

Abstract
Tuberculosis (TB) is a major threat to global health, recently exacerbated by the emergence of highly drug-resistant forms of the disease-causing pathogen and synergy with HIV/AIDS. In 2006, the Stop TB Partnership published “The global plan to stop TB: 2006–2015,” which set out a vision of halving the prevalence of and mortality caused by the disease by 2015, followed by eliminating the disease as a public health problem by 2050. This vision depends on the development of improved diagnostics, simpler treatment, and more effective vaccination. Recently, active translational research pipelines directed toward each of these goals have been established, but improved understanding of the fundamental biology of this complex disease will prove to be the key to radical advances in TB control.
Introduction
Tuberculosis (TB) is today the second highest cause of death from an infectious disease worldwide, after HIV/AIDS (1), and is the biggest killer of people infected with HIV (reviewed in ref. 2). Much of the burden of this disease falls on those living in the developing world. A recent report on the TB pandemic (3) revealed that in 2005, there were almost 9 million new cases and 1.6 million deaths. The latest data indicate that the incidence rate has probably stabilized, but the challenge is to reverse the trend and reduce global mortality and, in the long term, to eliminate the disease.
Most humans infected with the pathogen that causes TB, Mycobacterium tuberculosis, remain asymptomatic, and only a small proportion develop active TB disease immediately. TB is usually a chronic, slowly progressing disease that often remains undiagnosed in patients for many years. The disease has many clinical manifestations and can affect many organs, but the most common form in adults is a chronic pulmonary disease (Figure 1). In most people, the bacterium establishes a latent infection, and the lifetime risk of developing disease is only 10% unless an individual becomes immunocompromised, at which time the risk increases to about 10% per year (4–6). Unfortunately, it has been estimated that a third of the world’s population is latently infected with M. tuberculosis, providing an enormous reservoir for future disease.
Figure 1. Pulmonary TB in a patient who was sputum smear positive.
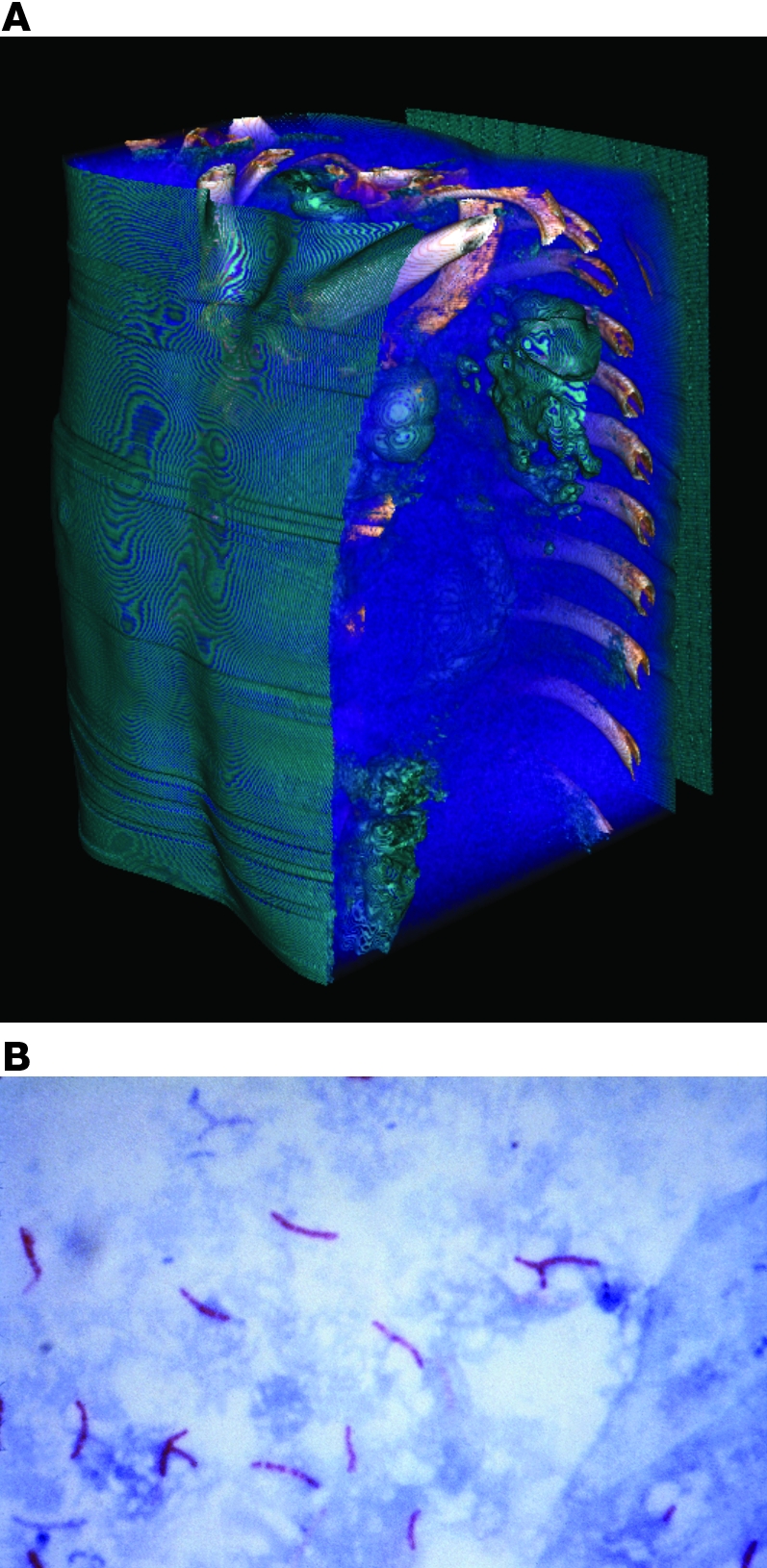
(A) TB has many clinical manifestations, but the most common form of the disease in adults is a chronic pulmonary disease. Shown here is a 3D reconstruction of a high-resolution tomography series of an individual with extensive TB disease. (B) The most common way to diagnose TB is by microscopic analysis of sputum to visualize M. tuberculosis; a positive diagnosis is obtained after spreading a sputum sample on a microscope slide, fixing, applying a stain, decolorizing with acid, counterstaining, and visualizing the acid-fast tubercle bacilli. However, this method is not very sensitive and only detects 60% of culture-positive patients with pulmonary TB. This delays early diagnosis and can result in substantial lung damage and transmission of the disease-causing pathogen before therapy is started. Original magnification, ×1,000.
“The global plan to stop TB: 2006-2015” (7, 8) was developed and endorsed in 2005 by the Stop TB Partnership, a network of international organizations, countries, donors from the public and private sectors, governmental and nongovernmental organizations, and individuals who expressed an interest in working together to achieve the goal of eliminating TB as a public health problem. It established a number of specific targets. The target for 2005 was to detect 70% of new sputum smear–positive cases (i.e., cases in which a positive diagnosis is obtained after spreading a sputum sample on a microscope slide, fixing, applying a stain, decolorizing with acid, counterstaining, and visualizing the acid-fast tubercle bacilli; Figure 1) and cure at least 85% of these cases (although this goal was not met, substantial progress toward it was made); for 2015, to sustain or exceed those indicators and reduce the prevalence and death rates of TB by 50% relative to the 1990 level; and for 2050, to eliminate TB as a public health problem. However, the current tools for diagnosing, treating, and preventing TB are inadequate for this task: latent and active TB are most commonly diagnosed using, respectively, a skin test that dates back to 1891 (9) and sputum-smear microscopy from the same era (10). The current “short-course” drug therapy consists of a cocktail of drugs taken over a period of at least six months, and despite its widespread use, the Mycobacterium bovis bacillus Calmette-Guérin (BCG) vaccine is largely ineffective, at least in preventing adult pulmonary disease. A growing problem that has had an impact on the success of treatment programs is the emergence of strains of M. tuberculosis resistant to the drugs used as first-line treatment, and around 400,000 cases of multidrug-resistant TB (MDR-TB), which is defined as resistance to isoniazid and rifampicin, occur per year (11). Furthermore, essentially untreatable outbreaks of extensively drug-resistant TB (XDR-TB), which is defined as MDR-TB plus resistance to a quinolone and one of the second-line anti-TB injectable drugs (Amikacin, Kanamycin, and Capreomycin), have begun to appear. In HIV-positive individuals, XDR-TB has been associated with very high mortality (12).
Achieving the goals in the Stop TB plan will require the development of new tools that are substantially better than those available today. This, in turn, will require an advance in our knowledge of the disease and the biology of M. tuberculosis as well as the application of innovative new approaches and technologies. Much progress has been made in the past decade, and a number of new tools are under development (13–22). However, much more needs to be done. In this Review, we look ahead to how the major challenges that must be overcome in the three areas of diagnosis, treatment, and prevention can be addressed to make a quantum leap forward (Table 1). Progress in each of these areas would also be accelerated by the identification and validation of biomarkers, biological markers that correlate with the disease status of the host or the response to intervention.
Table 1 .
Key issues that need to be addressed if radical advances in the control of TB are to be made

Diagnostics
On the face of it, diagnosing TB should be a trivial matter. Disease is often localized to the lung, and by the time patients present to a diagnostic facility, a marked proportion excrete so many bacilli in the sputum that they can readily be seen with a microscope using inexpensive stains that differentiate acid-fast bacilli (AFB) from routine bacterial flora (i.e., they are sputum smear positive; Figure 1). The only potential confounder, nontuberculous mycobacterial species, is an infrequent cause of positive sputum smear tests in countries where TB is endemic (14). There has been a great deal of progress recently in developing better tools to detect latent TB (23–26), and understanding the role and relevance of these assays is an area of active research. However, we have chosen to focus here on detection of active disease, as these new tools will have the greatest impact initially on the countries where the burden of disease is highest.
One would think that these aspects of TB would make it possible to achieve rapid and substantial gains in controlling TB globally with a standardized application of existing diagnostic methods. This indeed was one of the assumptions underpinning the DOTS (directly observed therapy, short course) TB control strategy launched by the WHO as a highly cost-effective health intervention in 1994 (27). DOTS has conventionally relied on passive detection of microscopy smear–positive samples from patients presenting to health clinics to detect cases and initiate therapy. In the absence of an effective vaccine, this has been the cornerstone of TB control.
Although DOTS has been very successful at standardizing care practices and increasing cure rates, case-detection targets have been more difficult to achieve (1). Furthermore, overall TB control, as measured by a decline in incidence, especially in settings where drug resistance or HIV coinfection are prevalent, has been limited (1). The core problem is that existing microscopy methods to diagnose TB are both technically and practically inadequate for use in high-burden countries. Microscopy is too complex to implement and too slow to perform to be used as a point-of-care test. The primary clinics where most patients with symptoms of TB first seek care rarely have AFB microscopy available, and patients must commonly wait until they are referred to a specialty clinic or microscopy center. Even then, most TB cases will be missed, both because of the inherent limits of the sensitivity of the test, which detects only 60% of culture-positive pulmonary TB patients even in clinical trial settings, and because of the logistical difficulties of making quality-assured microscopy widely and freely available. Moreover, microscopy misses exactly the patients, those with early disease, that one would like to detect and put on treatment to block transmission before it has begun. Delays in diagnosis of 3–6 months are common because of the lack of sensitivity of microscopy (28, 29), during which time the disease can progress and cause severe destruction of the airways (Figure 1) and transmission continues.
Mycobacterial culture is much more sensitive than microscopy but requires equipment and training beyond what is available at peripheral public health laboratories. Most countries with a high burden of TB have no more than a handful of laboratories with culture capability, so most patients never have access to this technology. When they do, results are often so slow as to be of little clinical relevance. This has proven especially true in patients with advanced HIV infection, where undetected or improperly treated TB can have a very high and early mortality, with survival measured in days or weeks (30–32). Conventional indirect drug-susceptibility testing (DST) on solid media is slower still and requires laboratory infrastructure that is so rarely available that only 3% of the 6.5 million TB cases arising annually in high-burden countries of Asia and Africa have access to DST (33).
A broad effort to develop and make available better diagnostics for TB is underway, and substantial gains have already been made. The feasibility and cost effectiveness of using rapid liquid culture systems for case detection and DST has recently been demonstrated in a number of high-burden countries. Their broader implementation has been facilitated by price negotiation with manufacturers and by the recent development of a simple and inexpensive method for species identification of culture isolates by immunocapture of the TB-specific protein MTB64 on lateral flow (34). Inexpensive and robust fluorescence microscopy platforms based on LED excitation have also been developed to speed TB detection in microscopy laboratories (35, 36). Although gains will be made with improved diagnostics, access to primary health care by patients is also a critical factor in early detection of TB, and further improvements in this are also required to maximize the impact on the epidemic.
Beyond conventional microbiologic methods, there are three molecular assays that are expected to be available to the public sector in the coming 6 to 18 months that can dramatically speed case detection and MDR screening without the need for biosafety precautions associated with phenotypic methods. One is a reference-level test employing manual PCR followed by line-probe hybridization to detect resistance-associated mutations (37). The other two molecular assays use automation (38, 39) that should allow them to be used in microscopy laboratories or similar settings.
Progress in developing true point-of-care tests, however, has been slower. Our ability to develop the types of tests that are most needed and in general to improve the control of TB through better diagnosis, is hampered by knowledge gaps related to the epidemiology and natural history of tuberculous disease, host and pathogen biology, and biodetection technologies. Some examples of these knowledge gaps follow.
Epidemiology and natural history of TB.
Although careful studies of transmission of the TB-causing pathogen in defined settings, such as during long-haul airplane flights (40), in school classrooms (41), and on ships (42), have been used to make assumptions about the duration and intensity of exposure to M. tuberculosis needed to generate secondary cases, little is known about the kinetics of transmission in the community. Specifically, the fraction of transmission that occurs (in settings of both high and low HIV prevalence) before index cases first report to the formal health system and what fraction occurs during the diagnostic process is unknown. This information would be useful in determining the importance of speed of testing and in understanding the degree to which active case-finding efforts in the community should be considered in different settings.
Although detection of individuals who have TB but are not smear positive has not been prioritized by national TB control programs in most countries, our assumptions related to the importance of such cases in disease transmission have not been well tested. Although there is an extensive literature describing the relatively low level of transmission from patients who are smear negative compared with their smear-positive counterparts (43, 44), little is known about the transmission impact of smear-negative TB cases in settings where prevalent HIV infection increases susceptibility to infection with M. tuberculosis and disease. Furthermore, it is not known what fraction of patients with smear-negative TB go on to develop smear-positive disease nor in what time frame.
Biology.
M. tuberculosis causes a range of pathology in humans and has a complex and highly evolved interaction with the immune system. Unfortunately, our understanding of the biology of the pathogen and of host-pathogen interactions in different disease states is incomplete, despite an extensive amount of work in this area. This biological uncertainty has largely frustrated attempts to exploit the host response as an effective diagnostic tool. We do not currently have any biomarkers, for example, that can be used to definitively distinguish individuals with prior, cured infection from those infected with dormant bacilli or from those with bacterial replication or incipient disease.
Laboratory studies have demonstrated that specific proteins can be upregulated in specific in vivo growth conditions, for example, within phagosomes (45). However, we know little about gene expression levels in M. tuberculosis as it is found in pulmonary cavities, inside granulomas, or even circulating in the blood of individuals with TB (46). More fundamental, from a diagnostic point of view, is our lack of information about the concentration of potentially detectable M. tuberculosis antigens or other moieties in clinical matrices such as urine, serum, sputum, or saliva. Published studies describing the potential for detecting mycobacterial antigens in clinical samples have, in large part, not been systematic, and most have focused on individual proteins or glycolipids abundant in culture filtrate such as MPT32 (47), the antigen 85 complex (48–50), the 38 kDa protein (51), and lipoarabinomannan (LAM) (52–56). Detection of mycobacterial antigens is an attractive approach to TB diagnosis, with the theoretical benefits of high specificity, correlation to mycobacterial burden, and independence from immune function. A number of proteomic approaches to the discovery of novel diagnostic target projects have been carried out (57, 58), but none has yet identified a readily detectable marker with demonstrated clinical utility.
As mentioned earlier, the development of simple molecular approaches to the detection of drug-sensitive and drug-resistant M. tuberculosis is a promising development. The development of more complete molecular methods for drug-resistance testing, which is both faster and safer than phenotypic methods, is hampered by incomplete knowledge of the molecular determinants of resistance for many of the important second-line agents used in treating MDR disease.
Technology.
At present, there are only three methods with proven clinical utility for the diagnosis of TB: microscopy, culture, and nucleic acid amplification. None of these approaches is simple enough to use in peripheral health care settings such as primary care clinics in the developing world. In fact, the only diagnostic technologies that have been widely used in lower levels of the health system for any purpose are enzymatic test strips for glucose monitoring in diabetes and immunochromatographic tests for pregnancy and various infectious diseases. Thus, developing simple point-of-care tests for TB requires either the dramatic simplification of one of the three current TB diagnostic methods or the successful development of tests based on the detection of diagnostic antibodies or TB-specific antigens. Both of these options present important technical challenges.
Lateral flow tests, such as commonly used for pregnancy testing, and other similar formats, suffer from limitations of sensitivity and are unlikely to detect mycobacterial proteins in the concentrations in which they are likely to be found in serum. Fortunately, a number of diagnostic companies are competing to develop point-of-care technologies that can detect low-abundance molecules in the pg/ml range and below.
TB moieties are likely to be much more abundant in sputum than in urine or serum. Sputum, however, is exceedingly difficult to work with as a diagnostic matrix, being viscous, inhibitory to many types of reactions, and highly variable in both nature and quantity. A range of processing protocols have been developed to liquefy or otherwise prepare sputum for use in diagnostic tests (59), but unfortunately, most of the protocols require laboratory procedures and equipment and are more complex than the detection methods they are meant to support. Although specimen-processing research is not attractive to research funding institutes, simple sample-processing methods that allow detection of DNA, RNA, or unaltered antigens are critically needed to accompany novel product development. Such translational research across all aspects of diagnostic research is now in full swing, and there is real hope for rapid point-of-care tests to diagnose both infection state and drug susceptibility.
Drugs
The current approach to treatment of TB comprises a cocktail of drugs taken daily over a period of at least six months, initially in a two-month “intensive” phase with four drugs — isoniazid, rifampicin, pyrazinamide, and ethambutol — followed by a four-month “continuation” phase with isoniazid and rifampicin. The last decade has seen a revival of drug development activity, and a number of drug candidates have been identified and are undergoing clinical evaluation (18) (Table 2).
Table 2 .
New drugs in development for treatment of TB

Toward an ultra-short treatment regimen.
M. tuberculosis bacilli are AFB that adapt well to growth in vitro in defined media with relatively simple requirements: a stable pH, an adequate carbon source, abundant oxygen, a surfactant to counteract the natural tendency of the organism to aggregate, a buffer for toxic lipids (typically serum albumin), and sufficient micronutrients to support essential enzymatic functions. The past two decades have seen the sequencing of the M. tuberculosis genome (60) and the development of ample genetic tools to manipulate the tubercle bacillus almost as casually as Escherichia coli (61). These tools have been employed to map essential genes (62–64) and define the biochemical function of many potential enzyme targets in the tubercle bacillus (65). In theory, the development of new drugs against M. tuberculosis would seem to be simply a matter of recapitulating the formalisms of conventional antibacterial programs — identify an essential target, preferably informed by a genomic analysis, develop potent inhibitors of the selected target, and chemically modify such inhibitors to confer drug-like qualities such as good oral bioavailability and low toxicity for the human host.
Unfortunately, this simple model of drug discovery has numerous pitfalls even when applied to more typical bacterial pathogens (66). Identifying hits can be surprisingly difficult for unusual bacterial enzymes due to a lack of biologically relevant chemical diversity, and translating successful enzyme inhibitors into compounds with activity against whole cells often fails because of poorly understood rules for bacterial cell penetration. Even when potent enzyme inhibitors penetrate cells effectively, failure to understand the biological “system” in sufficient detail to identify not only essential, but also rate-limiting, biochemical transformations often translates into failure. Worse, when applied to M. tuberculosis, even this imperfect model of drug discovery, and at times it seems any rational model of drug discovery, completely collapses beneath the complexities and peculiarities of the disease.
By the time an accurate diagnosis of TB is made, there is a substantial amount of tissue destruction spread among multiple lesions — each of which operates as an independent infection. Some lesions, such as those on the surface of cavities (often spherical lesions with air-liquid cores open to the bronchi and leaking mycobacteria to the pulmonary surface), contain actively replicating bacilli, whereas others (nodules or consolidations of various sizes) are still successfully containing their mycobacterial invaders in highly structured collections of immune cells known as granulomas (67). This heterogeneity of mycobacterial populations means either that a particular target must be vulnerable across all of the physiological states occupied by the bacilli or that the agents in a cocktail used must individually have targets that collectively cover all such states (68). Lesions containing mycobacteria in nonreplicating states are particularly problematic, as such mycobacteria have an intrinsically low basal metabolism that renders them much less susceptible to conventional mycobactericidal agents. In addition, the very structures created by the immune system to isolate and contain the mycobacteria (i.e., the granulomas) are poorly vascularized, dense structures, often with necrotic, acellular cores that may represent a substantial barrier to drug penetration from the bloodstream (see the extensive consolidation in the patient in Figure 1 for example). Finally, the cell envelope of M. tuberculosis represents a barrier for drug penetration more formidable than virtually any other bacterium in nature (69).
The conventional whole-cell mode of antibacterial discovery offers one path forward for the discovery of new drugs that target M. tuberculosis. With screens targeting replicating cells, however, such a path is likely to uncover only compounds with properties similar to those we have today and is unlikely to result in the quantum leap forward necessary to achieve rapid sterilization, shorten treatment times, and therefore improve global health. This is not to say that such strategies should be abandoned; MDR- and XDR-TB are important public health threats that deserve considerable effort, but the root cause of the global problem with drug-resistant TB-causing pathogens is the armamentarium of slowly acting agents currently used to treat drug-susceptible disease. Various strategies, which are discussed in detail below, are being engaged to combine the best of conventional methodology with the latest conceptual advances to transform TB drug discovery from lottery to logic.
Targeting nonreplicators — the “hypoxia hypothesis” and beyond.
Researchers in the TB field have learned some important lessons from lung cancer, another chronic disease with many similar pathological features. For example, anoxic lesions are a hallmark of some tumors, and there are many reasons to think that such anoxia is also a feature of some tuberculous lesions (70). M. tuberculosis is exquisitely adapted to respond to hypoxia and has a dedicated transcriptional program to realign metabolism for survival under hypoxic conditions (71). There are also in vitro models for generating hypoxia-induced nonreplicating cells, and these cells are refractory to killing by conventional antibiotics yet become susceptible to killing by anaerobe-specific agents like metronidazole (72). That observation has spurred an ongoing human clinical trial of the activity of metronidazole in MDR-TB patients (73). In addition, the hypoxia hypothesis underlies the excitement surrounding the development of nitroimidazoles such as PA-824 and OPC-67683, which are currently in clinical development for the treatment of TB and which have activity against both replicating and nonreplicating anaerobic cells (74–76). It is perhaps more telling that virtually any new agent developed for the treatment of TB today is screened for activity against anaerobic nonreplicating cells. Primary screens to identify lead compounds against such cells are part of several TB drug development programs.
Hypoxia, of course, is just one feature of the microenvironment in which the tubercle bacillus is embedded during disease. The lesional pH, the source of carbon, and the abundance of iron and various vitamins and cofactors are all measurable variables in different niches occupied by M. tuberculosis (77). Understanding the other essential features of the microenvironments present in the spectrum of lesions within patients with both active and latent TB infection would both identify novel targets and allow the reconstruction of in vitro conditions that reproduce these environments exactly — in other words, predictive models of drug efficacy. Current efforts to understand the biology of disease in increasingly realistic higher animal models are also essential to ground experimental hypotheses and test the effect of interrupting specific drug targets during established disease (78).
My kingdom for a target — the chemical genomics revolution.
There is deep uncertainty regarding how relevant a target identified as essential for M. tuberculosis growth under aerobic conditions on glucose as a primary carbon source (63) is to growth under conditions encountered by the organism in the host. It is therefore difficult to identify which targets, when interrupted by a small molecule during human disease, will result in a therapeutically beneficial effect. Perhaps even knowing which pathways are engaged by the bacillus during infection does not necessarily identify the step in the pathway that would make a suitable target for small molecule inhibition. In part, this is because some steps in a given pathway are naturally not rate limiting; thus, even a small amount of enzymatic activity may be sufficient to allow enough metabolic flux through a pathway in which even 99.9% inhibition would be insufficient to restrict mycobacterial growth or affect viability.
An attractive combination of conventional approaches using whole-cell screening and more modern genomics techniques is “chemical genomics,” which has been used to explore other areas of biology (79–82). In the case of TB, this technique aims to identify metabolic vulnerabilities of the mycobacteria under defined conditions. The technique at first glance looks like a normal whole-cell compound-based screen for activity, but by manipulating the in vitro assay conditions, one can select hits that are selective for one set of conditions or that are general for many sets. These hits are then coupled with genome-scale biology tools such as transcriptional profiles and/or genome resequencing technologies to identify the specific target being affected by the particular hit. The output from this strategy is a pharmacologically validated target that can then be the starting point for a conventional drug development program starting from high-throughput enzyme-based screening and extending through medicinal chemistry and lead optimization.
What will it take to get to a “Z-pack” for TB?
A patient with a bacterial upper respiratory tract infection caused by Streptococcus pyogenes often receives 5 days worth of curative therapy with azithromycin in a single package of 6 pills known as a Z-pack. Two weeks of therapy to cure a TB patient has an almost magical, transformative feeling to it. Scientifically, is this even a reasonable goal? The heart of the issue is really the fundamental killing kinetics of both replicating and nonreplicating cells. A culture containing more than 106 replicating cells/ml can be killed by a mixture of isoniazid and gatifloxacin in less than three hours in vitro (83), and although the killing kinetics are slower for stationary-phase cells, it still takes only four days to achieve similar kill with isoniazid and rifampicin. It is unclear why it then takes six months of therapy to cure a TB patient if these rates of kill apply in the patient. In the past, an issue like kill-rate would be a dissociated observation, made long after the commitment to a particular target and or molecular series was firm. This is an inevitable consequence of the old way of screening that puts the compound first and thinks about the biology later. Although this can be occasionally successful, as the development of the ATP synthase inhibitor R207910 clearly demonstrates (84), it doesn’t offer a sustainable way forward since the only real target validation, utility in human disease, must await the results of clinical trials of this agent.
At this point in time, there is much scientific uncertainty in understanding the fundamental difference between the in vitro and the in vivo kill rate, although there is considerable attention and effort directed at this phenomenon. The problem can at least be restated productively as understanding the effect of perturbing one step in a dynamic system encompassing both host and pathogen. From the vantage point of “systems biology,” at least the shape of the solution can be seen. Systems biology is roughly defined as the intersection between experimental and computational biology, and although in its infancy, this emerging alliance of biologists, programmers, and mathematicians holds considerable future promise for revolutionizing the search for TB drugs. Currently it is possible to describe discrete biochemical pathways as “systems” and integrate genetic and biochemical information to explore the effect of perturbing a step in such a pathway and predict which interventions would disrupt the pathway (85). Robust and predictive in silico models of even a single bacterial cell are still elusive but will undoubtedly emerge in the near term. In the future, what we need to do is integrate these models and pathways and reconstruct the physiology of both the organism and the host. Such models will allow us to predict which pathways would provide therapeutically useful responses.
Another equally important strategy is the development and validation of predictive animal models that more faithfully reflect human disease and an understanding of the effect of current agents in detail in these models. Nearly all of the current drugs in use have never been examined in anything other than mice, which develop little of the characteristic pathology of human disease. Without understanding the strengths and weaknesses of our current agents, we cannot begin to understand where they fall short. Likewise, most of the major TB drug trials were done in an era where there were few tools for looking in detail at the response to therapy both as reflected in the human immune response and even physically in terms of high-resolution imaging techniques. The cycle of a major expensive development program followed by a lengthy phase III clinical trial with only relapse as an end point must be broken. We must learn more from fewer patients and we must learn these things much faster if the results are to feed back into the quality of our model systems (whether animal or in silico) and inform the next generation of targets taken forward.
Vaccines
How close are we?
Considering the success of the natural immune response in controlling most TB infections, it is attractive to propose that a vaccine that can be delivered in advance of exposure to the pathogen and that mimics the natural immune response will further reduce the incidence of disease. Running counter to this “classical” vaccine approach is the fact that a major portion of cases of TB — particularly the sputum smear–positive cases that contribute most to the further spread of infection — arise from a population of individuals who successfully controlled an initial infection but then failed to mount a protective response to a subsequent reinfection or reactivation. In contrast to a typical vaccine-preventable disease, individuals whose immune system has been stimulated by a prior bout of active TB remain highly susceptible to subsequent reinfection (86). The conventional vaccination model, in which the adaptive immune response learns by repeated stimulation, seems to run backwards in the case of TB.
These two lines of argument offer radically differing prognoses for TB vaccine development. According to the first, we should learn the characteristics of the natural response and reproduce them using subunit and attenuated microbe tools of conventional vaccinology. From the second viewpoint, we should search for deficiencies in the natural response and repair these using some alternative form of immune manipulation. The classical vaccinology approach offers the potential for immediate practical application and is clearly a major priority for global health research and development. In parallel, it is, however, prudent to explore the fundamental biology required to underpin development of alternative vaccine approaches.
Front-line candidates and the IFN-γ hypothesis.
Albert Calmette and Camille Guérin followed Pasteur’s strategy of mimicking the natural response to infection using an attenuated pathogen to produce the BCG vaccine (87). An extensive literature attests to the fact that prior exposure to BCG vaccine accelerates development of the adaptive immune response to M. tuberculosis in animal models, conferring advantages in terms of reduced mycobacterial load and increased time to death (88–91). The same pattern can be seen in humans in the context of severe forms of childhood TB (92). However, BCG has variable efficacy against adult forms of TB, showing least effect where it is needed most (93). This is generally ascribed to an inherently weak immunogenicity, to adverse immune-mediated interactions with environmental mycobacteria, and to waning of immune memory (94). A logical approach to overcoming these problems is to boost BCG by priming an additional pool of mycobacteria-reactive T cells. This can be achieved by delivering M. tuberculosis antigens as purified proteins in adjuvant or in the form of recombinant viral vaccine vectors. Four such vaccines are lead candidates in current human clinical trials (22) (Table 3).
Table 3 .
New TB vaccine candidates in clinical trials

Antigens.
In spite of intensive efforts in experimental models, there are few guidelines for rational selection of vaccine antigens. An ability to elicit a recall response in healthy infected individuals provides evidence that a particular antigen is available as an immune target and is used as a common criterion. Proteins secreted from live mycobacteria become available for antigen presentation before those released by mycobacterial lysis and may have an advantage in triggering an earlier immune response. While this seems logical, rigorous proof for preferential recognition of secreted proteins is lacking and both secreted and nonsecreted antigens are included in current candidates (22) (Table 3). In general, collective effort has favored a family of mycolyl transferases found in the growth medium of mycobacterial cultures (Ag85A and Ag85B) and a family of low molecular weight proteins transported by specialized secretion systems (ESAT6, TB10.4) (95). With increasing use of ESAT6 in diagnostic tests, it would seem preferable to avoid its use in vaccines, as microbial antigens that are under selective pressure as targets of a protective immune response often undergo genetic variation, although there is no evidence of this in the case of the commonly studied M. tuberculosis antigens (96). It might be that protective immune responses in TB target a very broad repertoire of antigens, or it might be that we have yet to identify the key protective antigens.
Cytokines.
Protection against TB in mice is critically dependent on the Th1 immune response, an interplay among DCs, T cells, and macrophages that is promoted by IL-12 and IFN-γ (97, 98). Similarly, in humans, rare genetic defects that impair the IL-12–IFN-γ axis are associated with hypersusceptibility to mycobacterial disease (99). The central role of IFN-γ in stimulating antimicrobial functions of macrophages has encouraged its use as a marker for the beneficial immune response induced by vaccination (100). Although it is clear that some level of IFN-γ is essential for protection, the hypothesis that more IFN-γ equates with better protection remains to be proven. Clinical trials of the current front-line vaccine candidates will test this “more-better” hypothesis.
A multiplicity of mechanisms.
Although the IFN-γ hypothesis provides a simple marker for monotheistic vaccine development, many other immune parameters are affected by vaccination and might make an important contribution to efficacy (Figure 2). Multicolor flow cytometry provides a powerful tool for analysis of cytokine production and, by analogy with other infections, polyfunctional T cells that produce a combination of IFN-γ and IL-2 might be relevant to protection (101). It will be important to maximize the information gathered from current vaccine trials by including measurement of a broad range of immune parameters.
Figure 2. Mycobacterial infection activates a broad spectrum of immune responses.

Evidence from experimental models, from rare human genetic conditions, and from HIV-associated TB demonstrate that the Th1-mediated IL-12–IFN-γ axis is essential for prevention of progressive disease. Priming this response provides the major focus for the vaccine candidates currently being assessed in clinical trials. A broader view of the immune response suggests multiple alternative strategies that may influence the outcome of infection, including delivery of IFN-γ by alternative lymphocyte subsets, release of bacteria by cytolysis of poorly microbicidal cells, and activation of IFN-γ–independent pathways of mycobacterial killing. It is probable that all of these activities are subject to regulatory control mechanisms in vivo, and successful vaccination may ultimately depend on establishing or resetting multicellular immune networks rather than overactivation of a single pathway.
The particular cell type responsible for production of IFN-γ might be important. CD4+ T cells produce IFN-γ in response to antigens presented by MHC class II molecules. By responding to antigens presented by MHC class I molecules, CD8+ T cells might trigger responses to different cells at different stages of the infection process. BCG is less effective than M. tuberculosis in promoting presentation of antigens by the MHC class I pathway, due to reduced antigen transfer to adjacent cells (“cross-priming”) and perhaps to differences in lysis of phagosomal membranes (102). Recombinant BCG engineered to express a membrane-damaging hemolysin might have enhanced vaccine properties as a result of increased MHC class I expression (103). Similarly, CD1-restricted and γδ T cells that are specialized in recognizing nonprotein antigens from mycobacteria might offer further support to the overall protective response (104), although their potential for priming by vaccination is uncertain.
In addition to IFN-γ stimulation of antimicrobial functions, CD8+ T cells have the ability to kill infected cells by injection of cytotoxic granules. Depending on coordination with other immune cells, this could have a detrimental effect on promoting dissemination of the M. tuberculosis infection or a beneficial effect on exposing bacteria to freshly activated macrophages. Cytotoxic granules might also include antimicrobial peptides that have a direct effect on the intracellular mycobacteria. This could provide an important killing mechanism that is relevant for neutrophils and NKT cells, in addition to CD8+ T cells, that is complementary to IFN-γ activation, and that involves alternative signaling molecules such as vitamin D metabolites (105).
The range of immune mechanisms that might be involved in protection is increasing as our knowledge of immunology expands. A subset of T cell expressing the cytokine IL-17 is activated during mycobacterial infection. However, it remains to be determined whether this response plays a beneficial role — for example, by stimulating early recruitment of neutrophils — or a detrimental role in promoting pathology (106, 107).
Regulation and collateral damage.
The toxic effector molecules of the immune response are released within localized environments to maximize their chances of causing damage to the pathogen rather than to the host. Prolonged activation inevitably results in damage to surrounding tissues, however, and the ability to turn off an aggressive response is as fundamental to survival as the ability to turn it on. The importance of regulation of the immune response to TB is seen in the immune reconstitution inflammatory syndrome (IRIS), which causes acute pathology when an unregulated immune response is restored by antiretroviral therapy in individuals coinfected with HIV and M. tuberculosis (108). Dissection of regulatory mechanisms has proven a major challenge to immunology. Current interest focuses on a subset of CD4+CD25+ T cells that express the Foxp3 transcription factor, with cytokines IL-10 and TGF-β contributing to regulatory control (109). Perhaps rather than building a stronger Th1 response, a TB vaccine should aim to enhance the effectiveness of the preexisting response by resetting regulatory circuits.
Presumably, the limitation in the natural immune response — the reason that only 90% of the population is protected and that individuals who contain an initial infection remain at risk of secondary disease — is that it must balance the benefit of attacking the pathogen against the risk of unacceptable self damage. This equation might vary with the individual’s age. The first few years of life are associated with high susceptibility to TB, but very little disease is seen in older children (110). Susceptibility reemerges after adolescence and might be further enhanced in old age. From an evolutionary perspective, it can be argued that the natural immune response to TB has been selected to maximize the chance of reaching child-bearing age, leaving the researcher with potential scope for artificial enhancement in later life.
Immune subversion.
The lungs of a patient with pulmonary TB are transformed into an aerosol generator, and this is central to effective transmission. This transformation is dependent on an active Th1 immune response, comprising the same cells and cytokines that are required for protection. A successful disease cycle for M. tuberculosis involves an ability to withstand the initial impact of the immune response to establish an infection together with the ability to elicit a subsequent immune response that is sufficiently aggressive to promote transmission. Does M. tuberculosis actively program the host immune response in its own favor? It is attractive to imagine delivery of some inhibitor of the inflammatory response that is effective when mycobacterial numbers are low together with an inhibitor of the regulatory response that is effective at high population densities. Hints that M. tuberculosis might engage in constructive dialogue with the immune system are beginning to emerge from studies of the diversity among clinical isolates of M. tuberculosis (111). At least in animal models, subtle changes in the surface components expressed by M. tuberculosis can have radical effects on subsequent pathogenesis. For example, phenolic glycolipid produced by a subset of M. tuberculosis isolates belonging to the W-Beijing family show “hyperlethality” in murine disease models. Disruption of phenolic glycolipid synthesis results in loss of this hypervirulent phenotype without significantly affecting bacterial load during disease (112).
One form of immune subversion that has been proposed for M. tuberculosis is the induction of a Th2 response — measured in terms of production of IL-4 or its splice variants — analogous to that mounted in response to infection with intestinal parasites and during allergy (113). Th2 cytokines are certainly detectable in TB patients and probably contribute to disease-associated immunopathology. It remains to be determined, however, whether Th2 responses (triggered either by M. tuberculosis or by coinfecting parasites) influence earlier stages of infection.
The importance of immune subversion in the context of vaccine development is that, rather than trying to reproduce all of the details of the natural response to M. tuberculosis, there might be certain elements of the response that we should try and eliminate. Perhaps BCG is suboptimal because it is too close to the natural pathogen? An ideal vaccine strain might be one in which attenuation is mediated, not by loss of some physiological function necessary for survival, but as a consequence of induction of a stronger immune response. The phenotype of such a strain might include rapid clearance during infection of immunocompetent animals.
Homeostasis and forgetting.
It is easy to understand that HIV infection is associated with susceptibility to TB (114), but the trigger for secondary disease in the absence of overt immune suppression is not clear. Memory T cell pools primed during the initial infection might become depleted as a result of repeated stimulation by exposure to infection, driving cells toward terminal effector function and IFN-γ–mediated apoptosis (64). The mycobacteria might undergo some change that facilitates immune escape; there is every likelihood that M. tuberculosis oscillates between a range of replicating and nonreplicating phenotypes during infection and that this is accompanied by fluctuations in antigen profile. It might be that the mycobacteria disappear from the immunological radar when they are walled off within a granulomatous structure and that immune memory has faded by the time such structures disintegrate years later. Perhaps, after an initial period of conflict, host and pathogen adjust to some state of truce or equilibrium in which a small residual population of mycobacteria is accorded the tolerance normally associated with commensal flora? Comparisons with commensal bacteria might be as informative as comparisons with professionally aggressive pathogens in understanding the fundamental biology and immunology of our long-standing interaction with the tubercle bacillus.
Concluding remarks
There are formidable obstacles to development of point-of-care TB diagnostic tests, drugs that will make it possible to treat a patient with TB in weeks instead of months, and a vaccine that will provide long-term protection from infection. As the scientific enterprise struggles toward an ever-expanding knowledge base of both the physiology and the host-pathogen dynamic, the disease marches steadily on, killing millions each year. Fortunately, never in history has more brain power been focused on the scientific issues surrounding TB. Never has there been a higher chance that a real breakthrough in detection, treatment, and prevention tools will transform global health.
Acknowledgments
The authors wish to thank Adriana Alejandro-Osorio for her help in preparing the manuscript.
Footnotes
Nonstandard abbreviations used: AFB, acid-fast bacilli; BCG, bacillus Calmette-Guérin; DOTS, directly observed therapy, short course; DST, drug susceptibility testing; MDR-TB, multidrug-resistant TB; TB, tuberculosis; XDR-TB, extensively drug-resistant TB.
Conflict of interest: C.E. Barry III has declared that no conflict of interest exists. M.D. Perkins is an employee of FIND, which receives funding from the Bill and Melinda Gates Foundation. D.B. Young receives funding from the Bill and Melinda Gates Foundation and is a member of the Board of Directors of the Aeras Global TB Vaccine Foundation. K. Duncan is a shareholder of GlaxoSmithKline.
Citation for this article: J. Clin. Invest. 118:1255–1265 (2008). doi:10.1172/JCI34614.
References
- 1.[Anonymous]. 2004. World health report 2004: changing history. WHO. Geneva, Switzerland. http://www.who.int/whr/2004/en/report04_en.pdf. [Google Scholar]
- 2.Corbett E.L., et al. The growing burden of tuberculosis: global trends and interactions with the HIV epidemic. Arch. Intern. Med. 2003;163:1009–1021. doi: 10.1001/archinte.163.9.1009. [DOI] [PubMed] [Google Scholar]
- 3.[Anonymous]. 2007. Global tuberculosis control — surveillance, planning, financing. WHO. Geneva, Switzerland. http://www.who.int/tb/publications/global_report/2007/download_centre/en/index.html. [Google Scholar]
- 4.Girardi E., Raviglione M.C., Antonucci G., Godfrey-Faussett P., Ippolito G. Impact of the HIV epidemic on the spread of other diseases: the case of tuberculosis. AIDS. 2000;14(Suppl. 3):S47–S56. [PubMed] [Google Scholar]
- 5.Selwyn P.A., et al. A prospective study of the risk of tuberculosis among intravenous drug users with human immunodeficiency virus infection. . N. Engl. J. Med. 1989;320:545–550. doi: 10.1056/NEJM198903023200901. [DOI] [PubMed] [Google Scholar]
- 6.Bucher H.C., et al. Isoniazid prophylaxis for tuberculosis in HIV infection: a meta-analysis of randomized controlled trials. AIDS. 1999;13:501–507. doi: 10.1097/00002030-199903110-00009. [DOI] [PubMed] [Google Scholar]
- 7.[Anonymous]. 2006. The global plan to stop TB: 2006–2015. WHO. www.stoptb.org/globalplan/assets/documents/GlobalPlanFinal.pdf. [Google Scholar]
- 8.Raviglione M.C., Uplekar M.W. WHO’s new Stop TB Strategy. Lancet. 2006;367:952–955. doi: 10.1016/S0140-6736(06)68392-X. [DOI] [PubMed] [Google Scholar]
- 9.Koch R. Fortsetzung der mittheilungen uber ein heilmittel gegen tuberculose. Dtsch. Med. Wochenschr. 1891;17:101–102. [Google Scholar]
- 10.Ehrlich P. Aus dem Verein fur innere Medicin zu Berlin — Sitzung vom 1 Mai. Dtsch. Med. Wochenschr. 1882;8:269–270. [Google Scholar]
- 11.Matteelli A., et al. Multidrug-resistant and extensively drug-resistant Mycobacterium tuberculosis: epidemiology and control. Expert Rev. Anti Infect. Ther. 2007;5:857–871. doi: 10.1586/14787210.5.5.857. [DOI] [PubMed] [Google Scholar]
- 12.Gandhi N.R., et al. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet. 2006;368:1575–1580. doi: 10.1016/S0140-6736(06)69573-1. [DOI] [PubMed] [Google Scholar]
- 13.Perkins M.D. New diagnostic tools for tuberculosis. Int. J. Tuberc. Lung Dis. 2000;4:S182–S188. [PubMed] [Google Scholar]
- 14.Perkins M.D., Cunningham J. Facing the crisis: improving the diagnosis of tuberculosis in the HIV era. J. Infect. Dis. 2007;196(Suppl. 1):S15–S27. doi: 10.1086/518656. [DOI] [PubMed] [Google Scholar]
- 15.Palomino J.C. Nonconventional and new methods in the diagnosis of tuberculosis: feasibility and applicability in the field. Eur. Respir. J. 2005;26:339–350. doi: 10.1183/09031936.05.00050305. [DOI] [PubMed] [Google Scholar]
- 16.Connell T.G., Rangaka M.X., Curtis N., Wilkinson R.J. QuantiFERON-TB Gold: state of the art for the diagnosis of tuberculosis infection? Expert Rev. Mol. Diagn. 2006;6:663–677. doi: 10.1586/14737159.6.5.663. [DOI] [PubMed] [Google Scholar]
- 17.Williams K.J., Duncan K. Current strategies for identifying and validating targets for new treatment-shortening drugs for TB. Curr. Mol. Med. 2007;7:297–307. doi: 10.2174/156652407780598575. [DOI] [PubMed] [Google Scholar]
- 18.Ginsberg A.M., Spigelman M. Challenges in tuberculosis drug research and development. Nat. Med. 2007;13:290–294. doi: 10.1038/nm0307-290. [DOI] [PubMed] [Google Scholar]
- 19.Protopopova M., et al. In search of new cures for tuberculosis. Med. Chem. 2007;3:301–316. doi: 10.2174/157340607780620626. [DOI] [PubMed] [Google Scholar]
- 20.Gupta U.D., Katoch V.M., McMurray D.N. Current status of TB vaccines. Vaccine. 2007;25:3742–3751. doi: 10.1016/j.vaccine.2007.01.112. [DOI] [PubMed] [Google Scholar]
- 21.Sander C., McShane H. Translational mini-review series on vaccines: Development and evaluation of improved vaccines against tuberculosis. Clin. Exp. Immunol. 2007;147:401–411. doi: 10.1111/j.1365-2249.2006.03306.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skeiky Y.A., Sadoff J.C. Advances in tuberculosis vaccine strategies. Nat. Rev. Microbiol. 2006;4:469–476. doi: 10.1038/nrmicro1419. [DOI] [PubMed] [Google Scholar]
- 23.Lalvani A. Diagnosing tuberculosis infection in the 21st century: new tools to tackle an old enemy. Chest. 2007;131:1898–1906. doi: 10.1378/chest.06-2471. [DOI] [PubMed] [Google Scholar]
- 24.Menzies D., Pai M., Comstock G. Meta-analysis: new tests for the diagnosis of latent tuberculosis infection: areas of uncertainty and recommendations for research. Ann. Intern. Med. 2007;146:340–354. doi: 10.7326/0003-4819-146-5-200703060-00006. [DOI] [PubMed] [Google Scholar]
- 25.Porsa E., Cheng L., Graviss E.A. Comparison of an ESAT-6/CFP-10 peptide-based enzyme-linked immunospot assay to a tuberculin skin test for screening of a population at moderate risk of contracting tuberculosis. Clin. Vaccine Immunol. 2007;14:714–719. doi: 10.1128/CVI.00073-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pai M., Kalantri S., Dheda K. New tools and emerging technologies for the diagnosis of tuberculosis: part I. Latent tuberculosis. Expert Rev. Mol. Diagn. 2006;6:413–422. doi: 10.1586/14737159.6.3.413. [DOI] [PubMed] [Google Scholar]
- 27.Frieden T.R., Munsiff S.S. The DOTS strategy for controlling the global tuberculosis epidemic. Clin. Chest Med. 2005;26:197–205. doi: 10.1016/j.ccm.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 28.Madebo T., Lindtjorn B. Delay in treatment of pulmonary tuberculosis: an analysis of symptom duration among Ethiopian patients. Med. Gen. Med. 1999:E6. [PubMed] [Google Scholar]
- 29.Liam C.K., Tang B.G. Delay in the diagnosis and treatment of pulmonary tuberculosis in patients attending a university teaching hospital. Int. J. Tuberc. Lung Dis. 1997;1:326–332. [PubMed] [Google Scholar]
- 30.Friend J.H., Mason S., Harries A.D., Salaniponi F.M., Neuhann F. Management and outcome of TB suspects admitted to the medical wards of a central hospital in Malawi. Trop. Doct. 2005;35:93–95. doi: 10.1258/0049475054037165. [DOI] [PubMed] [Google Scholar]
- 31.Moll, A., et al. 2006. Identification of a multi-drug resistant tuberculosis cluster as a cause of death among HIV co-infected patients in rural South Africa [abstract 795]. Paper presented at the 13th Conference on Retroviruses and Opportunistic Infections. February 5–8. Denver, Colorado, USA. [Google Scholar]
- 32.[Anonymous]. From the Centers for Disease Control. Nosocomial transmission of multidrug-resistant tuberculosis among HIV-infected persons — Florida and New York, 1988–1991. JAMA. 1991;266:1483–1485. doi: 10.1001/jama.266.11.1483. [DOI] [PubMed] [Google Scholar]
- 33.Cunningham, J., and Perkins, M. 2006. Diagnostics for tuberculosis: global demand and market potential. WHO. Geneva, Switzerland. http://whqlibdoc.who.int/publications/2006/9241563303_eng.pdf. [Google Scholar]
- 34.Hasegawa N., et al. New simple and rapid test for culture confirmation of Mycobacterium tuberculosis complex: a multicenter study. J. Clin. Microbiol. 2002;40:908–912. doi: 10.1128/JCM.40.3.908-912.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anthony R.M., Kolk A.H., Kuijper S., Klatser P.R. Light emitting diodes for auramine O fluorescence microscopic screening of Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 2006;10:1060–1062. [PubMed] [Google Scholar]
- 36.Foundation for Innovative New Diagnostics. 2007. FIND and Zeiss team up to develop an affordable fluorescence microscope for the diagnosis of TB and other infectious diseases [press release]. http://www.finddiagnostics.org/news/press/zeiss_nov07.shtml. [Google Scholar]
- 37.Marinus-Barnard M., Albert H., Coetzee G., O’Brien R., Bosman M.E. Implementation of rapid molecular screening for multi-drug resistant tuberculosis in a high volume public health laboratory in South Africa. Am. J. Respir. Crit. Care Med. 2008 doi: 10.1164/rccm.200709-1436OC. In press. [DOI] [PubMed] [Google Scholar]
- 38.Helb, D., et al. 2005. Mycobacterium tuberculosis isolation detection, quantitation, and susceptibility testing in a single hands-free step: Integrating rapid sputum processing with real-time PCR. Paper presented at Keystone Symposium — Tuberculosis: Integrating Host and Pathogen Biology. April 2–7, 2005. Whistler, British Columbia, Canada. [Google Scholar]
- 39.Boehme C.C., et al. Operational feasibility of using loop-mediated isothermal amplification for diagnosis of pulmonary tuberculosis in microscopy centers of developing countries. J. Clin. Microbiol. 2007;45:1936–1940. doi: 10.1128/JCM.02352-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller M.A., Valway S., Onorato I.M. Tuberculosis risk after exposure on airplanes. Tuber. Lung Dis. 1996;77:414–419. doi: 10.1016/S0962-8479(96)90113-6. [DOI] [PubMed] [Google Scholar]
- 41.Ewer K., et al. Comparison of T-cell-based assay with tuberculin skin test for diagnosis of Mycobacterium tuberculosis infection in a school tuberculosis outbreak. Lancet. 2003;361:1168–1173. doi: 10.1016/S0140-6736(03)12950-9. [DOI] [PubMed] [Google Scholar]
- 42.Centers for Disease Control and Prevention (CDC). . Latent Tuberculosis infection among sailors and civilians aboard U.S.S. Ronald Reagan — United States, January–July 2006. MMWR Morb. Mortal. Wkly. Rep. 2007;55:1381–1382. [PubMed] [Google Scholar]
- 43.Grzybowski S., Barnett G.D., Styblo K. Contacts of cases of active pulmonary tuberculosis. Bull. Int. Union Tuberc. 1975;50:90–106. [PubMed] [Google Scholar]
- 44.Behr M.A., et al. Transmission of Mycobacterium tuberculosis from patients smear-negative for acid-fast bacilli. Lancet. 1999;353:444–449. doi: 10.1016/S0140-6736(98)03406-0. [DOI] [PubMed] [Google Scholar]
- 45.Mattow J., et al. Proteins unique to intraphagosomally grown Mycobacterium tuberculosis. Proteomics. 2006;6:2485–2494. doi: 10.1002/pmic.200500547. [DOI] [PubMed] [Google Scholar]
- 46.Rachman H., et al. Unique transcriptome signature of Mycobacterium tuberculosis in pulmonary tuberculosis. Infect. Immun. 2006;74:1233–1242. doi: 10.1128/IAI.74.2.1233-1242.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chanteau S., et al. 45/47 kilodalton (APA) antigen capture and antibody detection assays for the diagnosis of tuberculosis. Int. J. Tuberc. Lung Dis. 2000;4:377–383. [PubMed] [Google Scholar]
- 48.Wallis R.S., et al. Induction of the antigen 85 complex of Mycobacterium tuberculosis in sputum: a determinant of outcome in pulmonary tuberculosis treatment. J. Infect. Dis. 1998;178:1115–1121. doi: 10.1086/515701. [DOI] [PubMed] [Google Scholar]
- 49.Landowski C.P., et al. Combinatorial use of antibodies to secreted mycobacterial proteins in a host immune system-independent test for tuberculosis. J. Clin. Microbiol. 2001;39:2418–2424. doi: 10.1128/JCM.39.7.2418-2424.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bentley-Hibbert S.I., Quan X., Newman T., Huygen K., Godfrey H.P. Pathophysiology of antigen 85 in patients with active tuberculosis: antigen 85 circulates as complexes with fibronectin and immunoglobulin G. Infect. Immun. 1999;67:581–588. doi: 10.1128/iai.67.2.581-588.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Radhakrishnan V.V., Mathai A. A dot-immunobinding assay for the laboratory diagnosis of tuberculous meningitis and its comparison with enzyme-linked immunosorbent assay. J. Appl. Bacteriol. 1991;71:428–433. doi: 10.1111/j.1365-2672.1991.tb03812.x. [DOI] [PubMed] [Google Scholar]
- 52.Hamasur B., et al. Rapid diagnosis of tuberculosis by detection of mycobacterial lipoarabinomannan in urine. J. Microbiol. Methods. 2001;45:41–52. doi: 10.1016/S0167-7012(01)00239-1. [DOI] [PubMed] [Google Scholar]
- 53.Tessema T.A., et al. Clinical and radiological features in relation to urinary excretion of lipoarabinomannan in Ethiopian tuberculosis patients. Scand. J. Infect. Dis. 2002;34:167–171. doi: 10.1080/00365540110077254. [DOI] [PubMed] [Google Scholar]
- 54.Tessema T.A., et al. Circulating antibodies to lipoarabinomannan in relation to sputum microscopy, clinical features and urinary anti-lipoarabinomannan detection in pulmonary tuberculosis. Scand. J. Infect. Dis. 2002;34:97–103. doi: 10.1080/00365540110077263. [DOI] [PubMed] [Google Scholar]
- 55.Tessema T.A., Hamasur B., Bjun G., Svenson S., Bjorvatn B. Diagnostic evaluation of urinary lipoarabinomannan at an Ethiopian tuberculosis centre. Scand. J. Infect. Dis. 2001;33:279–284. doi: 10.1080/003655401300077306. [DOI] [PubMed] [Google Scholar]
- 56.Boehme C., et al. Detection of mycobacterial lipoarabinomannan with an antigen-capture ELISA in unprocessed urine of Tanzanian patients with suspected tuberculosis. Trans. R. Soc. Trop. Med. Hyg. 2005;99:893–900. doi: 10.1016/j.trstmh.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 57.Agranoff D., et al. Identification of diagnostic markers for tuberculosis by proteomic fingerprinting of serum. Lancet. 2006;368:1012–1021. doi: 10.1016/S0140-6736(06)69342-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cole, R.A., et al. 2005. Identification of M. tuberculosis proteins in the body fluids of tuberculosis patients. Paper presented at the IUATLD World Congress of Lung Health. October 21. Madrid, Spain. [Google Scholar]
- 59.Steingart K.R., Ramsay A., Pai M. Optimizing sputum smear microscopy for the diagnosis of pulmonary tuberculosis. Expert Rev. Anti Infect. Ther. 2007;5:327–331. doi: 10.1586/14787210.5.3.327. [DOI] [PubMed] [Google Scholar]
- 60.Cole S.T., et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 61.Murry J.P., Rubin E.J. New genetic approaches shed light on TB virulence. Trends Microbiol. 2005;13:366–372. doi: 10.1016/j.tim.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 62.Sassetti C.M., Boyd D.H., Rubin E.J. Comprehensive identification of conditionally essential genes in mycobacteria. Proc. Natl. Acad. Sci. U. S. A. 2001;98:12712–12717. doi: 10.1073/pnas.231275498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sassetti C.M., Boyd D.H., Rubin E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003;48:77–84. doi: 10.1046/j.1365-2958.2003.03425.x. [DOI] [PubMed] [Google Scholar]
- 64.Sassetti C.M., Rubin E.J. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U. S. A. 2003;100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Balganesh T.S., Furr B.J. Molecular approaches to target discovery: evaluating targets for anti-tuberculosis drug discovery programmes. Infect. Disord. Drug Targets. 2007;7:120–126. doi: 10.2174/187152607781001826. [DOI] [PubMed] [Google Scholar]
- 66.Payne D.J., Gwynn M.N., Holmes D.J., Pompliano D.L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discov. 2007;6:29–40. doi: 10.1038/nrd2201. [DOI] [PubMed] [Google Scholar]
- 67.Kaplan G., et al. Mycobacterium tuberculosis growth at the cavity surface: a microenvironment with failed immunity. Infect. Immun. 2003;71:7099–7108. doi: 10.1128/IAI.71.12.7099-7108.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Duncan K. Identification and validation of novel drug targets in tuberculosis. Curr. Pharm. Des. 2004;10:3185–3194. doi: 10.2174/1381612043383223. [DOI] [PubMed] [Google Scholar]
- 69.Brennan P.J. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis (Edinb.). 2003;83:91–97. doi: 10.1016/S1472-9792(02)00089-6. [DOI] [PubMed] [Google Scholar]
- 70.Via L., et al. Tuberculous granulomas are hypoxic in guinea pigs, rabbits and non-human primates. J. Infect. Dis. 2008 doi: 10.1128/IAI.01515-07. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sherman D.R., et al. Regulation of the Mycobacterium tuberculosis hypoxic response gene encoding alpha -crystallin. Proc. Natl. Acad. Sci. U. S. A. 2001;98:7534–7539. doi: 10.1073/pnas.121172498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wayne L.G., Sramek H.A. Metronidazole is bactericidal to dormant cells of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1994;38:2054–2058. doi: 10.1128/aac.38.9.2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.National Institute of Allergy and Infectious Diseases (NIAID), and National Institutes of Health Clinical Center (CC). 2006. Metronidazole for pulmonary tuberculosis (South Korea). http://clinicaltrials.gov/ct/show/NCT00425113?order = 3. [Google Scholar]
- 74.Barry C.E., 3rd, Boshoff H.I., Dowd C.S. Prospects for clinical introduction of nitroimidazole antibiotics for the treatment of tuberculosis. Curr. Pharm. Des. 2004;10:3239–3262. doi: 10.2174/1381612043383214. [DOI] [PubMed] [Google Scholar]
- 75.Matsumoto M., et al. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 2006;3:e466. doi: 10.1371/journal.pmed.0030466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stover C.K., et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature. 2000;405:962–966. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 77.McKinney J.D. In vivo veritas: the search for TB drug targets goes live. Nat. Med. 2000;6:1330–1333. doi: 10.1038/82142. [DOI] [PubMed] [Google Scholar]
- 78.Flynn J.L. Lessons from experimental Mycobacterium tuberculosis infections. . Microbes Infect. 2006;8:1179–1188. doi: 10.1016/j.micinf.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 79.Stockwell B.R. Exploring biology with small organic molecules. Nature. 2004;432:846–854. doi: 10.1038/nature03196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lenz G.R., Nash H.M., Jindal S. Chemical ligands, genomics and drug discovery. Drug Discov. Today. 2000;5:145–156. doi: 10.1016/S1359-6446(00)01468-9. [DOI] [PubMed] [Google Scholar]
- 81.Smukste I., Stockwell B.R. Advances in chemical genetics. Annu. Rev. Genomics Hum. Genet. 2005;6:261–286. doi: 10.1146/annurev.genom.6.080604.162136. [DOI] [PubMed] [Google Scholar]
- 82.Inglese J., et al. High-throughput screening assays for the identification of chemical probes. Nat. Chem. Biol. 2007;3:466–479. doi: 10.1038/nchembio.2007.17. [DOI] [PubMed] [Google Scholar]
- 83.Paramasivan C.N., Sulochana S., Kubendiran G., Venkatesan P., Mitchison D.A. Bactericidal action of gatifloxacin, rifampin, and isoniazid on logarithmic- and stationary-phase cultures of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2005;49:627–631. doi: 10.1128/AAC.49.2.627-631.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Andries K., et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 85.Boshoff, H.I., and Barry, C.E., 3rd. 2007. Systems biological approaches to infectious diseases. Birkhauser. Basel, Switzerland. 349 pp. [Google Scholar]
- 86.van Rie A., et al. Exogenous reinfection as a cause of recurrent tuberculosis after curative treatment. N. Engl. J. Med. 1999;341:1174–1179. doi: 10.1056/NEJM199910143411602. [DOI] [PubMed] [Google Scholar]
- 87.Calmette A., Guérin C. Nouvelles recherches expérimentales sur la vaccination des bovidés contre la tuberculose. Ann. Inst. Pasteur. 1920;34:553–560. [Google Scholar]
- 88.Smith D.W., McMurray D.N., Wiegeshaus E.H., Grover A.A., Harding G.E. Host-parasite relationships in experimental airborne tuberculosis. IV. Early events in the course of infection in vaccinated and nonvaccinated guinea pigs. Am. Rev. Respir. Dis. 1970;102:937–949. doi: 10.1164/arrd.1970.102.6.937. [DOI] [PubMed] [Google Scholar]
- 89.Langermans J.A., et al. Divergent effect of bacillus Calmette-Guerin (BCG) vaccination on Mycobacterium tuberculosis infection in highly related macaque species: implications for primate models in tuberculosis vaccine research. Proc. Natl. Acad. Sci. U. S. A. 2001;98:11497–11502. doi: 10.1073/pnas.201404898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hewinson R.G., Vordermeier H.M., Buddle B.M. Use of the bovine model of tuberculosis for the development of improved vaccines and diagnostics. Tuberculosis (Edinb.). 2003;83:119–130. doi: 10.1016/S1472-9792(02)00062-8. [DOI] [PubMed] [Google Scholar]
- 91.Orme I.M. Immunity to mycobacteria. Curr. Opin. Immunol. 1993;5:497–502. doi: 10.1016/0952-7915(93)90029-R. [DOI] [PubMed] [Google Scholar]
- 92.Colditz G.A., et al. The efficacy of bacillus Calmette-Guerin vaccination of newborns and infants in the prevention of tuberculosis: meta-analyses of the published literature. Pediatrics. 1995;96:29–35. [PubMed] [Google Scholar]
- 93.Fine P.E. The BCG story: lessons from the past and implications for the future. Rev. Infect. Dis. 1989;11(Suppl. 2):S353–S359. doi: 10.1093/clinids/11.supplement_2.s353. [DOI] [PubMed] [Google Scholar]
- 94.Sterne J.A., Rodrigues L.C., Guedes I.N. Does the efficacy of BCG decline with time since vaccination? Int. J. Tuberc. Lung Dis. 1998;2:200–207. [PubMed] [Google Scholar]
- 95.DiGiuseppe Champion P.A., Cox J.S. Protein secretion systems in Mycobacteria. Cell. Microbiol. 2007;9:1376–1384. doi: 10.1111/j.1462-5822.2007.00943.x. [DOI] [PubMed] [Google Scholar]
- 96.Musser J.M., Amin A., Ramaswamy S. Negligible genetic diversity of mycobacterium tuberculosis host immune system protein targets: evidence of limited selective pressure. Genetics. 2000;155:7–16. doi: 10.1093/genetics/155.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Flynn J.L., et al. An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. J. Exp. Med. 1993;178:2249–2254. doi: 10.1084/jem.178.6.2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Flynn J.L., Chan J. Immunology of tuberculosis. Annu. Rev. Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 99.Casanova J.L., Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu. Rev. Immunol. 2002;20:581–620. doi: 10.1146/annurev.immunol.20.081501.125851. [DOI] [PubMed] [Google Scholar]
- 100.McShane H., et al. Recombinant modified vaccinia virus Ankara expressing antigen 85A boosts BCG-primed and naturally acquired antimycobacterial immunity in humans. Nat. Med. 2004;10:1240–1244. doi: 10.1038/nm1128. [DOI] [PubMed] [Google Scholar]
- 101.Foulds K.E., Wu C.Y., Seder R.A. Th1 memory: implications for vaccine development. Immunol. Rev. 2006;211:58–66. doi: 10.1111/j.0105-2896.2006.00400.x. [DOI] [PubMed] [Google Scholar]
- 102.van der Wel N., et al. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell. 2007;129:1287–1298. doi: 10.1016/j.cell.2007.05.059. [DOI] [PubMed] [Google Scholar]
- 103.Grode L., et al. Increased vaccine efficacy against tuberculosis of recombinant Mycobacterium bovis bacille Calmette-Guerin mutants that secrete listeriolysin. J. Clin. Invest. 2005;115:2472–2479. doi: 10.1172/JCI24617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Behar S.M., Porcelli S.A. CD1-restricted T cells in host defense to infectious diseases. Curr. Top. Microbiol. Immunol. 2007;314:215–250. doi: 10.1007/978-3-540-69511-0_9. [DOI] [PubMed] [Google Scholar]
- 105.Martineau A.R., et al. IFN-gamma- and TNF-independent vitamin D-inducible human suppression of mycobacteria: the role of cathelicidin LL-37. J. Immunol. 2007;178:7190–7198. doi: 10.4049/jimmunol.178.11.7190. [DOI] [PubMed] [Google Scholar]
- 106.Khader S.A., et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat. Immunol. 2007;8:369–377. doi: 10.1038/ni1449. [DOI] [PubMed] [Google Scholar]
- 107.Khader S.A., Cooper A.M. IL-23 and IL-17 in tuberculosis. Cytokine. 2008;41:79–83. doi: 10.1016/j.cyto.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McIlleron H., Meintjes G., Burman W.J., Maartens G. Complications of antiretroviral therapy in patients with tuberculosis: drug interactions, toxicity, and immune reconstitution inflammatory syndrome. J. Infect. Dis. 2007;196(Suppl. 1):S63–S75. doi: 10.1086/518655. [DOI] [PubMed] [Google Scholar]
- 109.Sakaguchi S., Powrie F. Emerging challenges in regulatory T cell function and biology. Science. 2007;317:627–629. doi: 10.1126/science.1142331. [DOI] [PubMed] [Google Scholar]
- 110. Dubos, R.J. 1952. The white plague: tuberculosis, man and society. Rutgers University Press. Piscataway, New Jersey, USA. 320 pp. [Google Scholar]
- 111.Gagneux S., Small P.M. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect. Dis. 2007;7:328–337. doi: 10.1016/S1473-3099(07)70108-1. [DOI] [PubMed] [Google Scholar]
- 112.Reed M.B., et al. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. 2004;431:84–87. doi: 10.1038/nature02837. [DOI] [PubMed] [Google Scholar]
- 113.Rook G.A., Dheda K., Zumla A. Immune responses to tuberculosis in developing countries: implications for new vaccines. Nat. Rev. Immunol. 2005;5:661–667. doi: 10.1038/nri1666. [DOI] [PubMed] [Google Scholar]
- 114.Hopewell P.C. Impact of human immunodeficiency virus infection on the epidemiology, clinical features, management, and control of tuberculosis. Clin. Infect. Dis. 1992;15:540–547. doi: 10.1093/clind/15.3.540. [DOI] [PubMed] [Google Scholar]
- 115. Arora, S.K., et al. 2004. Synthesis and in vitro anti-mycobacterial activity of a novel anti-TB composition LL4858 [abstract F-1115]. Paper presented at the 44th Interscience Conference on Antimicrobial Agents and Chemotherapy. October 30–November 2. Washington, DC, USA. [Google Scholar]
- 116. Sinha, R.K., et al. 2004. In vivo activity of LL4858 against Mycobacterium tuberculosis. Paper presented at the 44th Interscience Conference on Antimicrobial Agents and Chemotherapy. October 30–November 2. Washington, DC, USA. [Google Scholar]
- 117.Lee R.E., et al. Combinatorial lead optimization of [1,2]-diamines based on ethambutol as potential antituberculosis preclinical candidates. . J. Comb. Chem. 2003;5:172–187. doi: 10.1021/cc020071p. [DOI] [PubMed] [Google Scholar]
- 118.Jia L., et al. Pharmacodynamics and pharmacokinetics of SQ109, a new diamine-based antitubercular drug. Br. J. Pharmacol. 2005;144:80–87. doi: 10.1038/sj.bjp.0705984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Sequella Inc. SQ109 Therapeutic: phase I [proposal]. www.sequella.com/docs/Sequella_Licensing_SQ109_v8_(dist).pdf. [Google Scholar]
- 120. Otsuka Frankfurt Research Institute GmbH. 2007. Safety, efficacy and pharmacokinetics of OPC-67683 in patients with pulmonary tuberculosis. http://clinicaltrials.gov/ct2/show/NCT00401271?term=otsuka+opc-67683rank=1. [Google Scholar]
- 121.Stover C.K., et al. Complete genome sequence of Pseudomonas aeruginosa PA01, an opportunistic pathogen. Nature. 2000;406:959–964. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- 122. Global Alliance for TB Drug Development. 2007. TB alliance advances two drugs in clinical trials on path to faster, better tuberculosis treatments. http://www.tballiance.org/newscenter/view-brief.php?id=726 [Google Scholar]
- 123. Tibotec Pharmaceuticals Limited. What is TB and what is its cause? http://www.tibotec.com/bgdisplay.jhtml?itemname=TB_disease. [Google Scholar]
- 124.Miyazaki E., Miyazaki M., Chen J.M., Chaisson R.E., Bishai W.R. Moxifloxacin (BAY12-8039), a new 8-methoxyquinolone, is active in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 1999;43:85–89. doi: 10.1128/aac.43.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nuermberger E.L., et al. Moxifloxacin-containing regimens of reduced duration produce a stable cure in murine tuberculosis. Am. J. Respir. Crit. Care Med. 2004;170:1131–1134. doi: 10.1164/rccm.200407-885OC. [DOI] [PubMed] [Google Scholar]
- 126. The REMoxTB website. Rapid evaluation of moxifloxacin in the treatment of sputum smear positive tuberculosis. www.remoxtb.org. [Google Scholar]
- 127.Alvirez-Freites E.J., Carter J.L., Cynamon M.H. In vitro and in vivo activities of gatifloxacin against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2002;46:1022–1025. doi: 10.1128/AAC.46.4.1022-1025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. WHO. World Health Organization. 2005. New tuberculosis therapy offers potential shorter treatment [press release]. www.who.int/mediacentre/news/releases/2005/pr71/en/index.html. [Google Scholar]
- 129.Pathan A.A., et al. Boosting BCG with recombinant modified vaccinia ankara expressing antigen 85A: different boosting intervals and implications for efficacy trials. PLoS ONE. 2007;2:e1052. doi: 10.1371/journal.pone.0001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. McShane, H., and Hill, A.V. 2005. A Phase I study of the safety and immunogenicity of a recombinant MVA vaccine encoding a secreted antigen from M. Tuberculosis, antigen 85A, delivered intradermally by a needle injection in healthy volunteers who have previously received BCG. http://clinicaltrials.gov/ct2/show/NCT00427830?term=TB+vaccinesrank=23 [Google Scholar]
- 131.Radosevic K., et al. Protective immune responses to a recombinant adenovirus type 35 tuberculosis vaccine in two mouse strains: CD4 and CD8 T-cell epitope mapping and role of gamma interferon. Infect. Immun. 2007;75:4105–4115. doi: 10.1128/IAI.00004-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Crucell N.V., and Aeras Global TB Vaccine Foundation. 2007. Aeras and Crucell announce start of a tuberculosis vaccine clinical trial in the United States. Aeras commits a further USD 5 million to the development of the tuberculosis vaccine [press release]. http://preventb.org/news/documents/AerasandCrucellAnnounceStartofaTBVaccineClinicalTrial_FINAL_Dec212007.pdf. [Google Scholar]
- 133.Brandt L., et al. The protective effect of the Mycobacterium bovis BCG vaccine is increased by coadministration with the Mycobacterium tuberculosis 72-kilodalton fusion polyprotein Mtb72F in M. tuberculosis-infected guinea pigs. Infect. Immun. 2004;72:6622–6632. doi: 10.1128/IAI.72.11.6622-6632.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Clinical Trials and GlaxoSmithKline. 2008. Immunogenicity and safety of a candidate tuberculosis (TB) vaccine given to healthy adults in a TB-endemic region. http://clinicaltrials.gov/ct2/show/NCT00600782?term=TB+vaccinesrank=8. [Google Scholar]
- 135.Doherty T.M., et al. Comparative analysis of different vaccine constructs expressing defined antigens from Mycobacterium tuberculosis. J. Infect. Dis. 2004;190:2146–2153. doi: 10.1086/425931. [DOI] [PubMed] [Google Scholar]
- 136. Euro Adhoc and Statens Serum Institute. 2005. New TB vaccine enters clinical trials [press release]. http://ots.euroadhoc.com/irmeldung.php?schluessel=OTA_20051118_OTA0008&ag=OTA. [Google Scholar]
- 137.Agger E.M., et al. Protective immunity to tuberculosis with Ag85B-ESAT-6 in a synthetic cationic adjuvant system IC31. Vaccine. 2006;24:5452–5460. doi: 10.1016/j.vaccine.2006.03.072. [DOI] [PubMed] [Google Scholar]
- 138. Aeras Global TB Vaccine Foundation. 2007. Statens Serum Institut (SSI), Intercell (ICLL), and Aeras Global Tuberculosis Vaccine Foundation (Aeras) announce the initiation of a clinical trial for a novel vaccine candidate against tuberculosis (TB) [press release]. http://www.aeras.org/TB-SSI.html. [Google Scholar]
- 139.Horwitz M.A., Harth G. A new vaccine against tuberculosis affords greater survival after challenge than the current vaccine in the guinea pig model of pulmonary tuberculosis. Infect. Immun. 2003;71:1672–1679. doi: 10.1128/IAI.71.4.1672-1679.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Aeras Global TB Vaccine Foundation. 2004. Clinical trial of a new TB vaccine in the U.S. begins: new vaccine may prove more potent than 100-year-old current vaccine [press release]. http://www.aeras.org/news/releases/02172004.html. [Google Scholar]
- 141. Max Planck Institute for Infection Biology. 2004. Cooperation to create a new tuberculosis vaccine: Max Planck Institute for Infection Biology and MOLOGEN initiate development of a new tuberculosis subunit vaccine [press release]. http://www.mpg.de/english/illustrationsDocumentation/documentation/pressReleases/2004/pressRelease20041008/index.html. [Google Scholar]
- 142.Stanford J.L., et al. Immunotherapy with Mycobacterium vaccae as an adjunct to chemotherapy in the treatment of pulmonary tuberculosis. Tubercle. 1990;71:87–93. doi: 10.1016/0041-3879(90)90002-P. [DOI] [PubMed] [Google Scholar]
- 143.Katoch K., et al. Potential of Mw as a prophylactic vaccine against pulmonary tuberculosis. Vaccine. 2008 doi: 10.1016/j.vaccine.2007.12.025. In press. [DOI] [PubMed] [Google Scholar]
- 144. Sharma, S.K., et al. 2006. Ministry of Science and Technology, India Cadila Pharmaceuticals. http://clinicaltrials.gov/ct2/show/NCT00265226?term=TB+vaccinesrank=24. [Google Scholar]
- 145.Cardona P.J. RUTI: a new chance to shorten the treatment of latent tuberculosis infection. Tuberculosis (Edinb.). 2006;86:273–289. doi: 10.1016/j.tube.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 146. Cardona, P.J., and Germans Trias i Pujol Hospital. 2008. Tolerability and immunogenicity of RUTI antituberculous vaccine in healthy volunteers: Phase I clinical trial. http://clinicaltrials.gov/ct2/show/record/NCT00546273. [Google Scholar]