

Abstract
More than a century has elapsed since the description of Alois Alzheimer's patient Auguste D. Yet, the well-documented generation of β-amyloid aggregates and neurofibrillary tangles that define Alzheimer's disease is believed to represent only a portion of the cellular processes that can determine the course of Alzheimer's disease. Understanding of the complex nature of this disorder has evolved with an increased appreciation for pathways that involve the generation of reactive oxygen species and oxidative stress, apoptotic injury that leads to nuclear degradation in both neuronal and vascular populations, and the early loss of cellular membrane asymmetry that mitigates inflammation and vascular occlusion. Recent work has identified novel pathways, such as the Wnt pathway and the serine-threonine kinase Akt, as central modulators that oversee cellular apoptosis and the formation of neurofibrillary tangles through their downstream substrates that include glycogen synthase kinase-3β, Bad, and Bcl-xL. Other closely integrated pathways control microglial activation, release of inflammatory cytokines, and caspase and calpain activation for the processing of amyloid precursor protein, tau protein cleavage, and presenilin disposal. New therapeutic avenues that are just open to exploration, such as with nicotinamide adenine dinucleotide modulation, cell cycle modulation, metabotropic glutamate system modulation, and erythropoietin targeted expression, may provide both attractive and viable alternatives to treat Alzheimer's disease.
Keywords: β-Amyloid, Akt, Cysteine proteases, Erythropoietin, Metabotropic, Wnt
1. Introduction
1.1. Alzheimer's disease in the aging population
As the population continues to age, the cost of physician services, hospital and nursing home care, and medications continues to rise dramatically. In addition, these medical costs for neurodegenerative disease parallel a progressive loss of economic productivity with rising morbidity and mortality, ultimately resulting in an annual deficit to the economy that is greater than US$380 billion. Although the original patient description by Alois Alzheimer is believed to be consistent with an early-onset form of Alzheimer's disease, the most significant portion of this economic loss is composed of only a few neurodegenerative disease entities with Alzheimer's disease of the late-onset sporadic form representing a significant portion. For example, the annual cost per patient with Alzheimer's disease is estimated at US$174,000 with an annual aggregate cost of US$100 billion [149,153].
Alzheimer's disease leads to a progressive deterioration of cognitive function with loss of memory. Neuronal injury presents in regions of the brain that involve the hippocampus and the cortex. Alzheimer's disease is characterized by two pathologic hallmarks that consist of extracellular plaques (Fig. 1) of amyloid-β peptide aggregates and intracellular neurofibrillary tangles (Fig. 2) composed of hyperphosphorylated microtubular protein tau. The β-amyloid deposition that constitutes the plaques is composed of a 39−42 amino acid peptide (Aβ), which is the proteolytic product of the amyloid precursor protein (APP) [141].
Fig. 1.

Representative clinical pathology of Alzheimer's disease. Microscopic evaluation of the cerebral cortex with a silver stain in a patient with Alzheimer's disease demonstrating “senile plaques” with neuronal degeneration. Image supplied by Daniel P. Perl, M.D., of Mount Sinai School of Medicine. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Fig. 2.
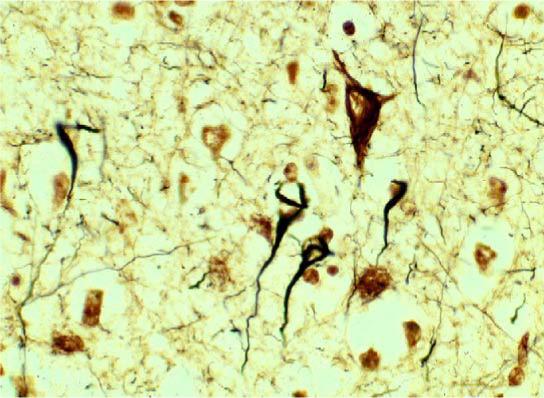
Images of neurofibrillary tangles in Alzheimer's disease. In this section of the cerebral cortex in a patient with Alzheimer's disease, several neurofibrillary tangles can be visualized with a silver stain. Image supplied by Daniel P. Perl, M.D., of Mount Sinai School of Medicine. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
1.2. Uncovering novel cellular pathways that may lead to Alzheimer's disease
Unfortunately, no definitive therapy for the prevention or resolution of Alzheimer's disease exists. Yet, it is clear that a variety of cellular mechanisms can lead to this debilitating disorder. Crucial to the task of preventing or potentially reducing neuronal injury during Alzheimer's disease is the ability to elucidate the cellular mechanisms that precipitate neuronal degeneration. Recent work has begun to focus on pathways of oxidative stress that involve a variety of cellular pathways in addition to the presentation of β-amyloid aggregates and neurofibrillary tangles. In this review, we discuss the role of oxidative stress and apoptotic injury in Alzheimer's disease. We further present for consideration novel cellular pathways that originate from the proto-oncogene Wnt and the serine-threonine kinase Akt and involve cellular pathways related to phagocytic microglia, glycogen synthase kinase-3β, Bad, Bcl-xL, and cysteine proteases. Further knowledge of these processes may ultimately identify unique therapeutic strategies linked to cellular metabolism, genomic DNA repair, cell cycle regulation, metabotropic glutamate modulation, and trophic factor expression.
2. Oxidative stress, apoptotic injury, and Alzheimer's disease
2.1. Generation of reactive oxygen species, DNA degradation, and membrane asymmetry
The overproduction of reactive oxygen species (ROS) leads to oxidative stress. ROS consist of oxygen free radicals and associated entities that include superoxide free radicals, hydrogen peroxide, singlet oxygen, nitric oxide (NO), and peroxynitrite. Oxygen free radicals result in the oxidation of other agents by accepting an electron to already unpaired electrons. The superoxide radical is an oxygen free radical that can lead to hydroxyl radical formation through hydrogen peroxide. Hydroxyl radicals are generated from hydrogen peroxide through the Haber–Weiss reaction in the presence of ferrous iron or via a chemical reaction between the superoxide radical and NO. Through its reaction with a superoxide radical, NO yields peroxynitrite that produces a nitrosyl radical. As a result, nitrosyl radicals subsequently break down to form hydroxyl radicals.
Oxidative stress represents a significant pathway that leads to the destruction of both neuronal and vascular cells.
The production of ROS, such as peroxynitrite and NO, can lead to cell injury through cell membrane lipid destruction and cleavage of DNA [234,241]. ROS result in the peroxidation of cellular membrane lipids [204], peroxidation of docosahexaenoic acid, a precursor of neuroprotective docosanoids [160], the cleavage of DNA during the hydroxylation of guanine and methylation of cytosine [116], and the oxidation of proteins that yield protein carbonyl derivatives and nitrotyrosine [2]. In addition to the detrimental effects to cellular integrity, ROS can inhibit complex enzymes in the electron transport chain of the mitochondria resulting in the blockade of mitochondrial respiration [250].
Oxidative stress precipitates apoptotic cellular injury that consists of both nuclear DNA degradation and membrane phosphatidylserine (PS) exposure [42,234,247]. Membrane PS exposure and DNA fragmentation are independent processes that lead to apoptosis, also known as programmed cell death (PCD). Several general biochemical and physiologic features of apoptosis have been identified and include nuclear chromatin condensation, DNA fragmentation, and the loss of plasma membrane asymmetry. The biological role of membrane PS externalization can vary in different cell populations. In many cell systems, membrane PS externalization can become a signal for the phagocytosis of cells [42,46,89,104]. In neurons, cells expressing externalized PS may be removed by microglia. In contrast, membrane PS exposure can also function in vascular cells to activate coagulation cascades. The externalization of membrane PS residues in endothelial cells (ECs) can promote the formation of a procoagulant surface [61,70,139]. Furthermore, oxidative stress can significantly increase chromosomal aberrations and micronuclei [26,39] and lead to the activation of apoptotic pathways in ECs [38,116,193]. ECs that are exposed to oxidative stress incur both DNA fragmentation and membrane PS externalization during exposure to insults, such as hypoxia, oxidants, and free radicals [9,28,38,39,124].
When one compares the early externalization of membrane PS residues to genomic DNA fragmentation, the cleavage of genomic DNA into fragments is considered to be a delayed event that occurs late during apoptosis [61,98,104,139]. Several enzymes responsible for DNA degradation have been differentiated based on their ionic sensitivities to zinc [223] and magnesium [210]. Calcium, a critical independent component that can determine cell survival [242], may also determine endonuclease activity through calcium/magnesium-dependent endonucleases such as DNase I [137]. Other enzymes that may disassemble DNA include the acidic, cation independent endonuclease (DNase II) [222], cyclophilins [158], and the 97 kDa magnesium-dependent endonuclease [169]. In the nervous system, three separate endonuclease activities are present that include a constitutive acidic cation-independent endonuclease, a constitutive calcium/magnesium-dependent endonuclease, and an inducible magnesium-dependent endonuclease [234]. The physiologic characteristics of the magnesium-dependent endonuclease, such as a pH range of 7.4−8.0, a dependence on magnesium, and a molecular weight of 95−108 kDa, are consistent with a recently described constitutive 97 kDa endonuclease in non-neuronal tissues, but the endonuclease in the nervous system is inducible rather than constitutive in nature.
Exposure to ROS can precipitate apoptosis in neurons and ECs through multiple cellular pathways. Oxidative stress, such as NO or hydrogen peroxide, results in nuclei condensation and DNA fragmentation [42,82,178,234]. In neurons, NO exposure produces apoptotic death in hippocampal and dopaminergic neurons [41,199,233,247]. Injury during NO exposure can also become synergistic with hydrogen peroxide to render neurons more sensitive to oxidative injury [57,241]. Hydrogen peroxide also results in neuronal injury through impaired mitochondrial function and increased levels of pro-apoptotic gene products, such as CD95/Fas [56,178,231]. Externalization of membrane PS residues also occurs in neurons during anoxia [38], NO exposure [44], or during the administration of agents that induce the production of ROS, such as 6-hydroxydopamine [194].
2.2. Evidence for apoptotic injury in Alzheimer's disease during oxidative stress
Oxidative stress is considered to play a significant role in the onset and progression of Alzheimer's disease. Transient hypoxia in sporadic Alzheimer's disease can lead to mitochondrial dysfunction, impaired membrane integrity, and APP cleavage [35]. During the progression of Alzheimer's disease, lipid peroxidation [30], protein oxidation [37], and DNA oxidation [131] have been reported. Furthermore, in mice overexpressing APP, the brain Aβ deposits that are characteristically found in Alzheimer's disease co-localize with an array of oxidative stress markers [206], suggesting that there exists a close correlation between oxidative stress and Aβ deposition.
Aβ has been found to result in the generation of ROS, such as hydrogen peroxide, through metal ion reduction and to lead to oxidative toxicity in neurons [92]. Free radical generation by Aβ is strongly influenced by the aggregational state of the peptides [157]. Given the link between Aβ deposition and oxidative stress, agents that modulate ROS may be potentially useful in the therapy of Alzheimer's disease. For example, application of the free radical antioxidant vitamin E has been demonstrated to prevent neurotoxicity from Aβ [209].
Other evidence exists that suggests cellular injury during Alzheimer's disease may result from not only increased insults from oxidative stress, but also from impaired cellular repair mechanisms following oxidative injury. In one study, 8-hydroxy-2′-deoxyguanosine (8-OHdG), a marker of oxidative damage in intact DNA and as a “free” repair product during DNA repair mechanisms, was examined in the cerebrospinal fluid of Alzheimer's patients. Significant elevations of 8-OHdG linked to intact DNA were observed in the cerebrospinal fluid of Alzheimer's patients, suggesting that these patients suffer from impaired DNA mechanisms. Yet, levels of free 8-OHdG, which are generated during normal cellular repair mechanisms, were found to be significantly depleted in the cerebrospinal fluid of Alzheimer's patients, further supporting the premise of deficient DNA repair mechanisms in these patients [131].
Cellular oxidative pathways that proceed through apoptosis appear to be a predominant factor in the cell loss observed during Alzheimer's disease. Accumulating evidence has been obtained from human and in vitro models of Alzheimer's disease suggesting that PCD contributes to the neuronal loss during the disease. Data from in situ terminal deoxynucleotidyl transferase nick-end labeling (TUNEL) assays of brain tissues from Alzheimer's patients demonstrate neuronal demise consistent with apoptotic cell death. A correlation between the incidence of TUNEL-positive cells and plaque density was also observed [52]. Levels of the apoptotic marker prostate apoptosis response-4 (Par-4) has also been shown to be significantly increased in the brains of patients with Alzheimer's disease [83]. Other lines of evidence link apoptotic cellular injury with APP and its proteolytic product Aβ. In in vitro studies, expression of familial Alzheimer's disease mutants of APP results in apoptotic neuronal injury [150]. It is the cytoplasmic domain of APP that can lead to sustained apoptosis through c-Jun N-terminal kinase pathways [85]. Additional studies have illustrated that direct application of Aβ to neuronal cells can lead to chromatin condensation, DNA fragmentation, and membrane PS exposure characteristic of apoptosis in cultured neurons (Fig. 3).
Fig. 3.
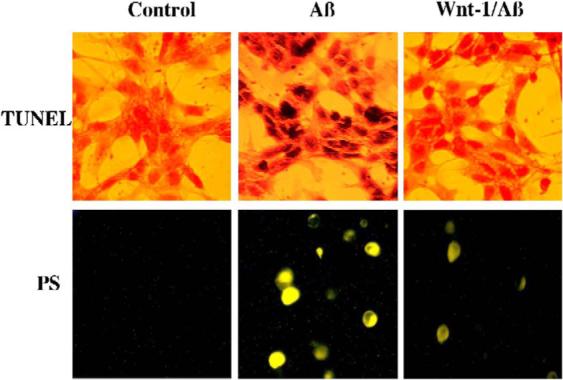
Over-expression of Wnt-1 prevents β-amyloid (Aβ)-induced DNA fragmentation and membrane phosphatidylserine (PS) exposure in human neuroblastoma SH-SY5Y cells. Representative images of SH-SY5Y cells illustrate DNA fragmentation with terminal deoxynucleotidyl transferase nick end labeling (TUNEL) and phosphatidylserine (PS) exposure with annexin V phycoerythrin labeling in SH-SY5Y cells 24 h after administration of Aβ (20 μM). Aβ yielded significant DNA fragmentation and membrane PS exposure when compared to control untreated cultures. In contrast, overexpression of Wnt-1 results in a significant reduction in DNA fragmentation and PS exposure.
3. Microglial inflammatory cell injury in Alzheimer's disease
3.1. Cellular mechanisms and mediators that lead to microglial activation in the nervous system
Microglia are monocyte-derived immunocompetent cells that enter the central nervous system during embryonic development and function similar to peripheral macrophages for the phagocytic removal of apoptotic cells. There exist several potential mechanisms that may regulate the phagocytosis of cells that have entered the apoptotic pathway. Some studies identify the generation of annexin I and membrane PS exposure that appears to be necessary to connect an apoptotic cell with a phagocyte [12]. Secreted factors by either apoptotic or phagocytic cells, such as milk fat globule-EGF-factor 8 [84], fractalkine [86], and lipid lysophosphosphatidylcholine [113], have also been shown to assist with the phagocytic removal of injured cells.
The translocation of membrane PS residues from the inner cellular membrane to the outer surface appears to be critical for the removal of apoptotic cells [70,104,139]. The phospholipids of the plasma membrane are normally in an asymmetric pattern with the outer leaflet of the plasma membrane consisting primarily of choline-containing lipids, such as phosphatidylcholine and sphingomyelin, and the inner leaflets consisting of aminophospholipids that include phosphatidylethanolamine and PS. The loss of membrane phospholipid asymmetry leads to the externalization of membrane PS residues and serves to identify cells for phagocytosis [43,89,104,140].
Expression of the phosphatidylserine receptor (PSR) on microglia works in concert with cellular membrane PS externalization to activate microglia. Cells, such as neurons or ECs, exposed to ROS can lead to the induction of both microglial activation and microglial PSR expression (Fig. 4). Treatment with an anti-PSR neutralizing antibody in microglia prevents this microglial activation [42,103], and application of PS directly results in microglial activation that can be blocked by a PSR neutralizing antibody [42,104], suggesting that both PS exposure in target cells and PSR expression in microglia are necessary for microglial recognition of apoptotic cells in the nervous system. Recognition of cellular membrane PS by the PS-specific receptors on microglia may require cofactors, such as Gas6 [164] or other agents, such as integrin and lectin [247].
Fig. 4.

Oxidative stress in primary hippocampal neurons leads to microglia activation and proliferation. Pure microglial cultures were treated for 3 h with media from rat hippocampal neuronal cultures conditioned with 24 h of oxidative stress (nitric oxide, 300 μM). Representative images illustrate the significant expression of proliferating cell nuclear antigen (PCNA) (microglial activation) or the uptake of bromodeoxyuridine (BrdU) (microglial proliferation) in microglia treated with media from neurons exposed to oxidative stress when compared to control cultures. In all cases, control = treated with media from neurons not exposed to oxidative stress.
3.2. A potential detrimental role for microglia during Alzheimer's disease
Several lines of evidence indicate that microglial activation may be involved in the pathogenesis of Alzheimer's disease. In addition to assisting with the removal of injured cells and cellular debris, microglia may sometimes aggravate a cellular insult. Studies with microglia stimulated by phorbol myristate acetate have demonstrated the release of superoxide radicals. Application of scavenger agents for ROS, such as superoxide dismutase or deferoxamine mesylate, in the presence of activated microglia can prevent cellular injury. These studies suggest that oxidative stress generated by microglia can be responsible for cellular injury [215].
Microglia may lead to cellular damage in Alzheimer's disease not only through the generation of ROS products [195], but also through the production of cytokines and the demise of neighboring neurons and ECs [18,152]. Microglia promote the production of pro-inflammatory and cytokines, such as tumor necrosis factor-α (TNF-α) and interleukin-1β, free radicals such as NO and superoxide [195], and fatty acid metabolites such as eicosanoids that can precipitate cell death [128]. TNF-α production by microglia may be linked to neurodegeneration by increasing the sensitivity of neurons to free radical exposure [53].
Microglial activation in Alzheimer's disease patients has been identified through glial cultures in autopsy specimens [135]. Expression of markers that are indicative of micro-glial activation was found to be significantly increased in patients with Alzheimer's disease [189]. Application of a position emission tomography marker [11C](R)-PK11195 for microglial activation in patients with mild and early Alzheimer's disease has also demonstrated microglial activation in regions of the entorhinal, parietal, and cingulate cortex, suggesting that microglial activation is an early event in the pathogenesis of the disease [31].
One of the major pathogens of Alzheimer's disease, namely Aβ, can lead to inflammatory cell injury through a variety of routes. Aβ cannot only precipitate a significant inflammatory response with microglial activation and the secretion of TNF-α [24], but also Aβ can elicit the neuronal expression of inducible nitric oxide synthase, peroxinitrite production, and neuronal apoptosis during an acute inflammatory response [53]. Interestingly, in patients with Alzheimer's disease, microglial cells co-localize with the perivascular deposits of Aβ. Microglial activation has been observed to occur in concert with the evolution of amyloid plaques [201]. Ultrastructural three-dimensional reconstruction of human classical plaques in different stages of development illustrates that the number of microglia in the amyloid plague parallels a progressive increase in fibrillar deposition and the size of fibrillar plague [243]. The generation of oxidative stress by microglia during Aβ deposition suggests that microglia may play an important role in the pathogenesis of Alzheimer's disease.
4. A novel role for the Wnt pathway in Alzheimer's disease
4.1. Origin and function of the Wnt gene family during cell survival
The Wnt gene family and its signal transduction pathways are important during embryonic development and oncogenesis [105,136]. The Wnt gene family encodes a group of secreted glycoproteins that are categorized into two groups named canonical and noncanonical Wnt. Canonical Wnts include Wnt-1, Wnt-3a, and Wnt-8, and function through β-catenin-dependent pathways. The noncanonical Wnts consist of Wnt-4, Wnt-5a, and Wnt-11, and function through non-β-catenin-dependent pathways, such as the planar cell polarity pathway and the Wnt-calcium-dependent pathway [205,211].
As one of the best characterized members of the Wnt family, Wnt-1 was first identified as a proto-oncogene in mammary carcinomas, but has recently been illustrated to play a critical role in neuronal development [216]. Wnt functions by binding to the transmembrane receptor Frizzled and the co-receptor lipoprotein related protein 5 and 6 (LRP-5/6) [244] followed by recruitment of dishevelled, the cytoplasmic bridging molecule, to inhibit glycogen synthase kinase (GSK-3β) [93,170]. The inhibition of GSK-3β prevents phosphorylation of β-catenin and its degradation. The free β-catenin translocates to the nucleus where it activates lymphocyte enhancer factor (Lef) and T cell factor (Tcf) [96], leading to stimulation of Wnt-response genes.
In some cell systems, Wnt-1 signaling has been associated with the control of apoptosis (Fig. 5). Wnt-1 prevents apoptosis through β-catenin/Tcf transcription-mediated pathways [34,186]. Over-expression of exogenous Wnt-1 results in the protection of cells against c-myc-induced apoptosis through induction of β-catenin, cyclooxygenase-2, and Wnt-1-induced secreted protein (WISP-1) [253]. Wnt-1 signaling can also inhibit apoptosis through prevention of cytochrome c release from mitochondria and the subsequent inhibition of caspase 9 activation [34]. The adenomatous polyposis coli (APC) gene, a member of the Wnt pathway, appears to represent another mechanism that regulates PCD. The APC gene functions to cleave β-catenin leading to the down-regulation of transactivation of Tcf/Lef [220]. Without Tcf/Lef activity, APC is then permitted to increase the activities of caspase 3, caspase 7, and caspase 9, and lead to the cleavage of poly (ADP-ribose) polymerase (PARP) to enhance the vulnerability of cells to apoptosis [36].
Fig. 5.

Wnt and Akt signaling pathways in Alzheimer's disease. The common downstream target of Wnt and Akt, glycogen synthase kinase-3 β (GSK-3β), phosphorylates tau protein facilitating the formation of neurofibrillary tangles. GSK-3β also phosphorylates (P) β-catenin leading to its degradation and subsequent induction of apoptosis. GSK-3β may also influence the amyloid precursor protein (APP) processing resulting in an increase in the production of Aβ, which conversely increases the activity of GSK-3β. Akt inhibits GSK-3β through phosphorylation and also phosphorylates tau protein at the site of Ser214 to prevent formation of neurofibrillary tangles. Wnt activates Akt directly or through Wnt-1-induced secreted protein (WISP-1). The phosphorylation and inactivation of GSK-3β by Wnt may occur through protein kinase C (PKC) or through Akt activation.
4.2. Restoration of Wnt activity may foster cellular survival during neurodegenerative disease
Loss of Wnt activity may precipitate a host of neurodegenerative disorders. Wnt-1 expression has been demonstrated in the brains of individuals affected by neuropsychiatric disorders [155]. Furthermore, retinal degeneration during retinitis pigmentosa with the progressive loss of photoreceptors has been associated with increased secretion of Frizzled-related protein-2, a Wnt inhibitory protein, suggesting that loss of Wnt signaling may contribute to retinal neurodegeneration [100]. In contrast, enhanced Wnt-1 activity may function to prevent apoptosis during neuronal or vascular injury in the central nervous system. Recent work illustrates that Wnt signaling may foster specific protection against cellular destruction and inflammatory injury by maintaining genomic DNA integrity and cellular membrane PS asymmetry [46,139]. For example, Wnt-1 overexpression in primary hippocampal neurons protects cells against oxidative stress or Aβ toxicity that increases cell survival and prevents PS exposure and DNA degradation [46] (Fig. 3).
Loss of Wnt signaling also appears to play a role in Alzheimer's disease (Fig. 3). Neurotoxicity of β-amyloid deposition of the 39−42 amino acid peptide (Aβ) in hippocampal neurons during Alzheimer's disease has been linked to increased levels of GSK-3β and loss of β-catenin. Decreased production of Aβ can occur during the enhancement of protein kinase C (PKC) activity [197] which may be controlled by the Wnt pathway [78].
The proteolytic processing of APP during Alzheimer's disease has been closely linked to the Wnt pathway through presenilin 1 (PS1) and dishevelled. PS1 is required for the processing of APP and has been shown to down-regulate Wnt signaling and interact with β-catenin to promote its turnover [207]. Dishevelled, a known downstream transducer of Wnt signaling pathway, can also regulate the α-secretase cleavage of APP through PKC/ mitogen-activated protein kinase-dependent pathways, increasing soluble production of APP (sAPP) [159]. Overexpression of mouse dishevelled-1 and −2 inhibits GSK-3β-mediated phosphorylation of tau protein and may thus prevent formation of neurofibrillary tangles during Alzheimer's disease [239]. Thus, dishevelled may increase neuronal protection during neurodegenerative disorders through sAPP production and reduction in tau phosphorylation. As a result, modulation of the Wnt pathway as well as its downstream constituents may offer novel therapeutic approaches to tackle neurodegeneration in Alzheimer's disease.
5. Akt and its substrates form a central regulatory platform in Alzheimer's disease
5.1. Expression of Akt occurs throughout mammalian tissues, but is robustly upregulated during cellular compromise
Protein kinase B (PKB), also known as Akt after the oncogene v-Akt, has been identified as a central component in a variety of pathways to promote cell survival and block apoptotic degradation [48]. In mammals, three family members of PKB have been identified, which are termed PKBα or Akt1, PKBβ or Akt2, and PKBγ or Akt3. Akt belongs to the AGC (cAMP-dependent kinase/protein kinase G/protein kinase C) superfamily of protein kinases and consists of three functionally domains [16,73]. The N-terminal pleckstrin homology (PH) domain provides binding sites for membrane phospholipids, which are involved in the recruitment of Akt to the plasma membrane. The catalytic domain of Akt has specificity for serine or threonine residues of several Akt substrates. The C-terminal hydrophobic motif (HM) functions to provide a docking site for the activation of kinases.
In the central nervous system, the gene expression for Akt1 and Akt2 is present at high levels in the brain during development, but is gradually decreased during postnatal periods [167]. In the adult, gene expression for Akt1 and Akt2 is normally subdued, but quickly increased in response to a variety of environmental factors. Following stimulation by agents such as trophic factors or cytokines, phosphoinositide 3 kinase (PI 3-K) is recruited to the plasma membrane, phosphorylates glycerophospholipid phosphatidylinositol 4,5-bisphosphate, and results in the production of phosphatidylinositol 3,4 bisphosphate (PIP2) and phosphatidylinositol 3,4, 5 trisphosphate (PIP3). As a cytosolic protein, Akt then translocates to the cell membrane after its binding to PIP2 and PIP3 through their respective PH domains, and subsequently becomes activated through phosphorylation by phosphoinositide-dependent kinase 1 [245].
Once activated, Akt can provide protection against cellular injury. Maximal activity of Akt is achieved through phosphorylation by phosphoinositide-dependent kinase 1 at Ser473 to confer protection against genomic DNA degradation [38,246,249] and membrane PS exposure [38,42,104]. During a number of injury paradigms, such as toxic insults involving excitotoxicity [106], free radical exposure [45,42,148], hypoxia [38], or trauma [162], Akt is phosphorylated leading to increased activity and protection against PCD induction (Fig. 5).
5.2. The forkhead transcription factor, glycogen synthase kinase-3β, and Bad are essential substrates of Akt during cell injury and Alzheimer's disease
The forkhead transcription factor (FOXO3a, FKHRL1) represents one cellular pathway that is centrally controlled by Akt. Activation of FOXO3a can result in apoptotic cellular degeneration in a transcription-dependent manner following its translocation to the nucleus [27,60,80]. FOXO3a activation has been demonstrated to disrupt mitochondrial membrane permeability (Δψm) and may result in cytochrome c release [254]. Akt inhibition of FOXO3a requires its phosphorylation that results in the association of FOXO3a with 14−3−3 protein and retention of FOXO3a in the cytoplasm, rendering it ineffective to target genes in the nucleus and thus blocking apoptosis. During periods of oxidative stress in the nervous system, an initial inhibitory phosphorylation of FOXO3a at regulatory phosphorylation sites (Thr32 and Ser253) [27,185] can occur [47]. However, loss of phosphorylated FOXO3a expression appears to subsequently result over a 12-h period, possibly by caspase degradation, which can potentially enhance the vulnerability of neurons to apoptotic injury during neuro-degenerative disorders, such as Alzheimer's disease [47].
Protein kinase B also modulates the activity of glycogen synthase kinase-3β (GSK-3β), a serine/threonine kinase that is specifically expressed in the central nervous system. Although phosphorylation of GSK-3β at Ser9 by Akt results in its inactivation, it is important to note that phosphorylation of GSK-3β at Thr216 results in an enhanced activity of the enzyme, which can occur during neuronal degeneration [20]. Interestingly, GSK-3β has been shown to be involved with the neurotoxicity of Aβ during Alzheimer's disease. In brains of Alzheimer's patients, GSK-3β expression is present in the cytoplasm of pretangle neurons and its expression coincides with the development of neurofibrillary changes [175]. On a cellular level, Aβ exposure in cultured hippocampal neurons can activate GSK-3β [214]. In addition, application of antisense oligonucleotides against GSK-3β and the GSK-3β inhibitor, lithium, can prevent cellular injury mediated by Aβ [4,213].
GSK-3β can also regulate APP processing and the phosphorylation of tau. GSK-3β facilitates Aβ release by increasing the cellular maturation of APP [10], a process believed to occur during the early onset of Alzheimer's disease [51]. GSK-3β is also one of the protein kinase candidates that can phosphorylate the tau protein. Hyperphosphorylated tau is the major component of neurofibrillary tangles that consist of paired helical filaments. GSK-3β appears to be necessary for sequential phosphorylation of tau at sites that are required for the formation of paired-helical-filaments [256]. Overexpression of GSK-3β in transgenic mice results in the hyperphosphorylation of tau in hippocampal neurons and pre-tangle-like somatodendritic localization of tau [134]. If one removes GSK-3β activity through the inhibitor lithium, hyperphosphorylation of tau is blocked and tau binding to microtubules is promoted to yield microtubule assembly [90,161]. Tau hyperphosphorylation of GSK-3β may require caspase 3 cleavage of APP (Fig. 5). The generation of the C-terminal fragment C31 results from the cleavage of APP at the caspase site D720 of the C-terminus by caspase 3 [165]. Once generated, C31 enhances glycogen synthase kinase-3β expression and tau protein phosphorylation [107].
Akt can also inactivate Bad, a pro-apoptotic Bcl-2 family member, through phosphorylation of its serine residues. Bad is a Bcl-2 homology 3 (BH3)-only subfamily member of Bcl-2 proteins that are associated with the regulation of apoptosis. Three phosphorylated serine sites have been identified on Bad, including serine112, serine136, and serine155. Akt preferentially phosphorylates the residue serine136 of Bad [55]. A fourth phosphorylation site of Bad has recently been identified at serine170 that also results in the blockade of pro-apoptotic activity of Bad [64]. The endogenous de-phosphorylated Bad is localized in the outer mitochondrial membrane and binds to the anti-apoptotic Bcl-2 family member Bcl-xL through its BH3 domain. Subsequent phosphorylation of Bad by Akt leads to the binding of Bad with the cytosolic protein 14−3−3 to release Bcl-xL and allow it to block apoptosis [119]. Bcl-2 and Bcl-xL prevent Bax translocation to the mitochondria, maintain the mitochondrial membrane potential, and prevent the release of cytochrome c from the mitochondria [179].
Interestingly, a number of Bcl-2 family members have been linked to cellular injury during Alzheimer's disease. Apoptotic injury as a result of Aβ (1−42) administration is associated with an increase of Bax and caspase 3 activation [109]. Furthermore, work that has infused Aβ (1−40) into the cerebral ventricles of rats results in the up-regulation of Bax as well as the down-regulation of Bcl-2 in cortical and hippocampal regions bordering the lateral ventricle [240]. Similarly, in primary human cultured neurons, application of Aβ (1−42) produces sustained induction of Bax and a reduction in Bcl-2 expression [171]. Additional evidence for the ability of Bcl-2 proteins to modulate neuronal survival during Alzheimer's disease is demonstrated with microinjection of a human cDNA expression construct of Bcl-2 that can block cell injury during Aβ exposure [255].
6. Execution of cell injury during Alzheimer's disease can be mediated through caspase activity
6.1. Onset of apoptotic injury by caspases and their association with Alzheimer's disease
Caspases are a family of cysteine proteases that cleave their substrates after aspartic residues. They are usually synthesized as inactive zymogens that are proteolytically cleaved into subunits at the onset of apoptosis and function as active caspases after reconstitution to molecular heterodimers. Caspases are composed of three domains including an N-terminal prodomain, a large subunit, and a small subunit [66]. The apoptotic-associated caspases include initiator caspases, such as caspases 2, 8, 9, and 10, that activate downstream executioner caspases, resulting in an amplification of cascade activity. The initiator caspases consist of long N-terminal prodomains that contain caspase recruitment domains (CARDs) in caspase 2 and caspase 9, or death effector domains (DEDs) in caspase 8 and caspase 10 [88]. Another set of caspases, termed the executioner caspases, consists of caspases 3, 6, and 7 that function to directly cleave crucial cellular protein substrates that result in cell destruction. The executioner caspases contain short prodomains or have no prodomains.
Apoptotic injury during Alzheimer's disease may require caspase-mediated pathways [163,227] (Fig. 6). A strong body of evidence supports the premise that caspase activation is involved in the pathological process of Alzheimer's disease. The elevation of caspase genes including caspases 1, 2, 3, 5, 6, 7, 8, and 9 has been observed in human postmortem brains from Alzheimer's disease patients [176]. In the brains of Alzheimer's patients, single neurons with DNA fragmentation have been shown to contain cytoplasmic immunoreactivity for active caspase 3, implying that apoptotic injury results during Alzheimer's disease. In addition, activation of caspase 3 was found to occur in the parahippocampal gyrus in brains from patients with mild forms of Alzheimer's disease. Caspase 3 immunoreactivity was also co-localized with paired helical filaments in neurons, suggesting that caspase 3 activation may contribute to the formation of neurofibrillary tangles [79]. This premise was further supported by a study that demonstrated the existence of fodrin caspase-cleavage product in the hippocampus of Alzheimer's patients, which was co-localized with neurofibrillary tangles [190]. Additional work in cell culture experiments has demonstrated that treatment with Aβ directly results in the activation of caspase 1 [101], caspase 2, and caspase 3 [226].
Fig. 6.

Signaling pathways that are involved in caspase activation during Alzheimer's disease. Caspase activation during Alzheimer's disease results in the cleavage of presenilin leading to an increase in the susceptibility of neurons to apoptosis with loss of β-catenin, poly(ADP-ribose)polymerase (PARP), and Bcl-2. Caspases can cleave amyloid precursor protein (APP) and the resulting C-terminal fragment C31 to produce hyperphosphorylation of tau protein (p-tau) as well as activation of glycogen synthase kinase-3β (GSK-3β). C31 and β-amyloid (Aβ) promotes the activation of caspases. Caspases also directly cleave tau protein to contribute to the formation of neurofibrillary tangles.
6.2. Induction of intrinsic and extrinsic caspase pathways for cell destruction
Activation of caspases proceeds through two separate “cellular corridors” identified as the extrinsic and intrinsic pathways. The extrinsic pathway is initiated by death receptor activation at the cell surface, resulting in the recruitment and activation of the initiator caspase 8 upon apoptotic stimuli [13]. The intracellular death domain of death receptors, such as the TNF superfamily, CD95/Fas/Apo-1, and the death receptor 3 (DR3), undergoes conformational change upon binding to extracellular ligands and forms an intracellular death-inducing signaling complex (DISC) following recruitment of adaptor molecules, such as the Fas-associated death domain (FADD). FADD recruits caspase 8 through its DED domain and leads to caspase 8 activation [102,230]. Caspase 8 can subsequently activate caspase 3. In addition, caspase 8 activation may also result in the cleavage of Bid, a pro-apoptotic member of Bcl-2 family, allowing the truncated Bid (tBid) to translocate to the mitochondria [118]. This leads to cytochrome c release through Bax resulting in the subsequent activation of executioner caspases [252].
The intrinsic caspase pathway involves mitochondrial dysfunction. The mitochondrial pathway is associated with the release of cytochrome c and subsequent activation of caspase 9 followed by activation of caspase 3 [127]. The process is regulated by the Bcl-2 subfamily BH3-only proteins, which are normally located in cellular compartments other than mitochondria, but translocate to the mitochondria in response to apoptotic stimuli [54]. The translocation of these proteins delivers an apoptotic signal to mitochondria through the interaction with Bax to induce the release of cytochrome c that then binds to apoptotic protease activating factor-1 (Apaf-1). Apaf-1 consists of three different domains that include CARD, repeats of tryptophan and aspartate residues (WD-40 repeats), and a nucleotide-binding domain CED-4. Binding of cytochrome c to Apaf-1 results in the removal of the WD-40 domain, masking the CED-4 and CARD domains, and leads to the oligomerization of Apaf-1 under the assistance of dATP/ATP [91]. The oligomerization of Apaf-1 promotes the allosteric activation of caspase 9 by forming the Apaf-1 apoptosome [117]. Caspase 9 can subsequently activate caspase 3 [117] as well as caspase 1 through the intermediary caspase 8 [212]. Together, caspase 1 and caspase 3 lead to both DNA fragmentation and membrane PS exposure [38,117,139].
6.3. Processing of APP by secretases and caspases
Processing of the transmembrane glycoprotein APP to result in β-amyloid deposition is determined by three secretases, termed α, β, and γ [168]. α-secretases cleave APP within the Aβ region resulting in the release of soluble fragments of α-APPs into the extracellular space along with a C-terminal fragment containing 83 residues (C83) that remains bound to the cellular membrane [69]. β-Secretases cleave APP to generate β-APPs and a remaining fragment that contains 99 C-terminal residues (C99) [198]. Both the C83 and C99 fragments are substrates for γ-secretase. Aβ is generated from C99 proteolysis through a transmembrane domain. The Aβ produced includes a 40-residue peptide (Aβ40) and a 42-residue peptide (Aβ42). Although Aβ42 is produced in much smaller quantities than Aβ40, it more readily forms fibrils and cerebral plaque than Aβ40. As a result, Aβ42 is considered to be the β-amyloid product that most directly contributes to the pathogenesis of Alzheimer's disease and apoptotic injury.
In addition to secretases, caspase activation is also necessary for the processing of APP (Fig. 6). Caspases cleave APP at three major caspase recognition sites, one at the C-terminus, D720, and two at the N-terminus, D197 and D219. Caspase activation results in the increased production of Aβ. Yet, in some cases, Aβ generation may not be entirely dependent upon the cleavage of APP at its C-terminal (D720) and/or N-terminal caspase sites. For example, during etoposide-induced apoptosis, ablation of caspase-dependent cleavage at D720, D197, and D219 (by site-directed mutagenesis) does not prevent enhanced Aβ production [219]. It is conceivable that APP may lead to cell injury through a more direct route that involves the generation of the C-terminal fragment C31. Production of C31 is a result of APP cleavage at the caspase site D720 of the C-terminus. Following caspase 3 activation, caspase 3 generates the carboxyl-terminally truncated fragment C31 from APP, which has been shown to be capable of apoptotic injury independent of caspase 3 [165]. Furthermore, caspase-dependent APP cleavage at D720 has also been observed in brains of Alzheimer's disease patients through demonstration of C31 expression [132].
6.4. Multiple pathways for caspase activity to influence the toxicity of tau and presenilins
Caspase processing of the tau protein may contribute to PCD and the formation of intracellular neurofibrillary tangles that are composed of the microtubule-associated protein tau (Fig. 6). Tau can be cleaved at Asp421 by caspases 1, 3, 7, and 8 resulting in the generation of a truncated tau protein (1−421) that is more readily assembling into tau filaments [76]. The C-terminal peptide (422−441) produced by caspase cleavage at Asp421 inhibits polymerization of the tau protein or the truncated tau protein (1−421). Removal of the C-terminal peptide, an inhibitory control element, by caspase cleavage of tau enhances its polymerization and results in the formation of neurofibrillary tangles [19].
The cleaved fragments of the tau protein have also been linked to apoptotic injury. As a neuronal microtubule-associated protein, tau has a central role in the formation of neuronal architecture. During the cleavage of tau in Alzheimer's disease, a soluble dephosphorylated tau fragment of 17 kDa is produced that cannot associate with microtubules. The generation of this fragment is blocked by caspase inhibitors, and without its accumulation in cells, it is hypothesized that apoptotic injury may not result [32]. Several other fragments of the tau protein also have been intimately linked to PCD. In an in vitro study, a 50-kDa fragment of tau protein produced by caspase 3 promoted cell death in mouse cortical neurons [50]. Overexpression of the tau fragment 152−391 also leads to apoptotic morphological changes, membrane blebbing, and nuclear pyknosis [71]. Additionally, the tau fragments 1−422 and 22−422 can precipitate PCD [71].
In the brains of Alzheimer's disease patients, the truncated tau protein has been identified by immunohistochemistry, suggesting that caspase cleavage of tau is involved in the pathology of Alzheimer's disease [76]. In cases of frontotemporal dementia, an increase in tau degradation was observed that was histologically associated with DNA fragmentation and caspase 3 activation in neurons [208]. Taken together with the biochemical data for the caspase processing of tau, these results suggest that tau cleavage may be a significant contributory factor in the pathogenesis of Alzheimer's disease.
The cleavage of presenilins by caspases has also been suggested to contribute to the progression of Alzheimer's disease. Presenilins are proteins that contain eight transmembrane domains. Both presenilin-1 (PS1) and presenilin-2 (PS2) are expressed in neurons throughout the brain. At the subcellular level, PS1 and PS2 are primarily located in the endoplasmic reticulum, Golgi apparatus, nuclear envelope, cell membrane, and the inner membrane of mitochondria [8,110,112].
Presenilins are proteolytically processed by caspases (Fig. 6). The endoproteolytical cleavage of PS1 and PS2 yields 27−35 kDa N-terminal fragments (NTFs) and 15−24 kDa C-terminal fragments (CTFs). This cleavage of PS1 or PS2 can be prevented by blockade of caspase 1 or 3 activity or the mutation of caspase cleave sites, such as Asp345/Ser346 for PS1 and Asp329/Ser330 for PS2 [129]. It has been demonstrated that multiple caspases possess the ability to cleave presenilins. Caspases 8 and 3 exert proteolytical activity on both PS1 and PS2, whereas caspases 1, 6, and 7 predominantly cleave PS2 [229]. During PCD, cleavage of presenilins, such as PS1, may foster a cell's demise. It is believed that the association of PS1 with β-catenin is necessary to prevent apoptosis by providing stability to β-catenin. Cleavage of β-catenin by caspases can disrupt cellular cytoskeleton components [25]. In addition, it has been demonstrated that overexpression of CTFs of PS2 promotes apoptosis by promoting Aβ production, caspase 3 activity, cleavage of PARP, and loss of the “anti-apoptotic” protein Bcl-2 [6].
7. Calpains and their intimate links to neuronal injury during Alzheimer's disease
7.1. Cellular structure and activity of calpains
Calpains are part of an intracellular family of cysteine proteases that are independent from caspases. At least 15 mammalian calpains have been identified, with two of these calpains, calpain 1 (μ-calpain) and calpain 2 (m-calpain), expressed primarily in the central nervous system. μ-calpain and m-calpain are heterodimeric proteins with a large 70−80 kDa catalytic subunit and a 29 kDa regulated subunit. In the nervous system, μ-calpain is predominantly distributed in dendrites and the bodies of neurons while m-calpain is expressed in axons and in glia [166].
Calpain activation is initiated by calcium with limited autolysis [156]. μ-calpain has a relatively high binding affinity to calcium and is activated by micromolar concentrations of calcium, while m-calpain binds to calcium with lower affinity and requires a higher concentration of calcium (millimolar) for activation [147,172,200]. The activity of calpains is also regulated by the endogenous inhibitor calpastatin and the state of phosphorylation. For example, phosphorylation at Ser369 by protein kinase A (PKA) blocks the activation of m-calpain [202].
7.2. Integral role of calpains during cellular integrity and injury
Calpains function not only as key regulators in cytoskeletal remodeling, but also in initiating cell injury. Calpains cleave plasma-membrane-associated proteins, such as the epidermal growth factor receptor, the platelet-derived growth factor receptor, and soluble kinases that include protein kinase C [224]. In addition, cell structure can be modified through calpains through their substrates that involve the microtubule-associated proteins tau, neurofilament, and actin [14,177]. On the converse side, calpains can lead to cell injury through the induction of apoptotic pathways. For example, in neuronal cell lines, calpain activation was found to increase with an elevation in intracellular calcium during oxidative stress [95]. Additional work has illustrated that free radical injury with associated mitochondrial dysfunction [238] as well as apoptotic injury following spinal cord trauma [181] could be prevented by calpain inhibition.
Calpains function through a number of pathways in the apoptotic cascade. Bax, a member of the Bcl-2 family, has been identified as a target of calpain [248]. The generation of a pro-apoptotic 18-kDa fragment of Bax by calpain results in the induction of cytochrome c release, caspase 3 activation, and subsequent induction of apoptosis [77]. Calpains can also directly modulate the activity of caspases. m-Calpain cleaves caspase 3 producing a 29-kDa fragment, which further facilitates the subsequent cleavage of caspase 3 into active forms [23]. In addition, calpain can directly activate caspase 7 and caspase 12 [163,191]. Although calpains may enhance caspase activity, calpains can also function to block the activation of caspases. Calpains can cleave caspase 9 rendering it incapable of activating caspase 3 and preventing the subsequent release of cytochrome c [49].
Given the broad function of calpains during both the maintenance of cellular cytoskeleton integrity and the induction of cell injury, it is not surprising that calpains have a significant role in the pathogenesis of Alzheimer's disease. For example, calpain activation has been found in clinical brain specimens of Alzheimer's disease [217], calpain 2 was demonstrated to be present in approximately 75% of neurofibrillary tangles [1], and calpains may promote cell cycle activation, a potential source of cell injury in Alzheimer's disease, through the activation of cyclin-dependent kinase 5 [217]. Further work have closely tied calpain activity to the toxicity of Aβ and presenilins. In rat hippocampal neurons, calpain appears to be required for the induction of apoptosis during Aβ application [101]. Over-expression of APP in neurons leads to calpain and caspase 3 activity, but activation of these pathways is lost in neurons that express an APP mutant defective in the Aβ domain [111]. In regards to presenilin activity, mutations in PS1 have been associated with increased activity of m-calpain and neuronal dysfunction [33], while m-calpain and μ-calpain have been shown to regulate PS1 activity by cleaving this protein [146].
8. Therapeutic avenues that employ novel cellular pathways against apoptotic and inflammatory injury in Alzheimer's disease
Therapeutic regimens for Alzheimer's disease currently focus on a limited number of strategies. For example, present treatments include the potentiation of cholinergic transmission in brain regions such as in the nucleus basalis to compensate for presynaptic cholinergic deficiency [114]. Although four cholinesterase inhibitors are presently available for the treatment of Alzheimer's disease, more recent entries into the field concentrate on the use of donepezil, rivastigamine, and galantamine which offer a longer duration of action with reduced side effects [62]. Prevention of N-methyl-d-aspartate (NMDA) receptor activity during cognitive loss is another approach for the treatment of Alzheimer's disease. Memantine, an antagonist of the NMDA receptor, can lead to cognitive improvement in patients with moderate to severe forms of Alzheimer's disease [183], but the mechanism of action for this agent is unclear [184]. Evidence for the efficacy of non-steroidal anti-inflammatory agents [3] or for estrogen replacement therapy [87] for the treatment of Alzheimer's disease is presently lacking. Unfortunately, present therapies are limited in nature and may provide only marginal symptomatic relief. As we move forward in understanding the multiple mechanisms that may contribute to Alzheimer's disease, new investigative avenues may provide not only attractive alternative therapies, but also viable treatments to either prevent or conceivably reverse the course of the disease. Several unique pathways are now under consideration that involve the modulation of cellular metabolic activity, regulation of attempted cell cycle induction in post-mitotic neurons, manipulation of the metabotropic glutamate system, and trophic factor and cytokine management.
8.1. Nicotinamide adenine dinucleotide (NAD+) and its precursor nicotinamide
The coenzyme nicotinamide adenine dinucleotide (NAD+) is closely tied to cellular metabolism and genomic DNA repair. During a cellular insult that affects DNA integrity, PARP catalyses the synthesis of poly(ADP-ribose) from its substrate NAD+, which stimulates the process of DNA repair [196]. Increased activation of PARP leads to an extensive turnover of NAD+ and a significant reduction in NAD+ levels. This can trigger the loss of NAD+ and ATP, leading to the death of a cell. Furthermore, oxidative stress can trigger the opening of mitochondrial membrane permeability transition pore [41,59,104,122] and subsequently result in the release of NAD+ from mitochondria [59]. During conditions of oxidative stress and energy depletion in neurons, poly(ADP-ribosylation) activation and loss of NAD+ stores in mitochondria have been shown to lead to apoptotic injury. Restoration of NAD+ content in mitochondria with liposomal NAD+ prevents neuronal injury [65].
Given the detrimental cellular ramifications of NAD+ depletion, both acute and chronic neurodegenerative diseases have been linked to the loss of NAD+ stores. In particular, in patients with Alzheimer's disease, PARP and poly(ADP-ribose) can be detected in the frontal and temporal cortex more frequently than in controls, suggesting that increased levels of functional PARP enzyme are present to result in a significant consumption of NAD+ stores [130]. Interestingly, a limited pilot study suggested that administration of nicotinamide adenine dinucleotide (NADH) in patients with Alzheimer's disease may show improvement in their cognitive function [21].
Yet, one must approach restorative therapy for NAD+ with caution, since a darker side may exist that involves the NAD+ precursor nicotinamide with cellular aging. In fact, recent work that employs transcriptional profiling of the human frontal cortex in the aging brain suggests that altered expression of a variety of genes may promote oxidative stress, DNA damage, and impaired mitochondrial function [133]. If one examines the aging process on more specific cellular terms, agents such as nicotinamide, an NAD+ precursor that is intimately tied to cell survival during acute apoptotic injury [47,120,140,144], may negatively influence the life span of cells through the regulation of the Sir2 gene [126]. The Sir2 gene belongs to a family of genes which is a highly conserved group in the genomes of organisms ranging from archaebacteria to eukaryotes [74,232]. Interestingly, SIRT1 (Sir2α), as a human homologue of Sir2, is intimately linked with the modulation of cellular apoptotic pathways. The Sir 2 protein is associated with nicotinamide and pyrazinamidase/nicotinamidase 1 (PNC1), an enzyme that deaminates nicotinamide. Nicotinamide appears to be capable of decreasing cell longevity through Sir2. Nicotinamide blocks cellular Sir2 by intercepting an ADP-ribosyl-enzyme-acetyl peptide intermediate with the regeneration of NAD+ (transglycosidation) [97]. Physiological concentrations of nicotinamide noncompetitively inhibit both Sir2 and SIRT1 in vitro, suggesting that nicotinamide is a physiologically relevant regulator of Sir2 enzymes [22].
Alternative concepts for the treatment of Alzheimer's disease may focus on the prevention of intracellular nicotinamide accumulation. During nicotinamide depletion, Sir2 is activated and employs PNC1 to regulate cell longevity. Increased expression of PNC1 has been found to be both necessary and sufficient for life span extension during calorie restriction in Saccharomyces cerevisiae [7]. Nicotinamide and PCN1 are closely linked in controlling cell life span. PNC1 can stimulate Sir2 histone deactylase activity by preventing the accumulation of nicotinamide through its conversion to nicotinic acid in the NAD+ salvage pathway. Overexpression of PNC1 has been demonstrated to suppress the inhibitory effect of exogenous nicotinamide on silencing, life span, and transcriptional repression of Sir2. As a result, PNC1 can positively regulate Sir2-mediated silencing and longevity by preventing the accumulation of intracellular nicotinamide [75]. Although significant further work is required to link these studies to higher organisms, it is possible that nicotinamide may control similar processes during neurodegenerative disorders.
8.2. Aberrant induction of the cell cycle in post-mitotic neurons
Attempted cell cycle induction also appears to be an important factor in neuronal cell loss during Alzheimer's disease [11,29,138,180]. In pathological specimens of brains from Alzheimer's patients, the cell cycle regulators P16 and CDK4 have increased expression in regions such as the hippocampus [151]. In addition, aberrant expression of other components of the cell cycle, such as cyclin D, Cdk4, proliferating cell nuclear antigen (PCNA), and cyclin B1, has been shown to be present in the hippocampus, subiculum, locus coeruleus, and dorsal raphe nuclei. Staining for cell cycle proteins has been shown to be absent in brain regions without neuronal injury of Alzheimer's patients and in age-matched brains [29]. Increased accumulation of cell cycle kinases, such as Cdk5, has also been found in neurons that are developing neurofibrillary tangles [174]. Interestingly, in patients with mild cognitive impairment, many of which can progress to develop Alzheimer's disease [17], cell cycle proteins, such as cyclin D, cyclin B, and PCNA, are significantly increased in the hippocampus and basal nucleus [251].
Results in experimental models have provided further evidence that cell cycle induction in post-mitotic neurons can activate cellular mechanisms that lead to neuronal apoptosis [68,94,108,123,187,221]. For example, application of Aβ (1−40), Aβ (1−42), and its active fragment Aβ (25−35) in neurons can result in the induction of cyclin D1, cyclin E and A, and the phosphorylation of the retinoblastoma protein. The activation of the upstream cyclin-dependent kinases (Cdk)4/5/6 appears to be required for the induction of apoptosis in neurons by Aβ, since inhibition of Cdks can prevent Aβ-induced neuronal apoptosis [5,81]. In addition, expression of familial Alzheimer's disease mutants of APP precipitate apoptotic injury through cell cycle induction and p21-mediated pathways [150]. Cell cycle proteins may also contribute to neurofibrillary tangle development. Cdk5 has been identified as a critical regulator of tau protein which is a primary component of neurofibrillary tangles. Cdk5 can phosphorylate tau directly [72]. Phosphorylation of tau by Aβ can be blocked by treatment with antisense against p35, a protein that yields the potent Cdk5 activator p25 following cleavage, suggesting that Aβ potently activates Cdk5 and subsequent tau phosphorylation that is dependent on the cleavage of p35 [225]. In addition, the cleavage of p35 to p25 has been shown to occur in Alzheimer's disease patients and the p25/Cdk5 complex has been demonstrated to hyperphosphorylate tau and induce apoptosis in primary neurons [173]. Taken together, these studies, in addition to clinical observations, suggest that prevention of cell cycle deregulation in Alzheimer's disease may block neuronal loss.
8.3. Modulation of the metabotropic glutamate system
In work that has studied metabotropic glutamate receptors (mGluRs) in Alzheimer's disease, a down-regulation of mGluR binding sites has been reported [58]. Current interest has focused on the protective role of the mGluR system in the nervous system. mGluR activation prevents and, in some cases, reverses genomic DNA degradation [236], modulates endonuclease activation [237], and maintains cellular membrane asymmetry [235]. Cytoprotection by the mGluR system is believed to act at or below the level of free radical generation and oxidative stress [143,192,236]. More recent work has suggested that mGluR offers similar protective capacity to the vascular system by preventing endothelial cell DNA degradation, caspase activity, and inhibiting a thrombotic state through the maintenance of membrane asymmetry [125,124,142].
Other studies that involve Alzheimer's disease illustrate that group I mGluRs can regulate the metabolism of APP and accelerate the processing of APP into non-amyloidogenic APP [115]. Activation of group I/II mGluRs can enhance the secretion of APP [99]. The process is blocked by the administration of (+)-α-methyl-4-carboxyphenylglycine, a non-selective antagonist of group I/II mGluRs [228]. Activation of group III mGluRs has also been shown to protect neurons against microglial neurotoxicity during Aβ application [218] that may be a result of the regulation of caspase activity [43,124]. Under some circumstances, diminished activity of mGluRs may prove useful for cellular protection. For example, inhibition of group II mGluRs can attenuate microglial activation and subsequent neurotoxicity during toxic stimuli such as chromogranin A [218], a protein up-regulated in Alzheimer's disease. mGluRs are also believed to be necessary for the processing of learning and memory [188]. Given the ability of mGluRs to prevent cellular toxicity and to alter memory function, agents that modulate the activation of mGluRs may be viable during Alzheimer's disease.
8.4. Cytokines and trophic factors as alternate strategies for cell rescue
Cytokines and trophic factors may be another viable option that can specifically target cellular pathways in Alzheimer's disease that have been implicated in cell injury. For example, cytokines that were previously thought to have no role in the brain, such as erythropoietin (EPO), have recently been demonstrated to regulate pathways that involve Akt and the Bcl-2 family member Bcl-xL [121,145]. EPO appears to significantly enhance the activity of Akt during oxidative stress [38,40] and prevent inflammatory activation of microglia [42]. In addition, EPO can enhance the expression of Bcl-xL. EPO may require Bcl-xL expression for cytoprotection, since without EPO, Bcl-xL is not expressed and apoptotic cell death results in hematopoietic cells [203]. Similar results in neurons and ECs illustrate that up-regulation of Bcl-xL by EPO may be necessary for the prevention of PCD [41] in combination with the modulation of Apaf-1 expression and cytochrome c release, similar to other “anti-apoptotic” proteins, such as heat shock proteins [15,67]. Growth factors may also be relevant during Alzheimer's disease. During the progression of Alzheimer's disease, brain-derived neurotrophic factor (BDNF) has been demonstrated to be decreased in the parietal cortex [154]. Application of BDNF in conjunction with sonic hedgehog can increase the number of cholinergic neurons in culture [182]. Insulin-like growth factor 1 may also be neuroprotective, since it can rescue neurons from Aβ toxicity [63].
It is clear that the cellular pathways responsible for the precipitation of Alzheimer's disease are complex in nature and extend beyond the production of extracellular plaques of β-amyloid aggregates and intracellular neurofibrillary tangles. Oxidative stress with the generation of free radicals plays a critical role in PCD and cell demise. Central to cellular integrity and phagocytic disposal of injured cells are the proto-oncogene Wnt and the serine-threonine kinase Akt that orchestrate closely aligned cellular pathways that involve components such as inflammatory cytokines, glycogen synthase kinase-3β, Bad, Bcl-xL, caspase activation, and calpain regulation. Effective translation of knowledge of the cellular mechanisms responsible for Alzheimer's disease into novel therapeutic modalities that may not conform to current consensus may require a level of dedication and enthusiasm that allowed Alois Alzheimer to bring forth his formidable observations and identification of Alzheimer's disease as a defined clinical entity.
Acknowledgments
We greatly appreciate the generosity of Daniel P. Perl, MD, of Mount Sinai School of Medicine for supplying us with images of clinical specimens of Alzheimer's disease for Figs. 1 and 2. This research was supported by the following grants (KM): American Heart Association (National), Janssen Neuroscience Award, Johnson and Johnson Focused Investigator Award, LEARN Foundation Award, MI Life Sciences Challenge Award, and NIH NIEHS (P30 ES06639).
References
- 1.Adamec E, Mohan P, Vonsattel JP, Nixon RA. Calpain activation in neurodegenerative diseases: confocal immunofluorescence study with antibodies specifically recognizing the active form of calpain 2. Acta Neuropathol. (Berl) 2002;104:92–104. doi: 10.1007/s00401-002-0528-6. [DOI] [PubMed] [Google Scholar]
- 2.Adams S, Green P, Claxton R, Simcox S, Williams MV, Walsh K, Leeuwenburgh C. Reactive carbonyl formation by oxidative and non-oxidative pathways. Front. Biosci. 2001;6:A17–A24. doi: 10.2741/adams. [DOI] [PubMed] [Google Scholar]
- 3.Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ. Effects of rofecoxib or naproxen vs. placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289:2819–2826. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez G, Munoz-Montano JR, Satrustegui J, Avila J, Bogonez E, Diaz-Nido J. Lithium protects cultured neurons against beta-amyloid-induced neurodegeneration. FEBS Lett. 1999;453:260–264. doi: 10.1016/s0014-5793(99)00685-7. [DOI] [PubMed] [Google Scholar]
- 5.Alvarez A, Munoz JP, Maccioni RB. A Cdk5-p35 stable complex is involved in the beta-amyloid-induced deregulation of Cdk5 activity in hippocampal neurons. Exp. Cell Res. 2001;264:266–274. doi: 10.1006/excr.2001.5152. [DOI] [PubMed] [Google Scholar]
- 6.Alves da Costa C, Mattson MP, Ancolio K, Checler F. The C-terminal fragment of presenilin 2 triggers p53-mediated staurosporine-induced apoptosis, a function independent of the presenilinase-derived N-terminal counterpart. J. Biol. Chem. 2003;278:12064–12069. doi: 10.1074/jbc.M212379200. [DOI] [PubMed] [Google Scholar]
- 7.Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Sinclair DA. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature. 2003;423:181–185. doi: 10.1038/nature01578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ankarcrona M, Hultenby K. Presenilin-1 is located in rat mitochondria. Biochem. Biophys. Res. Commun. 2002;295:766–770. doi: 10.1016/s0006-291x(02)00735-0. [DOI] [PubMed] [Google Scholar]
- 9.Aoki M, Nata T, Morishita R, Matsushita H, Nakagami H, Yamamoto K, Yamazaki K, Nakabayashi M, Ogihara T, Kaneda Y. Endothelial apoptosis induced by oxidative stress through activation of nf-kappab: antiapoptotic effect of antioxidant agents on endothelial cells. Hypertension. 2001;38:48–55. doi: 10.1161/01.hyp.38.1.48. [DOI] [PubMed] [Google Scholar]
- 10.Aplin AE, Jacobsen JS, Anderton BH, Gallo JM. Effect of increased glycogen synthase kinase-3 activity upon the maturation of the amyloid precursor protein in transfected cells. NeuroReport. 1997;8:639–643. doi: 10.1097/00001756-199702100-00012. [DOI] [PubMed] [Google Scholar]
- 11.Arendt T, Holzer M, Stobe A, Gartner U, Luth HJ, Bruckner MK, Ueberham U. Activated mitogenic signaling induces a process of dedifferentiation in Alzheimer's disease that eventually results in cell death. Ann. N. Y. Acad. Sci. 2000;920:249–255. doi: 10.1111/j.1749-6632.2000.tb06931.x. [DOI] [PubMed] [Google Scholar]
- 12.Arur S, Uche UE, Rezaul K, Fong M, Scranton V, Cowan AE, Mohler W, Han DK. Annexin I is an endogenous ligand that mediates apoptotic cell engulfment. Dev. Cell. 2003;4:587–598. doi: 10.1016/s1534-5807(03)00090-x. [DOI] [PubMed] [Google Scholar]
- 13.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 14.Banik NL, Matzelle D, Gantt-Wilford G, Hogan EL. Role of calpain and its inhibitors in tissue degeneration and neuroprotection in spinal cord injury. Ann. N. Y. Acad. Sci. 1997;825:120–127. doi: 10.1111/j.1749-6632.1997.tb48421.x. [DOI] [PubMed] [Google Scholar]
- 15.Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat. Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- 16.Bellacosa A, Testa JR, Staal SP, Tsichlis PN. A retroviral oncogene, akt, encoding a serine-threonine kinase containing an SH2-like region. Science. 1991;254:274–277. doi: 10.1126/science.254.5029.274. [DOI] [PubMed] [Google Scholar]
- 17.Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J. Natural history of mild cognitive impairment in older persons. Neurology. 2002;59:198–205. doi: 10.1212/wnl.59.2.198. [DOI] [PubMed] [Google Scholar]
- 18.Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, Brunden KR. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol. Aging. 1999;20:581–589. doi: 10.1016/s0197-4580(99)00065-2. [DOI] [PubMed] [Google Scholar]
- 19.Berry RW, Abraha A, Lagalwar S, LaPointe N, Gamblin TC, Cryns VL, Binder LI. Inhibition of tau polymerization by its carboxy-terminal caspase cleavage fragment. Biochemistry. 2003;42:8325–8331. doi: 10.1021/bi027348m. [DOI] [PubMed] [Google Scholar]
- 20.Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, Lee CM. Regulation and localization of tyrosine216 phosphorylation of glycogen synthase kinase-3beta in cellular and animal models of neuronal degeneration. Proc. Natl. Acad. Sci. U. S. A. 2000;97:11074–11079. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Birkmayer JG. Coenzyme nicotinamide adenine dinucleotide: new therapeutic approach for improving dementia of the Alzheimer type. Ann. Clin. Lab. Sci. 1996;26:1–9. [PubMed] [Google Scholar]
- 22.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 23.Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA, Mallard C, Hagberg H. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J. Biol. Chem. 2001;276:10191–10198. doi: 10.1074/jbc.M007807200. [DOI] [PubMed] [Google Scholar]
- 24.Bornemann KD, Wiederhold KH, Pauli C, Ermini F, Stalder M, Schnell L, Sommer B, Jucker M, Staufenbiel M. Abeta-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am. J. Pathol. 2001;158:63–73. doi: 10.1016/s0002-9440(10)63945-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brancolini C, Lazarevic D, Rodriguez J, Schneider C. Dismantling cell–cell contacts during apoptosis is coupled to a caspase-dependent proteolytic cleavage of beta-catenin. J. Cell Biol. 1997;139:759–771. doi: 10.1083/jcb.139.3.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bresgen N, Karlhuber G, Krizbai I, Bauer H, Bauer HC, Eckl PM. Oxidative stress in cultured cerebral endothelial cells induces chromosomal aberrations, micronuclei, and apoptosis. J. Neurosci. Res. 2003;72:327–333. doi: 10.1002/jnr.10582. [DOI] [PubMed] [Google Scholar]
- 27.Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. doi: 10.1016/s0092-8674(00)80595-4. [DOI] [PubMed] [Google Scholar]
- 28.Burlacu A, Jinga V, Gafencu AV, Simionescu M. Severity of oxidative stress generates different mechanisms of endothelial cell death. Cell Tissue Res. 2001;306:409–416. doi: 10.1007/s004410100424. [DOI] [PubMed] [Google Scholar]
- 29.Busser J, Geldmacher DS, Herrup K. Ectopic cell cycle proteins predict the sites of neuronal cell death in Alzheimer's disease brain. J. Neurosci. 1998;18:2801–2807. doi: 10.1523/JNEUROSCI.18-08-02801.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Butterfield DA, Castegna A, Lauderback CM, Drake J. Evidence that amyloid beta-peptide-induced lipid peroxidation and its sequelae in Alzheimer's disease brain contribute to neuronal death. Neurobiol. Aging. 2002;23:655–664. doi: 10.1016/s0197-4580(01)00340-2. [DOI] [PubMed] [Google Scholar]
- 31.Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- 32.Canu N, Dus L, Barbato C, Ciotti MT, Brancolini C, Rinaldi AM, Novak M, Cattaneo A, Bradbury A, Calissano P. Tau cleavage and dephosphorylation in cerebellar granule neurons undergoing apoptosis. J. Neurosci. 1998;18:7061–7074. doi: 10.1523/JNEUROSCI.18-18-07061.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chan SL, Culmsee C, Haughey N, Klapper W, Mattson MP. Presenilin-1 mutations sensitize neurons to DNA damage-induced death by a mechanism involving perturbed calcium homeostasis and activation of calpains and caspase-12. Neurobiol. Dis. 2002;11:2–19. doi: 10.1006/nbdi.2002.0542. [DOI] [PubMed] [Google Scholar]
- 34.Chen S, Guttridge DC, You Z, Zhang Z, Fribley A, Mayo MW, Kitajewski J, Wang CY. Wnt-1 signaling inhibits apoptosis by activating beta-catenin/T cell factor-mediated transcription. J. Cell Biol. 2001;152:87–96. doi: 10.1083/jcb.152.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen GJ, Xu J, Lahousse SA, Caggiano NL, de la Monte SM. Transient hypoxia causes Alzheimer-type molecular and biochemical abnormalities in cortical neurons: potential strategies for neuroprotection. J. Alzheimer's Dis. 2003;5:209–228. doi: 10.3233/jad-2003-5305. [DOI] [PubMed] [Google Scholar]
- 36.Chen T, Yang I, Irby R, Shain KH, Wang HG, Quackenbush J, Coppola D, Cheng JQ, Yeatman TJ. Regulation of caspase expression and apoptosis by adenomatous polyposis coli. Cancer Res. 2003;63:4368–4374. [PubMed] [Google Scholar]
- 37.Choi J, Malakowsky CA, Talent JM, Conrad CC, Carroll CA, Weintraub ST, Gracy RW. Anti-apoptotic proteins are oxidized by Abeta25−35 in Alzheimer's fibroblasts. Biochim. Biophys. Acta. 2003;1637:135–141. doi: 10.1016/s0925-4439(02)00227-2. [DOI] [PubMed] [Google Scholar]
- 38.Chong ZZ, Kang JQ, Maiese K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation. 2002;106:2973–2979. doi: 10.1161/01.cir.0000039103.58920.1f. [DOI] [PubMed] [Google Scholar]
- 39.Chong ZZ, Lin SH, Maiese K. Nicotinamide modulates mitochondrial membrane potential and cysteine protease activity during cerebral vascular endothelial cell injury. J. Vasc. Res. 2002;39:131–147. doi: 10.1159/000057762. [DOI] [PubMed] [Google Scholar]
- 40.Chong ZZ, Kang J, Maiese K. Erythropoietin: cytoprotection in vascular and neuronal cells. Curr. Drug Targets-Cardiovasc. Hematol. Dis. 2003;3:141–154. doi: 10.2174/1568006033481483. [DOI] [PubMed] [Google Scholar]
- 41.Chong ZZ, Kang JQ, Maiese K. Apaf-1, Bcl-xL, cytochrome c, and caspase-9 form the critical elements for cerebral vascular protection by erythropoietin. J. Cereb. Blood Flow Metab. 2003;23:320–330. doi: 10.1097/01.WCB.0000050061.57184.AE. [DOI] [PubMed] [Google Scholar]
- 42.Chong ZZ, Kang JQ, Maiese K. Erythropoietin fosters both intrinsic and extrinsic neuronal protection through modulation of microglia, Akt1, Bad, and caspase-mediated pathways. Br. J. Pharmacol. 2003;138:1107–1118. doi: 10.1038/sj.bjp.0705161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chong ZZ, Kang JQ, Maiese K. Metabotropic glutamate receptors promote neuronal and vascular plasticity through novel intracellular pathways. Histol. Histopathol. 2003;18:173–189. doi: 10.14670/HH-18.173. [DOI] [PubMed] [Google Scholar]
- 44.Chong ZZ, Lin S-H, Kang J, Maiese K. The tyrosine phosphatase SHP2 modulates MAP kinase p38 and caspase 1 and 3 to foster neuronal survival. Cell Mol. Neurobiol. 2003;23(4−5):561–578. doi: 10.1023/A:1025158314016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chong ZZ, Kang JQ, Maiese K. Akt1 drives endothelial cell membrane asymmetry and microglial activation through Bcl-x(L) and caspase 1, 3, and 9. Exp. Cell Res. 2004;296:196–207. doi: 10.1016/j.yexcr.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 46.Chong ZZ, Kang JQ, Maiese K. Essential cellular regulatory elements of oxidative stress in early and late phases of apoptosis in the central nervous system. Antioxid. Redox Signal. 2004;6:277–287. doi: 10.1089/152308604322899341. [DOI] [PubMed] [Google Scholar]
- 47.Chong ZZ, Lin S-H, Maiese K. The NAD+ precursor nicotinamide governs neuronal survival during oxidative stress through protein kinase B coupled to FOXO3a and mitochondrial membrane potential. J. Cereb. Blood Flow. Metab. 2004;24:728–743. doi: 10.1097/01.WCB.0000122746.72175.0E. [DOI] [PubMed] [Google Scholar]
- 48.Chong ZZ, Li F, Maiese K. Activating Akt and the brain's resources to drive cellular survival and prevent inflammatory injury. Histol. Histopathol. 2005;20:299–315. doi: 10.14670/hh-20.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chua BT, Guo K, Li P. Direct cleavage by the calcium-activated protease calpain can lead to inactivation of caspases. J. Biol. Chem. 2000;275:5131–5135. doi: 10.1074/jbc.275.7.5131. [DOI] [PubMed] [Google Scholar]
- 50.Chung CW, Song YH, Kim IK, Yoon WJ, Ryu BR, Jo DG, Woo HN, Kwon YK, Kim HH, Gwag BJ, Mook-Jung IH, Jung YK. Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol. Dis. 2001;8:162–172. doi: 10.1006/nbdi.2000.0335. [DOI] [PubMed] [Google Scholar]
- 51.Citron M, Vigo-Pelfrey C, Teplow DB, Miller C, Schenk D, Johnston J, Winblad B, Venizelos N, Lannfelt L, Selkoe DJ. Excessive production of amyloid beta-protein by peripheral cells of symptomatic and presymptomatic patients carrying the Swedish familial Alzheimer disease mutation. Proc. Natl. Acad. Sci. U. S. A. 1994;91:11993–11997. doi: 10.1073/pnas.91.25.11993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colurso GJ, Nilson JE, Vervoort LG. Quantitative assessment of DNA fragmentation and beta-amyloid deposition in insular cortex and midfrontal gyrus from patients with Alzheimer's disease. Life Sci. 2003;73:1795–1803. doi: 10.1016/s0024-3205(03)00512-5. [DOI] [PubMed] [Google Scholar]
- 53.Combs CK, Karlo JC, Kao SC, Landreth GE. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 2001;21:1179–1188. doi: 10.1523/JNEUROSCI.21-04-01179.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cosulich SC, Worrall V, Hedge PJ, Green S, Clarke PR. Regulation of apoptosis by BH3 domains in a cell-free system. Curr. Biol. 1997;7:913–920. doi: 10.1016/s0960-9822(06)00410-6. [DOI] [PubMed] [Google Scholar]
- 55.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 56.de la Monte SM, Lu BX, Sohn YK, Etienne D, Kraft J, Ganju N, Wands JR. Aberrant expression of nitric oxide synthase III in Alzheimer's disease: relevance to cerebral vasculopathy and neurodegeneration. Neurobiol. Aging. 2000;21:309–319. doi: 10.1016/s0197-4580(99)00108-6. [DOI] [PubMed] [Google Scholar]
- 57.de la Monte SM, Chiche J, von dem Bussche A, Sanyal S, Lahousse SA, Janssens SP, Bloch KD. Nitric oxide synthase-3 overexpression causes apoptosis and impairs neuronal mitochondrial function: relevance to Alzheimer's-type neurodegeneration. Lab. Invest. 2003;83:287–298. doi: 10.1097/01.lab.0000056995.07053.c0. [DOI] [PubMed] [Google Scholar]
- 58.Dewar D, Chalmers DT, Graham DI, McCulloch J. Glutamate metabotropic and AMPA binding sites are reduced in Alzheimer's disease: an autoradiographic study of the hippocampus. Brain Res. 1991;553:58–64. doi: 10.1016/0006-8993(91)90230-s. [DOI] [PubMed] [Google Scholar]
- 59.Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J. Biol. Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- 60.Dijkers PF, Birkenkamp KU, Lam EW, Thomas NS, Lammers JW, Koenderman L, Coffer PJ. FKHR-L1 can act as a critical effector of cell death induced by cytokine withdrawal: protein kinase B-enhanced cell survival through maintenance of mitochondrial integrity. J. Cell Biol. 2002;156:531–542. doi: 10.1083/jcb.200108084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dombroski D, Balasubramanian K, Schroit AJ. Phosphatidylserine expression on cell surfaces promotes antibody-dependent aggregation and thrombosis in beta2-glycoprotein I-immune mice. J. Autoimmun. 2000;14:221–229. doi: 10.1006/jaut.2000.0365. [DOI] [PubMed] [Google Scholar]
- 62.Doody RS. Current treatments for Alzheimer's disease: cholinesterase inhibitors. J. Clin. Psychiatry. 2003;64(Suppl 9):11–17. [PubMed] [Google Scholar]
- 63.Dore S, Kar S, Quirion R. Insulin-like growth factor I protects and rescues hippocampal neurons against beta-amyloid- and human amylin-induced toxicity. Proc. Natl. Acad. Sci. U. S. A. 1997;94:4772–4777. doi: 10.1073/pnas.94.9.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dramsi S, Scheid MP, Maiti A, Hojabrpour P, Chen X, Schubert K, Goodlett DR, Aebersold R, Duronio V. Identification of a novel phosphorylation site, Ser-170, as a regulator of bad proapoptotic activity. J. Biol. Chem. 2002;277:6399–6405. doi: 10.1074/jbc.M109990200. [DOI] [PubMed] [Google Scholar]
- 65.Du L, Zhang X, Han YY, Burke NA, Kochanek PM, Watkins SC, Graham SH, Carcillo JA, Szabo C, Clark RS. Intramitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J. Biol. Chem. 2003;278:18426–18433. doi: 10.1074/jbc.M301295200. [DOI] [PubMed] [Google Scholar]
- 66.Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 67.Ekshyyan O, Aw TY. Apoptosis: a key in neurodegenerative disorders? Curr. Neurovasc. Res. 2004;1:355–371. doi: 10.2174/1567202043362018. [DOI] [PubMed] [Google Scholar]
- 68.El-Khodor BF, Frances Oo T, Kholodilov N, Burke RE. Ectopic expression of cell cycle markers in models of induced programmed cell death in dopamine neurons of the rat substantia nigra pars compacta. Exp. Neurol. 2003;179:17–27. doi: 10.1006/exnr.2002.8047. [DOI] [PubMed] [Google Scholar]
- 69.Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, McClure D, Ward PJ. Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science. 1990;248:1122–1124. doi: 10.1126/science.2111583. [DOI] [PubMed] [Google Scholar]
- 70.Fadok VA, de Cathelineau A, Daleke DL, Henson PM, Bratton DL. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J. Biol. Chem. 2001;276:1071–1077. doi: 10.1074/jbc.M003649200. [DOI] [PubMed] [Google Scholar]
- 71.Fasulo L, Ugolini G, Visintin M, Bradbury A, Brancolini C, Verzillo V, Novak M, Cattaneo A. The neuronal microtubule-associated protein tau is a substrate for caspase-3 and an effector of apoptosis. J. Neurochem. 2000;75:624–633. doi: 10.1046/j.1471-4159.2000.0750624.x. [DOI] [PubMed] [Google Scholar]
- 72.Flaherty DB, Soria JP, Tomasiewicz HG, Wood JG. Phosphorylation of human tau protein by microtubule-associated kinases: GSK3beta and cdk5 are key participants. J. Neurosci. Res. 2000;62:463–472. doi: 10.1002/1097-4547(20001101)62:3<463::AID-JNR16>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 73.Franke TF, Tartof KD, Tsichlis PN. The SH2-like Akt homology (AH) domain of c-akt is present in multiple copies in the genome of vertebrate and invertebrate eucaryotes. Cloning and characterization of the Drosophila melanogaster c-akt homolog Dakt1. Oncogene. 1994;9:141–148. [PubMed] [Google Scholar]
- 74.Frye RA. Phylogenetic classification of prokaryotic and eukaryotic Sir2-like proteins. Biochem. Biophys. Res. Commun. 2000;273:793–798. doi: 10.1006/bbrc.2000.3000. [DOI] [PubMed] [Google Scholar]
- 75.Gallo CM, Smith DL, Jr., Smith JS. Nicotinamide clearance by Pnc1 directly regulates Sir2-mediated silencing and longevity. Mol. Cell. Biol. 2004;24:1301–1312. doi: 10.1128/MCB.24.3.1301-1312.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gamblin TC, Chen F, Zambrano A, Abraha A, Lagalwar S, Guillozet AL, Lu M, Fu Y, Garcia-Sierra F, LaPointe N, Miller R, Berry RW, Binder LI, Cryns VL. Caspase cleavage of tau: linking amyloid and neurofibrillary tangles in Alzheimer's disease. Proc. Natl. Acad. Sci. U. S. A. 2003;100:10032–10037. doi: 10.1073/pnas.1630428100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gao G, Dou QP. N-terminal cleavage of bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes bcl-2-independent cytochrome C release and apoptotic cell death. J. Cell. Biochem. 2000;80:53–72. doi: 10.1002/1097-4644(20010101)80:1<53::aid-jcb60>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 78.Garrido JL, Godoy JA, Alvarez A, Bronfman M, Inestrosa NC. Protein kinase C inhibits amyloid beta peptide neurotoxicity by acting on members of the Wnt pathway. FASEB J. 2002;16:1982–1984. doi: 10.1096/fj.02-0327fje. [DOI] [PubMed] [Google Scholar]
- 79.Gastard MC, Troncoso JC, Koliatsos VE. Caspase activation in the limbic cortex of subjects with early Alzheimer's disease. Ann. Neurol. 2003;54:393–398. doi: 10.1002/ana.10680. [DOI] [PubMed] [Google Scholar]
- 80.Gilley J, Coffer PJ, Ham J. FOXO transcription factors directly activate bim gene expression and promote apoptosis in sympathetic neurons. J. Cell Biol. 2003;162:613–622. doi: 10.1083/jcb.200303026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Giovanni A, Wirtz-Brugger F, Keramaris E, Slack R, Park DS. Involvement of cell cycle elements, cyclin-dependent kinases, pRb, and E2F × DP, in B-amyloid-induced neuronal death. J. Biol. Chem. 1999;274:19011–19016. doi: 10.1074/jbc.274.27.19011. [DOI] [PubMed] [Google Scholar]
- 82.Goldshmit Y, Erlich S, Pinkas-Kramarski R. Neuregulin rescues PC12-ErbB4 cells from cell death induced by H(2)O(2). Regulation of reactive oxygen species levels by phosphatidylinositol 3-kinase. J. Biol. Chem. 2001;276:46379–46385. doi: 10.1074/jbc.M105637200. [DOI] [PubMed] [Google Scholar]
- 83.Guo Q, Fu W, Xie J, Luo H, Sells SF, Geddes JW, Bondada V, Rangnekar VM, Mattson MP. Par-4 is a mediator of neuronal degeneration associated with the pathogenesis of Alzheimer disease. Nat. Med. 1998;4:957–962. doi: 10.1038/nm0898-957. [DOI] [PubMed] [Google Scholar]
- 84.Hanayama R, Tanaka M, Miyasaka K, Aozasa K, Koike M, Uchiyama Y, Nagata S. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–1150. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 85.Hashimoto Y, Niikura T, Chiba T, Tsukamoto E, Kadowaki H, Nishitoh H, Yamagishi Y, Ishizaka M, Yamada M, Nawa M, Terashita K, Aiso S, Ichijo H, Nishimoto I. The cytoplasmic domain of Alzheimer's amyloid-beta protein precursor causes sustained apoptosis signal-regulating kinase 1/c-Jun NH2-terminal kinase-mediated neurotoxic signal via dimerization. J. Pharmacol. Exp. Ther. 2003;306:889–902. doi: 10.1124/jpet.103.051383. [DOI] [PubMed] [Google Scholar]
- 86.Hatori K, Nagai A, Heisel R, Ryu JK, Kim SU. Fractalkine and fractalkine receptors in human neurons and glial cells. J. Neurosci. Res. 2002;69:418–426. doi: 10.1002/jnr.10304. [DOI] [PubMed] [Google Scholar]
- 87.Henderson VW. Hormone therapy and Alzheimer's disease: benefit or harm? Expert Opin. Pharmacother. 2004;5:389–406. doi: 10.1517/14656566.5.2.389. [DOI] [PubMed] [Google Scholar]
- 88.Hofmann K, Bucher P, Tschopp J. The CARD domain: a new apoptotic signalling motif. Trends Biochem. Sci. 1997;22:155–156. doi: 10.1016/s0968-0004(97)01043-8. [DOI] [PubMed] [Google Scholar]
- 89.Hoffmann PR, deCathelineau AM, Ogden CA, Leverrier Y, Bratton DL, Daleke DL, Ridley AJ, Fadok VA, Henson PM. Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J. Cell Biol. 2001;155:649–659. doi: 10.1083/jcb.200108080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hong M, Chen DC, Klein PS, Lee VM. Lithium reduces tau phosphorylation by inhibition of glycogen synthase kinase-3. J. Biol. Chem. 1997;272:25326–25332. doi: 10.1074/jbc.272.40.25326. [DOI] [PubMed] [Google Scholar]
- 91.Hu Y, Benedict MA, Ding L, Nunez G. Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis. EMBO J. 1999;18:3586–3595. doi: 10.1093/emboj/18.13.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, Cuajungco MP, Gray DN, Lim J, Moir RD, Tanzi RE, Bush AI. The A beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 93.Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 1998;17:1371–1384. doi: 10.1093/emboj/17.5.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ino H, Chiba T. Cyclin-dependent kinase 4 and cyclin D1 are required for excitotoxin-induced neuronal cell death in vivo. J. Neurosci. 2001;21:6086–6094. doi: 10.1523/JNEUROSCI.21-16-06086.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ishihara I, Minami Y, Nishizaki T, Matsuoka T, Yamamura H. Activation of calpain precedes morphological alterations during hydrogen peroxide-induced apoptosis in neuronally differentiated mouse embryonal carcinoma P19 cell line. Neurosci. Lett. 2000;279:97–100. doi: 10.1016/s0304-3940(99)00960-x. [DOI] [PubMed] [Google Scholar]
- 96.Ishitani T, Ninomiya-Tsuji J, Matsumoto K. Regulation of lymphoid enhancer factor 1/T-cell factor by mitogen-activated protein kinase-related Nemo-like kinase-dependent phosphorylation in Wnt/beta-catenin signaling. Mol. Cell. Biol. 2003;23:1379–1389. doi: 10.1128/MCB.23.4.1379-1389.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Jackson MD, Schmidt MT, Oppenheimer NJ, Denu JM. Mechanism of nicotinamide inhibition and transglycosidation by Sir2 histone/protein deacetylases. J. Biol. Chem. 2003;278:50985–50998. doi: 10.1074/jbc.M306552200. [DOI] [PubMed] [Google Scholar]
- 98.Jessel R, Haertel S, Socaciu C, Tykhonova S, Diehl HA. Kinetics of apoptotic markers in exogeneously induced apoptosis of EL4 cells. J. Cell Mol. Med. 2002;6:82–92. doi: 10.1111/j.1582-4934.2002.tb00313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jolly-Tornetta C, Gao ZY, Lee VM, Wolf BA. Regulation of amyloid precursor protein secretion by glutamate receptors in human Ntera 2 neurons. J. Biol. Chem. 1998;273:14015–14021. doi: 10.1074/jbc.273.22.14015. [DOI] [PubMed] [Google Scholar]
- 100.Jones SE, Jomary C, Grist J, Stewart HJ, Neal MJ. Modulated expression of secreted frizzled-related proteins in human retinal degeneration. NeuroReport. 2000;11:3963–3967. doi: 10.1097/00001756-200012180-00012. [DOI] [PubMed] [Google Scholar]
- 101.Jordan J, Galindo MF, Miller RJ. Role of calpain- and interleukin-1 beta converting enzyme-like proteases in the beta-amyloid-induced death of rat hippocampal neurons in culture. J. Neurochem. 1997;68:1612–1621. doi: 10.1046/j.1471-4159.1997.68041612.x. [DOI] [PubMed] [Google Scholar]
- 102.Juo P, Kuo CJ, Yuan J, Blenis J. Essential requirement for caspase-8/FLICE in the initiation of the Fas-induced apoptotic cascade. Curr. Biol. 1998;8:1001–1008. doi: 10.1016/s0960-9822(07)00420-4. [DOI] [PubMed] [Google Scholar]
- 103.Kang JQ, Chong ZZ, Maiese K. Akt1 protects against inflammatory microglial activation through maintenance of membrane asymmetry and modulation of cysteine protease activity. J. Neurosci. Res. 2003;74:37–51. doi: 10.1002/jnr.10740. [DOI] [PubMed] [Google Scholar]
- 104.Kang JQ, Chong ZZ, Maiese K. Critical role for Akt1 in the modulation of apoptotic phosphatidylserine exposure and microglial activation. Mol. Pharmacol. 2003;64:557–569. doi: 10.1124/mol.64.3.557. [DOI] [PubMed] [Google Scholar]
- 105.Kawakami Y, Capdevila J, Buscher D, Itoh T, Rodriguez Esteban C, Izpisua Belmonte JC. WNT signals control FGF-dependent limb initiation and AER induction in the chick embryo. Cell. 2001;104:891–900. doi: 10.1016/s0092-8674(01)00285-9. [DOI] [PubMed] [Google Scholar]
- 106.Kim AH, Yano H, Cho H, Meyer D, Monks B, Margolis B, Birnbaum MJ, Chao MV. Akt1 regulates a JNK scaffold during excitotoxic apoptosis. Neuron. 2002;35:697–709. doi: 10.1016/s0896-6273(02)00821-8. [DOI] [PubMed] [Google Scholar]
- 107.Kim HS, Kim EM, Lee JP, Park CH, Kim S, Seo JH, Chang KA, Yu E, Jeong SJ, Chong YH, Suh YH. C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3beta expression. FASEB J. 2003;15:15. doi: 10.1096/fj.03-0106fje. [DOI] [PubMed] [Google Scholar]
- 108.Konishi Y, Bonni A. The E2F-Cdc2 cell-cycle pathway specifically mediates activity deprivation-induced apoptosis of postmitotic neurons. J. Neurosci. 2003;23:1649–1658. doi: 10.1523/JNEUROSCI.23-05-01649.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Koriyama Y, Chiba K, Mohri T. Propentofylline protects beta-amyloid protein-induced apoptosis in cultured rat hippocampal neurons. Eur. J. Pharmacol. 2003;458:235–241. doi: 10.1016/s0014-2999(02)02789-9. [DOI] [PubMed] [Google Scholar]
- 110.Kovacs DM, Fausett HJ, Page KJ, Kim TW, Moir RD, Merriam DE, Hollister RD, Hallmark OG, Mancini R, Felsenstein KM, Hyman BT, Tanzi RE, Wasco W. Alzheimer-associated presenilins 1 and 2: neuronal expression in brain and localization to intracellular membranes in mammalian cells. Nat. Med. 1996;2:224–229. doi: 10.1038/nm0296-224. [DOI] [PubMed] [Google Scholar]
- 111.Kuwako K, Nishimura I, Uetsuki T, Saido TC, Yoshikawa K. Activation of calpain in cultured neurons overexpressing Alzheimer amyloid precursor protein. Brain Res. Mol. Brain Res. 2002;107:166–175. doi: 10.1016/s0169-328x(02)00489-8. [DOI] [PubMed] [Google Scholar]
- 112.Lah JJ, Heilman CJ, Nash NR, Rees HD, Yi H, Counts SE, Levey AI. Light and electron microscopic localization of presenilin-1 in primate brain. J. Neurosci. 1997;17:1971–1980. doi: 10.1523/JNEUROSCI.17-06-01971.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lauber K, Bohn E, Krober SM, Xiao YJ, Blumenthal SG, Lindemann RK, Marini P, Wiedig C, Zobywalski A, Baksh S, Xu Y, Autenrieth IB, Schulze-Osthoff K, Belka C, Stuhler G, Wesselborg S. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113:717–730. doi: 10.1016/s0092-8674(03)00422-7. [DOI] [PubMed] [Google Scholar]
- 114.Lazareno S, Popham A, Birdsall NJ. Progress toward a high-affinity allosteric enhancer at muscarinic M1 receptors. J. Mol. Neurosci. 2003;20:363–367. doi: 10.1385/JMN:20:3:363. [DOI] [PubMed] [Google Scholar]
- 115.Lee RK, Jimenez J, Cox AJ, Wurtman RJ. Metabotropic glutamate receptors regulate APP processing in hippocampal neurons and cortical astrocytes derived from fetal rats. Ann. N. Y. Acad. Sci. 1996;777:338–343. doi: 10.1111/j.1749-6632.1996.tb34443.x. [DOI] [PubMed] [Google Scholar]
- 116.Lee DH, O'Connor TR, Pfeifer GP. Oxidative DNA damage induced by copper and hydrogen peroxide promotes CG → TT tandem mutations at methylated CpG dinucleotides in nucleotide excision repair-deficient cells. Nucleic Acids Res. 2002;30:3566–3573. doi: 10.1093/nar/gkf478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 118.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 119.Li Y, Tennekoon GI, Birnbaum M, Marchionni MA, Rutkowski JL. Neuregulin signaling through a PI3K/Akt/Bad pathway in Schwann cell survival. Mol. Cell Neurosci. 2001;17:761–767. doi: 10.1006/mcne.2000.0967. [DOI] [PubMed] [Google Scholar]
- 120.Li F, Chong ZZ, Maiese K. Navigating novel mechanisms of cellular plasticity with the NAD+ precursor and nutrient nicotinamide. Front. Biosci. 2004;9:2500–2520. doi: 10.2741/1412. [DOI] [PubMed] [Google Scholar]
- 121.Li F, Chong ZZ, Maiese K. Erythropoietin on a tightrope: balancing neuronal and vascular protection between intrinsic and extrinsic pathways. NeuroSignals. 2004;13:265–289. doi: 10.1159/000081963. [DOI] [PubMed] [Google Scholar]
- 122.Lin SH, Vincent A, Shaw T, Maynard KI, Maiese K. Prevention of nitric oxide-induced neuronal injury through the modulation of independent pathways of programmed cell death. J. Cereb. Blood Flow Metab. 2000;20:1380–1391. doi: 10.1097/00004647-200009000-00013. [DOI] [PubMed] [Google Scholar]
- 123.Lin S, Chong ZZ, Maiese K. Cell cycle induction in post-mitotic neurons proceeds in concert with the initial phase of programmed cell death in rat. Neurosci. Lett. 2001;310:173–177. doi: 10.1016/s0304-3940(01)02118-8. [DOI] [PubMed] [Google Scholar]
- 124.Lin SH, Maiese K. The metabotropic glutamate receptor system protects against ischemic free radical programmed cell death in rat brain endothelial cells. J. Cereb. Blood Flow Metab. 2001;21:262–275. doi: 10.1097/00004647-200103000-00010. [DOI] [PubMed] [Google Scholar]
- 125.Lin S-H, Chong ZZ, Maiese K. The metabotropic glutamate receptor system: G-protein mediated pathways that modulate neuronal and vascular cellular injury. Curr. Med. Chem. -Central Nervous System Agents. 2002;2:17–28. [Google Scholar]
- 126.Lin SJ, Guarente L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr. Opin. Cell Biol. 2003;15:241–246. doi: 10.1016/s0955-0674(03)00006-1. [DOI] [PubMed] [Google Scholar]
- 127.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 128.Liu B, Hong JS. Role of microglia in inflammation-mediated neurodegenerative diseases: mechanisms and strategies for therapeutic intervention. J. Pharmacol. Exp. Ther. 2003;304:1–7. doi: 10.1124/jpet.102.035048. [DOI] [PubMed] [Google Scholar]
- 129.Loetscher H, Deuschle U, Brockhaus M, Reinhardt D, Nelboeck P, Mous J, Grunberg J, Haass C, Jacobsen H. Presenilins are processed by caspase-type proteases. J. Biol. Chem. 1997;272:20655–20659. doi: 10.1074/jbc.272.33.20655. [DOI] [PubMed] [Google Scholar]
- 130.Love S, Barber R, Wilcock GK. Increased poly(ADP-ribosyl)ation of nuclear proteins in Alzheimer's disease. Brain. 1999;122(Pt 2):247–253. doi: 10.1093/brain/122.2.247. [DOI] [PubMed] [Google Scholar]
- 131.Lovell MA, Gabbita SP, Markesbery WR. Increased DNA oxidation and decreased levels of repair products in Alzheimer's disease ventricular CSF. J. Neurochem. 1999;72:771–776. doi: 10.1046/j.1471-4159.1999.0720771.x. [DOI] [PubMed] [Google Scholar]
- 132.Lu DC, Rabizadeh S, Chandra S, Shayya RF, Ellerby LM, Ye X, Salvesen GS, Koo EH, Bredesen DE. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat. Med. 2000;6:397–404. doi: 10.1038/74656. [DOI] [PubMed] [Google Scholar]
- 133.Lu T, Pan Y, Kao SY, Li C, Kohane I, Chan J, Yankner BA. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 134.Lucas JJ, Hernandez F, Gomez-Ramos P, Moran MA, Hen R, Avila J. Decreased nuclear beta-catenin, tau hyperphosphorylation and neurodegeneration in GSK-3beta conditional transgenic mice. EMBO J. 2001;20:27–39. doi: 10.1093/emboj/20.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lue LF, Brachova L, Walker DG, Rogers J. Characterization of glial cultures from rapid autopsies of Alzheimer's and control patients. Neurobiol. Aging. 1996;17:421–429. doi: 10.1016/0197-4580(96)00006-1. [DOI] [PubMed] [Google Scholar]
- 136.Lustig B, Behrens J. The Wnt signaling pathway and its role in tumor development. J. Cancer Res. Clin. Oncol. 2003;129:199–221. doi: 10.1007/s00432-003-0431-0. [DOI] [PubMed] [Google Scholar]
- 137.Madaio MP, Fabbi M, Tiso M, Daga A, Puccetti A. Spontaneously produced anti-DNA/DNase I autoantibodies modulate nuclear apoptosis in living cells. Eur. J. Immunol. 1996;26:3035–3041. doi: 10.1002/eji.1830261232. [DOI] [PubMed] [Google Scholar]
- 138.Maiese K. The dynamics of cellular injury: transformation into neuronal and vascular protection. Histol. Histopathol. 2001;16:633–644. doi: 10.14670/HH-16.633. [DOI] [PubMed] [Google Scholar]
- 139.Maiese K, Vincent AM. Membrane asymmetry and DNA degradation: functionally distinct determinants of neuronal programmed cell death. J. Neurosci. Res. 2000;59:568–580. doi: 10.1002/(SICI)1097-4547(20000215)59:4<568::AID-JNR13>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 140.Maiese K, Chong ZZ. Nicotinamide: necessary nutrient emerges as a novel cytoprotectant for the brain. Trends Pharmacol. Sci. 2003;24:228–232. doi: 10.1016/S0165-6147(03)00078-6. [DOI] [PubMed] [Google Scholar]
- 141.Maiese K, Chong ZZ. Insights into oxidative stress and potential novel therapeutic targets for Alzheimer disease. Restor. Neurol. Neurosci. 2004;22:87–104. [PubMed] [Google Scholar]
- 142.Maiese K, Chong ZZ. Impacting neuronal and vascular cellular signal transduction through the metabotropic glutamate receptor system. Medicinal Chemistry Reviews. in press. [Google Scholar]
- 143.Maiese K, Swiriduk M, TenBroeke M. Cellular mechanisms of protection by metabotropic glutamate receptors during anoxia and nitric oxide toxicity. J. Neurochem. 1996;66:2419–2428. doi: 10.1046/j.1471-4159.1996.66062419.x. [DOI] [PubMed] [Google Scholar]
- 144.Maiese K, Lin S, Chong ZZ. Elucidating neuronal and vascular injury through the cytoprotective agent nicotinamide. Curr. Med. Chem.-Imm., Endocr. Metab. Agents. 2001;1:257–267. [Google Scholar]
- 145.Maiese K, Faqi L, Chong ZZ. Erythropoietin in the brain: can the promise to protect be fulfilled? Trends Pharmacol. Sci. 2004;25:577–583. doi: 10.1016/j.tips.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 146.Maruyama K, Usami M, Kametani F, Tomita T, Iwatsubo T, Saido TC, Mori H, Ishiura S. Molecular interactions between presenilin and calpain: inhibition of m-calpain protease activity by presenilin-1, 2 and cleavage of presenilin-1 by m-, mu-calpain. Int. J. Mol. Med. 2000;5:269–273. doi: 10.3892/ijmm.5.3.269. [DOI] [PubMed] [Google Scholar]
- 147.Matsumura Y, Saeki E, Otsu K, Morita T, Takeda H, Kuzuya T, Hori M, Kusuoka H. Intracellular calcium level required for calpain activation in a single myocardial cell. J. Mol. Cell. Cardiol. 2001;33:1133–1142. doi: 10.1006/jmcc.2001.1373. [DOI] [PubMed] [Google Scholar]
- 148.Matsuzaki H, Tamatani M, Mitsuda N, Namikawa K, Kiyama H, Miyake S, Tohyama M. Activation of Akt kinase inhibits apoptosis and changes in Bcl-2 and Bax expression induced by nitric oxide in primary hippocampal neurons. J. Neurochem. 1999;73:2037–2046. [PubMed] [Google Scholar]
- 149.McCormick WC, Hardy J, Kukull WA, Bowen JD, Teri L, Zitzer S, Larson EB. Healthcare utilization and costs in managed care patients with Alzheimer's disease during the last few years of life. J. Am. Geriatr. Soc. 2001;49:1156–1160. doi: 10.1046/j.1532-5415.2001.49231.x. [DOI] [PubMed] [Google Scholar]
- 150.McPhie DL, Coopersmith R, Hines-Peralta A, Chen Y, Ivins KJ, Manly SP, Kozlowski MR, Neve KA, Neve RL. DNA synthesis and neuronal apoptosis caused by familial Alzheimer disease mutants of the amyloid precursor protein are mediated by the p21 activated kinase PAK3. J. Neurosci. 2003;23:6914–6927. doi: 10.1523/JNEUROSCI.23-17-06914.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.McShea A, Harris PL, Webster KR, Wahl AF, Smith MA. Abnormal expression of the cell cycle regulators P16 and CDK4 in Alzheimer's disease. Am. J. Pathol. 1997;150:1933–1939. [PMC free article] [PubMed] [Google Scholar]
- 152.Mehlhorn G, Hollborn M, Schliebs R. Induction of cytokines in glial cells surrounding cortical beta-amyloid plaques in transgenic Tg2576 mice with Alzheimer pathology. Int. J. Dev. Neurosci. 2000;18:423–431. doi: 10.1016/s0736-5748(00)00012-5. [DOI] [PubMed] [Google Scholar]
- 153.Mendiondo MS, Kryscio RJ, Schmitt FA. Models of progression in AD: predicting disability and costs. Neurology. 2001;57:943–944. doi: 10.1212/wnl.57.6.943. [DOI] [PubMed] [Google Scholar]
- 154.Michalski B, Fahnestock M. Pro-brain-derived neurotrophic factor is decreased in parietal cortex in Alzheimer's disease. Brain Res. Mol. Brain Res. 2003;111:148–154. doi: 10.1016/s0169-328x(03)00003-2. [DOI] [PubMed] [Google Scholar]
- 155.Miyaoka T, Seno H, Ishino H. Increased expression of Wnt-1 in schizophrenic brains. Schizophr. Res. 1999;38:1–6. doi: 10.1016/s0920-9964(98)00179-0. [DOI] [PubMed] [Google Scholar]
- 156.Moldoveanu T, Hosfield CM, Lim D, Elce JS, Jia Z, Davies PL. A Ca(2+) switch aligns the active site of calpain. Cell. 2002;108:649–660. doi: 10.1016/s0092-8674(02)00659-1. [DOI] [PubMed] [Google Scholar]
- 157.Monji A, Utsumi H, Ueda T, Imoto T, Yoshida I, Hashioka S, Tashiro K, Tashiro N. The relationship between the aggregational state of the amyloid-beta peptides and free radical generation by the peptides. J. Neurochem. 2001;77:1425–1432. doi: 10.1046/j.1471-4159.2001.00392.x. [DOI] [PubMed] [Google Scholar]
- 158.Montague JW, Hughes F, Jr., Cidlowski JA. Native recombinant cyclophilins A, B, and C degrade DNA independently of peptidylprolyl cis–trans-isomerase activity. Potential roles of cyclophilins in apoptosis. J. Biol. Chem. 1997;272:6677–6684. doi: 10.1074/jbc.272.10.6677. [DOI] [PubMed] [Google Scholar]
- 159.Mudher A, Chapman S, Richardson J, Asuni A, Gibb G, Pollard C, Killick R, Iqbal T, Raymond L, Varndell I, Sheppard P, Makoff A, Gower E, Soden PE, Lewis P, Murphy M, Golde TE, Rupniak HT, Anderton BH, Lovestone S. Dishevelled regulates the metabolism of amyloid precursor protein via protein kinase C/mitogen-activated protein kinase and c-Jun terminal kinase. J. Neurosci. 2001;21:4987–4995. doi: 10.1523/JNEUROSCI.21-14-04987.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Mukherjee PK, Marcheselli VL, Serhan CN, Bazan NG. Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc. Natl. Acad. Sci. U. S. A. 2004;101:8491–8496. doi: 10.1073/pnas.0402531101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Munoz-Montano JR, Moreno FJ, Avila J, Diaz-Nido J. Lithium inhibits Alzheimer's disease-like tau protein phosphorylation in neurons. FEBS Lett. 1997;411:183–188. doi: 10.1016/s0014-5793(97)00688-1. [DOI] [PubMed] [Google Scholar]
- 162.Murashov AK, Ul Haq I, Hill C, Park E, Smith M, Wang X, Goldberg DJ, Wolgemuth DJ. Crosstalk between p38, Hsp25 and Akt in spinal motor neurons after sciatic nerve injury. Brain Res. Mol. Brain Res. 2001;93:199–208. doi: 10.1016/s0169-328x(01)00212-1. [DOI] [PubMed] [Google Scholar]
- 163.Nakagawa T, Zhu H, Morishima N, Li E, Xu J, Yankner BA, Yuan J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- 164.Nakano T, Ishimoto Y, Kishino J, Umeda M, Inoue K, Nagata K, Ohashi K, Mizuno K, Arita H. Cell adhesion to phosphatidylserine mediated by a product of growth arrest-specific gene 6. J. Biol. Chem. 1997;272:29411–29414. doi: 10.1074/jbc.272.47.29411. [DOI] [PubMed] [Google Scholar]
- 165.Nishimura I, Uetsuki T, Kuwako K, Hara T, Kawakami T, Aimoto S, Yoshikawa K. Cell death induced by a caspase-cleaved transmembrane fragment of the Alzheimer amyloid precursor protein. Cell Death Differ. 2002;9:199–208. doi: 10.1038/sj.cdd.4400931. [DOI] [PubMed] [Google Scholar]
- 166.Onizuka K, Kunimatsu M, Ozaki Y, Muramatsu K, Sasaki M, Nishino H. Distribution of mu-calpain proenzyme in the brain and other neural tissues in the rat. Brain Res. 1995;697:179–186. doi: 10.1016/0006-8993(95)00838-h. [DOI] [PubMed] [Google Scholar]
- 167.Owada Y, Utsunomiya A, Yoshimoto T, Kondo H. Expression of mRNA for Akt, serine-threonine protein kinase, in the brain during development and its transient enhancement following axotomy of hypoglossal nerve. J. Mol. Neurosci. 1997;9:27–33. doi: 10.1007/BF02789392. [DOI] [PubMed] [Google Scholar]
- 168.Panchal M, Rholam M, Brakch N. Abnormalities of peptide metabolism in Alzheimer disease. Curr. Neurovasc. Res. 2004;1:317–323. doi: 10.2174/1567202043362117. [DOI] [PubMed] [Google Scholar]
- 169.Pandey S, Walker PR, Sikorska M. Identification of a novel 97 kDa endonuclease capable of internucleosomal DNA cleavage. Biochemistry. 1997;36:711–720. doi: 10.1021/bi962387h. [DOI] [PubMed] [Google Scholar]
- 170.Papkoff J, Aikawa M. WNT-1 and HGF regulate GSK3 beta activity and beta-catenin signaling in mammary epithelial cells. Biochem. Biophys. Res. Commun. 1998;247:851–858. doi: 10.1006/bbrc.1998.8888. [DOI] [PubMed] [Google Scholar]
- 171.Paradis E, Douillard H, Koutroumanis M, Goodyer C, LeBlanc A. Amyloid beta peptide of Alzheimer's disease downregulates Bcl-2 and upregulates bax expression in human neurons. J. Neurosci. 1996;16:7533–7539. doi: 10.1523/JNEUROSCI.16-23-07533.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Pasquet JM, Dachary-Prigent J, Nurden AT. Calcium influx is a determining factor of calpain activation and microparticle formation in platelets. Eur. J. Biochem. 1996;239:647–654. doi: 10.1111/j.1432-1033.1996.0647u.x. [DOI] [PubMed] [Google Scholar]
- 173.Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 174.Pei JJ, Grundke-Iqbal I, Iqbal K, Bogdanovic N, Winblad B, Cowburn RF. Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer's disease neurofibrillary degeneration. Brain Res. 1998;797:267–277. doi: 10.1016/s0006-8993(98)00296-0. [DOI] [PubMed] [Google Scholar]
- 175.Pei JJ, Braak E, Braak H, Grundke-Iqbal I, Iqbal K, Winblad B, Cowburn RF. Distribution of active glycogen synthase kinase 3beta (GSK-3beta) in brains staged for Alzheimer disease neurofibrillary changes. J. Neuropathol. Exp. Neurol. 1999;58:1010–1019. doi: 10.1097/00005072-199909000-00011. [DOI] [PubMed] [Google Scholar]
- 176.Pompl PN, Yemul S, Xiang Z, Ho L, Haroutunian V, Purohit D, Mohs R, Pasinetti GM. Caspase gene expression in the brain as a function of the clinical progression of Alzheimer disease. Arch. Neurol. 2003;60:369–376. doi: 10.1001/archneur.60.3.369. [DOI] [PubMed] [Google Scholar]
- 177.Potter DA, Tirnauer JS, Janssen R, Croall DE, Hughes CN, Fiacco KA, Mier JW, Maki M, Herman IM. Calpain regulates actin remodeling during cell spreading. J. Cell Biol. 1998;141:647–662. doi: 10.1083/jcb.141.3.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Pugazhenthi S, Nesterova A, Jambal P, Audesirk G, Kern M, Cabell L, Eves E, Rosner MR, Boxer LM, Reusch JE. Oxidative stress-mediated down-regulation of bcl-2 promoter in hippocampal neurons. J. Neurochem. 2003;84:982–996. doi: 10.1046/j.1471-4159.2003.01606.x. [DOI] [PubMed] [Google Scholar]
- 179.Putcha GV, Deshmukh M, Johnson EM., Jr. BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J. Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Raina AK, Zhu X, Rottkamp CA, Monteiro M, Takeda A, Smith MA. Cyclin' toward dementia: cell cycle abnormalities and abortive oncogenesis in Alzheimer disease. J. Neurosci. Res. 2000;61:128–133. doi: 10.1002/1097-4547(20000715)61:2<128::AID-JNR2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 181.Ray SK, Matzelle DD, Sribnick EA, Guyton MK, Wingrave JM, Banik NL. Calpain inhibitor prevented apoptosis and maintained transcription of proteolipid protein and myelin basic protein genes in rat spinal cord injury. J. Chem. Neuroanat. 2003;26:119–124. doi: 10.1016/s0891-0618(03)00044-9. [DOI] [PubMed] [Google Scholar]
- 182.Reilly JO, Karavanova ID, Williams KP, Mahanthappa NK, Allendoerfer KL. Cooperative effects of Sonic Hedgehog and NGF on basal forebrain cholinergic neurons. Mol. Cell Neurosci. 2002;19:88–96. doi: 10.1006/mcne.2001.1063. [DOI] [PubMed] [Google Scholar]
- 183.Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ. Memantine in moderate-to-severe Alzheimer's disease. N. Engl. J. Med. 2003;348:1333–1341. doi: 10.1056/NEJMoa013128. [DOI] [PubMed] [Google Scholar]
- 184.Religa D, Winblad B. Therapeutic strategies for Alzheimer's disease based on new molecular mechanisms. Acta Neurobiol. Exp. (Wars) 2003;63:393–396. doi: 10.55782/ane-2003-1480. [DOI] [PubMed] [Google Scholar]
- 185.Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J. Biol. Chem. 1999;274:17179–17183. doi: 10.1074/jbc.274.24.17179. [DOI] [PubMed] [Google Scholar]
- 186.Rhee CS, Sen M, Lu D, Wu C, Leoni L, Rubin J, Corr M, Carson DA. Wnt and frizzled receptors as potential targets for immunotherapy in head and neck squamous cell carcinomas. Oncogene. 2002;21:6598–6605. doi: 10.1038/sj.onc.1205920. [DOI] [PubMed] [Google Scholar]
- 187.Rideout HJ, Wang Q, Park DS, Stefanis L. Cyclin-dependent kinase activity is required for apoptotic death but not inclusion formation in cortical neurons after proteasomal inhibition. J. Neurosci. 2003;23:1237–1245. doi: 10.1523/JNEUROSCI.23-04-01237.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Riedel G, Opitz T, Reymann KG. Blockade of metabotropic glutamate receptors protects hippocampal neurons from hypoxia-induced cell death in rat in vivo. Prog. Neuro-psychopharmacol. Biol. Psychiatry. 1996;20:1253–1263. doi: 10.1016/s0278-5846(96)00110-8. [DOI] [PubMed] [Google Scholar]
- 189.Rogers J, Lue LF. Microglial chemotaxis, activation, and phagocytosis of amyloid beta-peptide as linked phenomena in Alzheimer's disease. Neurochem. Int. 2001;39:333–340. doi: 10.1016/s0197-0186(01)00040-7. [DOI] [PubMed] [Google Scholar]
- 190.Rohn TT, Head E, Su JH, Anderson AJ, Bahr BA, Cotman CW, Cribbs DH. Correlation between caspase activation and neurofibrillary tangle formation in Alzheimer's disease. Am. J. Pathol. 2001;158:189–198. doi: 10.1016/S0002-9440(10)63957-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ruiz-Vela A, Gonzalez de Buitrago G, Martinez AC. Implication of calpain in caspase activation during B cell clonal deletion. EMBO J. 1999;18:4988–4998. doi: 10.1093/emboj/18.18.4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sagara Y, Schubert D. The activation of metabotropic glutamate receptors protects nerve cells from oxidative stress. J. Neurosci. 1998;18:6662–6671. doi: 10.1523/JNEUROSCI.18-17-06662.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Sakamaki K. Regulation of endothelial cell death and its role in angiogenesis and vascular regression. Curr. Neurovasc. Res. 2004;1:305–315. doi: 10.2174/1567202043362072. [DOI] [PubMed] [Google Scholar]
- 194.Salinas M, Diaz R, Abraham NG, Ruiz de Galarreta CM, Cuadrado A. Nerve growth factor protects against 6-hydroxydopamine-induced oxidative stress by increasing expression of heme oxygenase-1 in a phosphatidylinositol 3-kinase-dependent manner. J. Biol. Chem. 2003;278:13898–13904. doi: 10.1074/jbc.M209164200. [DOI] [PubMed] [Google Scholar]
- 195.Sankarapandi S, Zweier JL, Mukherjee G, Quinn MT, Huso DL. Measurement and characterization of superoxide generation in microglial cells: evidence for an NADPH oxidase-dependent pathway. Arch. Biochem. Biophys. 1998;353:312–321. doi: 10.1006/abbi.1998.0658. [DOI] [PubMed] [Google Scholar]
- 196.Satoh MS, Lindahl T. Role of poly(ADP-ribose) formation in DNA repair. Nature. 1992;356:356–358. doi: 10.1038/356356a0. [DOI] [PubMed] [Google Scholar]
- 197.Savage MJ, Trusko SP, Howland DS, Pinsker LR, Mistretta S, Reaume AG, Greenberg BD, Siman R, Scott RW. Turnover of amyloid beta-protein in mouse brain and acute reduction of its level by phorbol ester. J. Neurosci. 1998;18:1743–1752. doi: 10.1523/JNEUROSCI.18-05-01743.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Seubert P, Oltersdorf T, Lee MG, Barbour R, Blomquist C, Davis DL, Bryant K, Fritz LC, Galasko D, Thal LJ, et al. Secretion of beta-amyloid precursor protein cleaved at the amino terminus of the beta-amyloid peptide. Nature. 1993;361:260–263. doi: 10.1038/361260a0. [DOI] [PubMed] [Google Scholar]
- 199.Sharma SK, Ebadi M. Metallothionein attenuates 3-morpholinosydnonimine (SIN-1)-induced oxidative stress in dopaminergic neurons. Antioxid. Redox Signal. 2003;5:251–264. doi: 10.1089/152308603322110832. [DOI] [PubMed] [Google Scholar]
- 200.Shea TB. Restriction of microM-calcium-requiring calpain activation to the plasma membrane in human neuroblastoma cells: evidence for regionalized influence of a calpain activator protein. J. Neurosci. Res. 1997;48:543–550. [PubMed] [Google Scholar]
- 201.Sheng JG, Mrak RE, Griffin WS. Neuritic plaque evolution in Alzheimer's disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathol. (Berl) 1997;94:1–5. doi: 10.1007/s004010050664. [DOI] [PubMed] [Google Scholar]
- 202.Shiraha H, Glading A, Gupta K, Wells A. IP-10 inhibits epidermal growth factor-induced motility by decreasing epidermal growth factor receptor-mediated calpain activity. J. Cell Biol. 1999;146:243–254. doi: 10.1083/jcb.146.1.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Silva M, Grillot D, Benito A, Richard C, Nunez G, Fernandez-Luna JL. Erythropoietin can promote erythroid progenitor survival by repressing apoptosis through Bcl-XL and Bcl-2. Blood. 1996;88:1576–1582. [PubMed] [Google Scholar]
- 204.Siu AW, To CH. Nitric oxide and hydroxyl radical-induced retinal lipid peroxidation in vitro. Clin. Exp. Optom. 2002;85:378–382. [PubMed] [Google Scholar]
- 205.Slusarski DC, Corces VG, Moon RT. Interaction of Wnt and a Frizzled homologue triggers G-protein-linked phosphatidylinositol signalling. Nature. 1997;390:410–413. doi: 10.1038/37138. [DOI] [PubMed] [Google Scholar]
- 206.Smith MA, Hirai K, Hsiao K, Pappolla MA, Harris PL, Siedlak SL, Tabaton M, Perry G. Amyloid-beta deposition in Alzheimer transgenic mice is associated with oxidative stress. J. Neurochem. 1998;70:2212–2215. doi: 10.1046/j.1471-4159.1998.70052212.x. [DOI] [PubMed] [Google Scholar]
- 207.Soriano S, Kang DE, Fu M, Pestell R, Chevallier N, Zheng H, Koo EH. Presenilin 1 negatively regulates beta-catenin/T cell factor/lymphoid enhancer factor-1 signaling independently of beta-amyloid precursor protein and notch processing. J. Cell Biol. 2001;152:785–794. doi: 10.1083/jcb.152.4.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Stanford PM, Shepherd CE, Halliday GM, Brooks WS, Schofield PW, Brodaty H, Martins RN, Kwok JB, Schofield PR. Mutations in the tau gene that cause an increase in three repeat tau and frontotemporal dementia. Brain. 2003;126:814–826. doi: 10.1093/brain/awg090. [DOI] [PubMed] [Google Scholar]
- 209.Subramaniam R, Koppal T, Green M, Yatin S, Jordan B, Drake J, Butterfield DA. The free radical antioxidant vitamin E protects cortical synaptosomal membranes from amyloid beta-peptide(25−35) toxicity but not from hydroxynonenal toxicity: relevance to the free radical hypothesis of Alzheimer's disease. Neurochem. Res. 1998;23:1403–1410. doi: 10.1023/a:1020754807671. [DOI] [PubMed] [Google Scholar]
- 210.Sun XM, Cohen GM. Mg(2+)-dependent cleavage of DNA into kilobase pair fragments is responsible for the initial degradation of DNA in apoptosis. J. Biol. Chem. 1994;269:14857–14860. [PubMed] [Google Scholar]
- 211.Tada M, Smith JC. Xwnt11 is a target of Xenopus Brachyury: regulation of gastrulation movements via Dishevelled, but not through the canonical Wnt pathway. Development. 2000;127:2227–2238. doi: 10.1242/dev.127.10.2227. [DOI] [PubMed] [Google Scholar]
- 212.Takahashi H, Nakamura S, Asano K, Kinouchi M, Ishida-Yamamoto A, Iizuka H. Fas antigen modulates ultraviolet B-induced apoptosis of SVHK cells: sequential activation of caspases 8, 3, and 1 in the apoptotic process. Exp. Cell Res. 1999;249:291–298. doi: 10.1006/excr.1999.4476. [DOI] [PubMed] [Google Scholar]
- 213.Takashima A, Noguchi K, Sato K, Hoshino T, Imahori K. Tau protein kinase I is essential for amyloid beta-protein-induced neurotoxicity. Proc. Natl. Acad. Sci. U. S. A. 1993;90:7789–7793. doi: 10.1073/pnas.90.16.7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Takashima A, Honda T, Yasutake K, Michel G, Murayama O, Murayama M, Ishiguro K, Yamaguchi H. Activation of tau protein kinase I/glycogen synthase kinase-3beta by amyloid beta peptide (25−35) enhances phosphorylation of tau in hippocampal neurons. Neurosci. Res. 1998;31:317–323. doi: 10.1016/s0168-0102(98)00061-3. [DOI] [PubMed] [Google Scholar]
- 215.Tanaka M, Sotomatsu A, Yoshida T, Hirai S, Nishida A. Detection of superoxide production by activated microglia using a sensitive and specific chemiluminescence assay and microglia-mediated PC12h cell death. J. Neurochem. 1994;63:266–270. doi: 10.1046/j.1471-4159.1994.63010266.x. [DOI] [PubMed] [Google Scholar]
- 216.Tang K, Yang J, Gao X, Wang C, Liu L, Kitani H, Atsumi T, Jing N. Wnt-1 promotes neuronal differentiation and inhibits gliogenesis in P19 cells. Biochem. Biophys. Res. Commun. 2002;293:167–173. doi: 10.1016/S0006-291X(02)00215-2. [DOI] [PubMed] [Google Scholar]
- 217.Taniguchi S, Fujita Y, Hayashi S, Kakita A, Takahashi H, Murayama S, Saido TC, Hisanaga S, Iwatsubo T, Hasegawa M. Calpain-mediated degradation of p35 to p25 in postmortem human and rat brains. FEBS Lett. 2001;489:46–50. doi: 10.1016/s0014-5793(00)02431-5. [DOI] [PubMed] [Google Scholar]
- 218.Taylor DL, Diemel LT, Pocock JM. Activation of microglial group III metabotropic glutamate receptors protects neurons against microglial neurotoxicity. J. Neurosci. 2003;23:2150–2160. doi: 10.1523/JNEUROSCI.23-06-02150.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Tesco G, Koh YH, Tanzi RE. Caspase activation increases Abeta generation independently of caspase cleavage of APP. J. Biol. Chem. 2003;5:5. doi: 10.1074/jbc.M307809200. [DOI] [PubMed] [Google Scholar]
- 220.Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422–426. doi: 10.1038/18884. [DOI] [PubMed] [Google Scholar]
- 221.Timsit S, Rivera S, Ouaghi P, Guischard F, Tremblay E, Ben-Ari Y, Khrestchatisky M. Increased cyclin D1 in vulnerable neurons in the hippocampus after ischaemia and epilepsy: a modulator of in vivo programmed cell death? Eur. J. Neurosci. 1999;11:263–278. doi: 10.1046/j.1460-9568.1999.00434.x. [DOI] [PubMed] [Google Scholar]
- 222.Torriglia A, Chaudun E, Chany-Fournier F, Jeanny JC, Courtois Y, Counis MF. Involvement of DNase II in nuclear degeneration during lens cell differentiation. J. Biol. Chem. 1995;270:28579–28585. doi: 10.1074/jbc.270.48.28579. [DOI] [PubMed] [Google Scholar]
- 223.Torriglia A, Chaudun E, Courtois Y, Counis MF. On the use of Zn2+ to discriminate endonucleases activated during apoptosis. Biochimie. 1997;79:435–438. doi: 10.1016/s0300-9084(97)86153-6. [DOI] [PubMed] [Google Scholar]
- 224.Touyarot K, Poussard S, Verret C, Aragon B, Cottin P, Nogues X, Micheau J. Calpain-PKC inter-relations in mouse hippocampus: a biochemical approach. Neurochem. Res. 2000;25:781–790. doi: 10.1023/a:1007509322362. [DOI] [PubMed] [Google Scholar]
- 225.Town T, Zolton J, Shaffner R, Schnell B, Crescentini R, Wu Y, Zeng J, DelleDonne A, Obregon D, Tan J, Mullan M. p35/Cdk5 pathway mediates soluble amyloid-beta peptide-induced tau phosphorylation in vitro. J. Neurosci. Res. 2002;69:362–372. doi: 10.1002/jnr.10299. [DOI] [PubMed] [Google Scholar]
- 226.Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML. Caspase-2 mediates neuronal cell death induced by beta-amyloid. J. Neurosci. 2000;20:1386–1392. doi: 10.1523/JNEUROSCI.20-04-01386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Troy CM, Rabacchi SA, Xu Z, Maroney AC, Connors TJ, Shelanski ML, Greene LA. beta-Amyloid-induced neuronal apoptosis requires c-Jun N-terminal kinase activation. J. Neurochem. 2001;77:157–164. doi: 10.1046/j.1471-4159.2001.t01-1-00218.x. [DOI] [PubMed] [Google Scholar]
- 228.Ulus IH, Wurtman RJ. Metabotropic glutamate receptor agonists increase release of soluble amyloid precursor protein derivatives from rat brain cortical and hippocampal slices. J. Pharmacol. Exp. Ther. 1997;281:149–154. [PubMed] [Google Scholar]
- 229.van de Craen M, de Jonghe C, van den Brande I, Declercq W, van Gassen G, van Criekinge W, Vanderhoeven I, Fiers W, van Broeckhoven C, Hendriks L, Vandenabeele P. Identification of caspases that cleave presenilin-1 and presenilin-2. Five presenilin-1 (PS1) mutations do not alter the sensitivity of PS1 to caspases. FEBS Lett. 1999;445:149–154. doi: 10.1016/s0014-5793(99)00108-8. [DOI] [PubMed] [Google Scholar]
- 230.Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- 231.Vaudry D, Pamantung TF, Basille M, Rousselle C, Fournier A, Vaudry H, Beauvillain JC, Gonzalez BJ. PACAP protects cerebellar granule neurons against oxidative stress-induced apoptosis. Eur. J. Neurosci. 2002;15:1451–1460. doi: 10.1046/j.1460-9568.2002.01981.x. [DOI] [PubMed] [Google Scholar]
- 232.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 233.Vincent AM, Maiese K. Direct temporal analysis of apoptosis induction in living adherent neurons. J. Histochem. Cytochem. 1999;47:661–672. doi: 10.1177/002215549904700508. [DOI] [PubMed] [Google Scholar]
- 234.Vincent AM, Maiese K. Nitric oxide induction of neuronal endonuclease activity in programmed cell death. Exp. Cell Res. 1999;246:290–300. doi: 10.1006/excr.1998.4282. [DOI] [PubMed] [Google Scholar]
- 235.Vincent AM, Maiese K. The metabotropic glutamate system promotes neuronal survival through distinct pathways of programmed cell death. Exp. Neurol. 2000;166:65–82. doi: 10.1006/exnr.2000.7487. [DOI] [PubMed] [Google Scholar]
- 236.Vincent AM, Mohammad Y, Ahmad I, Greenberg R, Maiese K. Metabotropic glutamate receptors prevent nitric oxide induced programmed cell death. J. Neurosci. Res. 1997;50:549–564. doi: 10.1002/(SICI)1097-4547(19971115)50:4<549::AID-JNR6>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 237.Vincent AM, TenBroeke M, Maiese K. Metabotropic glutamate receptors prevent programmed cell death through the modulation of neuronal endonuclease activity and intracellular pH. Exp. Neurol. 1999;155:79–94. doi: 10.1006/exnr.1998.6966. [DOI] [PubMed] [Google Scholar]
- 238.Volbracht C, Fava E, Leist M, Nicotera P. Calpain inhibitors prevent nitric oxide-triggered excitotoxic apoptosis. NeuroReport. 2001;12:3645–3648. doi: 10.1097/00001756-200112040-00008. [DOI] [PubMed] [Google Scholar]
- 239.Wagner U, Brownlees J, Irving NG, Lucas FR, Salinas PC, Miller CC. Overexpression of the mouse dishevelled-1 protein inhibits GSK-3beta-mediated phosphorylation of tau in transfected mammalian cells. FEBS Lett. 1997;411:369–372. doi: 10.1016/s0014-5793(97)00733-3. [DOI] [PubMed] [Google Scholar]
- 240.Wang R, Zhang HY, Tang XC. Huperzine A attenuates cognitive dysfunction and neuronal degeneration caused by beta-amyloid protein-(1−40) in rat. Eur. J. Pharmacol. 2001;421:149–156. doi: 10.1016/s0014-2999(01)01030-5. [DOI] [PubMed] [Google Scholar]
- 241.Wang JY, Shum AY, Ho YJ. Oxidative neurotoxicity in rat cerebral cortex neurons: synergistic effects of H2O2 and NO on apoptosis involving activation of p38 mitogen-activated protein kinase and caspase-3. J. Neurosci. Res. 2003;72:508–519. doi: 10.1002/jnr.10597. [DOI] [PubMed] [Google Scholar]
- 242.Weber J. Calcium homeostasis following traumatic neuronal injury. Curr. Neurovasc. Res. 2004;1:151–171. doi: 10.2174/1567202043480134. [DOI] [PubMed] [Google Scholar]
- 243.Wegiel J, Wang KC, Tarnawski M, Lach B. Microglia cells are the driving force in fibrillar plaque formation, whereas astrocytes are a leading factor in plague degradation. Acta Neuropathol. (Berl) 2000;100:356–364. doi: 10.1007/s004010000199. [DOI] [PubMed] [Google Scholar]
- 244.Wehrli M, Dougan ST, Caldwell K, O'Keefe L, Schwartz S, Vaizel-Ohayon D, Schejter E, Tomlinson A, DiNardo S. arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature. 2000;407:527–530. doi: 10.1038/35035110. [DOI] [PubMed] [Google Scholar]
- 245.Wick MJ, Dong LQ, Riojas RA, Ramos FJ, Liu F. Mechanism of phosphorylation of protein kinase B/Akt by a constitutively active 3-phosphoinositide-dependent protein kinase-1. J. Biol. Chem. 2000;275:40400–40406. doi: 10.1074/jbc.M003937200. [DOI] [PubMed] [Google Scholar]
- 246.Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J. Neurosci. 2002;22:6401–6407. doi: 10.1523/JNEUROSCI.22-15-06401.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Witting A, Muller P, Herrmann A, Kettenmann H, Nolte C. Phagocytic clearance of apoptotic neurons by Microglia/Brain macrophages in vitro: involvement of lectin-, integrin-, and phosphatidylserine-mediated recognition. J. Neurochem. 2000;75:1060–1070. doi: 10.1046/j.1471-4159.2000.0751060.x. [DOI] [PubMed] [Google Scholar]
- 248.Wood DE, Thomas A, Devi LA, Berman Y, Beavis RC, Reed JC, Newcomb EW. Bax cleavage is mediated by calpain during drug-induced apoptosis. Oncogene. 1998;17:1069–1078. doi: 10.1038/sj.onc.1202034. [DOI] [PubMed] [Google Scholar]
- 249.Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J. Biol. Chem. 2001;276:5256–5264. doi: 10.1074/jbc.M008552200. [DOI] [PubMed] [Google Scholar]
- 250.Yamamoto T, Maruyama W, Kato Y, Yi H, Shamoto-Nagai M, Tanaka M, Sato Y, Naoi M. Selective nitration of mitochondrial complex I by peroxynitrite: involvement in mitochondria dysfunction and cell death of dopaminergic SH-SY5Y cells. J. Neural Transm. 2002;109:1–13. doi: 10.1007/s702-002-8232-1. [DOI] [PubMed] [Google Scholar]
- 251.Yang Y, Mufson EJ, Herrup K. Neuronal cell death is preceded by cell cycle events at all stages of Alzheimer's disease. J. Neurosci. 2003;23:2557–2563. doi: 10.1523/JNEUROSCI.23-07-02557.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Yin XM, Luo Y, Cao G, Bai L, Pei W, Kuharsky DK, Chen J. Bid-mediated mitochondrial pathway is critical to ischemic neuronal apoptosis and focal cerebral ischemia. J. Biol. Chem. 2002;277:42074–42081. doi: 10.1074/jbc.M204991200. [DOI] [PubMed] [Google Scholar]
- 253.You Z, Saims D, Chen S, Zhang Z, Guttridge DC, Guan KL, MacDougald OA, Brown AM, Evan G, Kitajewski J, Wang CY. Wnt signaling promotes oncogenic transformation by inhibiting c-Myc-induced apoptosis. J. Cell Biol. 2002;157:429–440. doi: 10.1083/jcb.200201110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Yu C, Rahmani M, Dai Y, Conrad D, Krystal G, Dent P, Grant S. The lethal effects of pharmacological cyclin-dependent kinase inhibitors in human leukemia cells proceed through a phosphatidylinositol 3-kinase/Akt-dependent process. Cancer Res. 2003;63:1822–1833. [PubMed] [Google Scholar]
- 255.Zhang Y, McLaughlin R, Goodyer C, LeBlanc A. Selective cytotoxicity of intracellular amyloid beta peptide1−42 through p53 and Bax in cultured primary human neurons. J. Cell Biol. 2002;156:519–529. doi: 10.1083/jcb.200110119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Zheng-Fischhofer Q, Biernat J, Mandelkow EM, Illenberger S, Godemann R, Mandelkow E. Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur. J. Biochem. 1998;252:542–552. doi: 10.1046/j.1432-1327.1998.2520542.x. [DOI] [PubMed] [Google Scholar]