can be completely prevented by the neutralizing antibody of Fas ligand, suggesting that p27KIP1 induces apoptosis through Fas mediated pathways (Yamamoto et al., 2003). Furthermore, transfection of the full-length human p27 cDNA results in apoptotic cell death in retinoblastoma protein (Rb) expressing cells, but not in cells which do not express Rb, suggesting that some level of Rb expression is required for the induction of apoptosis by p27 (Naruse et al., 2000). FOXO3a results in a striking increase in p27KIP1 promoter activity with the induction of the apoptosis (Dijkers et al., 2000). Interestingly, new work has suggested that in cases in which the Sir2 homolog SIRT1 can form a complex with FOXO3 during oxidative stress, cell longevity is fostered rather than inhibited by promoting cell cycle arrest and preventing apoptosis (Brunet et al., 2004).
Akt, GSK-3ß, and apoptotic neurovascular cell injury
Glycogen synthase kinase-3 is a serine/threonine kinase that also is a substrate of Akt (Fig. 2). In mammalian cells, there are two GSK-3 isoforms termed GSK-3α and GSK-3ß, which have a mass of 51 kDa and 47 kD respectively. Of the two isoforms of GSK-3, GSK-3ß is specifically expressed in the CNS. GSK-3 is a constitutively active kinase in cells and is primarily regulated through inhibition of its activity. Akt can phosphorylate GSK-3ß at Ser9 and inactivate the enzyme (Shaw et al., 1997). In contrast, phosphorylation of GSK-3ß at Thr216 results in an enhanced activity of the enzyme, which can occur during neuronal degeneration (Bhat et al., 2000).
GSK-3ß plays a significant role in the regulation of apoptosis. Apoptotic injury is enhanced by the over-expression of GSK-3ß, suggesting that the activity of GSK-3ß contributes to cellular injury (Crowder and Freeman, 2000). During oxidative stress, GSK-3ß can lead to caspase 3 activation and cytochrome c release (Koh et al., 2003). In erythroid progenitors, the expression of a constitutively active mutant of GSK-3ß during serum deprivation promotes cellular PCD (Somervaille et al., 2001). GSK-3ß also can lead to apoptotic injury in vascular smooth muscle cells (Loberg et al., 2002) and cardiomyocytes (Yin et al., 2004). Yet, the inactivation of GSK-3ß can result in the prevention or reduction in apoptotic injury in neurons (Alvarez et al., 1999), vascular smooth muscle cells (Loberg et al., 2002), and cardiomyocytes (Yin et al., 2004).
GSK-3ß is involved in the pathological process of neurodegenerative disorders, such as Alzheimer's disease (Jope and Johnson, 2004; Maiese and Chong, 2004). In Alzheimer's patients, GSK-3ß expression is present in the cytoplasm of pretangle neurons and its expression coincides with the development of neurofibrillary changes (Pei et al., 1999). On a cellular level, Aß exposure in cultured hippocampal neurons can activate GSK-3ß (Li et al., 2004; Maiese and Chong, 2004). In addition, application of antisense oligonucleotides against GSK-3ß or the GSK-3ß inhibitor, lithium, can prevent cellular injury mediated by Aß (Li et al., 2004; Maiese and Chong, 2004).
GSK-3ß also can regulate amyloid precursor protein (APP) processing and the phosphorylation of tau. GSK-3ß facilitates Aß release by increasing the cellular maturation of APP (Maiese and Chong, 2004). GSK-3ß is also one of the protein kinase candidates that can phosphorylate the tau protein. Hyperphosphorylated tau is the major component of neurofibrillary tangles that consist of paired helical filaments. GSK-3ß can be necessary for sequential phosphorylation of tau at sites that are required for the formation of a paired-helical-filaments (Zheng-Fischhofer et al., 1998). Over-expression of GSK-3ß in transgenic mice results in the hyperphosphorylation of tau in hippocampal neurons and pre-tangle-like somatodendritic localization of tau (Lucas et al., 2001). If one removes GSK-3ß activity through the inhibitor lithium, hyperphosphorylation of tau is blocked and tau binding to microtubules is promoted to yield microtubule assembly (Hong et al., 1997).
Point mutations in the presenilin-1 (PS1) gene and its gene product also have been tied to tau phosphorylation and GSK-3ß activity. Mutations in PS1 account for approximately fifty percent of all cases of familial Alzheimer's disease. PS1 is a 43-48 kDa membrane bound protein with 6-8 membrane-spanning domains and is expressed in neurons throughout the brain (Maiese and Chong, 2004). Presenilin proteins that are mutated in Alzheimer's disease are believed to accelerate the production of amyloid plaques as well as possibly activate apoptotic genes. In addition, mutations in PS1 are thought to promote the phosphorylation of tau through GSK-3ß mediated pathways. PS1 can bind GSK-3ß and tau at the same region of residues 250-298 on the PS1 protein. Through this binding, PS1 may regulate the interaction of GSK-3ß with tau by bringing them into close proximity (Takashima et al., 1998). Point mutations of PS1 also have been shown to enhance the binding of PS1 to GSK-3ß and result in increased phosphorylation of tau (Takashima et al., 1998).
GSK-3ß and the substrates ß-catenin, eIF2B, c-Jun, CREB
GSK-3ß mediates cellular survival through the regulation of its multiple substrates. ß-catenin appears to one of its important targets and may be relevant during both neuronal and vascular injury models (Fig. 2). For example, in many cell systems that include ECs and cancer cells, inhibition of apoptosis has been suggested to be regulated through the stabilization and activation of ß-catenin (Chen et al., 2001; Wu et al., 2003). It has been demonstrated that loss of ß-catenin signaling in neurons increases their vulnerability to apoptotic injury in the presence of Aß (Zhang et al., 1998). Furthermore, GSK-3ß and PS1 may have an additional relationship through ß-catenin. PS1 can promote the stability of ß-catenin, a substrate of GSK-3ß. GSK-3ß negatively regulates ß-catenin through phosphorylation and degradation. In the PS1 hydrophilic loop domain, three GSK-3ß consensus phosphorylation sites are present that phosphorylate PS1 to abolish the ability of PS1 to stabilize and prevent the degradation of ß-catenin (Kirschenbaum et al., 2001).
ß-catenin also is a critical component of the Wnt signaling pathway (Chong and Maiese, 2004). Wnt can block the activity of GSK-3ß by binding to the transmembrane receptor Frizzled and the co-receptor lipoprotein related protein 5 and 6 (LRP-5/6) that leads to the recruitment of disheveled, a cytoplasmic bridging molecule (Wehrli et al., 2000). In the absence of Wnt activity, GSK-3ß phosphorylates ß-catenin at serine or threonine residues of the N-terminal region to predispose degradation of ß-catenin through ubiquination. GSK-3ß dependent phosphorylation of ß-catenin can be promoted through phosphorylation of Axin, a negative regulator of Wnt (Yamamoto et al., 1999).
The translation initiation factor 2B (eIF2B) is another phosphorylation target of GSK-3ß that is associated with regulation of cell survival. Expression of eIF2B mutants in cells that lack GSK-3ß phosphorylation or priming sites is sufficient to prevent apoptosis during GSK-3ß over-expression, PI 3-K inhibition, or growth factor deprivation (Pap and Cooper, 2002). These mutations in eIF2B also can prevent cytochrome c release resulting from PI 3-K inhibition (Pap and Cooper, 2002), suggesting that eIF2B protects cells by functioning upstream of mitochondrial cytochrome c release.
GSK-3 can lead to cell death through the expression of the c-Jun protein. c-Jun is a critical transcription factor in response to cellular stress. For example, in neurons, c-Jun triggers apoptosis through transcription activation of some pro-apoptotic genes, such as Bim (Whitfield et al., 2001). An increase in c-Jun expression occurs during trophic factor withdrawal (Hongisto et al., 2003), administration of DNA damaging agents (Besirli and Johnson, 2003), or in the presence of low potassium (Yamagishi et al., 2003). Genetic deletion of c-Jun or application of neutralizing antibodies against c-Jun results in the resistance of neurons to apoptotic stimuli (Watson et al., 1998; Behrens et al., 1999), suggesting that c-Jun plays an important role in the generation of apoptosis. In vascular ECs, specific expression of c-Jun is sufficient for induction of apoptosis in ECs. Yet, over-expression of a dominant-negative mutant of c-Jun will attenuate hydrogen peroxide cell injury in ECs (Wang et al., 1999), indicating that c-Jun also is an important cell survival regulator in ECs.
Given the roles that c-Jun plays in apoptosis, recent work has suggested that GSK-3 mediates PCD through the modulation of c-Jun activity. Application of indirubin, an inhibitor of GSK-3, can reduce c-Jun expression during trophic factor deprivation in cerebellar granule neurons. Similarly, the physiological inhibitor of GSK-3ß, FRAT1 (frequently rearranged in advanced T-cell lymphoma type 1) also prevents c-Jun activity and blocks neuronal injury during trophic factor deprivation (Hongisto et al., 2003). The more specific inhibitor of GSK-3ß, lithium, also has been shown to prevent both cJun expression and neuronal injury, suggesting that c-Jun is one of the executioners of GSK-3ß to lead to apoptotic injury.
In addition to ß-catenin, eIF2B, and c-Jun, the cAMP-response element-binding protein (CREB) also emerges as a transcriptional target of GSK-3ß for the regulation of apoptosis. GSK-3ß phosphorylates CREB at serine133 and serine129 to lead to CREB transcription and activation (Salas et al., 2003). CREB appears to have a dual role in the regulation of apoptosis. The involvement of CREB as a protectant against PCD has been well documented in a variety of cell systems. In neurons, staurosporine induced apoptosis in neuroblastoma cells is accompanied by reduced levels of CREB (Francois et al., 2000). In contrast, over-expression of CREB can protect cell lines from okadaic acid-induced apoptosis (Walton et al., 1999). In addition, some growth factors can prevent neuronal apoptosis through CREB activation and the subsequent increase in Bcl-2 expression (Pugazhenthi et al., 1999). In vascular ECs, CREB also has been demonstrated to promote expression of human heme oxygenase-1, which is a potent regulator of inflammation and can protect cells from oxidative stress (Kronke et al., 2003).
On the other hand, CREB has been reported to promote apoptotic injury. In some cell systems and under specific experimental conditions, activation of CREB promotes the development of apoptosis. For example, CREB activation functioned to foster apoptotic injury in human microvascular ECs during 2,2'4,6,6'-pentachlorobiphenyl administration (Lee et al., 2003). Furthermore, additional studies with human amnion FL cells, simian COS-7 cells, and Chinese hamster ovary cells illustrate that CREB over-expression can act to enhance apoptotic cell death (Saeki et al., 1999).
Akt and Bad
Akt also can inactivate Bad, a pro-apoptotic Bcl-2 family member, through phosphorylation of its serine residues. Bad is a Bcl-2 homology 3 (BH3)-only subfamily member of Bcl-2 proteins that are associated with the regulation of PCD. The activity of Bad is mediated through phosphorylation on its serine residues. Three phosphorylated serine sites have been identified on Bad, including serine112, serine136, and serine155. Serine112 can be phosphorylated by a number of kinases such as mitogen-activated protein kinase-activated protein kinase 1 (MAPKAP-K1), protein kinase A (PKA), and p21-activated kinase. Serine155 is phosphorylated by PKA while Akt preferentially phosphorylates the residue serine136 of Bad. A fourth phosphorylation site of Bad has recently been identified at serine170 that also results in the blockade of pro-apoptotic activity of Bad (Dramsi et al., 2002). The endogenous de-phosphorylated Bad is localized in the outer mitochondrial membrane and binds to the anti-apoptotic Bcl-2 family member Bcl-xL through its BH3 domain. Subsequent phosphorylation of Bad by Akt leads to the binding of Bad with the cytosolic protein 14-3-3 to release Bcl-xL and allows it to block PCD (Li et al., 2001). Bcl-2 and Bcl-xL prevent Bax translocation to the mitochondria, maintain the mitochondrial membrane potential, and prevent the release of cytochrome c from the mitochondria (Putcha et al., 1999; Chong et al., 2003a).
Akt promotes cellular survival in many injury systems through its phosphorylation and inhibition of Bad (Fig. 2). Akt can block neuronal apoptosis that is mediated through the phosphorylation of Bad at serine136 (Datta et al., 1997). In addition, ectopic over-expression of constitutively active Akt increases the survival of adult rat ventricular myocytes during hypoxia with reoxygenation and leads to the phosphorylation of Bad, suggesting that Akt phosphorylation of Bad is involved in cell survival mechanisms (Uchiyama et al., 2004). During hypoxia, activation of platelet-derived growth factor ß in the dorsocaudal brain stem also has been shown to block apoptosis through Akt activation and the subsequent phosphorylation of Bad (Simakajornboon et al., 2001). The complement complex C5b-9 consisting of C5b, C6, C7, C8, and C9 proteins, which play a significant role in the pathogenesis of neurodegenerative diseases, can protect oligodendrocytes from mitochondrial dysfunction, cytochrome c release, and caspase 3 activation (Soane et al., 2001). This protective capacity of C5b-9 is controlled through the combined activation of Akt with the phosphorylation of Bad, resulting in dissociation of Bad from the Bad/Bcl-xL complex (Soane et al., 2001). In the vascular system, the protection of ECs during growth factor deprivation or oxidative stress by some agents also can fostered by activation of Akt and the direct inactivation of Bad (Chong et al., 2003b; Chen et al., 2004). In cell systems that have examined neoplastic growth, estradiol has been shown to prevent apoptosis in breast cancer cells through phosphorylation of Bad that is linked to Akt activation (Fernando and Wimalasena, 2004).
Akt and IκB kinase
IκB kinase (IKK) is another downstream target of Akt that is intimately associated with cell survival (Fig. 2). IKKα and IKKß are catalytic subunits of IKK that possess serine/threonine kinase activity. IKKγ is a regulatory unit that is essential for IKK function. Akt can target IKKα activity and modulate NF-κB function. Akt appears to require IKK to efficiently stimulate the transactivation domain of the p65 subunit of NF-κB.
NF-κB is activated in response to cytokines and cellular stress (Chong et al., 2002c, 2003c). In resting cells, NF-κB is held captive by proteins of the IκB family and sequestered in the cytoplasm. Stimulation by TNF-α or oxidative stress results in the activation of the IKK complex, which phosphorylate IκB, ensuring that it is ubiquitinated by the addition of a ubiquitin group, degraded, and has released the bound NF-κB. The liberated NF-κB can then translocate to the nucleus and transcriptionally activate target genes (Chen et al., 1996).
Akt-transformed cells have been shown to be subsequently dependent upon NF-κB to suppress apoptosis during cell injury (Madrid et al., 2000). NF-κB functions as an anti-apoptotic factor through its induction of genes that inhibit PCD. NF-κB has been shown to induce the expression of the inhibitor of apoptosis (IAP) protein family (c-IAP1, c-IAP2, and x-chromosome-linked IAP (xIAP)). These proteins inhibit caspase 3, caspase 7, and caspase 9 activities (Chong et al., 2002c, 2003c). Induction of c-IAP1 and c-IAP2 activity by NF-κB also suppresses TNF-α initiated apoptosis through the inhibition of caspase 8 activity (Wang et al., 1998) as well as through the Gadd45ß protein (De Smaele et al., 2001). NF-κB also may prevent apoptosis through the direct activation of Bcl-XL (Chen et al., 2000).
Akt and caspase activation
Caspases are a family of cysteine proteases that are initially synthesized as inactive zymogens. These proteins are then proteolytically cleaved into subunits at the onset of PCD and function as active caspases after reconstitution to molecular heterodimers. Caspases are composed of three domains including an N-terminal prodomain, a large subunit, and a small subunit. The apoptotic-associated caspases include initiator caspases, such as caspase 2, 8, 9, and 10, that activate downstream executioner caspases, resulting in an amplification of cascade activity. The initiator caspases consist of long N-terminal prodomains that contain caspase recruitment domains (CARDs) in caspase 2 and caspase 9, or death effector domains (DEDs) in caspase 8 and caspase 10. Another set of caspases, termed the executioner caspases, consist of caspase 3, 6, and 7 that function to directly cleave crucial cellular protein substrates that result in cell destruction. The executioner caspases contain short prodomains or have no prodomains (Li et al., 2004; Maiese and Chong, 2004).
Activation of caspases proceeds through an extrinsic pathway and an intrinsic pathway. The extrinsic pathway is initiated by death receptor activation at the cell surface, resulting in the recruitment and activation of the initiator caspase 8 upon apoptotic stimuli (Ashkenazi and Dixit, 1998). The intracellular death domain of death receptors, such as the TNF superfamily, CD95/Fas/Apo-1, and the death receptor 3 (DR3), undergoes conformational change upon binding to extracellular ligands and forms an intracellular death-inducing signaling complex (DISC) following recruitment of adaptor molecules, such as the Fas associated death domain (FADD). FADD recruits caspase 8 through its DED domain and leads to caspase 8 activation (Juo et al., 1998). Caspase 8 can subsequently activate caspase 3. In addition, caspase 8 activation also may result in the cleavage of Bid, a pro-apoptotic member of Bcl-2 family, allowing the truncated Bid (tBid) to translocate to the mitochondria. This leads to cytochrome c release through Bax resulting in the subsequent activation of executioner caspases.
The intrinsic caspase pathway involves mitochondrial dysfunction. The mitochondrial pathway is associated with the release of cytochrome c and subsequent activation of caspase 9 followed by activation of caspase 3 (Liu et al., 1996). The process is regulated by the Bcl-2 subfamily BH3-only proteins, which are normally located in cellular compartments other than mitochondria, but translocate to the mitochondria in response to apoptotic stimuli (Cosulich et al., 1997). The translocation of these proteins delivers an apoptotic signal to mitochondria through the interaction with Bax to induce the release of cytochrome c that then binds to apoptotic protease activating factor-1 (Apaf-1). Apaf-1 consists of three different domains that include a CARD, repeats of tryptophan and aspartate residues (WD-40 repeats), and a nucleotide-binding domain CED-4. Binding of cytochrome c to Apaf-1 results in the removal of the WD-40 domain, masking the CED-4 and CARD domains, and leads to the oligomerization of Apaf-1 under the assistance of dATP/ATP (Hu et al., 1999). The oligomerization of Apaf-1 promotes the allosteric activation of caspase 9 by forming the Apaf-1 apoptosome (Li et al., 1997). Caspase 9 can subsequently activate caspase 3 as well as caspase 1 through the intermediary caspase 8. Together, caspase 1 and caspase 3 lead to both DNA fragmentation and membrane PS exposure (Li et al., 1997; Maiese and Vincent, 2000; Chong et al., 2002b).
Akt may prevent the induction of caspase activity in the CNS either through direct inhibition or through the modulation of its substrates (Fig. 2). In neuronal cell populations, Akt1 can maintain nuclear DNA integrity and membrane PS exposure through the specific inhibition of caspase 3-, 8-, and 9-like activities (Kang et al., 2003a,b). Furthermore, Akt can directly modulate the activity of caspase 9. Akt phosphorylates caspase 9 on its serine196 residue and inhibits its protease activity (Chong, 2005). Over-expression of Akt has been shown to block microglial activation and proliferation that requires the inhibition of caspase 9 (Kang et al., 2003a). In addition, Akt enhances cell survival in neurons or ECs through modulation of caspase 9 during oxidative stress (Kang, 2003; Chong, 2004, 2005). Insulin also has been demonstrated to protect ECs against TNF-α injury as a result of Akt activation and the subsequent phosphorylation of caspase 9 (Hermann et al., 2000).
Through its substrate FOXO, Akt can indirectly control cell survival through the modulation of caspases. Fas-associated death domain-like interleukins 1ß-converting enzyme (FLICE)-like inhibitory protein (FLIP) is a homolog of caspase 8 and functions as a dominant-negative inhibitor of caspase 8. A recent study has demonstrated that transduction of active FOXO3a can result in the down-regulation of FLIP, an increase of caspase 8 activity, and the promotion of apoptosis in ECs (Skurk et al., 2004). In contrast, over-expression of a dominant-negative FOXO3a increased FLIP expression, down-regulated caspase 8 activity, and blocked apoptosis during serum deprivation (Skurk et al., 2004).
Interestingly, caspase 3 also may be responsible for the degradation of FOXO3a. The integrity of FOXO may function as a significant precipitant of cell injury. During periods of oxidative stress in the nervous system, an initial inhibitory phosphorylation of FOXO3a at the regulatory phosphorylation sites (Thr32 and Ser253) can occur (Chong, 2005). However, loss of phosphorylated FOXO3a expression appears to subsequently result over a twelve hour period, possibly by caspase 3 cleavage, which potentially can enhance the vulnerability of cells to apoptotic injury (Chong, 2005). FOXO3a proteolysis occurs during cell injury yielding an amino-terminal (Nt) fragment that can become biologically active (Charvet et al., 2003). During cell injury and caspase-dependent cleavage of FOXO3a, it is the activation of FOXO3a Nt fragments that become available and result in apoptotic cellular injury.
Akt and the tumor suppressor protein p53
Modulation of cell function, integrity and survival can be dependent upon the activation of Akt and the control of the tumor suppressor protein p53 (Fig. 2). The protein p53 plays an important role in the induction of apoptosis that is precipitated by DNA damage. In several cell populations, studies suggest that p53 can directly lead to PCD. For example, over-expression of p53 can lead to widespread neuronal cell death through apoptosis (Jordan et al., 1997). The induction of spinal motor neuronal apoptosis following nerve avulsion also is triggered by p53 activation (Martin and Liu, 2002). In vascular ECs, over-expression of p53 also induces apoptosis in normoxic condition and accelerates apoptosis during hypoxia (Stempien-Otero et al., 1999).
Akt can phosphorylate murine double minute (Mdm2), an ubiquitin ligase, to control p53 ubiquitination and degradation. Akt has been demonstrated to phosphorylate Mdm2 at serine166 and serine186 which increases the nuclear localization of Mdm2 (Zhou et al., 2001). Mdm2 can prevent PCD by controlling the activity of the pro-apoptotic protein p53. In the nucleus, Mdm2 targets p53 for ubiquitination resulting in the translocation of p53 to the cytoplasm and subsequent degradation of p53. Once activated, Akt places Mdm2 into action to block p53 dependent apoptosis. Akt may promote cell survival through the inhibition of p53 transcriptional activity (Yamaguchi et al., 2001) that may come under control by the nicotinamide adenine dinucleotide (NAD+) precursor nicotinamide (Maiese and Chong, 2003; Chong et al., 2004c). Activation of p53 can promote the expression of Bax to result in apoptotic cell death. Nicotinamide has been shown to either directly limit the expression of p53 (Sonee et al., 2003) or prevent an NAD-dependent p53 deacetylation induced by Sir2α (Luo et al., 2001). In addition to nicotinamide, Ras has been suggested to protect sympathetic neurons by suppressing the p53 apoptotic pathways through activation of PI-3 K/Akt pathway (Mazzoni et al., 1999).
Akt and JIPs
JIPs, which are also known as islet-brain proteins, are scaffold proteins that are present in the islets of Langerhans and in the nervous system (Moulin and Widmann, 2004). JIPs control a host of cellular mechanisms that include the mitogen-activated protein kinase (MAPK) pathways of JNK and p38. JIPs have been linked to several neurodegenerative processes such as Alzheimer's disease (Moulin and Widmann, 2004).
In particular, c-Jun N-terminal kinase interacting protein 1 (JIP-1) has been identified as a target of Akt to regulate apoptotic injury in the nervous system (Fig. 2). JIP-1 can bind to JNK to upregulate JNK activity that may be detrimental to cells (Whitmarsh et al., 2001). It is Akt1 that preferentially interacts with JIP-1 to prevent the association of JIP-1 with JNK pathways to block apoptotic injury (Kim et al., 2002). In a number of instances, activation of Akt appears to temper the actions of JNK. Over-expression of Akt in neuronal cell lines prevents the induction of JNK activity during insults, such as serum withdrawal or UV irradiation, that is accompanied by the induction of JIP-1 (Levresse et al., 2000). Yet, it is important to note that under some conditions, Akt may be ineffective to alter the association between JNK and JIP-1. Activation of Akt can result in enhanced JIP-1 expression in neurons (Levresse et al., 2000). In addition, toxic insults that include excitotoxicty have been shown to not only decrease the association between Akt1 and JIP1, but also increase the formation of JIP1-JNK complexes that lead to PCD (Kim et al., 2002).
Conclusion
As a potential therapeutic target, Akt as well as its downstream substrates, offer great promise for the development of therapeutics against a number of neurodegenerative disorders that may be acute in nature, such as stroke, or more sub-acute in duration, such as Alzheimer's disease. Initially believed to have cellular functions directed primarily toward cell development and maintenance, Akt is now seen as a potential broad cytoprotective agent. Akt can offer cellular protection not only through the repair of apoptotic DNA degradation, but also through the preservation of membrane PS asymmetry that applies to both vascular thrombosis and cellular inflammation. Akt drives cellular survival through a series of distinct pathways that involve the Forkhead family of transcription factors, GSK-3ß, ß-catenin, eIF2B, c-Jun, CREB, Bad, IKK, p53, and JIPs. Interestingly, both extrinsic and intrinsic caspase pathways are intimately tied to the protective capacity of Akt to both foster genomic integrity as well as prevent microglial activation that leads to inflammatory cell demise. Yet, it is evident that further work that clarifies the cellular environment controlled by Akt will be of exceptional value to refine our knowledge of Akt and to maximize the potential of this protein as a therapeutic agent.
Acknowledgements
This research was supported by the following grants (KM): American Heart Association (National), Janssen Neuroscience Award, Johnson and Johnson Focused Investigator Award, LEARN Foundation Award, MI Life Sciences Challenge Award, and NIH NIEHS (P30 ES06639).
References
- Altomare DA, Guo K, Cheng JQ, Sonoda G, Walsh K, Testa JR. Cloning, chromosomal localization and expression analysis of the mouse Akt2 oncogene. Oncogene. 1995;11:1055–1060. [PubMed] [Google Scholar]
- Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, Diaz-Nido J. Lithium protects cultured neurons against beta-amyloid-induced neurodegeneration. FEBS Lett. 1999;453:260–264. doi: 10.1016/s0014-5793(99)00685-7. [DOI] [PubMed] [Google Scholar]
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Balendran A, Casamayor A, Deak M, Paterson A, Gaffney P, Currie R, Downes CP, Alessi DR. PDK1 acquires PDK2 activity in the presence of a synthetic peptide derived from the carboxyl terminus of PRK2. Curr. Biol. 1999;9:393–404. doi: 10.1016/s0960-9822(99)80186-9. [DOI] [PubMed] [Google Scholar]
- Basso AD, Solit DB, Chiosis G, Giri B, Tsichlis P, Rosen N. Akt forms an intracellular complex with heat shock protein 90 (Hsp90) and Cdc37 and is destabilized by inhibitors of Hsp90 function. J. Biol. Chem. 2002;277:39858–39866. doi: 10.1074/jbc.M206322200. [DOI] [PubMed] [Google Scholar]
- Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17:313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- Besirli CG, Johnson EM., Jr. JNK-independent activation of c-Jun during neuronal apoptosis induced by multiple DNA-damaging agents. J. Biol. Chem. 2003;278:22357–22366. doi: 10.1074/jbc.M300742200. [DOI] [PubMed] [Google Scholar]
- Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, Lee CM. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. 2000;97:11074–11079. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Cai Z, Manalo DJ, Wei G, Rodriguez ER, Fox-Talbot K, Lu H, Zweier JL, Semenza GL. Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation. 2003;108:79–85. doi: 10.1161/01.CIR.0000078635.89229.8A. [DOI] [PubMed] [Google Scholar]
- Carver DJ, Aman MJ, Ravichandran KS. SHIP inhibits Akt activation in B cells through regulation of Akt membrane localization. Blood. 2000;96:1449–1456. [PubMed] [Google Scholar]
- Charvet C, Alberti I, Luciano F, Jacquel A, Bernard A, Auberger P, Deckert M. Proteolytic regulation of Forkhead transcription factor FOXO3α by caspase-3-like proteases. Oncogene. 2003;22:4557–4568. doi: 10.1038/sj.onc.1206778. [DOI] [PubMed] [Google Scholar]
- Chen C, Edelstein LC, Gelinas C. The Rel/NF-kappaB family directly activates expression of the apoptosis inhibitor Bcl-x(L) Mol. Cell. Biol. 2000;20:2687–2695. doi: 10.1128/mcb.20.8.2687-2695.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JH, Hsiao G, Lee AR, Wu CC, Yen MH. Andrographolide suppresses endothelial cell apoptosis via activation of phosphatidyl inositol-3-kinase/Akt pathway. Biochem. Pharmacol. 2004;67:1337–1345. doi: 10.1016/j.bcp.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Chen S, Guttridge DC, You Z, Zhang Z, Fribley A, Mayo MW, Kitajewski J, Wang CY. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J. Cell. Biol. 2001;152:87–96. doi: 10.1083/jcb.152.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, Parent L, Maniatis T. Site-specific phosphorylation of IkappaBalpha by a novel ubiquitination-dependent protein kinase activity. Cell. 1996;84:853–862. doi: 10.1016/s0092-8674(00)81064-8. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Maiese K. Targeting WNT, protein kinase B, and mitochondrial membrane integrity to foster cellular survival in the nervous system. Histol. Histopathol. 2004;19:495–504. doi: 10.14670/hh-19.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Angiogenesis and plasticity: role of erythropoietin in vascular systems. J. Hematother. Stem. Cell Res. 2002a;11:863–871. doi: 10.1089/152581602321080529. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation. 2002b;106:2973–2979. doi: 10.1161/01.cir.0000039103.58920.1f. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Hematopoietic Factor Erythropoietin Fosters Neuroprotection Through Novel Signal Transduction Cascades. J. Cereb. Blood Flow Metab. 2002c;22:503–514. doi: 10.1097/00004647-200205000-00001. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Apaf-1, Bcl-xL, Cytochrome c, and Caspase-9 Form the Critical Elements for Cerebral Vascular Protection by Erythropoietin. J. Cereb. Blood Flow Metab. 2003a;23:320–330. doi: 10.1097/01.WCB.0000050061.57184.AE. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br. J. Pharmacol. 2003b;138:1107–1118. doi: 10.1038/sj.bjp.0705161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Erythropoietin: cytoprotection in vascular and neuronal cells. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2003c;3:141–154. doi: 10.2174/1568006033481483. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Kang JQ, Maiese K. Erythropoietin prevents early and late neuronal demise through modulation of Akt1 and induction of caspase 1, 3, and 8. J. Neurosci. Res. 2003d;71:659–669. doi: 10.1002/jnr.10528. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Kang JQ, Maiese K. The tyrosine phosphatase SHP2 modulates MAP kinase p38 and caspase 1 and 3 to foster neuronal survival. Cell. Mol. Neurobiol. 2003e;23:561–578. doi: 10.1023/A:1025158314016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Akt1 drives endothelial cell membrane asymmetry and microglial activation through Bcl-x(L) and caspase 1, 3, and 9. Exp. Cell Res. 2004a;296:196–207. doi: 10.1016/j.yexcr.2004.01.021. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Kang JQ, Maiese K. Essential cellular regulatory elements of oxidative stress in early and late phases of apoptosis in the central nervous system. Antioxid. Redox. Signal. 2004b;6:277–287. doi: 10.1089/152308604322899341. [DOI] [PubMed] [Google Scholar]
- Chong ZZ, Lin SH, Maiese K. The NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3α and mitochondrial membrane potential. J. Cereb. Blood Flow Metab. 2004c;24:728–743. doi: 10.1097/01.WCB.0000122746.72175.0E. [DOI] [PubMed] [Google Scholar]
- Conery AR, Cao Y, Thompson EA, Townsend CM, Jr., Ko TC, Luo K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat. Cell Biol. 2004;6:366–372. doi: 10.1038/ncb1117. [DOI] [PubMed] [Google Scholar]
- Cosulich SC, Worrall V, Hedge PJ, Green S, Clarke PR. Regulation of apoptosis by BH3 domains in a cell-free system. Curr. Biol. 1997;7:913–920. doi: 10.1016/s0960-9822(06)00410-6. [DOI] [PubMed] [Google Scholar]
- Crowder RJ, Freeman RS. Glycogen synthase kinase-3 beta activity is critical for neuronal death caused by inhibiting phosphatidylinositol 3-kinase or Akt but not for death caused by nerve growth factor withdrawal. J. Biol. Chem. 2000;275:34266–34271. doi: 10.1074/jbc.M006160200. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature. 2001;414:308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EW, Burgering BM, Raaijmakers JA, Lammers JW, Koenderman L, Coffer PJ. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1) Mol. Cell. Biol. 2000;20:9138–9148. doi: 10.1128/mcb.20.24.9138-9148.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkers PF, Birkenkamp KU, Lam EW, Thomas NS, Lammers JW, Koenderman L, Coffer PJ. FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J. Cell Biol. 2002;156:531–542. doi: 10.1083/jcb.200108084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dramsi S, Scheid MP, Maiti A, Hojabrpour P, Chen X, Schubert K, Goodlett DR, Aebersold R, Duronio V. Identification of a novel phosphorylation site, Ser-170, as a regulator of bad pro-apoptotic activity. J. Biol. Chem. 2002;277:6399–6405. doi: 10.1074/jbc.M109990200. [DOI] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Fernando RI, Wimalasena J. Estradiol abrogates apoptosis in breast cancer cells through inactivation of BAD: Ras dependent non-genomic pathways requiring signaling through ERK and Akt. Mol. Biol. Cell. 2004;25:3266–3284. doi: 10.1091/mbc.E03-11-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine V, Mohand-Said S, Hanoteau N, Fuchs C, Pfizenmaier K, Eisel U. Neurodegenerative and neuroprotective effects of tumor Necrosis factor (TNF) in retinal ischemia: opposite roles of TNF receptor 1 and TNF receptor 2. J. Neurosci. 2002;22:RC216. doi: 10.1523/JNEUROSCI.22-07-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois F, Godinho MJ, Grimes ML. CREB is cleaved by caspases during neural cell apoptosis. FEBS Lett. 2000;486:281–284. doi: 10.1016/s0014-5793(00)02316-4. [DOI] [PubMed] [Google Scholar]
- Frodin M, Antal TL, Dummler BA, Jensen CJ, Deak M, Gammeltoft S, Biondi RM. A phosphoserine/threonine-binding pocket in AGC kinases and PDK1 mediates activation by hydrophobic motif phosphorylation. EMBO J. 2002;21:5396–5407. doi: 10.1093/emboj/cdf551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell Biol. 2003;162:613–622. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatsugai N, Kuroyanagi M, Yamada K, Meshi T, Tsuda S, Kondo M, Nishimura M, Hara-Nishimura I. A plant vacuolar protease, VPE, mediates virus-induced hypersensitive cell death. Science. 2004;305:855–858. doi: 10.1126/science.1099859. [DOI] [PubMed] [Google Scholar]
- Henry MK, Lynch JT, Eapen AK, Quelle FW. DNA damage-induced cell-cycle arrest of hematopoietic cells is overridden by activation of the PI-3 kinase/Akt signaling pathway. Blood. 2001;98:834–841. doi: 10.1182/blood.v98.3.834. [DOI] [PubMed] [Google Scholar]
- Hermann C, Assmus B, Urbich C, Zeiher AM, Dimmeler S. Insulin-mediated stimulation of protein kinase Akt: A potent survival signaling cascade for endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2000;20:402–409. doi: 10.1161/01.atv.20.2.402. [DOI] [PubMed] [Google Scholar]
- Hong M, Chen DC, Klein PS, Lee VM. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J. Biol. Chem. 1997;272:25326–25332. doi: 10.1074/jbc.272.40.25326. [DOI] [PubMed] [Google Scholar]
- Hongisto V, Smeds N, Brecht S, Herdegen T, Courtney MJ, Coffey ET. Lithium blocks the c-Jun stress response and protects neurons via its action on glycogen synthase kinase 3. Mol. Cell. Biol. 2003;23:6027–6036. doi: 10.1128/MCB.23.17.6027-6036.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Benedict MA, Ding L, Nunez G. Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase- 9 activation and apoptosis. EMBO J. 1999;18:3586–3595. doi: 10.1093/emboj/18.13.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivins Zito C, Kontaridis MI, Fornaro M, Feng GS, Bennett AM. SHP-2 regulates the phosphatidylinositide 3′-kinase/Akt pathway and suppresses caspase 3-mediated apoptosis. J. Cell. Physiol. 2004;199:227–236. doi: 10.1002/jcp.10446. [DOI] [PubMed] [Google Scholar]
- Jessel R, Haertel S, Socaciu C, Tykhonova S, Diehl HA. Kinetics of apoptotic markers in exogeneously induced apoptosis of EL4 cells. J. Cell. Mol. Med. 2002;6:82–92. doi: 10.1111/j.1582-4934.2002.tb00313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonassen AK, Sack MN, Mjos OD, Yellon DM. Myocardial protection by insulin at reperfusion requires early administration and is mediated via Akt and p70s6 kinase cell-survival signaling. Circ. Res. 2001;89:1191–1198. doi: 10.1161/hh2401.101385. [DOI] [PubMed] [Google Scholar]
- Jope RS, Johnson GV. The glamour and gloom of glycogen synthase kinase-3. Trends Biochem. Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Jordan J, Galindo MF, Prehn JH, Weichselbaum RR, Beckett M, Ghadge GD, Roos RP, Leiden JM, Miller RJ. p53 expression induces apoptosis in hippocampal pyramidal neuron cultures. J. Neurosci. 1997;17:1397–1405. doi: 10.1523/JNEUROSCI.17-04-01397.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juo P, Kuo CJ, Yuan J, Blenis J. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr. Biol. 1998;8:1001–1008. doi: 10.1016/s0960-9822(07)00420-4. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Chong ZZ, Maiese K. Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J. Neurosci. Res. 2003a;74:37–51. doi: 10.1002/jnr.10740. [DOI] [PubMed] [Google Scholar]
- Kang JQ, Chong ZZ, Maiese K. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol. Pharmacol. 2003b;64:557–569. doi: 10.1124/mol.64.3.557. [DOI] [PubMed] [Google Scholar]
- Katner AL, Hoang QB, Gootam P, Jaruga E, Ma Q, Gnarra J, Rayford W. Induction of cell cycle arrest and apoptosis in human prostate carcinoma cells by a recombinant adenovirus expressing p27(Kip1) Prostate. 2002;53:77–87. doi: 10.1002/pros.10124. [DOI] [PubMed] [Google Scholar]
- Kermer P, Klocker N, Labes M, Bahr M. Insulin-like growth factor-I protects axotomized rat retinal ganglion cells from secondary death via PI3-K-dependent Akt phosphorylation and inhibition of caspase-3 In vivo. J. Neurosci. 2000;20:2–8. [PubMed] [Google Scholar]
- Kim AH, Yano H, Cho H, Meyer D, Monks B, Margolis B, Birnbaum MJ, Chao MV. Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron. 2002;35:697–709. doi: 10.1016/s0896-6273(02)00821-8. [DOI] [PubMed] [Google Scholar]
- Kirschenbaum F, Hsu SC, Cordell B, McCarthy JV. Glycogen synthase kinase-3beta regulates presenilin 1 C-terminal fragment levels. J. Biol. Chem. 2001;276:30701–30707. doi: 10.1074/jbc.M102849200. [DOI] [PubMed] [Google Scholar]
- Koh SH, Kim SH, Kwon H, Park Y, Kim KS, Song CW, Kim J, Kim MH, Yu HJ, Henkel JS, Jung HK. Epigallocatechin gallate protects nerve growth factor differentiated PC12 cells from oxidative-radical-stress-induced apoptosis through its effect on phosphoinositide 3-kinase/Akt and glycogen synthase kinase-3. Brain Res. Mol. Brain Res. 2003;118:72–81. doi: 10.1016/j.molbrainres.2003.07.003. [DOI] [PubMed] [Google Scholar]
- Kops GJ, Dansen TB, Polderman PE, Saarloos I, Wirtz KW, Coffer PJ, Huang TT, Bos JL, Medema RH, Burgering BM. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature. 2002;419:316–321. doi: 10.1038/nature01036. [DOI] [PubMed] [Google Scholar]
- Kronke G, Bochkov VN, Huber J, Gruber F, Bluml S, Furnkranz A, Kadl A, Binder BR, Leitinger N. Oxidized phospholipids induce expression of human heme oxygenase-1 involving activation of cAMP-responsive element-binding protein. J. Biol. Chem. 2003;278:51006–51014. doi: 10.1074/jbc.M304103200. [DOI] [PubMed] [Google Scholar]
- Laine J, Kunstle G, Obata T, Sha M, Noguchi M. The protooncogene TCL1 is an Akt kinase coactivator. Mol. Cell. 2000;6:395–407. doi: 10.1016/s1097-2765(00)00039-3. [DOI] [PubMed] [Google Scholar]
- Latchman D. Protective effect of heat shock proteins in the nervous system. Curr. Neurovasc. Res. 2004;1:21–27. doi: 10.2174/1567202043480206. [DOI] [PubMed] [Google Scholar]
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP. Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- Lee YW, Park HJ, Son KW, Hennig B, Robertson LW, Toborek M. 2,2′,4,6,6′-pentachlorobiphenyl (PCB 104) induces apoptosis of human microvascular endothelial cells through the caspase-dependent activation of CREB. Toxicol. Appl. Pharmacol. 2003;189:1–10. doi: 10.1016/s0041-008x(03)00084-x. [DOI] [PubMed] [Google Scholar]
- Levresse V, Butterfield L, Zentrich E, Heasley LE. Akt negatively regulates the cJun N-terminal kinase pathway in PC12 cells. J. Neurosci. Res. 2000;62:799–808. doi: 10.1002/1097-4547(20001215)62:6<799::AID-JNR6>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Li F, Chong ZZ, Maiese K. Navigating novel mechanisms of cellular plasticity with the NAD+ precursor and nutrient nicotinamide. Front. Biosci. 2004;9:2500–2520. doi: 10.2741/1412. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Li Y, Tennekoon GI, Birnbaum M, Marchionni MA, Rutkowski JL. Neuregulin signaling through a PI3K/Akt/Bad pathway in Schwann cell survival. Mol. Cell. Neurosci. 2001;17:761–767. doi: 10.1006/mcne.2000.0967. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Loberg RD, Vesely E, Brosius FC., 3rd. Enhanced glycogen synthase kinase-3beta activity mediates hypoxia-induced apoptosis of vascular smooth muscle cells and is prevented by glucose transport and metabolism. J. Biol. Chem. 2002;277:41667–41673. doi: 10.1074/jbc.M206405200. [DOI] [PubMed] [Google Scholar]
- Lu Y, Yu Q, Liu JH, Zhang J, Wang H, Koul D, McMurray JS, Fang X, Yung WK, Siminovitch KA, Mills GB. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J. Biol. Chem. 2003;278:40057–40066. doi: 10.1074/jbc.M303621200. [DOI] [PubMed] [Google Scholar]
- Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3ßeta conditional transgenic mice. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- Madrid LV, Wang CY, Guttridge DC, Schottelius AJ, Baldwin AS, Jr., Mayo MW. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-kappaB. Mol. Cell. Biol. 2000;20:1626–1638. doi: 10.1128/mcb.20.5.1626-1638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ. Nicotinamide: necessary nutrient emerges as a novel cytoprotectant for the brain. Trends Pharmacol. Sci. 2003;24:228–232. doi: 10.1016/S0165-6147(03)00078-6. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ. Insights into oxidative stress and potential novel therapeutic targets for Alzheimer disease. Restor. Neurol. Neurosci. 2004;22:87–104. [PubMed] [Google Scholar]
- Maiese K, Vincent AM. Membrane asymmetry and DNA degradation: functionally distinct determinants of neuronal programmed cell death. J. Neurosci. Res. 2000;59:568–580. doi: 10.1002/(SICI)1097-4547(20000215)59:4<568::AID-JNR13>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ, Kang J. Transformation into treatment: Novel therapeutics that begin within the cell. In: Maiese K, editor. Neuronal and vascular plasticity: Elucidating basic cellular mechanisms for future therapeutic discovery. Kluwer Academic Publishers; Norwell, MA: 2003. pp. 1–26. [Google Scholar]
- Maiese K, Faqi L, Chong ZZ. Erythropoietin in the brain: Can the promise to protect be fulfilled? Trends. Pharmacol. Sci. 2004;25:577–583. doi: 10.1016/j.tips.2004.09.006. [DOI] [PubMed] [Google Scholar]
- Maira SM, Galetic I, Brazil DP, Kaech S, Ingley E, Thelen M, Hemmings BA. Carboxyl-terminal modulator protein (CTMP), a negative regulator of PKB/Akt and v-Akt at the plasma membrane. Science. 2001;294:374–380. doi: 10.1126/science.1062030. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Liu Z. Injury-induced spinal motor neuron apoptosis is preceded by DNA single-strand breaks and is p53- and Bax-dependent. J. Neurobiol. 2002;50:181–197. doi: 10.1002/neu.10026. [DOI] [PubMed] [Google Scholar]
- Matsuzaki H, Tamatani M, Mitsuda N, Namikawa K, Kiyama H, Miyake S, Tohyama M. Activation of Akt kinase inhibits apoptosis and changes in Bcl-2 and Bax expression induced by nitric oxide in primary hippocampal neurons. J. Neurochem. 1999;73:2037–2046. [PubMed] [Google Scholar]
- Mazzoni IE, Said FA, Aloyz R, Miller FD, Kaplan D. Ras regulates sympathetic neuron survival by suppressing the p53-mediated cell death pathway. J. Neurosci. 1999;19:9716–9727. doi: 10.1523/JNEUROSCI.19-22-09716.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature. 2000;404:782–787. doi: 10.1038/35008115. [DOI] [PubMed] [Google Scholar]
- Moulin N, Widmann C. Islet-brain (IB)/JNK-interacting proteins (JIPs): Future targets for the treatment of neurodegenerative diseases? Curr. Neurovasc. Res. 2004;1:111–127. doi: 10.2174/1567202043480161. [DOI] [PubMed] [Google Scholar]
- Nakatani K, Sakaue H, Thompson DA, Weigel RJ, Roth RA. Identification of a human Akt3 (protein kinase B gamma) which contains the regulatory serine phosphorylation site. Biochem. Biophys. Res. Commun. 1999;257:906–910. doi: 10.1006/bbrc.1999.0559. [DOI] [PubMed] [Google Scholar]
- Naruse I, Hoshino H, Dobashi K, Minato K, Saito R, Mori M. Over-expression of p27kip1 induces growth arrest and apoptosis mediated by changes of pRb expression in lung cancer cell lines. Int. J. Cancer. 2000;88:377–383. [PubMed] [Google Scholar]
- Obal I, Jakab JS, Siklos L, Engelhardt JI. Recruitment of activated microglia cells in the spinal cord of mice by ALS IgG. Neuroreport. 2001;12:2449–2452. doi: 10.1097/00001756-200108080-00032. [DOI] [PubMed] [Google Scholar]
- Owada Y, Utsunomiya A, Yoshimoto T, Kondo H. Expression of mRNA for Akt, serine-threonine protein kinase, in the brain during development and its transient enhancement following axotomy of hypoglossal nerve. J. Mol. Neurosci. 1997;9:27–33. doi: 10.1007/BF02789392. [DOI] [PubMed] [Google Scholar]
- Pap M, Cooper GM. Role of translation initiation factor 2B in control of cell survival by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta signaling pathway. Mol. Cell. Biol. 2002;22:578–586. doi: 10.1128/MCB.22.2.578-586.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papapetropoulos A, Fulton D, Mahboubi K, Kalb RG, O'Connor DS, Li F, Altieri DC, Sessa WC. Angiopoietin-1 inhibits endothelial cell apoptosis via the Akt/survivin pathway. J. Biol. Chem. 2000;275:9102–9105. doi: 10.1074/jbc.275.13.9102. [DOI] [PubMed] [Google Scholar]
- Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J. Neuropathol. Expl. Neurol. 1999;58:1010–1019. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
- Pekarsky Y, Koval A, Hallas C, Bichi R, Tresini M, Malstrom S, Russo G, Tsichlis P, Croce CM. Tcl1 enhances Akt kinase activity and mediates its nuclear translocation. Proc. Natl. Acad. Sci. 2000;97:3028–3033. doi: 10.1073/pnas.040557697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugazhenthi S, Miller E, Sable C, Young P, Heidenreich KA, Boxer LM, Reusch JE. Insulin-like growth factor-I induces bcl-2 promoter through the transcription factor cAMP-response element-binding protein. J. Biol. Chem. 1999;274:27529–27535. doi: 10.1074/jbc.274.39.27529. [DOI] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson EM., Jr. BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J. Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rena G, Prescott AR, Guo S, Cohen P, Unterman TG. Roles of the forkhead in rhabdomyosarcoma (FKHR) phosphorylation sites in regulating 14-3-3 binding, transactivation and nuclear targetting. Biochem. J. 2001;354:605–612. doi: 10.1042/0264-6021:3540605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rytomaa M, Lehmann K, Downward J. Matrix detachment induces caspase-dependent cytochrome c release from mitochondria: inhibition by PKB/Akt but not Raf signalling. Oncogene. 2000;19:4461–4468. doi: 10.1038/sj.onc.1203805. [DOI] [PubMed] [Google Scholar]
- Saeki K, Yuo A, Suzuki E, Yazaki Y, Takaku F. Aberrant expression of cAMP-response-element-binding protein (‘CREB’) induces apoptosis. Biochem. J. 1999;343(Pt 1):249–255. [PMC free article] [PubMed] [Google Scholar]
- Salas TR, Reddy SA, Clifford JL, Davis RJ, Kikuchi A, Lippman SM, Menter DG. Alleviating the suppression of glycogen synthase kinase-3beta by Akt leads to the phosphorylation of cAMP-response element-binding protein and its transactivation in intact cell nuclei. J. Biol. Chem. 2003;278:41338–41346. doi: 10.1074/jbc.M302972200. [DOI] [PubMed] [Google Scholar]
- Sasaoka T, Wada T, Fukui K, Murakami S, Ishihara H, Suzuki R, Tobe K, Kadowaki T, Kobayashi M. SH2-containing inositol phosphatase 2 predominantly regulates Akt2, and not Akt1, phosphorylation at the plasma membrane in response to insulin in 3T3-L1 adipocytes. J. Biol. Chem. 2004;279:14835–14843. doi: 10.1074/jbc.M311534200. [DOI] [PubMed] [Google Scholar]
- Sato S, Fujita N, Tsuruo T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. 2000;97:10832–10837. doi: 10.1073/pnas.170276797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Fernandez de Mattos S, van der Horst A, Klompmaker R, Kops GJ, Lam EW, Burgering BM, Medema RH. Cell cycle inhibition by FoxO forkhead transcription factors involves downregulation of cyclin D. Mol. Cell. Biol. 2002;22:7842–7852. doi: 10.1128/MCB.22.22.7842-7852.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw M, Cohen P, Alessi DR. Further evidence that the inhibition of glycogen synthase kinase-3beta by IGF-1 is mediated by PDK1/PKB-induced phosphorylation of Ser-9 and not by dephosphorylation of Tyr-216. FEBS Lett. 1997;416:307–311. doi: 10.1016/s0014-5793(97)01235-0. [DOI] [PubMed] [Google Scholar]
- Simak J, Holada K, Vostal JG. Release of annexin V-binding membrane microparticles from cultured human umbilical vein endothelial cells after treatment with camptothecin. BMC Cell. Biol. 2002;3:11. doi: 10.1186/1471-2121-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simakajornboon N, Szerlip NJ, Gozal E, Anonetapipat JW, Gozal D. In vivo PDGF beta receptor activation in the dorsocaudal brainstem of the rat prevents hypoxia-induced apoptosis via activation of Akt and BAD. Brain. Res. 2001;895:111–118. doi: 10.1016/s0006-8993(01)02054-6. [DOI] [PubMed] [Google Scholar]
- Singhrao SK, Neal JW, Morgan BP, Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington's disease. Exp. Neurol. 1999;159:362–376. doi: 10.1006/exnr.1999.7170. [DOI] [PubMed] [Google Scholar]
- Skurk C, Maatz H, Kim HS, Yang J, Abid MR, Aird WC, Walsh K. The Akt-regulated forkhead transcription factor FOXO3α controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J. Biol. Chem. 2004;279:1513–1525. doi: 10.1074/jbc.M304736200. [DOI] [PubMed] [Google Scholar]
- Soane L, Cho HJ, Niculescu F, Rus H, Shin ML. C5b-9 terminal complement complex protects oligodendrocytes from death by regulating Bad through phosphatidylinositol 3-kinase/Akt pathway. J. Immunol. 2001;167:2305–2311. doi: 10.4049/jimmunol.167.4.2305. [DOI] [PubMed] [Google Scholar]
- Somervaille TC, Linch DC, Khwaja A. Growth factor withdrawal from primary human erythroid progenitors induces apoptosis through a pathway involving glycogen synthase kinase-3 and Bax. Blood. 2001;98:1374–1381. doi: 10.1182/blood.v98.5.1374. [DOI] [PubMed] [Google Scholar]
- Sonee M, Martens JR, Evers MR, Mukherjee SK. The effect of tertiary butylhydroperoxide and nicotinamide on human cortical neurons. Neurotoxicology. 2003;24:443–448. doi: 10.1016/S0161-813X(03)00019-6. [DOI] [PubMed] [Google Scholar]
- Sordella R, Bell DW, Haber DA, Settleman J. Gefitinibsensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–1167. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- Stempien-Otero A, Karsan A, Cornejo CJ, Xiang H, Eunson T, Morrison RS, Kay M, Winn R, Harlan J. Mechanisms of hypoxia-induced endothelial cell death. Role of p53 in apoptosis. J. Biol. Chem. 1999;274:8039–8045. doi: 10.1074/jbc.274.12.8039. [DOI] [PubMed] [Google Scholar]
- Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PR, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT. Protein kinase B kinases that mediate phosphatidylinositol 3,4,5-trisphosphate-dependent activation of protein kinase B. Science. 1998;279:710–714. doi: 10.1126/science.279.5351.710. [DOI] [PubMed] [Google Scholar]
- Suhara T, Mano T, Oliveira BE, Walsh K. Phosphatidylinositol 3-kinase/Akt signaling controls endothelial cell sensitivity to Fas-mediated apoptosis via regulation of FLICE-inhibitory protein (FLIP) Circ. Res. 2001;89:13–19. doi: 10.1161/hh1301.092506. [DOI] [PubMed] [Google Scholar]
- Takashima A, Murayama M, Murayama O, Kohno T, Honda T, Yasutake K, Nihonmatsu N, Mercken M, Yamaguchi H, Sugihara S, Wolozin B. Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc. Natl. Acad. Sci. 1998;95:9637–9641. doi: 10.1073/pnas.95.16.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang ED, Nunez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J. Biol. Chem. 1999;274:16741–16746. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- Taub J, Lau JF, Ma C, Hahn JH, Hoque R, Rothblatt J, Chalfie M. A cytosolic catalase is needed to extend adult lifespan in C. elegans daf-C and clk-1 mutants. Nature. 1999;399:162–166. doi: 10.1038/20208. [DOI] [PubMed] [Google Scholar]
- Uchiyama T, Engelman RM, Maulik N, Das DK. Role of Akt signaling in mitochondrial survival pathway triggered by hypoxic preconditioning. Circulation. 2004;109:3042–3049. doi: 10.1161/01.CIR.0000130647.29030.90. [DOI] [PubMed] [Google Scholar]
- Vincent AM, Maiese K. Nitric oxide induction of neuronal endonuclease activity in programmed cell death. Exp. Cell. Res. 1999;246:290–300. doi: 10.1006/excr.1998.4282. [DOI] [PubMed] [Google Scholar]
- Walker KS, Deak M, Paterson A, Hudson K, Cohen P, Alessi DR. Activation of protein kinase B beta and gamma isoforms by insulin in vivo and by 3-phosphoinositide-dependent protein kinase-1 in vitro: comparison with protein kinase B alpha. Biochem. J. 1998;331:299–308. doi: 10.1042/bj3310299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton M, Woodgate AM, Muravlev A, Xu R, During MJ, Dragunow M. CREB phosphorylation promotes nerve cell survival. J. Neurochem. 1999;73:1836–1842. [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr. NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c- IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Wang N, Verna L, Hardy S, Zhu Y, Ma KS, Birrer MJ, Stemerman MB. c-Jun triggers apoptosis in human vascular endothelial cells. Circ. Res. 1999;85:387–393. doi: 10.1161/01.res.85.5.387. [DOI] [PubMed] [Google Scholar]
- Watson A, Eilers A, Lallemand D, Kyriakis J, Rubin LL, Ham J. Phosphorylation of c-Jun is necessary for apoptosis induced by survival signal withdrawal in cerebellar granule neurons. J. Neurosci. 1998;18:751–762. doi: 10.1523/JNEUROSCI.18-02-00751.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrli M, Dougan ST, Caldwell K, O'Keefe L, Schwartz S, Vaizel-Ohayon D, Schejter E, Tomlinson A, DiNardo S. Arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature. 2000;407:527–530. doi: 10.1038/35035110. [DOI] [PubMed] [Google Scholar]
- Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 2001;29:629–643. doi: 10.1016/s0896-6273(01)00239-2. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Kuan CY, Kennedy NJ, Kelkar N, Haydar TF, Mordes JP, Appel M, Rossini AA, Jones SN, Flavell RA, Rakic P, Davis RJ. Requirement of the JIP1 scaffold protein for stress-induced JNK activation. Genes Dev. 2001;15:2421–2432. doi: 10.1101/gad.922801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick MJ, Dong LQ, Riojas RA, Ramos FJ, Liu F. Mechanism of phosphorylation of protein kinase B/Akt by a constitutively active 3-phosphoinositide-dependent protein kinase-1. J. Biol. Chem. 2000;275:40400–40406. doi: 10.1074/jbc.M003937200. [DOI] [PubMed] [Google Scholar]
- Witthuhn BA, Quelle FW, Silvennoinen O, Yi T, Tang B, Miura O, Ihle JN. JAK2 associates with the erythropoietin receptor and is tyrosine phosphorylated and activated following stimulation with erythropoietin. Cell. 1993;74:227–236. doi: 10.1016/0092-8674(93)90414-l. [DOI] [PubMed] [Google Scholar]
- Wu WB, Peng HC, Huang TF. Disintegrin causes proteolysis of beta-catenin and apoptosis of endothelial cells. Involvement of cell-cell and cell-ECM interactions in regulating cell viability. Exp. Cell Res. 2003;286:115–127. doi: 10.1016/s0014-4827(03)00105-8. [DOI] [PubMed] [Google Scholar]
- Yaffe MB, Rittinger K, Volinia S, Caron PR, Aitken A, Leffers H, Gamblin SJ, Smerdon SJ, Cantley LC. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell. 1997;91:961–971. doi: 10.1016/s0092-8674(00)80487-0. [DOI] [PubMed] [Google Scholar]
- Yamagishi S, Matsumoto T, Yokomaku D, Hatanaka H, Shimoke K, Yamada M, Ikeuchi T. Comparison of inhibitory effects of brain-derived neurotrophic factor and insulin-like growth factor on low potassium-induced apoptosis and activation of p38 MAPK and c-Jun in cultured cerebellar granule neurons. Brain Res. Mol. Brain Res. 2003;119:184–191. doi: 10.1016/j.molbrainres.2003.09.009. [DOI] [PubMed] [Google Scholar]
- Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J. Biol. Chem. 2001;276:5256–5264. doi: 10.1074/jbc.M008552200. [DOI] [PubMed] [Google Scholar]
- Yamamoto H, Kishida S, Kishida M, Ikeda S, Takada S, Kikuchi A. Phosphorylation of axin, a Wnt signal negative regulator, by glycogen synthase kinase-3beta regulates its stability. J. Biol. Chem. 1999;274:10681–10684. doi: 10.1074/jbc.274.16.10681. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Katayose Y, Suzuki M, Unno M, Sasaki T, Mizuma M, Shiraso S, Ohtuka H, Cowan KH, Seth P, Matsuno S. Adenovirus expressing p27KIP1 induces apoptosis against cholangiocarcinoma cells by triggering Fas ligand on the cell surface. Hepatogastroenterology. 2003;50:1847–1853. [PubMed] [Google Scholar]
- Yin H, Chao L, Chao J. Adrenomedullin protects against myocardial apoptosis after ischemia/reperfusion through activation of Akt-GSK signaling. Hypertension. 2004;43:109–116. doi: 10.1161/01.HYP.0000103696.60047.55. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Hartmann H, Do VM, Abramowski D, Sturchler-Pierrat C, Staufenbiel M, Sommer B, van de Wetering M, Clevers H, Saftig P, De Strooper B, He X, Yankner BA. Destabilization of beta-catenin by mutations in presenilin-1 potentiates neuronal apoptosis. Nature. 1998;395:698–702. doi: 10.1038/27208. [DOI] [PubMed] [Google Scholar]
- Zheng-Fischhofer Q, Biernat J, Mandelkow EM, Illenberger S, Godemann R, Mandelkow E. Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur. J. Biochem. 1998;252:542–552. doi: 10.1046/j.1432-1327.1998.2520542.x. [DOI] [PubMed] [Google Scholar]
- Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell. Biol. 2001;3:973–982. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]