

Abstract
17β-Estradiol (E2), acting via estrogen receptor (ER)-α, inhibits feeding in animals. One mechanism apparently involves an increase in the satiating potency of cholecystokinin (CCK) released from the small intestine by ingested food. For example, the satiating potency of intraduodenal lipid infusions is increased by E2 in ovariectomized rats; this increased satiation is dependent on CCK, and it is accompanied by increases in the numbers of ERα-positive cells that express c-Fos in a subregion of the caudal nucleus tractus solitarius (cNTS) that receives abdominal vagal afferent projections. To test whether direct administration of E2 to this area of the hindbrain is sufficient to inhibit food intake, we first implanted 0.2 μg estradiol benzoate (EB) in cholesterol or cholesterol alone either sc or onto the surface of the hindbrain over the cNTS. Food intake was significantly reduced after hindbrain EB implants but not after sc EB implants. Next we verified that equimolar hindbrain implants of E2 and EB had similar feeding-inhibitory effects and determined that only small amounts of E2 reached brain areas outside the dorsal caudal hindbrain after hindbrain implants of 3H-labeled E2. Neither plasma estradiol concentration nor plasma inflammatory cytokine concentration was increased by either hindbrain or sc EB implants. Finally, hindbrain EB implants, but not sc implants, increased c-Fos in ERα-positive cells in the cNTS after ip injection of 4 μg/kg CCK-8. We conclude that E2, acting via ERα in cNTS neurons, including neurons stimulated by ip CCK, is sufficient to inhibit feeding.
ONE OF THE many biological actions of 17β-estradiol (E2) is its modulatory effect on eating. In both rats and women, daily food intake decreases during the periovulatory phase of the ovarian cycle (i.e. estrus in rats) (reviewed in Refs. 1,2,3). In addition, in rats, disruption of ovarian cycling by ovariectomy (OVX) chronically increases meal size and food intake, leading to increased adiposity (1,2,3). E2 is sufficient to account for these effects in rats because a near-physiological, cyclic E2 treatment regimen maintained normal patterns of food intake and body weight after OVX (4,5). E2 appears to inhibit feeding via estrogen receptor (ER)-α because OVX ERα-knockout mice did not eat less after E2 administration (6).
E2 inhibits feeding, at least in part, by increasing the potency of negative-feedback signals that control meal size, or satiation signals (1,2,3). One such satiation signal is cholecystokinin (CCK), which is released from the proximal small intestine during meals and produces a vagal satiation signal that is initially processed in the nucleus tractus solitarius (NTS) in the dorsal hindbrain (1,2,7,8,9,10). Evidence that E2 increases CCK satiation comes from demonstrations that, in OVX rats, E2 increases the satiating effect of exogenous CCK, the feeding-stimulatory effect of CCK-1 receptor antagonism, and the satiating potency of intraduodenal lipid infusions (1,2,5,11).
E2 appears to act in the brain to control feeding (1,2,3). The specific brain site(s) in which E2 acts to inhibit eating, however, remains unclear. Direct administration of E2 into several hypothalamic areas, including the ventromedial nucleus (VMN) (12), the paraventricular nuclei (PVN) (13,14), and the medial preoptic area (MPA) (15), has been reported to reduce food intake, and administration of E2 into the arcuate nucleus (Arc) has been reported to increase the feeding-inhibitory effect of exogenous leptin (16). Whether any of these sites is involved in the physiological action of E2 on feeding remains controversial (1,2,3,16,17).
E2-mediated changes in feeding-related neuronal activation, measured by c-Fos immunocytochemistry, have been used to identify potential sites of E2’s action on feeding. In OVX rats, E2 treatment increases both feeding- and CCK-induced c-Fos expression in the NTS, PVN, and central nucleus of the amygdala (CeA) (18,19). E2 treatment also increased the satiating potency of intraduodenal infusions of lipid in OVX rats, an effect mediated by endogenous CCK, and this increased satiation was associated with increased c-Fos expression in a circumscribed population of neurons in the caudal NTS (cNTS) that express ERα (11). Therefore, here we sought to determine whether ERα-positive cNTS neurons are sufficient to mediate the estrogenic inhibition of feeding. We report data supporting this hypothesis. Open surgical administration of 0.2 μg β-estradiol-3-benzoate (EB) onto the surface of the hindbrain over the cNTS, but not sc administration of the same EB dose, decreased food intake. Furthermore, the same EB treatment increased CCK-induced c-Fos expression in cNTS ERα cells, implicating CCK in the mechanism.
Materials and Methods
Subjects
Female Long-Evans rats (Centre d’Elevage R. Janvier; Le Genest-Saint-Isle, France) weighing 229 ± 2 g (mean ± sem) were housed individually in hanging cages with stainless steel wire-mesh floors (33 × 18 × 20 cm) in a room maintained at 22 ± 2 C with a 12-h light, 12-h dark cycle (lights on 0400 h). All rats had ad libitum access to pelleted standard laboratory chow (Provimi Kliba; Gossau, Switzerland) and tap water. Before OVX, rats were adapted to the housing conditions for 3 wk and handled 3 d/wk before surgery. All procedures were approved by the Canton of Zürich Veterinary Office.
OVX and E2 treatment
Rats were anesthetized with isoflurane (2.5–3% Attane; Minrad, Buffalo, NY) and bilaterally ovariectomized using an intraabdominal approach. Immediately after surgery, rats were sc injected with chloramphenicol (50 mg/kg, Septicol; Vetoquinol, Bern, Switzerland) for antibiotic prophylaxis and buprenorphine (1 mg/kg, Temgesic; Essex Chemie, Luzern, Switzerland) for analgesia.
E2 implants were prepared by pipetting 0.5 μl absolute ethanol (Fluka, Buchs, Switzerland) containing 9 μg/μl cholesterol (Sigma, St. Louis, MO) and 0, 0.4, or 4 μg/μl EB (Sigma) onto approximately 1-mm2 pieces of absorbable hemostat fabric (Collastas Aristavet, Ravensburg, Federal Republic of Germany), which were then air dried to yield implants containing 0, 0.2, or 2 μg EB, respectively.
Two weeks after OVX, rats were anesthetized with ip injections (1 ml/kg) of a mixture of 6 mg/kg xylazine (Rompun; Provet, Lyssach, Switzerland) and 60 mg/kg ketamine (Narketan, Vetoquinol) and placed in a stereotaxic instrument with the neck ventroflexed. A 15-mm midline skin incision was made above the occipitoatlantal joint, the neck muscles were retracted by blunt dissection, and the joint capsule was opened. A 2-mm incision was made in the dura, taking care not to damage the dorsal surface of the hindbrain, and an implant containing 0 or 0.2 μg EB was placed on the hindbrain, centered just caudal to the area postrema [AP; i.e. about 14.1 mm caudal to bregma (20)]. A photograph of the surgical site is shown in Fig. 1A. The neck muscles were repositioned, and a second implant containing 0, 0.2, or 2 μg EB was placed sc, just lateral to the skin incision, which was then sutured (the particular combinations used are described below). Brains from rats killed 1–7 d after implantation showed no evidence of gross pathology or alterations in cellular architecture as gauged by Nissl staining (Fig. 1, B and C). The dose of 0.2 μg EB was chosen based on pilot studies indicating that central implants of this dose did not produce cornification of vaginal mucosal cells or increased uterus weight, two peripheral estrogenic effects. The dose of 2 μg EB sc was chosen because this dose injected once every 4 d produces near-physiological cyclic patterns of plasma E2 concentration and normalizes food intake and body weight in OVX rats (5).
Figure 1.
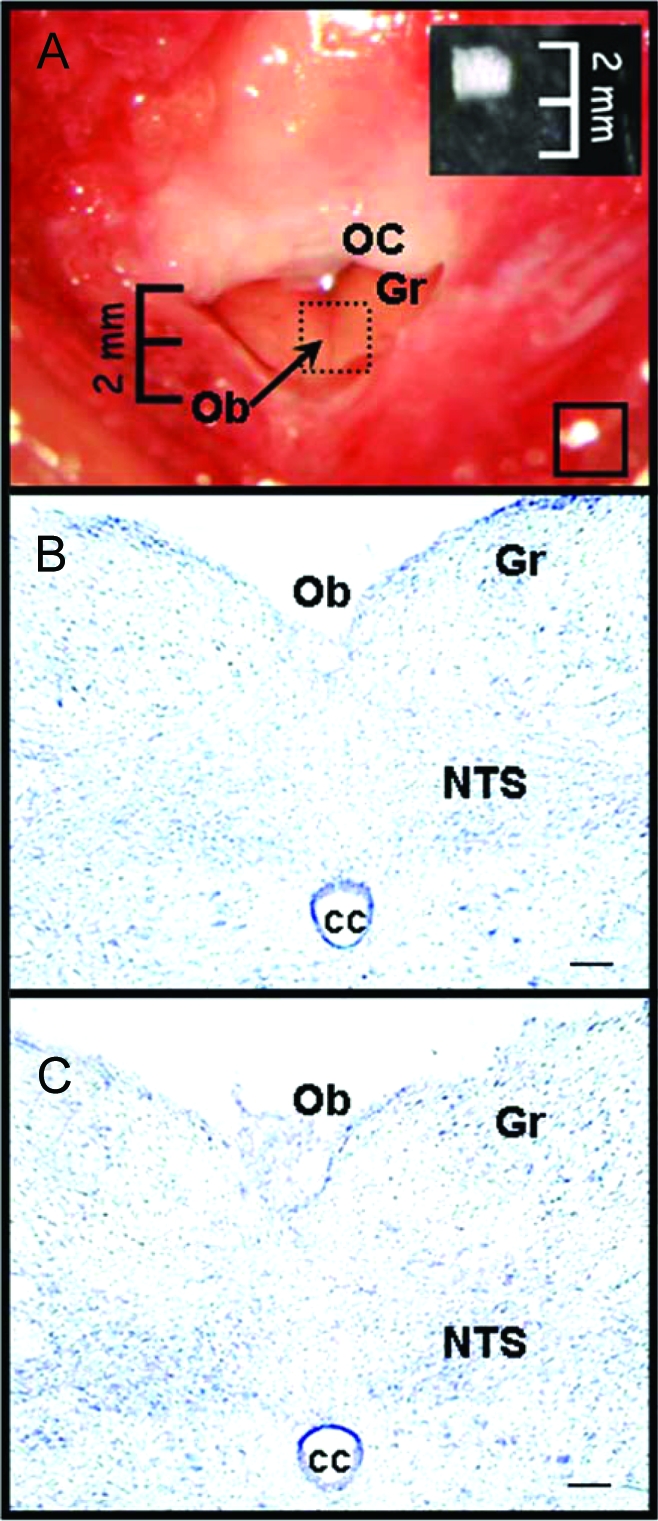
A, Photograph of the surgical window for implant placement. Note that the AP lies just rostral to the obex (Ob) and is obscured by the occipital crest (OC) in this image. Dotted-line square, Approximate location of hindbrain implants; solid-line square, approximate location of sc implants (between muscle layers). Inset, Absorbable hemostatic cloth used for implants, cut to size, and photographed at same magnification. B, Photomicrograph of Nissl-stained coronal section of the dorsal hindbrain at the level of the implant in an intact rat. C, Photomicrograph of Nissl-stained coronal section of the dorsal hindbrain at the level of the implant 3 d after implantation of 0.2 μg EB. Gr, Gracilis nucleus; CC, central canal.
Food intake and body weight
To determine whether local administration of E2 to the hindbrain is sufficient to inhibit feeding, we compared the effects of implants of cholesterol alone (control implants; n = 9), 0.2 μg EB on the hindbrain (n = 9), and 0.2 μg EB sc (n = 10) on daily food intake. Food intake (± 0.1 g, corrected for spillage) and body weight (± 1 g) were measured at 1000 h for 3 d before and 6 d after implantation. Data were analyzed with ANOVA (SAS general linear model procedure; SAS Institute, Cary, NC) followed by Tukey’s honestly significant difference contrasts between individual means and are reported as means ± sem, with the se of the difference (sed) given to indicate experiment-wide residual variability. Tests were two tailed, and minimum significance level was P < 0.05.
Implantation surgery alone had marked effects on food intake and body weight. One-way ANOVA of the means of the 3-d preoperative period (which did not vary significantly; F2,88 = 1.19 and 0.85, P > 0.05, for food intake and body weight, respectively) and the 6-d postimplant period showed that daily food intake did not recover fully until d 6 (F6,53 = 85.03, P < 0.0001, sed = 0.5 g) (Fig. 2A). Body weight also decreased transiently after surgery and recovered to the baseline level by d 5 (F6,54 = 28.05, P < 0.0001, sed = 1 g) (data not shown). Therefore, the effects of EB implants were analyzed in the context of the surgery effects by separate one-way ANOVA done on each of the 6 treatment days.
Figure 2.

Open-surgical hindbrain implants of 0.2 μg EB (H-EB), but not sc implants (sc-EB), decrease food intake in OVX rats. Food intake was measured daily at the middle of the light phase (dotted lines). Implants were done during the diurnal period of d 0 (arrow). Food intakes were not different among groups before implant surgery (d −2, −1, and 0) or after implantation on d 1 and 2, but food intake was decreased on postimplant d 3 and 4 in H-EB rats. A, Mean ± sem daily food intake in each group through the experiment. #, Different from control (CON) and sc-EB groups (P < 0.05). B, Food intake on d 3 and 4 after implantation in H-EB and sc-EB rats expressed as differences from control group intakes. The sed on both d 3 and 4 was 0.5 g. **, H-EB-control, different from sc-EB-control (P < 0.01).
Spread of implanted E2
The spread of E2 through the brain after hindbrain implantation was assessed. Ten rats received hindbrain implants of 0.2 μg E2 mixed with 6 ng of 3H-labeled E2 (0.33 μCi/ng; NEN Life Science Products, Perkin-Elmer, Waltham MA). Six, 12, 24, or 48 h later, one or two rats were deeply anesthetized with ip injections of sodium pentobarbital (Nembutal, 50 mg/kg; Abbott, Chicago, IL). The implants were removed, and the fresh brains were removed and snap frozen. Coronal cryosections (100 μm sections through 3 mm of hindbrain centered on the implant site and seven 500 μm sections through the hypothalamus) were cut on a cryostat (Leica CM3050S; Nussloch, Germany). Three other rats were killed 12 h after implantation. Their hindbrains were cut into 15 200-μm sections, which were then cut horizontally just ventral to the level of the central canal, and the dorsal and ventral parts were counted separately. Brain sections were dissolved in 6 m KOH and then neutralized with approximately 6 m HCl. After addition of 5 ml scintillation fluid (Quicksafe A; Zinsser Analytic, Berkshire, UK), radioactivity was counted in a β-counter (Packard 1600 TR; Canberra Packard, Zurich, Switzerland). Counts were corrected by subtracting the activity of corresponding brain samples from a rat not treated with 3H-labeled E2. Data are expressed as percent recovery of implanted 3H-labeled E2, using the counts of freshly prepared 3H-labeled E2 pads as a reference and as estimated counts per minute (cpm) per cubic millimeter tissue. Tissue volumes were estimated as the product of section thickness and the area of the corresponding sections in the atlas of Paxinos and Watson (20).
The kinetics of appearance of E2 in the brain after peripheral or central administration of EB have not been characterized. In one behavioral study, central implants of equimolar doses of EB and E2 had indistinguishable effects in ring doves (21). To ensure that EB and E2 have similar behavioral effects under our conditions, we compared the effects of hindbrain implants of cholesterol alone (control implants; n = 8), 0.2 μg EB (5.5 nm; n = 8), or 0.14 μg E2 (5.5 nm; n = 8) on food intake and body weight, as above.
Plasma E2 concentration
To determine whether hindbrain implants of 0.2 μg EB increased peripheral plasma E2 concentration, control implants (n = 16), hindbrain implants of 0.2 μg EB (n = 16), sc implants of 0.2 μg EB (n = 14), and sc implants of 2 μg EB (n = 17) were done. Rats were anesthetized with isoflurane 10 h after implantation, and 1.5 ml blood were collected from the retroorbital sinus. This time point was chosen because previously, plasma E2 levels were maximal 6–18 h after sc injection of 2 μg EB (5). Plasma E2 concentration was measured using a double-antibody RIA (Diagnostic Products, Los Angeles, CA). Two sets of standards were used, EB and E2, both diluted in serum from OVX rats. For both standards, 1-min counts between 18 and 3336 pmol/liter were linear on a logit-log plot (r2 = 0.998), and the intraassay variability was 3.2%. Standards less than 18 pmol/liter were indistinguishable and were considered nondetectable. Because rat plasma contains high levels of nonspecific steroidal esterases, which convert EB to E2 (22), we assumed that our assay results represent predominantly E2. Because the data were not normally distributed (Kolmogorov-Smirnov test, P < 0.001), they were analyzed using Kruskall-Wallis one-way ANOVA on ranks followed by Dunn’s test for pairwise comparisons and are reported as medians and interquartile ranges.
Plasma proinflammatory cytokine concentrations
E2 can increase the severity of anorexia produced by administration of bacterial pathogens or cytokines (23,24). Therefore, we measured the effects of hindbrain implants EB on plasma proinflammatory cytokine concentrations to investigate whether the feeding-inhibitory effect of this procedure might be due to an increased anorectic response to proinflammatory cytokines released after open-brain surgery. Samples of 1.5 ml blood were collected from the retroorbital sinus of rats receiving control implants (n = 6) or hindbrain implants of 0.2 μg EB rats (n = 6) 7 d before OVX, 7 d after OVX, during implantation surgery, 10 h after implants, and 2 d after implants. Plasma levels of interferon (IFN)-γ, TNF-α, IL-1β, and IL-6 were measured using a multiplex ELISA according to the manufacturer’s instructions (Bio-Plex cytokine assay system; Bio-Rad Laboratories, Ismaning, Germany). The detection limit was 50 pg/ml for each cytokine. These data were not normally distributed because many individual values in each assay were below the detection limit (Kolmogorov-Smirnov test, P < 0.001) so were analyzed using Kruskall-Wallis one-way ANOVA on ranks.
CCK-induced c-Fos expression
CCK-induced c-Fos expression was measured in rats after control implants (n = 10), hindbrain implants of 0.2 μg EB (n = 10), or sc implants of 2 μg EB (n = 10). Rats had been used previously for the plasma E2 assay. Three days after implant surgery, when the feeding-inhibitory effect of hindbrain E2 was largest (see Results), half of each group received ip injections of saline (1 ml/kg; 0.9% NaCl, ip) and half ip injections of 4 μg/kg sulfated CCK-8 (Bachem, Bubendorf, Switzerland) at dark onset. Ninety minutes later, the rats were deeply anesthetized with ip injections of sodium pentobarbital and transcardially perfused with ice-cold phosphate buffer [PB, 0.1 m (pH 7.4)] followed by 4% paraformaldehyde in 0.1 m PB. The brains were removed, postfixed at 4 C in the paraformaldehyde perfusion solution for 2 h and in 20% sucrose in 0.1 m PB for 2 d, and then cut into 40-μm sections on a freezing sliding microtome. Sets of each sixth hindbrain [∼17 to 11 mm posterior to bregma (20)] and forebrain [∼0.9 to 3.6 mm posterior to bregma (20)] sections were stored in cryoprotectant solution (a 4:3:3 mixture of 0.1 m PB, ethylene glycol, and glycerol; Sigma) at −20 C.
One set each of hindbrain and forebrain sections were processed for c-Fos expression using nickel-diaminobenzidine (DAB) immunocytochemistry. After a brief incubation in 0.5% H2O2 in 0.1 m PB, sections were incubated for 1 h in 1% normal goat serum (Vector Laboratories, Burlingame, CA) solution in 0.1 m PB + 0.3% Triton X-100 (Sigma) and then overnight, at room temperature, in primary antibody solution (1:20,000 rabbit polyclonal c-Fos antibody Ab-5; EMD Biosciences, Darmstadt, Germany, in 0.3% Triton X-100). Next, sections were incubated for 1 h with biotinylated antirabbit goat IgG (1:300; Vector Laboratories), stained using the streptavidin-nickel-DAB-peroxidase complex reaction (ABC, 1:500; Vector), mounted on gelatin-coated microscope slides, dehydrated in an increasing series of alcohols, defatted in xylene, coverslipped with Permount (Biomeda Corp., Foster City, CA), and air dried. Finally, the sections were digitally imaged (AxioCam; Carl Zeiss, Jena, Germany), and c-Fos positive cells were counted within the following areas of interest using templates based on the atlas of Paxinos and Watson (20) (NTS subregion nomenclature is our own; locations are millimeters caudal to bregma): cNTS (about 14.1–14.4 mm); subpostremal NTS (spNTS; about 13.7–14.0 mm); PVN (1.8–2.1 mm), Arc (2.8–3.1 mm), CeA (2.3–2.8 mm), raphe pallidus nucleus (RPa; 13.7–14.0 mm), and VMH (2.3–2.8 mm). Cells were considered c-Fos positive if their nuclei contained punctate blue-black immunolabeling and were counted using constant minimum and maximum OD and object size criteria, which were validated with visual counts. Data for each area were analyzed with two-way ANOVA, with E2 treatment and CCK treatment each between-subjects factors.
Colocalization of c-Fos and ERα
Double-label (ERα and c-Fos) immunofluorescence staining was used to determine whether EB hindbrain implants increased CCK-induced c-Fos expression in NTS cells expressing ERα. spNTS and cNTS sections from saline- and CCK-injected rats that had received hindbrain implants of 0.2 μg EB (n = 7 each) from the previous experiment were processed using an immunofluorescence procedure for separate detection of two primary antibodies raised in rabbits (25,26). Free-floating brain sections were incubated for 10 min each in 1% sodium borohydride and 0.5% H2O2 solutions, 1 h in 1% normal goat serum in 0.1 m PB + 0.3% Triton X-100, and then overnight with rabbit polyclonal ERα antibody (c1355, 1:100,000; Upstate Biotechnology, Lake Placid, NY). Sections were then incubated with biotinylated antirabbit goat antibody and avidin-biotin complex, for 1 h each, as described above. The avidin-biotin signal was amplified for 30 min with biotinylated tyramide solution (60 μl/10 ml PBS + 1:1000 H2O2), and ERα was visualized with Cy2-conjugated Streptavidin (1:400; Jackson ImmunoResearch Laboratory, West Grove, PA; peak emission 510 nm). Sections were washed and incubated for 1 h in 1% normal donkey serum in 0.1 m PB + 0.3% Triton X-100 and overnight with c-Fos primary antibody (1:10,000, rabbit polyclonal c-Fos antibody Ab-5; EMD Biosciences) in 0.1 m PB, 0.3% Triton X-100, and 1% normal donkey serum (Jackson ImmunoResearch Laboratory) and then with Cy3-conjugated AffiniPure donkey antirabbit IgG (Jackson ImmunoResearch Laboratory; peak emission 570 nm). Finally, sections were mounted on gelatinized microscope slides, dehydrated in a series of alcohols, defatted in xylene, coverslipped, dried, and digitally imaged. The numbers of cNTS cells expressing either ERα or c-Fos immunoreactivity were first counted with selective filters for Cy2 and Cy3 (sets 15, BP 515–565 nm, and 10, LP 570 nm, respectively; Carl Zeiss, Feldbach, Switzerland). The images were then overlayed, and cells expressing both labels were counted. Several controls for label specificity were performed. In some sections the c-Fos primary antibody was omitted from the run. When this was done, the Cy2 signal remained strong, but there was no Cy3 signal (data not shown). Furthermore, the Cy2 and Cy3 filters clearly revealed separate cells, both within individual brain areas (such as the cNTS, below) and between adjacent brain areas in which either ERα, but not c-Fos, or c-Fos, but not ERα, was expressed (i.e. in the CeA and medial nucleus of the amygdala (data not shown). Finally, examination of some samples with 1-μm confocal imaging indicated that counts done with light microscopic examination of the 40-μm sections revealed true cellular colocalization, not overlays of different cells (see below). Counts from saline- and CCK-treated rats for each label and each area were analyzed with unpaired t tests. Because the colocalization counts were not normally distributed (Kolmogorov-Smirnov test, P < 0.01), they were analyzed using Mann-Whitney rank sum tests and are reported as medians and interquartile ranges.
Results
Food intake and body weight
Administration of 0.2 μg EB onto the surface of the hindbrain over the cNTS, but not sc administration of 0.2 μg EB, decreased food intake (Fig. 2). Neither treatment produced detectable effects on d 1 and 2 after implantation (F2,27 = 1.50 and 0.91, P > 0.05, sed = 0.7 and 0.6 g, for d 1 and 2, respectively). On d 3 and 4 after implantation, however, food intake was significantly less in rats with hindbrain implants of 0.2 μg than in rats with either sc implants of 0.2 μg EB or control implants (F2,26 = 11.13 and 4.28, P < 0.0003 and 0.03, sed = 0.5 and 0.5 g, for d 3 and 4, respectively). Food intake returned to the control level on d 5 (F2,26 = 0.03 and 1.08, P > 0.05, sed = 0.7 and 0.6 g, for d 5 and 6, respectively). There were no differences in body weight among the groups on any of the 6 postimplant days (data not shown).
In the second experiment, administration of equimolar doses of EB and E2 (i.e. 0.2 μg EB and 0.14 μg E2, both 5.5 nm) onto the surface of the hindbrain decreased food intake similarly (Table 1). As previously, neither treatment produced detectable effects on d 1 and 2 after implantation (F2,21 = 2.86 and 0.04, P > 0.05, sed = 0.7 and 0.6 g, for d 1 and 2, respectively), but on d 3 after implantation, food intake was significantly less in both rats with EB and E2 implants, and there was no detectable difference between the effects of EB and E2 (F2,21 = 6.09, P < 0.01, sed = 0.6 g). Unlike the first experiment, no statistically reliable effects were detected on d 4 after implantation (F2,21 = 0.85, P > 0.05, sed = 1.0 g). As previously, there were no differences in body weight among the groups on any day (data not shown).
Table 1.
Hindbrain implants of E2 and EB have similar feeding inhibitory effects in OVX rats
| Group | Day
|
|||||
|---|---|---|---|---|---|---|
| B | 1 | 2 | 3 | 4 | 5 | |
| Control | 21.2 ± 0.4 | 14.7 ± 0.5 | 16.7 ± 0.7 | 19.1 ± 0.5 | 20.2 ± 0.8 | 19.8 ± 0.5 |
| H-EB | 20.0 ± 0.4 | 13.5 ± 1.1 | 16.8 ± 0.5 | 17.1 ± 0.5a | 19.0 ± 0.7 | 19.1 ± 0.6 |
| H-E2 | 21.1 ± 0.8 | 16.3 ± 0.8 | 17.0 ± 0.9 | 17.6 ± 0.3a | 20.1 ± 0.6 | 20.3 ± 0.6 |
Data are mean ± sem daily food intakes of OVX rats that received hindbrain implants of 5.5 nm of EB (H-EB; 0.2 μg) or 5.5 nm E2 (H-E2; 0.14 μg) in 4.5 μg cholesterol or cholesterol alone (Control). B, Preoperative baseline food intake.
Different from control (P < 0.02), Bonferroni-Holm tests after significant ANOVA [F(1,21) = 6.09, P < 0.01, sed = 0.6 g].
Spread of implanted E2
By 6 h after implant, less than 1% of implanted 3H-E2 remained in the implanted pads. At this time, approximately 0.1% of implanted 3H-E2, corresponding to approximately 200 pg total E2, was recovered from the hindbrain sections and approximately 0.0007% 3H-E2, corresponding to less than 2 pg total E2, from the hypothalamic sections. Recoveries were lower at later time points (data not shown). 3H-activity was markedly increased in the dorsal fractions of the 200 μm hindbrain sections from under the site of the implant (i.e. ∼13.5–15.0 mm caudal to bregma) and was near background levels more anteriorly or posteriorly (Fig. 3). In the ventral fractions, 3H-activity was near background in all sections, even directly under the implant.
Figure 3.

Radioactivity measured in the dorsal (unshaded bars) and ventral (shaded bars) parts of 200-μm coronal sections of the hindbrain 12 h after hindbrain implants of 0.2 μg E2 mixed with 6 ng 3H-labeled E2 (0.33 μCi/ng). Data are mean ± sem counts per minute (CPM) per cubic millimeter tissue (estimated from the cross sectional area of the sections in Ref. 20) from three rats. Initial activity of the implants was approximately 3,700,000 cpm. Black bar, Approximate location of implant pads.
Plasma E2 concentrations
In rats with control implants, hindbrain implants of 0.2 μg EB, and sc implants of 0.2 μg EB implants, plasma E2 levels were below the detection limit (18 pmol/liter) in more than 75% of samples (data not shown). In contrast, in rats with sc implants of 2 μg EB, plasma E2 concentration increased to the range of intact females during the proestrus phase (5,27) of the estrus cycle (median 105 pmol/liter, interquartile range 65–278 pmol/liter) and was significantly more than in any of the other groups (Dunn’s tests, all P < 0.05, after Kruskall-Wallis ANOVA on ranks, H3 = 42.5, P < 0.001).
Plasma proinflammatory cytokine concentrations
Plasma concentrations of TNF-α, IL-1 and IFN-γ were below the detection limit (50 pg/ml) at each time point in both rats receiving hindbrain implants of 0.2 μg EB and rats receiving control implants. Measurable concentrations of IL-6 were detected, but these were not different before vs. after implantation in either rats receiving hindbrain implants of 0.2 μg EB or control rats (H4 = 4.45 and 4.00, P = 0.35 and 0.41, respectively).
CCK-induced c-Fos expression
Hindbrain implants of 0.2 μg EB increased the expression of c-Fos in response to ip injection of 4 μg/kg CCK-8 in both cNTS and spNTS, as visualized by DAB staining (Fig. 4). In contrast, neither sc implants of 0.2 μg EB nor control implants increased CCK-elicited c-Fos in either area. Post hoc comparisons were based on ANOVA EB main effects and EB × CCK interaction effects (F1,22 = 7.53 and 8.10, P < 0.05 and P < 0.01, and F2,22 = 5.08 and 3.41, P < 0.02 and P = 0.051, sed = 1.2 and 1.2 cells/section, for the cNTS and spNTS, respectively).
Figure 4.

Open-surgical hindbrain implants of 0.2 μg EB (H-EB), but not sc implants of 0.2 μg EB (sc-EB), increase CCK-induced c-Fos expression, visualized with DAB staining, in the cNTS and spNTS, but not the Rpa, PVN, ventromedial nucleus, or Arc. Animals were killed on postimplant d 3, 90 min after ip injection of 4 μg/kg CCK-8 or saline. Data are number of c-Fos-positive cells per 40-μm section (mean ± sem). *, Number of c-Fos-positive cells different in CCK-injected H-EB rats from saline-injected H-EB rats (P < 0.05). +, Increase in number of c-Fos-positive cells in H-EB group (H-EB/CCK-H-EB/saline) different from sc-EB group (sc-EB/CCK-sc-EB/saline) or control group (control/CCK-control/saline) (P < 0.05).
No significant effects of either ip CCK or EB treatment on c-Fos expression was detected in any of the other brain areas tested (Fig. 4; range of main effects of CCK and E2, F1,24 = 0.11–2.22, P > 0.05; range of interaction effects, F2,24 = 0.71–2.15, Ps > 0.05).
Colocalization of c-Fos and ER-α
As previously reported (11,28,29,30), ERα was more abundantly expressed in the cNTS (∼15 cells/section) than the spNTS (∼five cells/section) (data not shown). As in the DAB-stained sections, CCK injection significantly increased c-Fos expression visualized by fluorescence staining in both the cNTS (Fig. 5A; t12 = 3.75, P < 0.01, sed = 3.7 cells/section) and the spNTS (9.5 ± 5.9 and 51.8 ± 9.7 cells/section for saline and CCK-injected rats, respectively, t12 = 3.44, P < 0.01, sed = 12.3 cells/section). In the cNTS, approximately 25% of the c-Fos-positive cells in CCK-injected rats also expressed ERα, whereas no c-Fos/ERα double-labeled cells were detected in the saline-injected rats (Fig. 5, A and B, Mann-Whitney rank test, U = 3.50, P < 0.01). In the spNTS, only one c-Fos/ERα double-labeled cell was detected in the seven CCK-injected rats, and no double-labeled cells were detected in the saline-injected rats.
Figure 5.

CCK activates cNTS ERα-positive cells in rats that received open-surgical hindbrain implants of 0.2 μg EB (H-EB). A, Numbers of single-labeled ERα-positive cells (left columns), single-labeled c-Fos-positive cells (middle columns), and double-labeled ERα/c-Fos-positive cells (right columns). Note all ERα/c-Fos values in saline-injected rats were 90 min after ip injection of saline or 4 μg/kg CCK-8 (CCK), visualized with immunofluorescence staining. In left and middle columns, data are mean ± sem numbers of ERα- or c-Fos-positive cells per section; in right columns, data are median number of ERα/c-Fos-positive cells per section; individual c-Fos single-labeling and ERα/c-Fos double-labeling data are shown in the middle and right CCK columns, with lines connecting each rat’s data. *, Number of c-Fos-positive cells different in saline- and CCK-injected rats (P < 0.05). +, Number of ERα/c-Fos-positive cells different in saline- and CCK-injected rats (P < 0.05). B, Optical micrographs of a 40-μm cNTS section showing immunofluorescence staining of ERα single labeling, c-Fos single labeling, and ERα/c-Fos double labeling (overlay) and a 1-μm confocal micrograph of ERα/c-Fos double labeling in area indicated by white box in overlay (confocal overlay). Asterisks indicate representative double-labeled cells. Scale bars, 60 μm in optical micrographs and 10 μm in confocal micrograph.
Discussion
We demonstrate here that in OVX rats, open-surgical administration of 0.2 μg EB or an equimolar dose (5.5 nm) of 0.14 μg E2 directly onto the surface of the hindbrain just caudal to the AP, i.e. just over the cNTS, elicited a transient decrease in food intake, i.e. 3–4 d later. The timing and duration of this decrease in feeding is similar to that elicited by sc administration of near-physiological doses of E2 (4,5). Furthermore, hindbrain EB implants appeared to induce only local increases in E2 concentration. Finally, hindbrain EB implants also increased the neuronal activation of cNTS ERα-expressing neurons that was induced by ip CCK, a satiation signal that previously has been shown to mediate the feeding-inhibitory effect of E2 (1,2,3,4,5,6,11). Our interpretation of these data is that E2 acts via ERα neurons in the cNTS to inhibit feeding by increasing satiation.
Several previous reports indicated that E2 acts in the brain, rather than peripherally, to inhibit eating but have not clearly disclosed the site(s) of the ER involved (1,2,3). The data reported here provide compelling evidence that one site in which E2 can act to inhibit feeding is the cNTS. The strongest support for a local action in the cNTS comes from our measurements of the effects of cNTS implants on brain and systemic E2 levels. First, neither hindbrain nor sc administration of 0.2 μg EB led to a detectable increase in systemic plasma E2 concentration, indicating that each of these E2 treatments produced only local increases in E2. Second, hindbrain implants containing 3H-E2 increased brain E2 concentrations to biologically significant levels only in the dorsal hindbrain just under the implant site, i.e. in and around the cNTS. The increase in radioactivity corresponded to a total amount of at most approximately 200 pg E2. The anterior-posterior spread of 3H-E2 in this study is similar to that reported by Davis et al. (31,32) to result from intrahypothalamic implants of about 3 ng E2 (i.e. a 1:300 mixture of E2 in cholesterol in a 28 gauge implant needle). Implanted into the VMH, and together with peripheral progesterone treatment, this amount of E2 increased sexual receptivity in OVX rats. In contrast, the smallest intrahypothalamic dose of E2 reported to inhibit feeding is approximately 30 times greater [i.e. the 1:10 E2-cholesterol mixtures used by Butera and colleagues (13,14); discussed further below]. Our hindbrain E2 implants increased E2 contents in more distal hindbrain sites by about an order of magnitude less than the increase in the immediate area of the cNTS and increased hypothalamic E2 contents about 2 orders of magnitude less. We regard these small increases as biologically insignificant. ERα in this part of the dorsal hindbrain are predominately localized in the cNTS (11,28,29,30), with sparse expression in the spinal trigeminal nucleus, an area not implicated in the control of feeding by gastrointestinal signals. Thus, we conclude that our hindbrain E2 implants acted in the cNTS to inhibit feeding.
Two additional observations provide indirect support for this conclusion. First, the same dose of 0.2 μg EB used for the hindbrain implants had no effect on food intake when implanted sc, adjacent to the hindbrain surgical site, indicating that this dose of E2 is not sufficient to inhibit eating when administered outside the brain. Second, pilot studies (data not shown) indicated that hindbrain administration of 0.2 μg EB did not produce the typical estrous pattern of cornified vaginal mucosal cells, a peripheral action, whereas doses of 0.5 μg EB or more did.
Our c-Fos study was based on our report (11) that sc injections of 10 μg EB increased the satiating potency of intraduodenal infusions of fat emulsions in OVX rats, that this was mediated by endogenous CCK, that it was associated with increased c-Fos expression only in the cNTS, and that many of the cNTS cells that expressed c-Fos also expressed ERα. The results here were similar. Hindbrain EB treatment decreased feeding; increased CCK-induced c-Fos expression only in the cNTS and spNTS (and not in forebrain areas related to the control of food intake, i.e. the PVN, VMN, and Arc); and increased c-Fos expression in ERα-positive neurons in the cNTS. The highly restricted rostral-caudal extent of c-Fos activation in the NTS here is especially interesting because this region seems to receive vagal projections from the intestines (33). Thus, it seems likely that intraduodenal lipid infusion in the previous experiment (11) led to activation of the same cNTS ERα-expressing cells that CCK injection did here and that, in turn, in both tests, these cells were part of the neuronal network mediating the estrogenic inhibition of feeding. Because hindbrain EB treatment was not sufficient to increase CCK-induced c-Fos in the hypothalamus, it is less likely that E2 acted there to inhibit eating.
Our data do not address whether endogenous E2 also acts in brain sites outside the NTS to inhibit feeding. Previous studies using intracranial administration of amounts of E2 that may have had only local effects (i.e. less than 0.5 μg) have implicated ER in three hypothalamic areas in the estrogenic inhibition of eating, but in each case the evidence is controversial (see Refs. 1,2,3for reviews). The most extensive data are those of Butera and colleagues (13,14), who reported that implantation of E2 into the PVN significantly decreased food intake. In their experiments, the distal 1 mm of a 28-gauge cannula was filled with about 1 μg cholesterol-E2 mixtures, so that the effective 3:1 and 10:1 cholesterol-E2 mixtures translate into doses of about 0.3 and 0.1 μg E2, respectively. The evidence that these doses worked locally, within the PVN, was that even undiluted E2 (1 μg) delivered into the MPA, VMH, or posterior hypothalamus failed to inhibit feeding and that, in comparison with undiluted E2, dilute E2 implants in the PVN produced fewer occurrences of cornification of the vaginal mucosa and, in combination with progesterone, significantly less lordosis (13). In addition, sc injections of E2 did not decrease feeding in rats with bilateral electrolytic lesions of the PVN (34). These data suggested that E2 could act in the PVN to inhibit eating. This conclusion remains controversial, however, because other laboratories have reported conflicting results. Dagnault and Richard (35) failed to find any decrease in the feeding-inhibitory potency of sc EB treatment in rats with similar electrolytic lesions of the PVN, and Hrupka et al. (17) failed to find any reliable eating-inhibitory effect of PVN EB implants similar to those that Butera and colleagues (13,14) reported to be effective.
Dagnault and Richard (15) also investigated the role of the MPA in the feeding-inhibitory action of E2. These investigators reported that food intake was reduced 2 h after injection of a water-soluble, cyclodextrin E2 preparation into the MPA but had recovered to control levels by 13 h after injection. The time course of this inhibition does not match that produced by peripheral injections of E2, which typically decrease food intake with a latency of more than 1 d, despite the fact that plasma E2 levels reach maximal levels in only about 6 h (i.e. Refs. 4,5). Because cyclodextrin has been shown to interfere with neuronal function (36,37), that Dagnault and Richard (15) used water rather than the cyclodextrin vehicle for control injections suggests that some or all of the effects that they reported may have been due to cyclodextrin rather than E2. Consistent with this possibility, other investigators have failed to detect any inhibition of feeding after MPA implants of E2 (13,17).
Two more recent reports have implicated the Arc in the estrogenic inhibition of eating. Clegg et al. (16) provided evidence that E2 may act in the Arc to inhibit feeding by increasing the potencies of the feeding-inhibitory adiposity hormones leptin and insulin. Despite affecting the potencies of exogenous insulin and leptin, however, Arc E2 implants alone did not inhibit feeding. It is possible that this was because the rats were tested several weeks after OVX, when the initial hyperphagia usually wanes, but this has not yet been verified. Gao et al. (38) reported that the feeding-inhibitory effects of intracerebroventricular E2 treatment are associated with increases in the number of excitatory proopiomelanocortin (POMC) synapses in the Arc. The physiological relevance of these data is questionable, however, because a central dose of 2 μg E2 was used and because the inhibition of feeding occurred much more rapidly than after more physiological E2 treatments (1,2,5). In summary, although E2 may act in brain sites outside the NTS to inhibit feeding, at present there is no compelling evidence for this.
In addition to eliminating the periovulatory decrease in eating, OVX also increases food intake to a level higher than that displayed at any phase of the normal estrus cycle. This suggests that ovarian function, specifically E2 secretion, exerts both a tonic and a phasic, or cyclic, inhibition of feeding (1,2,5,39,40). The tonic inhibitory effect on feeding is reflected in the higher level of food intake throughout the cycle in OVX rats, compared with intact rats, and the phasic effect is reflected in the cyclic, periovulatory decrease in food intake during the estrous cycle. The evidence that CCK mediates the phasic inhibitory effect is that the CCK-1 receptor antagonist devazepide increased meal size only in estrus but not diestrus, which suggests that endogenous CCK satiation varies across the estrous cycle (41). Our current demonstration that E2 acts in the hindbrain to transiently inhibit feeding and increase CCK-induced c-Fos expression, taken together with the previous observation that CCK selectively mediates the phasic estrogenic inhibition of feeding, suggests that E2 acts in the hindbrain to mediate the phasic estrogenic inhibition of feeding. It is important to note, however, that our data do not disclose whether E2 also acts on the hindbrain to mediate the tonic estrogenic inhibition of feeding.
E2 can increase the severity of illness anorexia produced by peripheral administration of bacterial pathogens or proinflammatory cytokines (23,24). Because open-surgical implantation of E2 onto the surface of the hindbrain poses the risk of postsurgical inflammation, we tested whether the inhibition of food intake that we observed 3–4 d after E2 administration might be related to increases in plasma proinflammatory cytokines. Plasma concentrations of the proinflammatory cytokines IL-1, IFN-γ, and TNF-α were below the detection limit both before and after implant surgery, and although IL-6 was detected in the plasma, its concentration did not increase after surgery. The low level of c-Fos expression in the posterior RPa also suggests the absence of postsurgical inflammation because c-Fos activation in this area has been linked to activation of the innate immune system by either peripheral or central inflammation (42,43,44). Finally, there were no signs of pathology in postmortem examinations of the brains. Thus, taken together, the data suggest that our rats did not suffer from systemic postsurgical inflammation. Nonetheless, we cannot definitely rule out any influence of inflammation on food intake under our conditions because we did not identify the cause of the postimplantation reduction in food intake in the control group and we did not determine whether our surgical approach produced local inflammation within the brain. More generally, we emphasize that because all our data were collected during a period of postoperative recovery, their relevance to normal physiological function requires further substantiation.
In conclusion, by demonstrating that hindbrain implants of 0.2 μg EB both inhibits feeding and activates ERα-containing neurons in the cNTS, the current experiments identify a new mechanism for the estrogenic control of feeding and provide a putative function for ERα in neurons in this region of the NTS. Furthermore, both these and previous data (11) suggest that one mechanism through which NTS ERα inhibits feeding is by increasing the neuronal processing of vagal afferent inputs encoding satiation signals initiated by intestinal CCK. An important next step in understanding this process would be to identify the neurotransmitter phenotypes and interneuronal connections of these neurons. Some details about these NTS neuronal networks have begun to emerge recently. In mice, ip CCK increases c-Fos expression in NTS cells, including the spNTS and cNTS cells, that receive monosynaptic vagal inputs and in many cases also express POMC, the precursor of the neuropeptide α-MSH (45,46). These cells appear to be a necessary part of CCK-satiation information processing because fourth ventricular administration of an antagonist to the relevant α-MSH receptors, the melanocortin-3 and -4 receptors, completely blocked the satiating action of ip CCK-8 (45). Appleyard et al. (45) also demonstrated that these POMC neurons were synaptically sensitive to opioids, glutamate, and CCK. Similarly, Berthoud et al. (47) demonstrated that NTS cells, again including those in the spNTS and cNTS, which expressed c-Fos after intraduodenal infusions of linoleic acid or glucose in most cases, also expressed both N-methyl-d-aspartate (NMDA)- and non-NMDA glutamate receptors. Future elaboration of the roles of ERα in these hindbrain neuronal networks controlling appetite has clear translational importance for the understanding and development of sex-specific therapies for disordered eating, which disproportionately affects women (1,2,3).
Acknowledgments
We thank Dr. Adrian Hehl (University of Zurich) for help with the confocal microscopy.
Footnotes
This work was supported by National Institutes of Health Research Grant DK 54523 (to N.G.) and Center for Integrative Human Physiology, University of Zürich (to T.A.L., S.T.).
Disclosure Statement: The authors have nothing to disclose.
First Published Online December 20, 2007
Abbreviations: Arc, Arcuate hypothalamic nucleus; AP, area postrema; CCK, cholecystokinin; CeA, central nucleus of the amygdala; cNTS, caudal nucleus tractus solitarius; DAB, diaminobenzidine; E2, 17β-estradiol; EB, β-estradiol 3-benzoate; ER, estrogen receptor; IFN, interferon; MPA, medial preoptic area; NTS, nucleus tractus solitarius; OVX, ovariectomy; PB, phosphate buffer; POMC, proopiomelanocortin; PVN, paraventricular hypothalamic nucleus; Rpa, raphe pallidus nucleus; sed, se of the difference; spNTS, subpostremal nucleus tractus solitarius; VMN, ventromedial nucleus.
References
- Asarian L, Geary N 2006 Modulation of appetite by gonadal steroid hormones. Philos Trans R Soc Lond B Biol Sci 361:1251–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geary N 2004 The estrogenic inhibition of eating. In: Stricker EM, Woods SC, eds. Handbook of behavioral neurobiology. Vol 14. New York: Kluwer Academic; 307–345 [Google Scholar]
- Geary N, Lovejoy L 2008 Sex differences in energy metabolism, obesity and eating behavior. In: Becker J, ed. Sex on the brain: from genes to behavior. New York: Oxford Publishers; 253–274 [Google Scholar]
- Geary N, Asarian L 1999 Cyclic estradiol treatment normalizes body weight and test meal size in ovariectomized rats. Physiol Behav 67:141–147 [DOI] [PubMed] [Google Scholar]
- Asarian L, Geary N 2002 Cyclic estradiol treatment normalizes body weight and restores physiological patterns of spontaneous feeding and sexual receptivity in ovariectomized rats. Horm Behav 42:461–471 [DOI] [PubMed] [Google Scholar]
- Geary N, Asarian L, Korach KS, Pfaff DW, Ogawa S 2001 Deficits in E2-dependent control of feeding, weight gain, and cholecystokinin satiation in ER-α null mice. Endocrinology 142:4751–4757 [DOI] [PubMed] [Google Scholar]
- Cummings DE, Overduin J 2007 Gastrointestinal regulation of food intake. J Clin Invest 117:13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beglinger C, Degen L 2004 Fat in the intestine as a regulator of appetite—role of CCK. Physiol Behav 83:617–621 [DOI] [PubMed] [Google Scholar]
- Moran TH 2006 Neural and hormonal controls of food intake and satiety. In: Johnson LR, ed. Physiology of the gastrointestinal tract. San Diego: Academic Press; 877–894 [Google Scholar]
- Ritter RC 2004 Gastrointestinal mechanisms of satiation for food. Physiol Behav 81:249–273 [DOI] [PubMed] [Google Scholar]
- Asarian L, Geary N 2007 Estradiol enhances CCK-dependent lipid-induced satiation and activates ERα-expressing cells in the NTS of ovariectomized rats. Endocrinology 148:5656–5666 [DOI] [PubMed] [Google Scholar]
- Nunez AA, Gray JM, Wade GN 1980 Food intake and adipose tissue lipoprotein lipase activity after hypothalamic estradiol benzoate implants in rats. Physiol Behav 25:595–598 [DOI] [PubMed] [Google Scholar]
- Butera PC, Beikirch RJ 1989 Central implants of diluted estradiol: independent effects on ingestive and reproductive behaviors of ovariectomized rats. Brain Res 491:266–273 [DOI] [PubMed] [Google Scholar]
- Butera PC, Xiong M, Davis RJ, Platania SP 1996 Central implants of dilute estradiol enhance the satiety effect of CCK-8. Behav Neurosci 110:823–830 [DOI] [PubMed] [Google Scholar]
- Dagnault A, Richard D 1997 Involvement of the medial preoptic area in the anorectic action of estrogens. Am J Physiol 272:R311–R317 [DOI] [PubMed] [Google Scholar]
- Clegg DJ, Brown LM, Woods SC, Benoit SC 2006 Gonadal hormones determine sensitivity to central leptin and insulin. Diabetes 55:978–987 [DOI] [PubMed] [Google Scholar]
- Hrupka BJ, Smith GP, Geary N 2002 Hypothalamic implants of dilute estradiol fail to reduce feeding in ovariectomized rats. Physiol Behav 77:233–241 [DOI] [PubMed] [Google Scholar]
- Eckel LA, Geary N 2001 Estradiol treatment increases feeding-induced c-Fos expression in the brains of ovariectomized rats. Am J Physiol Regul Integr Comp Physiol 281:R738–R746 [DOI] [PubMed] [Google Scholar]
- Eckel LA, Houpt TA, Geary N 2002 Estradiol treatment increases CCK-induced c-Fos expression in the brains of ovariectomized rats. Am J Physiol Regul Integr Comp Physiol 283:R1378–R1385 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C 1998 The rat brain stereotaxic coordinates. San Diego: Academic Press [Google Scholar]
- Cohen J, Cheng M-F 1981 The role of the midbrain in courtship behavior of the female ring dove (Streptopelia risoria): evidence from radiofrequency lesion and hormone implant studies. Brain Res 208:279–301 [DOI] [PubMed] [Google Scholar]
- Lund-Pero M, Jeppson B, Arneklo-Nobin B, Sjogren HO, Holmgren K, Pero RW 1994 Non-specific steroidal esterase activity and distribution in human and other mammalian tissues. Clin Chim Acta 224:9–20 [DOI] [PubMed] [Google Scholar]
- Geary N, Asarian L, Sheahan J, Langhans W 2004 Estradiol-mediated increases in the anorexia induced by intraperitoneal injection of bacterial lipopolysaccharide in female rats. Physiol Behav 82:251–261 [DOI] [PubMed] [Google Scholar]
- Butera PC, Doerflinger AL, Roberto F 2002 Cyclic estradiol treatment enhances the effects of interleukin-1β on food intake in female rats. Brain Behav Immun 16:275–281 [DOI] [PubMed] [Google Scholar]
- Karatsoreos IN, Yan L, Le Sauter J, Silver R 2004 Phenotype matters: identification of light-responsive cells in the mouse suprachiasmatic nucleus. J Neurosci 24:68–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegsfeld LJ, Korets R, Silver R 2003 Expression of the circadian clock gene Period 1 in neuroendocrine cells: an investigation using mice with a Per1:GFP transgene. Eur J Neurosci 17:212–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman ME 2006 Neuroendocrine control of the ovarian cycle of the rat. In: Neill JD, ed. Knobil and Neill’s physiology of reproduction. San Diego: Elsevier; 2327–2388 [Google Scholar]
- Haywood SA, Simonian SX, van der Beek EM, Bicknell RJ, Herbison AE 1999 Fluctuating estrogen and progesterone receptor expression in brainstem norepinephrine neurons through the rat estrous cycle. Endocrinology 140:3255–3263 [DOI] [PubMed] [Google Scholar]
- Shughrue PJ, Lane MV, Merchenthaler I 1997 Comparative distribution of estrogen receptor-α and -β in the rat central nervous system. J Comp Neurol 388:507–525 [DOI] [PubMed] [Google Scholar]
- Simonian SX, Herbison AE 1997 Differential expression of estrogen receptor α and β immunoreactivity by oxytocin neurons of rat paraventricular nucleus. J Neuroendocrinol 9:803–806 [DOI] [PubMed] [Google Scholar]
- Davis PG, Krieger MS, Barfield RJ, McEwen BS, Pfaff DW 1982 The site of action of intrahypothalmic estrogen implants in feminine sexual behavior: an autoradiographic analysis. Endocrinology 111:1581–1586 [DOI] [PubMed] [Google Scholar]
- Davis PG, McEwen BS, Pfaff DW 1979 Localized behavioral effects of tritiated estradiol implants in the ventromedial hypothalamus of female rats. Endocrinology 104:898–903 [DOI] [PubMed] [Google Scholar]
- Altschuler SM, Bao XM, Bieger D, Hopkins DA, Miselis RR 1989 Viscerotopic representation of the upper alimentary tract in the rat: sensory ganglia and nuclei of the solitary and spinal trigeminal tracts. J Comp Neurol 283:248–268 [DOI] [PubMed] [Google Scholar]
- Butera PC, Willard DM, Raymond SA 1992 Effects of PVN lesions on the responsiveness of female rats to estradiol. Brain Res 576:304–310 [DOI] [PubMed] [Google Scholar]
- Dagnault A, Richard D 1994 Lesions of hypothalamic paraventricular nuclei do not prevent the effect of estradiol on energy and fat balance. Am J Physiol 267:E32–E38 [DOI] [PubMed] [Google Scholar]
- Ottico E, Prinetti A, Prioni S, Giannotta C, Basso L, Chigorno V, Sonnino S 2003 Dynamics of membrane lipid domains in neuronal cells differentiated in culture. J Lipid Res 44:2142–2151 [DOI] [PubMed] [Google Scholar]
- Uekama K, Hirayama F, Irie T 1998 Cyclodextrin drug carrier systems. Chem Rev 98:2045–2076 [DOI] [PubMed] [Google Scholar]
- Gao Q, Mezei G, Nie Y, Rao Y, Choi CS, Bechman I, Leranth C, Tran-Allerand D, Priest CA, Roberts JL, Gao X-B, Mobbs C, Shulman GI, Diano S, Horvath TL 2007 Anorectic estradiol mimics leptin’s effects on the rewiring of melanocortin cells and Stat3 signaling in obese animals. Nat Med 13:89–94 [DOI] [PubMed] [Google Scholar]
- Drewett RF 1974 The meal patterns of the oestrous cycle and their motivational significance. Q J Exp Psychol 26:489–494 [DOI] [PubMed] [Google Scholar]
- Eckel LA, Houpt TA, Geary N 2000 Spontaneous meal patterns in female rats with and without access to running wheels. Physiol Behav 70:397–405 [DOI] [PubMed] [Google Scholar]
- Eckel LA, Geary N 1999 Endogenous cholecystokinin’s satiating action increases during estrus in female rats. Peptides 20:451–456 [DOI] [PubMed] [Google Scholar]
- Kopf BS, Geary N, Langhans W, Asarian L 2007 Intraperitoneal bacterial lipopolysaccharide (LPS) elicits rapid, graded increases in c-Fos expression in the raphe and central nucleus of the amygdala in male rats. Appetite 49:304 [Google Scholar]
- Nakamura K, Matsumura K, Hubschle T, Nakamura Y, Hioki H, Fujiyama F, Boldogkoi Z, Konig M, Thiel HJ, Gerstberger R, Kobayashi S, Kaneko T 2004 Identification of sympathetic premotor neurons in medullary raphe regions mediating fever and other thermoregulatory functions. J Neurosci 24:5370–5380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, McAllen RM 2005 A subsidiary fever center in the medullary raphe? Am J Physiol Regul Integr Comp Physiol 289:R1592–R1598 [DOI] [PubMed] [Google Scholar]
- Appleyard SM, Bailey TW, Doyle MW, Jin YH, Smart JL, Low MJ, Andresen MC 2005 Proopiomelanocortin neurons in nucleus tractus solitarius are activated by visceral afferents: regulation by cholecystokinin and opioids. J Neurosci 25:3578–3585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W, Ellacott KL, Halatchev IG, Takahashi K, Yu P, Cone RD 2004 Cholecystokinin-mediated suppression of feeding involves the brainstem melanocortin system. Nat Neurosci 7:335–336 [DOI] [PubMed] [Google Scholar]
- Berthoud HR, Earle T, Zheng H, Patterson LM, Phifer C 2001 Food-related gastrointestinal signals activate caudal brainstem neurons expressing both NMDA and AMPA receptors. Brain Res 915:143–154 [DOI] [PubMed] [Google Scholar]