

Abstract
We have recently shown that the pancreatic hormone glucagons-induced phosphorylation of mitogen-activated protein (MAP) kinase ERK 1/2 as well as growth and proliferation of rat glomerular mesangial cells (MCs) via activation of cAMP-dependent protein kinase A (PKA)-and phospholipase C (PLC)/Ca2+-mediated signaling pathways. Since circulating glucagon and tissue angiotensin II (Ang II) levels are inappropriately elevated in type 2 diabetes, we tested the hypothesis that glucagon induces phosphorylation of ERK 1/2 in MCs by interacting with Ang II receptor signaling. Stimulation of MCs by glucagon (10 nM) induced a marked increase in intracellular [Ca2+]i that was abolished by [Des-His1, Glu9]-glucagon (1 μM), a selective glucagon receptor antagonist. Both glucagon and Ang II-induced ERK 1/2 phosphorylation (glucagon: 214 ± 14%; Ang II: 174 ± 16%; p < 0.001 versus control), and these responses were inhibited by the AT1 receptor blocker losartan (glucagon + losartan: 77 ± 14%; Ang II + losartan: 84 ± 18%; p < 0.01 versus glucagon or Ang II) and the AT2 receptor blocker PD 123319 (glucagon + PD: 78 ± 7%; Ang II + PD: 87 ± 7%; p < 0.01 versus glucagon or Ang II). Inhibition of cAMP-dependent PKA with H89 (1 μM) or PLC with U73122 (1 μM) also markedly attenuated the phosphorylation of ERK 1/2 induced by glucagon (glucagon + U73122: 109 ± 15%; glucagon + H89: 113 ± 16%; p < 0.01 versus glucagon) or Ang II (Ang II + U73122: 111 ± 13%; Ang II + H89: 86 ± 10%; p < 0.01 versus Ang II). Wortmannin (1 μM), a selective PI 3-kinase inhibitor, also blocked glucagon- or Ang II-induced ERK 1/2 phosphorylation. These results suggest that AT1 receptor-activated cAMP-dependent PKA, PLC and PI 3-kinase signaling is involved in glucagon-induced MAP kinase ERK 1/2 phosphorylation in MCs. The inhibitory effect of PD 123319 on glucagon-induced ERK 1/2 phosphorylation further suggests that AT2 receptors also play a similar role in this response.
Keywords: Angiotensin II, Diabetes mellitus, Glucagon, Mesangial cells, Signaling cross-talk
1. Introduction
Diabetes mellitus is a major risk factor for the development of cardiovascular and renal target organ damage, leading to hypertension, atherosclerosis, ischemic heart disease and diabetic nephropathy [1,2]. Types 1 and 2 diabetes mellitus together affect more than 20 million Americans and rank as the sixth leading cause of disease-related death in the US. Type 1 diabetes is due to insulin deficiency, and therefore requires treatment with insulin or transplantation of pancreatic β cells [3]. By contrast, the majority of the diabetic population has type 2 diabetes, which is not insulin-dependent and in most case does not require insulin therapy[1,2]. Type 2 diabetes is characterized by persistent hyperglycemia, impaired glucose tolerance, glomerular hyperfiltration, and progression of albuminuria, ultimately leading to renal injury [1,2]. Although increased insulin resistance rather than insulin deficiency plays a key role in the development of type 2 diabetes [1], the factors that contribute to increased insulin resistance remain incompletely understood.
The pancreatic hormone glucagon [4–6] and the vasoactive peptide angiotensin II (Ang II) [7–9] are two important factors implicated in the development of insulin resistance in type 2 diabetes. Clinical studies have shown that an inappropriate increase in the hyperglycemic hormone glucagon, not deficiency of the hypoglycemic hormone insulin, is the key characteristic of type 2 diabetes [1,10]. Glucagon binds to Gs protein-coupled receptors, activating glycogenolytic and gluconeogenic pathways to induce hyperglycemia through cAMP-dependent protein kinase A (PKA)- and phospholipase C/IP3/Ca2+ signaling pathways [11–14]. Hyperglycemia promotes cell proliferation and growth (and/or hypertrophy) and induces tissue fibrosis by interacting with Ang II [15–17]. However, the role of glucagon in type 2 diabetic glomerular injury and the underlying cellular mechanisms are virtually unknown. By contrast, the role of Ang II in the development of diabetic nephropathy has been studied in humans using angiotensin-converting enzyme (ACE) inhibitors or Ang II type 1 receptor blockers [7–9]. There is evidence that the local rennin–angiotensin system is activated in the kidney of diabetic animals and humans[18,19]. In addition to acting as a vasoactive hormone, Ang II is also a glycogenolytic and gluconeogenic peptide [20,21] as well as a growth factor and proliferative cytokine [22,23]. In the kidney, Ang II, acting via AT1 receptors, causes contraction of vascular smooth muscle cells (VSMCs) and mesangial cells (MCs) and induces cell growth and proliferation through receptor-mediated PLC and cAMP signaling [22,24].
We have recently shown that glucagon stimulates MC growth and proliferation by inducing MAP kinase signaling via activation of cAMP-dependent PKA and phospholipase C [25]. However, we do not know whether these effects of glucagons involve interactions with Ang II or AT1/AT2 receptor signaling. Hyperglycemia has been shown to stimulate Ang II formation in MCs [19] and potentiate Ang II-induced growth and proliferation of VSMCs [15,17] and renal epithelial cells [16,26]. Glucagon may act on MCs either directly by activating its receptors or indirectly through hyperglycemia. We hypothesized that the pro-growth and proliferative effects of glucagon in MCs are mediated in part by receptor-activated MAP kinases ERK 1/2 and involve cross-talk or interactions with Ang II and/or its receptor signaling.
2. Materials and methods
2.1. Materials
Cultured glomerular MCs were obtained from American Type Culture Collection (ATCC). These cells are derived from the rat kidney and show both characteristics of glomerular MCs and typical responses to Ang II and glucagon [15,25,27]. RPMI-1640 medium, trypsin, heat-inactivated fetal bovine serum (FBS) and antibiotics (penicillin and streptomycin) were purchased from ATCC. Human Val5-Ang II, glucagon and [Des-His1-Glu9] glucagon and Ang II enzyme immunoassay kits were obtained from Biochem/Peninsula Laboratories. cAMP enzyme immunoassay kits were purchased from R&D. The AT1 receptor antagonist losartan was a gift from Merck Pharmaceuticals and the AT2 receptor antagonist PD 123319 was donated by Pfizer. A rabbit polyclonal AT1 receptor antibody targeting the N-terminal extracellular domain of the human AT1 receptor (N-10) was purchased from Santa Cruz. Western blot supplies were obtained from Amersham. U73122, H-89 and wortmannin were purchased from Sigma.
2.2. Mesangial cell culture
Rat glomerular MCs were maintained in RPMI-1640 medium (10 mM HEPES, 1 mM sodium pyruvate, 2 mM L-glutamine, 5 mM glucose, 1500 mg sodium bicarbonate/l; ATCC) containing 12% fetal calf serum (ATCC), penicillin (100 U/ml) and streptomycin (100 μg/ml) at 37 °C in 5% CO2/95% O2 as described previously [19,25,28]. MCs from passages 6 to 10 were sub-cultured in six-well plates containing the above growth medium until they reached 80% confluence, and maintained in serum-free medium for 24 h prior to the experiment. Unless otherwise specified, all experiments were performed with serum-free medium containing 5 mM glucose with or without glucagon or other blockers [19,25,28].
2.3. Measurement of intracellular calcium in MCs
To determine whether glucagon increases intracellular [Ca2+]i by activating its specific receptors, we measured [Ca2+]i responses to glucagon (10 nM) in MCs as described previously [14,25]. Sub-confluent MCs cultured on coverslips were loaded with Fura 2 (2 μM) for 30 min at 37 °C. After two washes with phosphate-buffered saline (PBS), coverslips were mounted on a perfusion chamber maintained at 37 °C, in turn mounted on a Nikon Eclipse TE 2000-U fluorescence microscope coupled with a Lambda DG4 illumination system (Sutter Instruments). Fura-2-loaded MCs were alternately excited at 340 and 380 nM every 3 s. Basal ratiometric calcium imaging was first recorded for 5–10 min with serum-free medium alone. MCs were then stimulated with glucagon (10 nM), and 340/380 ratiometric calcium images were continuously recorded at 3 s intervals for up to 10 min. A separate group of MCs was pretreated with the selective glucagon receptor antagonist [Des-His1-Glu9] glucagon (1 μM) for 30 min before glucagon stimulation. The effects of glucagon on [Ca2+]i were determined by calculating the average magnitude of the peak [Ca2+]i]i responses during the entire glucagon stimulation (200 s) [14,25].
2.4. Western blots of total and phosphorylated ERK 1/2 in MCs
Animal and clinical studies have shown that hyperglucagonemia plays an important role in the development of glomerular hyperfiltration and mesangial cell injury [4–6,29]. In rats, we found that glomerular hyperfiltration induced by glucagon was attenuated by inhibiting angiotensin-converting enzyme (ACE) with enalapril [30,31], together suggesting that glucagon interacts with Ang II to regulate glomerular filtration. To determine whether Ang II is involved in glucagon-induced phosphorylation of ERK 1/2 in MCs, sub-confluent MCs were treated with the ACE inhibitor captopril (10 μM) for 30–60 min before being stimulated with glucagon (1 nM). The specificity of glucagon-induced effects was verified using the glucagon receptor antagonist [Des-His1-Glu9]-glucagon (1 μM) [25,32].
After treatment, the medium was removed and MCs were washed twice with ice-cold PBS and lysed with Nonidet P-40 lysis buffer containing an inhibitor cocktail (Roche). Protein was measured using a BCA kit (Pierce, IL). A 10 μg protein from each sample was separated on 8–16% SDS/polyacrylamide gel and transferred semi-dry onto an Immobilon-P membrane (Millipore). Total and phosphorylated ERK 1/2 were detected by immunoblotting using a rabbit ERK 1/2 polyclonal antibody against the c-terminus of rat ERK 1/2 or a mouse anti-phosphorylated ERK 1/2 monoclonal antibody against an amino acid sequence containing phosphorylated Tyr-204 of human ERK 1/2 (Santa Cruz, CA) [25,33]. Western blot of β-actin (Sigma) was used to confirm equal protein loading. Immunoblots were visualized by enhanced chemiluminescence using horseradish peroxidase-conjugated secondary antibodies (Santa Cruz, CA) [15,25,33], scanned and analyzed using a microcomputer imaging device (MCID, Imaging Research, Ontario, CA).
2.5. Glucagon receptor-mediated increases in angiotensin II generation in MCs
Studies have shown that the local renin–angiotensin system, including renin, angiotensinogen and Ang II, is activated in the kidney of diabetic animals and humans [18,26]. Hyperglycemia purportedly plays a central role inthis process by increasing Ang II generation especially in MCs [19,34]; however, it is not known whether glucagon can increase Ang II formation directly in MCs. Clinical studies in type 2 diabetes have found inappropriately elevated plasma glucagon levels, which may be responsible for persistent hyperglycemia and increased insulin resistance. To determine whether glucagon stimulates Ang II formation in MCs, sub-confluent cells were treated with serum-free medium alone, glucagon (10 nM), or glucagon plus [Des-His1-Glu9]-glucagon (1 μM) or captopril (10 μM) for 24 h at 37 °C. After treatment, the medium was removed and the cells first washed twice with ice-cold PBS, then twice with ice-cold acid buffer (5 mM acetic acid, 150 mM NaCl, pH 2.5) to remove any remaining cell membrane-bound Ang II [25,35]. Ang II was extracted from MCs in a buffer containing 20 mM Tris–HCl, 10 mM EDTA, 5 mM EGTA, 5 mM mercaptoethanol, 50 g/ml PMSF, 1 μg/ml aprotinin, and 1 μg/ml pepstatin, and measured using a sensitive and specific Ang II enzyme immunoassay kit (Biochem/Peninsula) [35]. Protein concentration in each sample was used to calculate final Ang II levels, expressed as pg/mg protein [35].
2.6. Role of AT1 and AT2 receptors in glucagon-induced ERK 1/2 phosphorylation in MCs
Glomerular MCs express both AT1 and AT2 receptors (primarily AT1) and glucagon receptors [36]. In most cases, Ang II and glucagon stimulate MC growth and proliferation by activating AT1 and glucagon receptors, respectively, whereas the role of AT2 is not clear [22]. To determine their respective roles in mediating glucagon-induced ERK 1/2 phosphorylation, sub-confluent MCs were treated with serum-free medium alone, the AT1 antagonist losartan (10 μM), the AT2 antagonist PD 123319 (10 μM), or both AT1 and AT2 antagonists for 30 min before being stimulated with glucagon (1 nM) for 5 min. The antagonist(s) alone also served as a control. After treatment, MCs were washed, lysed and proteins extracted for Western blots of total and phosphorylated ERK 1/2 [25,33].
2.7. Role of AT1 and AT2 receptors in Ang II-induced ERK 1/2 phosphorylation in MCs
To determine whether Ang II induces ERK 1/2 phosphorylation in MCs by activating AT1 and AT2 signaling, sub-confluent MCs were treated with serum-free medium alone, losartan (10 μM), PD 123319 μM), or both losartan and PD 123319 for 30 min before being stimulated by Ang II (1 nM) for 5 min. After treatment, MCs were washed, lysed, and protein samples extracted for Western blots of total and phosphorylated ERK 1/2 [25,33].
2.8. Role of cAMP-dependent PKA, phospholipase C (PLC) and PI 3-kinase signaling in Ang II-induced ERK 1/2 phosphorylation in MCs
We recently showed that glucagon-induced ERK 1/2 phosphorylation involves activation of phospholipase C- and cAMP-dependent PKA signaling [25]. To determine whether activation of similar signaling pathways by Ang II induces ERK 1/2 phosphorylation, sub-confluent MCs were treated with serum-free medium alone (n = 6 wells), Ang II alone (1 nM), Ang II plus U73122 (1 μM), a selective PLC inhibitor, Ang II plus H-89 (1 μM), a selective PKA inhibitor, or the PI 3-kinase inhibitor wortmannin (1 μM) for 5 min. After treatment, protein samples were extracted for Western blot of total and phosphorylated ERK 1/2.
2.9. Statistical analysis
Where appropriate, results are expressed as mean ± S.E. Differences between groups were compared using one-way ANOVA followed by Dunnett’s post hoc test. P < 0.05 was considered significant.
3. Results
3.1. Glucagon receptor-mediated increases in intracellular calcium ([Ca2+]i) in MCs
Glucagon activates its Gs protein-coupled receptors to increase [Ca2+]i in hepatocytes and pancreatic cells [13,14], but its effect on [Ca2+]i in MCs has only recently been reported [25]. Fig. 1 shows that under basal conditions, 340/380 ratiometric calcium imaging revealed faint [Ca2+]i responses (panel A). Glucagon stimulation (10 nM) rapidly increased [Ca2+]i in MCs (panel B). Pretreating MCs with a selective glucagon receptor antagonist, [Des-His1-Glu9]-glucagon (1 μM), effectively prevented glucagon-induced [Ca2+]i responses, suggesting that the effect of glucagon was receptor-mediated.
Fig. 1.
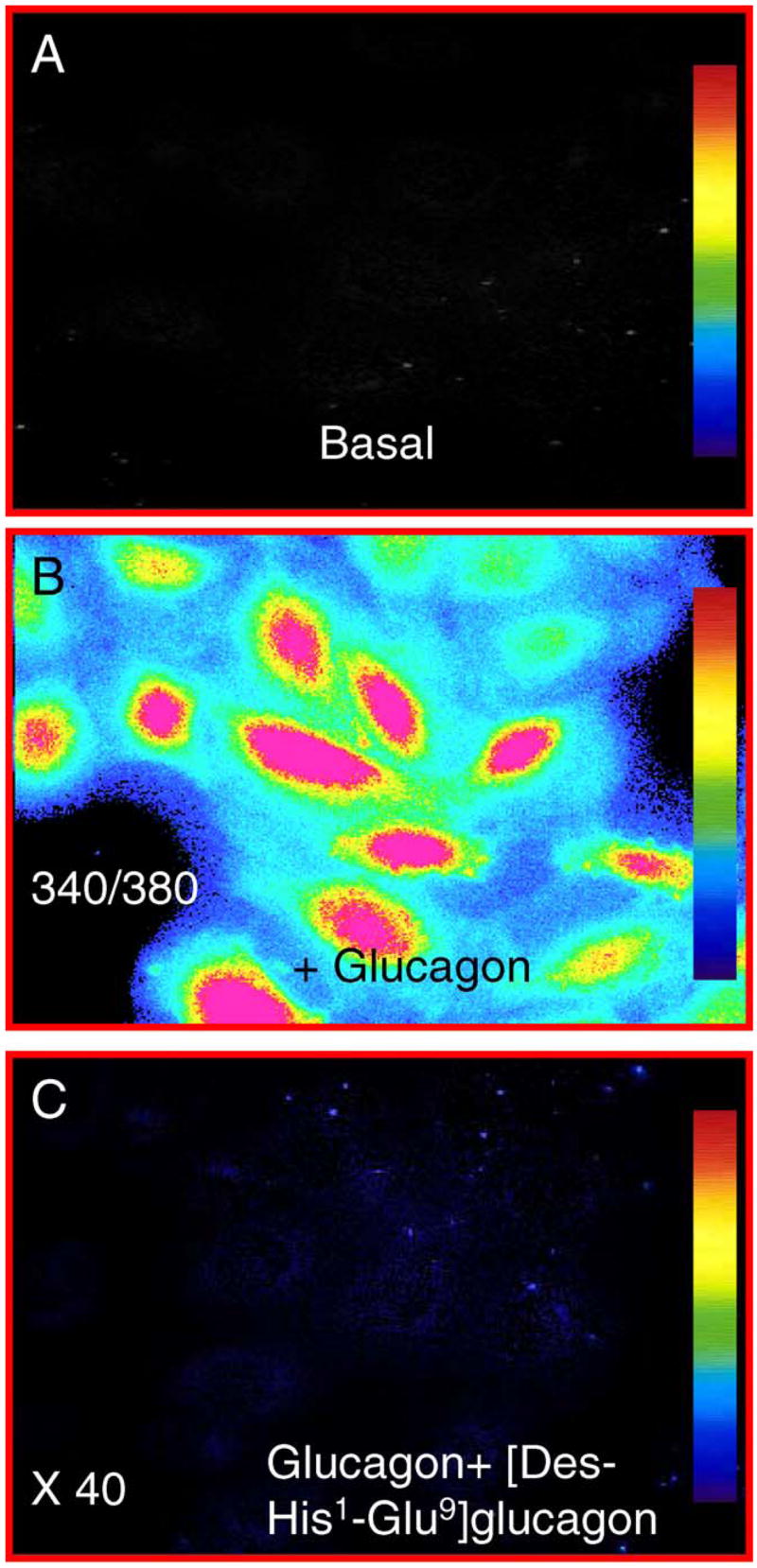
340/380 Ratiometric calcium imaging shows intracellular calcium ([Ca2+]i) responses in MCs under basal conditions (panel A) and during glucagon stimulation (10 nM, panel B). The glucagon-induced increase in [Ca2+]i was blocked by [Des-His1,Glu9] glucagon (1 μM, panel C). Intensity of [Ca2+]i responses is shown as color bars, with red representing the highest level and dark blue as background. Magnification: 40×.
3.2. Effect of Ang II on glucagon-induced ERK 1/2 phosphorylation in MCs
As we reported recently [25], exposure of MCs to glucagon for 5 min almost doubled phosphorylated ERK 1/2 in MCs (185 ± 19%; p < 0.001 versus control), and this stimulatory effect was blocked by the receptor antagonist [Des-His1-Glu9]-glucagon (1 μM) (118 ± 14%; p < 0.01 versus glucagon) (Fig. 2). We further determined whether glucagon induces ERK 1/2 phosphorylation by interacting with Ang II. Pretreating MCs with captopril to inhibit Ang II formation significantly attenuated glucagon-induced ERK 1/2 phosphorylation in MCs (125 ± 13%; p < 0.01 versus glucagon), whereas [Des-His1-Glu9]-glucagon (127 ± 12%; n.s.) or captopril alone (119 ± 17%; n.s.) had no significant effect. Total ERK 1/2 was not altered by any treatment.
Fig. 2.
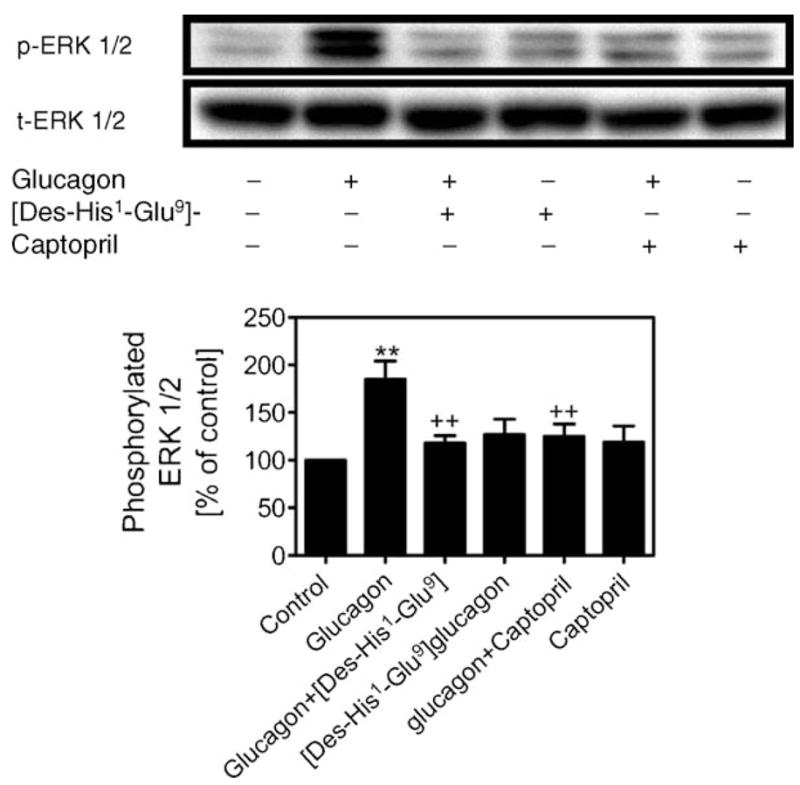
Effects of glucagon on MAP kinase ERK 1/2 phosphorylation in MCs. The top panel shows Western blots of phosphorylated and total ERK 1/2, and the bottom panel shows quantitative phosphorylated ERK 1/2 as percent of control. Glucagon-stimulated ERK 1/2 phosphorylation (1 nM) was blocked by [Des-His1,Glu9] glucagon (1 μM) and the ACE inhibitor captopril (10 μM), suggesting that the effect of glucagon is specific and involves interaction with Ang II. **p < 0.001 vs. control; ++p < 0.01 vs. glucagon. n = 4–6 per treatment.
3.3. Glucagon receptor-mediated increases in Ang II formation in MCs
High glucose has been shown to increase Ang II formation in MCs [19], but the effect of glucagon on Ang II formation in these cells has not been reported previously to our knowledge. Fig. 3 shows that basal Ang II was 788 ± 62 pg/mg protein, which was increased by 47% by glucagon (1160 ± 42 pg/mg protein; p < 0.001). Co-administration of glucagon with [Des-His1-Glu9]-glucagon (964 ± 38 pg/mg protein; p < 0.01) or captopril (854 ± 26 pg/mg protein; p < 0.001) significantly attenuated glucagon-increased Ang II formation.
Fig. 3.

Effects of glucagon on angiotensin II (Ang II) generation in MCs. Treatment of MCs with glucagon (10 nM) for 24 h increased Ang II by 47%, and this effect was attenuated by [Des-His1,Glu9] glucagon (1 μM) and captopril (10 μM). *p < 0.05, **p < 0.01 vs. control; ++p < 0.01 vs. glucagon. n = 6 per treatment.
3.4. Role of AT1 and AT2 receptors in glucagon-induced ERK 1/2 phosphorylation in MCs
The effects of AT1 or AT2 receptor blockade on glucagon-induced ERK 1/2 phosphorylation have not been reported previously in any renal cells to our knowledge. Fig. 4 shows that glucagon-induced ERK 1/2 phosphorylation in MCs was markedly attenuated by the AT1 antagonist losartan (glucagon: 189 14% versus glucagons + losartan: 77 ± 14%; p < 0.001) or the AT2 antagonist PD 123319 (78 ± 7%; p < 0.001 versus glucagon), suggesting that both AT1 and AT2 signaling are involved in glucagon-induced responses. Surprisingly, simultaneous blockade of AT1 and AT2 did not have an additive effect on glucagon-induced ERK 1/2 phosphorylation (107 ± 22%; n.s. versus control).
Fig. 4.

Effects of Ang II AT1 or AT2 receptor blockade on glucagon-induced ERK 1/2 phosphorylation in MCs. The top panel shows Western blots of phosphorylated and total ERK 1/2, and the bottom panel shows quantitative phosphorylated ERK 1/2 as percent of control. The AT1 receptor antagonist losartan (10 μM) and the AT2 receptor antagonist PD 123319 (10 μM) both blocked ERK 1/2 phosphorylation induced by glucagon (1 nM). However, losartan and PD 123319 did not have an additive effect suggesting that their effects may be mediated by similar signaling pathways. **p < 0.01 vs. control; ++p < 0.01 vs. glucagon. n = 6 per treatment.
3.5. Role of AT1 and AT2 receptors in Ang II-induced ERK 1/2 phosphorylation in MCs
The effects of losartan and PD 123319 on glucagon-induced ERK 1/2 phosphorylation suggest cross-talk between Ang II and glucagon receptor signaling in MCs. Although Ang II is known to induce MC growth and proliferation, the mechanisms responsible remain unclear. Fig. 5 shows that like glucagon, Ang II also caused ERK 1/2 phosphorylation, resulting in nearly an 80% increase (179 ± 16%; p < 0.001 versus control). Blockade of AT1 with losartan markedly attenuated the effect of Ang II below control (84 ± 18%; p < 0.001 versus Ang II), suggesting that AT1 receptors may have a tonic influence in MCs. Surprisingly, blockade of AT2 receptors with PD 123319 had similar inhibitory effects on Ang II-induced phosphorylation of ERK 1/2 (87 ± 7%; p < 0.001 versus Ang II). However, combined blockade of AT1 and AT2 had no additive effect on Ang II-induced ERK 1/2 phosphorylation (77 ± 5%; p < 0.01 versus Ang II). Losartan or PD 123319 alone did not alter either phosphorylated or total ERK 1/2.
Fig. 5.
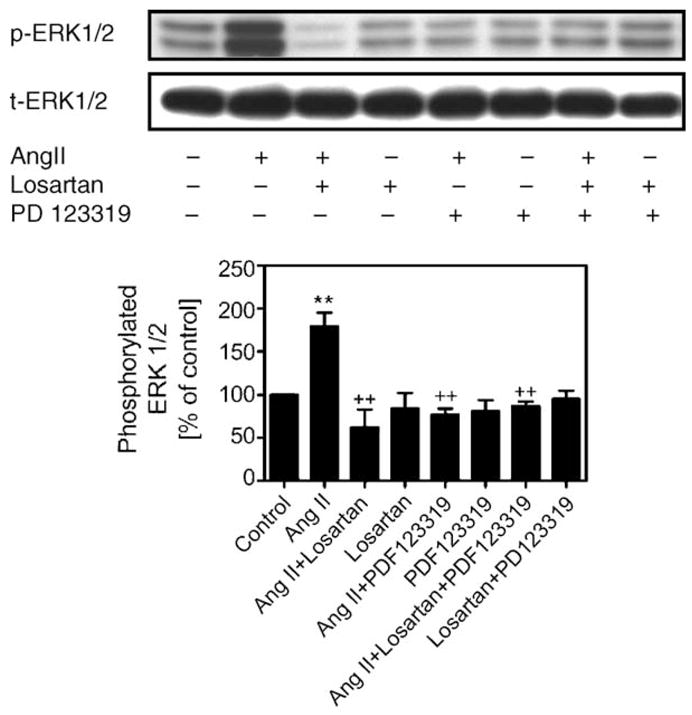
Effects of AT1 and AT2 receptor-mediated Ang II-induced ERK 1/2 phosphorylation in MCs. The top panel shows Western blots of phosphorylated and total ERK 1/2, and the bottom panel shows quantitative phosphorylated ERK 1/2 as percent of control. Ang II (1 nM) significantly increased phosphorylated ERK 1/2, and this response was completely blocked by pretreatment of MCs with losartan (10 μM) and PD 123319 (10 μM), respectively. **p < 0.001 vs. control; ++p < 0.01 vs. Ang II. n = 6 per treatment.
3.6. Role of cAMP-dependent PKA, phospholipase C (PLC) and PI 3-kinase signaling in Ang II-induced ERK 1/2 phosphorylation in MCs
We recently found that glucagons-induced ERK 1/2 phosphorylation by receptor-activated cAMP-dependent PKA and PLC/IP3/Ca2+ signaling [25]. Thus we questioned whether Ang II induces phosphorylation of ERK 1/2 by activating similar signaling transduction pathways. As shown in Fig. 6, blocking PLC with a selective inhibitor, U73122, significantly attenuated Ang II-increased phosphorylated ERK 1/2 (Ang II: 208 ± 8% versus Ang II + U73122: 111 ± 13%; p < 0.01). Blocking PKA with a selective inhibitor, H-89, likewise blunted the effect of Ang II (86 ± 10%; p < 0.001 versus Ang II). Furthermore, the PI 3-kinase-selective inhibitor wortmannin was also able to inhibit Ang II-induced ERK 1/2 phosphorylation (114 ± 8%; p < 0.01 versus Ang II). Since these inhibitors alone did not affect phosphorylated ERK 1/2, it would appear that Ang II can induce ERK 1/2 phosphorylation similarly via activation of cAMP-dependent PKA, PLC and PI 3-kinase signaling pathways.
Fig. 6.

Intracellular signaling mechanisms of Ang II-induced ERK 1/2 phosphorylation in MCs. The top panel shows Western blots of phosphorylated and total ERK 1/2, and the bottom panel shows quantitative phosphorylated ERK 1/2 as percent of control. Ang II-stimulated ERK 1/2 phosphorylation (1 nM) was significantly attenuated by pretreatment of MCs with the PLC-selective inhibitor U73122 (1 μM), the cAMP-dependent PKA inhibitor H-89 (1 μM), and the PI 3-kinase inhibitor wortmannin (1 μM), respectively. Inhibitor(s) alone had no effect on ERK 1/2 phosphorylation. **p < 0.001 vs. control; ++p < 0.01 vs. Ang II. n = 6 per treatment.
4. Discussion
Activation of MAP kinase ERK 1/2 by vasoactive peptides, growth factors or proliferative cytokines such as Ang II and glucagon plays a critical role in promoting cell growth, differentiation, and proliferation during the development of target organ damage in diabetes and other cardiovascular and renal diseases [23,25,34,37,38]. Although Ang II and glucagon bind to different G protein-coupled receptors, their intracellular signaling pathways appear to converge or interact with each other [13,22]. The present study demonstrates that crosstalk between glucagon- and Ang II-induced MAP kinase ERK 1/2 phosphorylation in MCs involves at least two important mechanisms, stimulation of Ang II formation and activation of AT1/AT2 receptor-mediated signaling. This conclusion is derived from the observations that incubation of MCs with glucagon (10 nM) for 24 h increased Ang II by almost 50%, and that the ACE inhibitor captopril, the AT1 antagonist losartan, and the AT2 antagonist PD 123319 all significantly attenuated glucagon-induced ERK 1/2 phosphorylation in MCs. Moreover, both Ang II and glucagon increased ERK 1/2 phosphorylation through similar receptor-mediated signaling pathways, including PLC/IP3/Ca2+, cAMP-dependent PKA and PI 3-kinase activation. Thus our results suggest that glucagon interacts with Ang II and its receptor signaling to regulate MC function, including growth, hypertrophy and proliferation.
Although it is well documented that serum glucagon is inappropriately elevated in diabetes mellitus, especially type 2 (non-insulin-dependent) [4–6] and that the RAS is activated in the glomeruli of diabetic kidneys [7–9], whether glucagon interacts with Ang II and/or Ang II receptor signaling has not been explored previously to our knowledge. Glucagon is a 29-amino acid hormone synthesized in and released from pancreatic α cells [13]. Its primary action is to counter the glucose-lowering effect of insulin released from β cells to raise serum glucose levels. The hyperglycemic effect of glucagon is mediated by binding to GTP-binding heterotrimeric Gs protein-coupled receptors, leading to activation of adenylate cyclase and increased intracellular cAMP production and PLC as well as mobilization of [Ca2+]i [13,14,39]. There is evidence that the Gi protein-activated signaling pathway may also play a role in glucagon-induced increases in [Ca2+]i [13]. Activation of cAMP-dependent PKA and increases in [Ca2+]i by glucagon are responsible for induction of MAP kinase ERK 1/2 phosphorylation, and hence cell growth, differentiation and proliferation. By contrast, the effects of Ang II are mediated by two receptors, AT1 and AT2 [22,36,40]. AT1 Rs belong to the GPCR superfamily and is coupled via pertussis toxin-insensitive G proteins to activation of phospholipase C and calcium signaling [22]. However, AT1 receptor is also coupled to other subunits of G proteins (Gq, Gi, Gq/11, etc.) to activate phospholipase D, phospholipase A2, adenylate cyclase and ion channels [22], along with intracellular signaling that regulates gene transcription and/or expression of growth factors and proliferative cytokines that induce Ang II-induced target organ damage [22,23,40]. The precise nature and role of AT2 receptor signaling are not well understood.
Although we have recently shown that glucagon can directly activate its G protein-coupled receptors to induce MAP kinase ERK 1/2 phosphorylation and growth and proliferation of MCs [25], our present results further suggest that these effects are mediated in part by stimulating Ang II formation and/or cross-talk with Ang II receptors. Indeed, pretreatment of MCs with the ACE inhibitor captopril to block local Ang II formation significantly inhibited glucagon-induced ERK 1/2 phosphorylation (Fig. 2). This finding was further supported by the observation that exposure of MCs to glucagon for 24 h increased Ang II generation by 47%, which was blocked by captopril (Fig. 3). The Ang II-stimulating effect of glucagon is specific, since it was blocked by the glucagon receptor antagonist [Des-His1-Glu9] glucagon [25,32]. However, we did not address the question of how glucagon increases Ang II formation in the present study. Hyperglycemia (or high glucose) reportedly increases the expression of renin and its substrate angiotensinogen (and therefore Ang II formation) in MCs [18,19,34]. In the present study, MCs were maintained in serum-free medium containing normal glucose (5 mM) before and during treatment, so that this effect was not likely mediated via hyperglycemia. One plausible explanation is that glucagon activates adenylate cyclase to increase cAMP production, since cAMP itself is a powerful stimulator of renin expression in the kidney. Thus the cAMP signaling pathway may play an important role in this response.
The present study shows that blockade of AT1 and/or AT2 receptors with losartan and PD 123319, respectively, prevented glucagon-induced phosphorylation of ERK 1/2 in MCs. This is a surprising finding and it may be interpreted in three different ways. First, both AT1 and AT2 receptors are activated either separately or simultaneously in order to induce ERK 1/2 phosphorylation. Second, the AT1 or AT2 receptor antagonist is not selective for its respective receptor. Third, both AT1 and AT2 receptor antagonists block Ang II- and glucagon-induced ERK 1/2 phosphorylation by interfering not at the receptor site but at a distal level such as intracellular signaling pathways. In the present study, we had no evidence that either of losartan or PD 123319 interferes with glucagon receptor binding, or conversely that glucagon administration significantly increases AT1 or AT2 receptor protein expression (unpublished observations). The second explanation is unlikely since losartan and PD 123319 are perhaps the best characterized and widely used AT1 or AT2 receptor blocker, respectively [22]. We also have extensive experience in using losartan and PD 123319 to characterize and localize AT1 and AT2 receptors, respectively, in the kidney, adrenals, heart, and brain [41]. Therefore, one plausible mechanism that could explain the effects of Ang II receptor blockers is cross-talk between intracellular signaling pathways for glucagon and Ang II. This may include activation of cAMP-dependent PKA, PLC/IP3/Ca2+ and PI 3-kinase signaling by both glucagon and Ang II. Glucagon receptors and Ang II AT1 receptors are [Ca2+]i-mobilizing and cAMP-modulating G protein-coupled receptors (GPCRs), and their activation by agonist(s) induces effects that often mimic growth factor and/or proliferative cytokine actions on MAP kinase ERK 1/2 phosphorylation [14,22,38,42–44]. Although upstream MAP kinase activation by glucagon and Ang II may be different following binding to their respective GPCRs, activation of their downstream ERK 1/2 in MCs appears to involve similar intracellular signaling pathways, namely cAMP-dependent PKA, PLC and PI 3-kinase [13,22]. For example, in further studies we found that pretreatment of MCs with losartan (10 μM) or U73112 (a PLC-selective antagonist), not PD 123319, for 30 min significantly attenuated glucagon-induced increases in intracellular calcium (unpublished observations). In VSMCs, AT1 receptor-activated MAP kinase ERK 1/2 phosphorylation may be mediated via a Gαq-PKC- or the Src-Ras-dependent mechanism, or via trans-activation of the epidermal growth factor receptor [22]. Whether glucagon-induced ERK 1/2 activation in MCs involves mechanisms similar to Ang II/AT1 receptors in VSMCs remains to be studied.
The effects of AT2 receptor blockade with PD 123319 on Ang II- and glucagon-induced ERK 1/2 phosphorylation in MCs were somewhat unexpected. Although we found that the AT1 receptor predominates in MCs with an AT1/AT2 receptor ratio of ~80% versus ~20%, the conventional view is that activation of AT2 receptors opposes the classic actions of AT1 receptors [22]. In the present study, PD 123319 blocked both glucagon-and Ang II-induced ERK 1/2 phosphorylation in MCs, suggesting that AT2 receptors may take part in these responses in a similar manner to AT1 receptors. Interestingly, we also found that PD 123319 blocked hyperglycemia-induced ERK 1/2 phosphorylation in MCs (unpublished observations). AT2 has no effect on either intracellular [Ca2+]i or cAMP, which are associated with classic activation of Gq protein-coupled receptors or Gs/Gi proteins, respectively [22]. Previous studies on AT2 receptors in other types of cells have revealed diverse signaling pathways involving activation of serine/threonine phosphatases or phospholipase A2 and changes in K+ and T-type Ca2+ channels [22]. AT2 may oppose AT1-mediated MAP kinase activation by inducing dephosphorylation or inactivation of MAP kinase ERK 1/2 [22]. However, the effects of AT2 on MAP kinase activation appear to depend on the specific type of cells or experimental conditions. For example, genetic deletion of AT2 receptors was found to prevent [45], whereas overexpression of cardiac AT2 receptors caused cardiac hypertrophy and heart failure in mice [46]. Furthermore, overexpression of AT2 receptors has been shown to increase MAP kinase activity in VSMCs [22] and PC12W cells [44] and induce severe hypertrophy in neonatal cardiac myocytes [48]. In VSMCs or epithelial cells, AT2 appears to induce nuclear factor-κB activation in a manner similar to AT1 [40]. Collectively, the AT2 receptor seems to positively regulate glucagon-,Ang II-, or hyperglycemia-induced activation of MAP kinase ERK 1/2, though the precise signaling mechanisms involved are not known.
In summary, the present study demonstrates that glucagon induces MAP kinase ERK 1/2 activation in part by stimulating local Ang II production and cross-talk with Ang II AT1/AT2 receptor-activated intracellular signaling pathways. The effects of glucagon were blocked by [Des-His1-Glu9] glucagon, a selective glucagon receptor antagonist, indicating a receptor-mediated event. These results are highly relevant to type 2 diabetic glomerular/MC injury, because proliferation, hypertrophy and mesangial expansion are hallmarks of early diabetic nephropathy. Since inappropriately elevated serum glucagon and activation of the intrarenal RAS are both seen during the development of type 2 diabetes, and since both glucagon and Ang II are glycogenolytic and gluconeogenic hormones that induce MAP kinase ERK 1/2 activation and promote growth and proliferation of MCs, we believe glucagon- and Ang II receptor-activated signaling pathways may serve as preventive and therapeutic targets in type 2 diabetic nephropathy and other cardiovascular diseases.
Acknowledgments
This work was supported in part by a National Institute of Diabetes & Digestive & Kidney Diseases RO1 grant (5RO1DK067299 to Zhuo) and a Henry Ford Health System institutional grant (A10217 to Zhuo). Dr. Oscar A. Carretero is supported by a National Heart, Lung and Blood Institute program project grant (HL-28982). Portions of this work were presented as abstracts at the 59th Annual Fall Conference and Scientific Sessions of the Council for High Blood Pressure Research in association with the Council on the Kidney in Cardiovascular Disease, American Heart Association, Washington, DC, USA, September 21–24, 2005, and at Cell Signaling World 2006, Luxembourg, January 25–28, 2006.
References
- 1.Matthaei S, Stumvoll M, Kellerer M, Haring H-U. Pathophysiology and pharmacological treatment of insulin resistance. Endocr Rev. 2000;21:585–618. doi: 10.1210/edrv.21.6.0413. [DOI] [PubMed] [Google Scholar]
- 2.Gaster B, Hirsch IB. The effects of improved glycemic control on complications in type 2 diabetes. Arch Intern Med. 1998;158:134–40. doi: 10.1001/archinte.158.2.134. [DOI] [PubMed] [Google Scholar]
- 3.Nett PC, Sollinger HW, Alam T. Hepatic insulin gene therapy in insulin-dependent diabetes mellitus. Am J Transplant. 2003;3:1197–203. doi: 10.1046/j.1600-6143.2003.00221.x. [DOI] [PubMed] [Google Scholar]
- 4.Unger RH, Orci L. The essential role of glucagon in the pathogenesis of diabetes mellitus. Lancet. 1975;4:14–6. doi: 10.1016/s0140-6736(75)92375-2. [DOI] [PubMed] [Google Scholar]
- 5.Shah P, Vella A, Basu A, Basu R, Schwenk WF, Rizza RA. Lack of suppression of glucagons contributes to postprandial hyperglycemia in subjects with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2000;85:4053–9. doi: 10.1210/jcem.85.11.6993. [DOI] [PubMed] [Google Scholar]
- 6.Orskov C, Jeppesen J, Madsbad S, Holst JJ. Proglucagon products in plasma of noninsulin-dependent diabetics and nondiabetic controls in the fasting state and after oral glucose and intravenous arginine. J Clin Invest. 1991;87:415–23. doi: 10.1172/JCI115012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barnett AH, Bain SC, Bouter P, Karlberg B, Madsbad S, Jervell J, et al. Angiotensin-receptor blockade versus converting-enzyme inhibition in type 2 diabetes and nephropathy. N Engl J Med. 2004;351:1952–61. doi: 10.1056/NEJMoa042274. [DOI] [PubMed] [Google Scholar]
- 8.Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–9. doi: 10.1056/NEJMoa011161. [DOI] [PubMed] [Google Scholar]
- 9.Ravid M, Savin H, Jutrin I, Bental T, Katz B, Lishner M. Long-term stabilizing effect of angiotensin-converting enzyme inhibition on plasma creatinine and on proteinuria in normotensive type 2 diabetic patients. Ann Intern Med. 1993;118:577–81. doi: 10.7326/0003-4819-118-8-199304150-00001. [DOI] [PubMed] [Google Scholar]
- 10.Reaven GM, Chen YD, Golay A, Swislocki AL, Jaspan JB. Documentation of hyperglucagonemia through the day in nonobese and obese patients with noninsulin-dependent diabetes mellitus. J Clin Endocrinol Metab. 1987;64:106–10. doi: 10.1210/jcem-64-1-106. [DOI] [PubMed] [Google Scholar]
- 11.Wakelam MJ, Murphy GJ, Hruby VJ, Houslay MD. Activation of two signal-transduction systems in hepatocytes by glucagon. Nature (Lond) 1986;323:68–71. doi: 10.1038/323068a0. [DOI] [PubMed] [Google Scholar]
- 12.Rodbell M, Birnbaumer L, Pohl SL, Krans HM. The glucagon-sensitive adenylyl cyclase system in plasma membranes of rat liver. V. An obligatory role of guanylnucleotides in glucagon action. J Biol Chem. 1971;246:1877–82. [PubMed] [Google Scholar]
- 13.Mayo KE, Miller LJ, Bataille D, Dalle S, Goke B, Thorens B, et al. International union of pharmacology. XXXV. The glucagon receptor family. Pharmacol Rev. 2003;55:167–94. doi: 10.1124/pr.55.1.6. [DOI] [PubMed] [Google Scholar]
- 14.Hansen LH, Gromada J, Bouchelouche P, Whitmore T, Jelinek LJ, Kingsvogel W, et al. Glucagon-mediated Ca2+ signaling in BHK cells expressing cloned human glucagon receptors. Am J Physiol. 1998;274:C1552–6. doi: 10.1152/ajpcell.1998.274.6.C1552. [DOI] [PubMed] [Google Scholar]
- 15.Amiri F, Shaw S, Wang X, Tang J, Waller JL, Eaton DC, et al. Angiotensin II activation of the JAK/STAT pathway in mesangial cells is altered by high glucose. Kidney Int. 2002;61:1605–16. doi: 10.1046/j.1523-1755.2002.00311.x. [DOI] [PubMed] [Google Scholar]
- 16.Park SH, Woo CH, Kim JH, Lee JH, Yang IS, Park KM, et al. High glucose down-regulates angiotensin II binding via the PKC-MAPK-cPLA2 signal cascade in renal proximal tubule cells. Kidney Int. 2002;61:913–25. doi: 10.1046/j.1523-1755.2002.00204.x. [DOI] [PubMed] [Google Scholar]
- 17.Amiri F, Venema VJ, Wang X, Ju H, Venema RC, Marrero MB. Hyperglycemia enhances angiotensin II-inducedjanus-activated kinase/STAT signaling in vascular smooth muscle cells. J Biol Chem. 1999;274:32382–6. doi: 10.1074/jbc.274.45.32382. [DOI] [PubMed] [Google Scholar]
- 18.Wilkes BM, Mento PF, Vernace MA. Angiotensin responsiveness in hyperfiltering and nonhyperfiltering diabetic rats. J Am Soc Nephrol. 1993;4:1346–53. doi: 10.1681/ASN.V461346. [DOI] [PubMed] [Google Scholar]
- 19.Singh R, Alavi N, Singh AK, Leehey DJ. Role of angiotensin II in glucose-induced inhibition of mesangial matrix degradation. Diabetes. 1999;48:2066–73. doi: 10.2337/diabetes.48.10.2066. [DOI] [PubMed] [Google Scholar]
- 20.Kraus-Friedmann N. Hormonal regulation of hepatic gluconeogenesis. Physiol Rev. 1984;64:170–259. doi: 10.1152/physrev.1984.64.1.170. [DOI] [PubMed] [Google Scholar]
- 21.Guder WG. Stimulation of renal gluconeogenesis by angiotensin II. Biochim Biophys Acta. 1979;584:507–19. doi: 10.1016/0304-4165(79)90123-5. [DOI] [PubMed] [Google Scholar]
- 22.De Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52:415–72. [PubMed] [Google Scholar]
- 23.Zhuo JL. Monocyte chemoattractant protein-1: a key mediator of angiotensin II-induced target organ damage in hypertensive heart disease. J Hypertens. 2004;22:451–514. doi: 10.1097/01.hjh.0000098211.37783.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mezzano S, Droguett A, Burgos ME, Ardiles LG, Flores CA, Aros CA, et al. Renin–angiotensin system activation and interstitial inflammation in human diabetic nephropathy. Kidney Int Suppl. 2003;86:S64–70. doi: 10.1046/j.1523-1755.64.s86.12.x. [DOI] [PubMed] [Google Scholar]
- 25.Li XC, Carretero OA, Shao Y, Zhuo JL. Glucagon receptor-mediated ERK 1/2 phosphorylation in rat mesangial cells: role of protein kinase A and phospholipase C. Hypertension. 2006;47:1–6. doi: 10.1161/01.HYP.0000197946.81754.0a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang S-L, To C, Chen X, Filep JG, Tang S-S, Ingelfinger JR, et al. Essential role(s) of the intrarenal renin–angiotensin system in transforming growth factor-beta1 gene expression and induction of hypertrophy of rat kidney proximal tubular cells in high glucose. J Am Soc Nephrol. 2002;13:302–12. doi: 10.1681/ASN.V132302. [DOI] [PubMed] [Google Scholar]
- 27.Douglas JG, Hopfer U. Novel aspect of angiotensin receptors and signal transduction in the kidney. Annu Rev Physiol. 1994;56:649–69. doi: 10.1146/annurev.ph.56.030194.003245. [DOI] [PubMed] [Google Scholar]
- 28.Tsiani E, Lekas P, Fantus IG, Dlugosz J, Whiteside C. High glucose-enhanced activation of mesangial cell p38 MAPK by ET-1, ANG II, and platelet-derived growth factor. Am J Physiol Endocrinol Metab. 2002;282:E161–9. doi: 10.1152/ajpendo.2002.282.1.E161. [DOI] [PubMed] [Google Scholar]
- 29.Ahloulay M, Dechaux M, Hassler C, Bouby N, Bankir L. Cyclic AMP is a hepatorenal link influencing natriuresis and contributing to glucagon-induced hyperfiltration in rats. J Clin Invest. 1996;98:2251–8. doi: 10.1172/JCI119035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris PJ, Skinner SL, Zhuo JL. The effects of atrial natriuretic peptide and glucagon on proximal glomerulotubular balance in anesthetized rats. J Physiol (Lond) 1988;402:29–42. doi: 10.1113/jphysiol.1988.sp017192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhuo JL, Harris PJ, Skinner SL. ACE inhibition prevents glucagon-induced glomerular hyperfiltration in anesthetized rats. Hypertension. 2003 Abstract. [Google Scholar]
- 32.Unson CG, Gurzenda EM, Merrifield RB. Biological activities of des-His1[Glu9] glucagons amide, a glucagon antagonist. Peptides. 1989;10:1171–7. doi: 10.1016/0196-9781(89)90010-7. [DOI] [PubMed] [Google Scholar]
- 33.Li XC, Campbell DJ, Ohishi M, Shao Y, Zhuo JL. AT1 receptor-activated signaling mediates Ang IV-induced renal cortical vasoconstriction in rats. Am J Physiol Renal Physiol. 2006;59:1–8. doi: 10.1152/ajprenal.00221.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leehey DJ, Singh AK, Alavi N, Singh R. Role of angiotensin II in diabetic nephropathy. Kidney Int Suppl. 2000;77:S93–8. doi: 10.1046/j.1523-1755.2000.07715.x. [DOI] [PubMed] [Google Scholar]
- 35.Li XC, Carretero OA, Navar LG, Zhuo JL. AT1a receptor-mediated accumulation of extracellular Ang II in proximal tubule cells: role of cytoskeleton microtubules and tyrosine phosphatase. Am J Physiol Renal Physiol. :59. doi: 10.1152/ajprenal.00405.2005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhuo JL, Ohishi M, Mendelsohn FAO. Roles of AT1 and AT2 receptors in the hypertensive Ren-2 gene transgenic rat kidney. Hypertension. 1999;33(1 Pt 2):347–53. doi: 10.1161/01.hyp.33.1.347. [DOI] [PubMed] [Google Scholar]
- 37.Kang MJ, Wu X, Ly H. Effect of glucose on stress-activated protein kinase activity in mesangial cells and diabetic glomeruli. Kidney Int. 1999;55:2203–14. doi: 10.1046/j.1523-1755.1999.00488.x. [DOI] [PubMed] [Google Scholar]
- 38.Sevetson BR, Kong X, Lawrence JC., Jr Increasing cAMP attenuates activation of mitogen-activated protein kinase. Proc Natl Acad Sci. 1993;90:10305–9. doi: 10.1073/pnas.90.21.10305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dalle S, Longuet C, Costes S, Broca C, Faruque O, Fontes G, et al. Glucagon promotes cAMP-response element-binding protein phosphorylation via activation of ERK [1/2] in MIN6 cell line and isolated islets of Langerhans. J Biol Chem. 2004;279:20345–5. doi: 10.1074/jbc.M312483200. [DOI] [PubMed] [Google Scholar]
- 40.Wolf G, Wenzel U, Burns KD, Harris RC, Stahl RA, Thaiss F. Angiotensin II activates nuclear transcription factor-κB through AT1 and AT2 receptors. Kidney Int. 2002;61:1986–95. doi: 10.1046/j.1523-1755.2002.00365.x. [DOI] [PubMed] [Google Scholar]
- 41.Zhuo JL, Allen AM, Alcorn D, MacGregor D, Aldred GP, Mendelsohn FAO. The distribution of angiotensin II receptors. In: Laragh JH, Brenner BM, editors. Hypertension: pathophysiology, diagnosis, and management. 2. New York: Raven Press; 1995. pp. 1739–62. [Google Scholar]
- 42.Tang EK, Houslay MD. Glucagon, vasopressin and angiotensin all elicit a rapid, transient increase in hepatocyte protein kinase C activity. Biochem J. 1992;283:341–6. doi: 10.1042/bj2830341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang Y, Cypess AM, Muse ED, Wu CR, Unson CG, Merrifield RB, et al. Glucagon receptor activates extracellular signal-regulated protein kinase 1/2 via cAMP-dependent protein kinase. Proc Natl Acad Sci. 2001;98:10102–7. doi: 10.1073/pnas.131200398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gomez E, Pritchard C, Herbert TP. cAMP-dependent protein kinase and Ca2+ influx through L-type voltage-gated calcium channels mediated raf-independent activation of extracellular regulated kinase in response to glucagon-like peptide-1 in pancreatic beta cells. J Biol Chem. 2002;277:48146–51. doi: 10.1074/jbc.M209165200. [DOI] [PubMed] [Google Scholar]
- 45.Ichihara S, Senbonmatsu T, Price E, Jr, Ichiki T, Gaffney FA, Inagami T. Angiotensin II type 2 receptor is essential for left ventricular hypertrophy and cardiac fibrosis in chronic angiotensin II-induced hypertension. Circulation. 2001;104:346–51. doi: 10.1161/01.cir.104.3.346. [DOI] [PubMed] [Google Scholar]
- 46.Yan X, Price RL, Nakayama M, Ito K, Schuldt AJ, Manning WJ, et al. Ventricular-specific expression of angiotensin II type 2 receptors causes dilated cardiomyopathy and heart failure in transgenic mice. Am J Physiol Heart Circ Physiol. 2003;285:H2179–87. doi: 10.1152/ajpheart.00361.2003. [DOI] [PubMed] [Google Scholar]
- 47.Stroth U, Blume A, Mielke K, Unger T. Angiotensin II AT2 receptor stimulates ERK1 and ERK2 in quiscent but inhibits ERK in NGF-stimulated PC12W cells. Mol Brain Res. 2000;78:175–80. doi: 10.1016/s0169-328x(00)00093-0. [DOI] [PubMed] [Google Scholar]
- 48.D’Amore A, Black MJ, Thomas WG. The angiotensin II type 2 receptor causes constitutive growth of cardiomyocytes and does not antagonize angiotensin II type 1 receptor-mediated hypertrophy. Hypertension. 2005;46:1347–754. doi: 10.1161/01.HYP.0000193504.51489.cf. [DOI] [PubMed] [Google Scholar]