

Abstract
Smad proteins are intracellular transducers for transforming growth factor-β (TGF-β signaling and play a critical role in differentiation, tissue repair and apoptosis of the central nervous system. Both TGF-β and its regulated gene, plasminogen activator inhibitor type-1 (PAI-1), have been implicated in the etiology and progression of neurodegenerative diseases and mood disorders. We previously reported that GSK-3β protein depletion suppresses Smad3/4-dependent gene transcription and causes a reduction in PAI-1 expression. Here, we provide evidence that lithium, the drug for the treatment and prophylaxis of bipolar disorder, inhibits Smad-dependent signaling by regulating cAMP-protein kinase A (PKA), AKT-glycogen synthase kinase-3β (GSK-3β), and CRE-dependent signaling pathways in neuron-enriched cerebral cortical cultures of rats. We demonstrate that lithium-induced activation of these pathways inhibits Smad3/4-dependent gene transcription through an increase in pCREBSer133 protein levels, an enhanced interaction between pCREBSer133 and p300/CBP, which causes Smad3/4-p300/CBP complex disruption and transcriptional suppression of Smad3/4-dependent genes. Therapeutic implications of our findings are discussed.
Keywords: Smad, TGF-beta, bipolar disorder, lithium, CBP, p300, AKT, PKA, GSK-3beta
Introduction
The mood stabilizer lithium has been used to treat bipolar mood disorder for over half a century, although its therapeutic mechanisms remain unclear (reviewed in Manji and Zarate, 2002). Recent clinical studies demonstrate that neuronal atrophy and loss of glial cells/neurons occur in discrete brain areas of bipolar patients, and suggest that lithium and other mood stabilizers may display their therapeutic effects in part via their neurotrophic and neuroprotective actions (reviewed in Manji and Zarate, 2002; Gurvich and Klein, 2002; Rowe and Chuang, 2004). For example, lithium treatment leads to an increase in the gray matter volume and N-acetyl-aspartate levels in the brain of bipolar patients (Moore et al., 2000a, 2000b). Lithium also normalizes the loss of volume in discrete brain areas of bipolar patients (Drevets, 2001). Preclinical studies demonstrated that lithium is neuroprotective and neurotrophic in cellular and animal models (reviewed in Chuang, 2004; Chuang and Priller, 2006), and that the protective/trophic effects involve alterations in signal transduction and gene expression. Hence, it has been proposed that lithium may have expanded use to treat a number of neurodegenerative disorders.
Glycogen synthase kinase-3 (GSK-3) is one of the targets directly inhibited by lithium (Klein and Melton, 1996; Stambolic et al., 1996). GSK-3 consists of α and β isoforms and is a key kinase involved in the regulation of an array of transcription factors (reviewed in Grimes and Jope, 2001a). Increasing evidence suggests that lithium elicits its neuroprotective/neurotrophic effects primarily through inhibition of GSK-3 (Gould et al., 2004; Liang and Chuang, 2007). Besides direct inhibition, lithium has been shown to indirectly inhibit GSK-3 through phosphorylation of GSK-3α at Ser21 and GSK-3β at Ser9 by multiple mechanisms including the activation of PKA (Jope, 1999a), phosphatidylinositol 3-kinase (PI3-K)-dependent AKT (Chalecka-Franaszek and Chuang, 1999), protein kinase C (PKC) (Kirshenboim et al., 2004), as well as the involvement of GSK-3 autoregulation (Zhang et al., 2003; Liang and Chuang, 2007). GSK-3 inhibition is also likely involved in the anti-depressant and anti-manic effects of lithium observed in rodent models (Gould et al., 2004; Kaidanovich-Beilin et al., 2004; O’Brien et al., 2004).
We have recently reported that the transcription factor Smad3/4 is a novel target for GSK-3 in neurons (Liang and Chuang, 2006). Smad3/4 is a downstream mediator of the signaling pathway triggered by transforming growth factor-beta (TGF-β) (Derynck and Zhang, 2003). Binding of TGF-β to its serine/threonine kinase receptor induces phosphorylation of Smad2 and Smad3. The phosphorylated Smad proteins then form a complex with Smad4 followed by translocation to the nucleus where they interact with other gene-specific transcription factors to regulate gene transcription (reviewed in Derynck and Zhang, 2003; Shi and Massague, 2003). Smad3/4 has a prominent role in regulating the expression of proteins involved in neuronal survival/death, differentiation, and plasticity (reviewed in Sanyal et al., 2004; Gomes et al., 2005). One of the Smad3/4-regulated protein targets is plasminogen activator inhibitor type-1 (PAI-1, also referred to as Serp1), which binds and inactivates tissue-type plasminogen activator (tPA), a multifaceted protein implicated in neurite outgrowth (Krystosek and Seeds, 1981), neuronal migration (Moonen et al., 1982; Seeds et al., 1999), learning (Qian et al., 1993; Seeds et al., 1995), excitotoxicity (Tsirka et al., 1995), synaptic plasticity (Baranes et al., 1998; Fiumelli et al., 1999; Neuhoff et al., 1999), stress response (Pawlak et al, 2003; 2005), and pathogenesis of mood disorders (Eskandari et al., 2005; Tsai, 2006). Using small interfering RNA (siRNA) and dominant negative mutants specific to GSK-3α and GSK-3β, we recently reported that GSK-3α inhibition causes an upregulation of Smad3/4-dependent transactivation and PAI-1 protein levels, while inhibition of GSK-3β induces a downregulation of Smad3/4 transactivation and PAI-1 expression (Liang and Chuang, 2006).
In addition to acting on GSK-3, lithium modulates the 3’,5’-cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA) signaling pathway. In general, lithium raises basal levels, but suppresses stimulus-induced increase in cAMP and PKA activity. This bimodal action of lithium has been proposed as the biochemical basis underlying lithium’s anti-depressant and anti-manic efficacy (Jope, 1999b). Consistent with this notion, alterations in the cAMP-PKA signaling system have been found in the brain of bipolar patients (Chang et al., 2003). Activation of PKA results in phosphorylation of cAMP-responsive element binding protein (CREB) at Ser133 and hence causes an increase in its transcriptional activity. Phosphorylated CREB in conjunction with CREB binding protein (CBP) or p300 has been shown to mediate the transcriptional activation of prominent neuroprotective proteins such as Bcl-2 and brain-derived neurotrophic factor (BDNF) (reviewed in Grimes and Jope, 2001a). CREB can also be activated through inhibition of GSK-3, which negatively regulates CREB through phosphorylation at Ser129 (reviewed in Grimes and Jope, 2001b).
The above mentioned observations underscore the complexity of the signaling pathways involved in the regulation of Smad3/4-dependent transcriptional activity. Lithium, which inhibits both GSK-3 isoforms, was thus investigated for its regulation of Smad/PAI-1 gene transcription possibly through CREB activity and the CRE-dependent signaling pathway. In this study, we first attempted to examine whether lithium modulates Smad3/4-dependent transactivation of target genes, notably PAI-1 and p21 in neuron-enriched cerebral cortical cultures from rats. We then investigated the role of lithium-induced activation of PKA and AKT, and inhibition of GKS-3 in regulating Smad3/4-dependent transcription and gene expression. Finally, we studied the role of p300/CBP and CREB in modulating lithium’s effects on Smad3/4-dependent transactivation.
Results
Lithium inhibits Smad3/4-dependent transcriptional activation and reduces Smad3/4-dependent gene promoter activity of PAI-1 and p21 in neuron-enriched cortical cultures
We previously reported that protein depletion of GSK-3 isoforms α and β through isoform-specific RNA interference caused an increase and decrease, respectively, in Smad3/4-dependent transcriptional activity in cultured cortical neurons (Liang and Chuang, 2006). Since lithium is an inhibitor of GSK-3, we examined the effects of lithium on Smad3/4-dependent transactivation by using Smad3/4-Luc, an artificial reporter construct consisting of repeats of the Smad3/4-specific recognition sequence. In rat neuron-enriched cortical cultures, treatment with LiCl for 72 h caused a concentration-dependent transcriptional suppression in the range tested (0.5-3 mM) (Fig. 1a). In addition, treatment with 1.5 mM lithium caused a time-dependent decrease in Smad3/4 transcriptional activity with a near maximum effect after 3 days (Supplementary Fig. 1a). In contrast, short-term treatment of the cortical cultures with 1, 3, or 5 mM lithium salt does not cause changes in Smad-dependent transcription for up to 16 h (Supplementary Fig. 1b). Moreover, lithium’s inhibitory effect on Smad3/4 transcriptional regulation was also observed in neurons stimulated with TGF-β at 0.5, 1, and 3 ng/ml concentrations (Fig. 1b).
Figure 1.
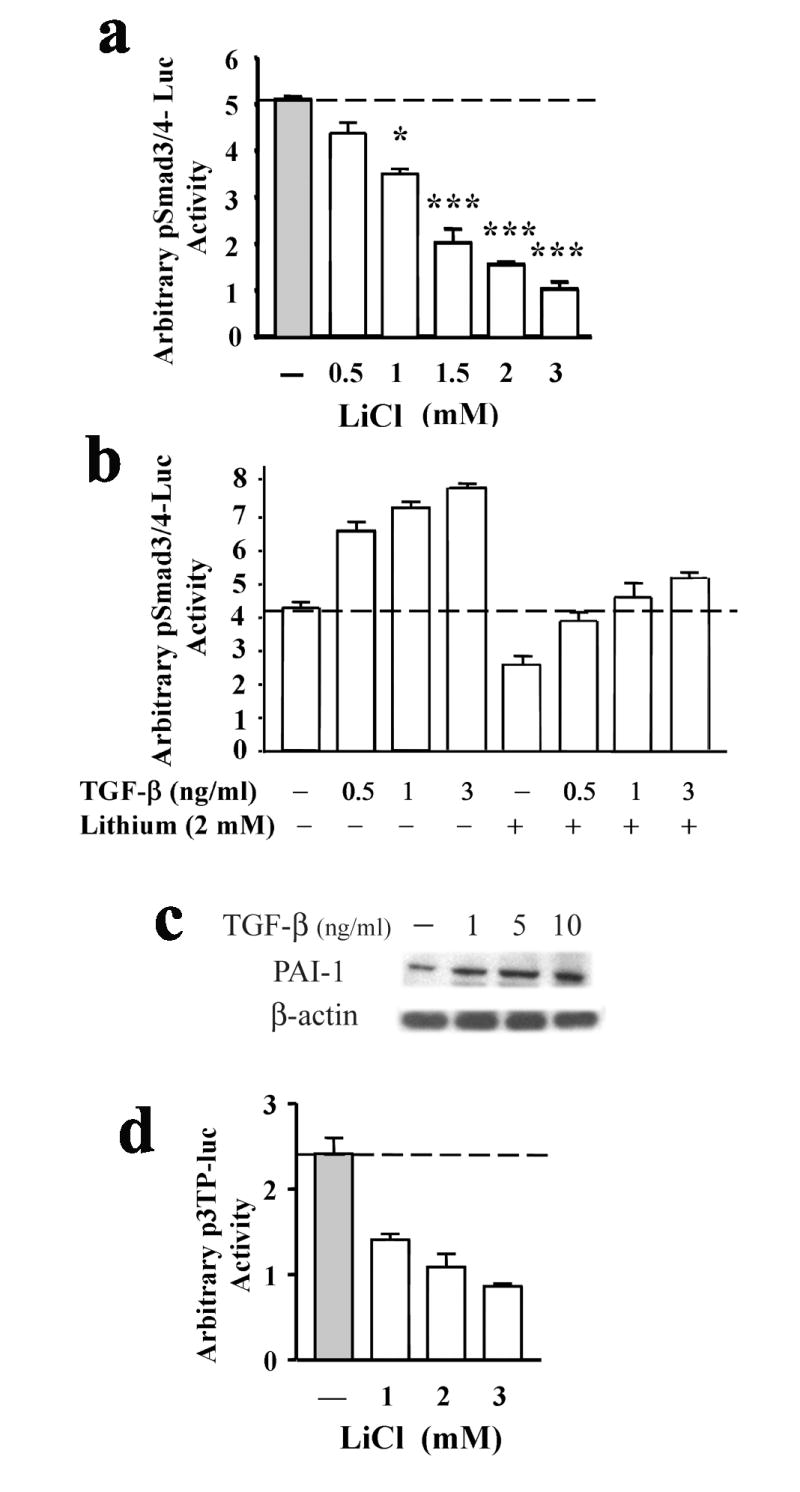
Lithium suppresses Smad3/4-dependent transactivation and reduces TGF-β-responsive Smad3/4-dependent gene promoter activity of PAI-1 gene. (a) Cultured cortical neurons were transfected with pSmad3/4-Luc reporter construct at the time of plating and treated with or without lithium on 8-DIV at various concentrations for 72 h. (b) Cultured cortical neurons were transfected with pSmad3/4-Luc reporter construct at the time of plating and at 9-DIV pre-treated without or with TGF-β (3 ng/ml) for 8 h followed by optional lithium treatment for an additional 40 h. (c) Representative Western blots of levels of PAI-1 in cortical neurons treated with TGF-β for 24 h at 10-DIV. Cell lysates were collected for Western blotting analysis at 11-DIV. β-actin was used as a loading control. (d) Cultured cortical neurons were transfected with p3TP-Luc reporter construct at the time of plating and treated with or without lithium on 9-DIV at various concentrations for 48 h. Luciferase activities were determined on 11-DIV. Representative results shown as arbitrary luciferase activities are means ± SEM of one set of data from three independent experiments. * p<0.05; *** p<0.001, compared to the untreated control using one-way ANOVA.
TGF-β stimulation of cortical cells for 24 h caused an increase in protein levels of PAI-1, a TGF-β-targeting gene (Fig. 1c). Lithium’s therapeutic effects in treating biplolar disorders and its neuroprotective functions in in vitro models are known to take effect only after chronic administration of the drug. Thus, our data suggest that lithium’s inhibition on Smad3/4 transcription is likely an indirect effect that is regulated through other signaling pathways. Additionally, due to potential toxicity of lithium salt in high concentrations, we consistently exposed cortical cells to 1.5 mM lithium salt for 48 h in this study. By using p3TP-Luc, a PAI-1 promoter reporter construct consisting of Smad3/4-TGF-β-responsive elements, we observed a dose-dependent decrease in p3TP gene promoter activity in neurons treated with lithium for 48 h (Fig. 1d). Similar results were observed in neurons transfected with other TGF-β-responsive gene promoter constructs, p21P-Luc and p15-Luc, followed by lithium treatment at various concentrations (Supplementary Fig. 1c and 1d). Cyclin-dependent kinase inhibitor p21WAF/Cip1, also referred to as cdkn1a, has been known to be activated by p53 (reviewed in Kumar et al., 2006; Maddika et al., 2007) or induced by TGF-β through a p53-independent mechanism (Datto et al., 1995). Together, these results indicate lithium’s inhibitory effects on Smad3/4-dependent TGF-β signaling.
Lithium suppresses Smad3/4-dependent transactivation partially through the PKA signaling pathway
To study the involvement of cAMP/PKA signaling in lithium’s inhibitory effect on Smad3/4-dependent gene expression, the PKA kinase assay was used to determine lithium’s ability to modulate basal PKA activity in cultured cortical neurons. LiCl induced a concentration-dependent activation of PKA with a 2.5-fold increase in activity after treatment with 1 mM for 24 h (Fig. 2a). The PKA activation was slightly decreased by treating the cultures with higher concentrations of this drug. In support of the previous report that lithium treatment increases intracellular levels of cAMP in human neuroblastoma cells (Montezinho et al, 2004), we found that cAMP levels were significantly increased to 140 ± 5.6%, 157 ± 4.7%, and 148 ± 7.2% of control after 24 h treatment of cortical cultures with 1.5 mM, 2 mM and 2.5 mM LiCl, respectively. The viability of cortical cells treated with various concentrations of LiCl determined by MTT staining or LDH release was not significantly affected under these experimental conditions (data not shown).
Figure 2.

Lithium’s inhibitory effects on Smad3/4 transactivation are partially mediated through the PKA signaling pathway. (a) Effects of lithium on PKA kinase activity in cultured cortical neurons. Cells (2000 cells/well in 96-well poly-D-lysine coated culture plates) were incubated with or without lithium at the indicated concentrations for 24 h. Non-radioactive PKA activity was determined as described in the Materials and Methods. Values are means ± SEM of % of control from three independent experiments. (b) Cortical neurons were transfected with pSmad3/4-Luc reporter construct at the time of plating and pretreated for 4 h with PKA specific inhibitors, KT5720 and H-89, at the indicated concentrations, followed by incubation with 2 mM LiCl on 9-DIV. Alternatively, pSmad3/4-Luc-transfected cultured cortical neurons were treated with 2 mM lithium and cAMP-elevating agents, 8-Br-cAMP and forskolin, on 9-DIV. Incubations were continued for 48 h, after which luciferase activities were determined on 11-DIV. Representative results shown as arbitrary luciferase activities are means ± SEM of triplicates of one set of data from three independent experiments. ** p<0.05; *** p<0.001, compared to the respective treatment controls using one-way ANOVA. (c) Representative Western blots of levels of PAI-1, pCREBSer133, CREB, and β-actin from three experiments. Cortical neurons were pretreated with H-89 (90 nM) for 4 h followed by lithium at the indicated concentrations on 9-DIV for 48 h. Cell lysates were collected on 11-DIV for immunoblotting analysis. (d) Representative Western blots of levels of PAI-1, pCREBSer133, CREB, and β-actin (used as a loading control) from three experiments. Cultured cortical neurons were pretreated with H-89 (90 nM) for 4 h on 9-DIV followed by forskolin (100 μM) treatment for 48 h. Cell lysates were collected on 11-DIV for immunoblotting analysis.
Two cAMP-elevating agents were tested for their ability to affect Smad3/4-dependent gene expression using Smad3/4-Luc. Pre-incubation of cortical cultures with the membrane-permeable cAMP analog, 8-Br-cAMP, or the adenylate cyclase (AC) activator, forskolin, for 24 h resulted in a concentration-dependent decrease of basal Smad3/4-dependent transcriptional activities (Supplementary Fig. 2a). These results strongly suggest that Smad3/4-specific gene expression is negatively regulated by the activation of PKA signaling. Conversely, treatment with PKA inhibitors such as KT5720, H-89 or PKI raised basal levels of Smad3/4-specific transcriptional activity in a dose-dependent manner (Supplementary Fig. 2b), further supporting a negative role of PKA signaling in Smad3/4-dependent transactivation. Under these experimental conditions, treatment with 2 mM lithium chloride for 48 h reduced Smad3/4-dependent transcriptional activity by about 55% (Supplementary Fig. 2a and b).
To verify the stimulus specificity of PKA in antagonizing Smad3/4-specific TGF-β signaling, cultured cortical neurons were transfected with the Smad3/4-Luc reporter construct, and then pretreated with the PKA inhibitors KT5720 or H-89 for 4 h, followed by LiCl incubation for 48 h. We found that both KT5720 and H-89 strongly, although not completely, blocked lithium-induced suppression of Smad3/4-dependent transactivation (Fig. 2b). Thus, lithium-induced inhibition of Smad3/4-dependent activity was reduced from 51% to 17% and 21% by the presence of KT5720 and H-89, respectively. These results suggest that lithium’s inhibitory effect on Smad3/4 signaling is in part mediated through cAMP-dependent PKA activation.
Treatment of cortical cultures with lithium resulted in a decrease in levels of a TGF-β-responsive Smad3/4-dependent gene, PAI-1, and an increase in levels of pCREBSer133 (Fig. 2c). Quantification of blots indicated that inhibition of PKA signaling with H-89 antagonized lithium’s regulatory effects on gene expression of PAI-1 and pCREBSer133 (Supplementary Fig. 2c). Similar to lithium, forskolin treatment resulted in decreased levels of PAI-1 and increased expression of pCREBSer133, and H-89 pretreatment antagonized these forskolin-induced effects (Fig. 2d).
The inhibition of AKT or PI3-K signaling partially blocked lithium’s inhibitory effects in Smad3/4 signaling
Our previous study demonstrates a downregulation of TGF-β/Smad3/4 signaling by GSK-3β protein depletion (Liang and Chuang, 2006). Lithium is known to inhibit GSK-3 indirectly through PI3-K/AKT signaling (Chalecka-Franaszek and Chuang, 1999). We thus examined whether the activation of AKT causes GSK-3β inhibition and results in changes in Smad3/4-dependent transcriptional activation. As shown in Fig. 3a, overexpression of a constitutively active form of AKT (AKT-CA), which serves as a GSK-3β inhibitor, suppressed Smad3/4 transcription to an extent similar to that elicited by lithium (2 mM for 48 h). Additional inhibitory effects were observed in cells transfected with AKT-CA and further treated with lithium. Similar additional inhibitory effects were observed in neuronal cultures overexpressing AKT-CA followed by 100 μM forskolin or 8-Br-cAMP treatment ( Supplementary Fig. 3a). Western blotting analysis demonstrated that AKT-CA overexpression resulted in an increase and decrease in β-catenin and PAI-1 expression, respectively (Supplementary Fig. 3b). Of interest, AKT-CA overexpression also caused an increase in levels of pAKTThr308, similar to the effect of lithium.
Figure 3.
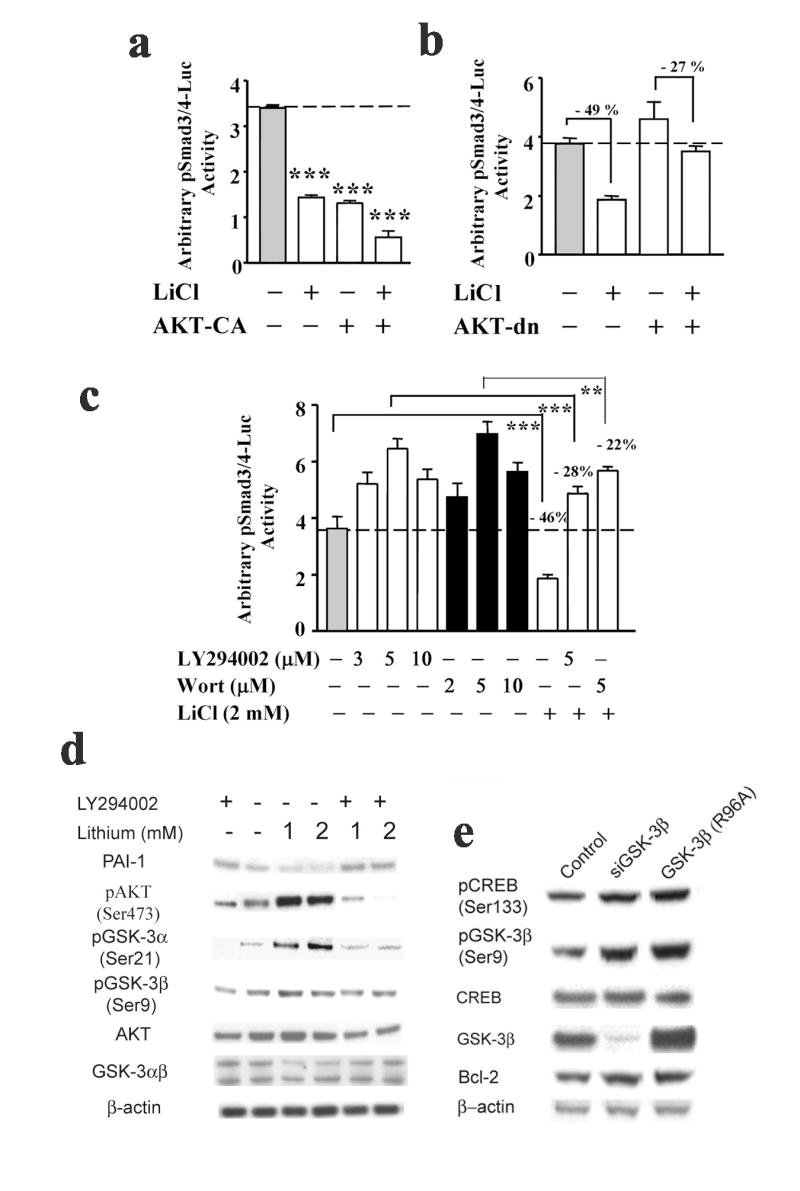
Lithium’s negative regulation in Smad3/4-dependent transcription is partially blocked by the inhibition of AKT and PI3-K signaling pathways. (a) At the time of plating, cortical neurons were either co-transfected with 4 μg of pSmad3/4-Luc reporter construct and the expression vector of AKT constitutively active mutant or transfected with pSmad3/4-Luc reporter construct followed by treatment with lithium for 48 h on 9-DIV. The luciferase activities of cell lysates were determined on 11-DIV. (b) At the time of plating, cortical neurons were either co-transfected with 4 μg of pSmad3/4-Luc reporter construct and expression vector for the dominant negative form of AKT, or transfected with pSmad3/4-Luc reporter construct followed by treatment with lithium for 72 h on 8-DIV. The luciferase activities of cell lysates were determined on 11-DIV. (c) Cortical neurons were transfected with 4 μg of pSmad3/4-Luc reporter construct at the time of plating and followed by 4 h pretreatment with PI3-K inhibitors, LY294002 or Wortmannin (Wort), at the indicated concentrations, followed by lithium treatment on 9-DIV for 48 h. The luciferase activities of cell lysates were determined on 11-DIV. Representative results shown as arbitrary luciferase activities are means ± SEM of triplicates of one set of data from three independent experiments. ** p< 0.01; *** p<0.001, compared to the respective controls using one-way ANOVA. (d) Representative Western blots of levels of PAI-1, pAKT (Ser473), pGSK-3α (Ser21), pGSK-3β (Ser9), AKT, and GSK3αβ from three experiments. Cultured cortical neurons were pretreated with 5 μM LY294002 on 9-DIV for 4 h followed by lithium treatment at the indicated concentrations for 48 h. Cell lysates were collected on 11-DIV for immunoblotting analysis. (e) Representative Western blotting analysis of levels of pCREB (Ser133), pGSK-3β (Ser9), CREB, GSK-3β, and Bcl-2 from three experiments. Cultured cortical neurons were transfected with either GSK-3β isoform-specific siRNAs on 9-DIV or GSK-3β dominant negative mutant (R96A) at the time of plating. Cell lysates were collected on 11-DIV for immunoblotting analysis. β-actin was used as a loading control.
Overexpression of AKT-dn, a dominant negative mutant form of AKT, reduced lithium-induced Smad3/4-dependent transcriptional suppression by 22% (49% for lithium’s effect vs 27% for the effects of AKT-dn plus lithium; Fig. 3b). These results showed that the inactivation of AKT signaling partially antagonized lithium’s negative regulation of Smad3/4-dependent transcriptional activation. Similarly, cortical cultures treated with the PI3-K inhibitors LY-294002 and Wortmannin dose-dependently increased Smad3/4 transcriptional activity and partially blocked lithium-induced inhibitory effects on Smad3/4-dependent gene transcription (− 28% for LY-294002 plus lithium and − 22 % for Wortmannin plus lithium vs 46% for lithium alone; Fig. 3c). Our results to date indicate that lithium’s inhibitory regulation on Smad3/4 transcription is mediated through both PKA and PI3-K-dependent AKT signaling pathways.
Additionally, lithium-induced activation of these two signaling pathways resulted in an increase in the protein level of pCREBSer133, which is indicative of increased CRE-dependent transcription. Western blotting analyses further demonstrated that lithium treatment induced an increase in protein levels of pAKTSer473 and pGSK-3αβSer21/9 and a decrease in PAI-1 protein levels (Fig. 3d). Importantly, these inducible effects were partially blocked by pretreatment with LY-294002 (5 μM) (Fig. 3d), suggesting an involvement of PI3-K activation. Conversely, GSK-3β inhibition by isoform-specific GSK-3β protein depletion and overexpression of GSK-3β dominant negative mutant (R96A) resulted in a modest increase in protein levels of pCREBSer133, pGSK-3βSer9 and anti-apoptotic protein Bcl-2 (Fig. 3e). This data further supports the commonly recognized concept that activated AKT inhibits GSK-3β to induce upregulation of pGSK-3βSer9 and pCREBSer133 (reviewed in Rowe and Chuang, 2004). Together, our results strongly suggest that activation of both AKT and PKA signaling is involved in lithium’s inhibitory regulation on Smad3/4-dependent transcription through an increase in the levels of pGSK-3βSer9 and pCREBSer133.
Lithium-activated CRE-dependent response is partially blocked by the inhibition of PKA and AKT signaling
We previously reported that GSK-3β gene silencing activates cAMP-responsive gene transactivation by 3-fold in cultured cortical neurons (Liang and Chuang, 2006). In addition, treatment of cortical cultures with 1 mM LiCl for 5 days resulted in a 1.8-fold increase in CRE-dependent transactivation (unpublished data). The ability of PKA activation to induce cAMP-dependent transactivation in cortical cultures was thus examined by transfecting cortical cells with pCRE-Luc reporter construct and then treating cultures with lithium or forskolin at various concentrations for 48 h. As shown in Fig. 4a, both lithium and forskolin treatments activated CRE-dependent transcription in a concentration-dependent manner. Treatment of cultures with 1.5 mM lithium salt and 50 mM forskolin yielded a marked increase in CRE-dependent transcriptional activity above basal values (Fig. 4a). On the other hand, H-89 treatment caused a 53% reduction in CRE-dependent transcriptional activity. Inactivation of PKA by H-89 inhibited lithium-induced CRE-dependent transcriptional upregulation by 23%. However, PKA inhibition attenuated forskolin-induced CRE-responsive transactivation to a greater extent (41%). These results imply involvement of additional signaling pathways in lithium’s effects on CRE-dependent transactivation. In addition, lithium treatment caused a dose-dependent increase in protein levels of pCREBSer133, Bcl-2, Bcl-xL and to a lesser extent total CREB, but a decrease in the expression of the pro-apoptotic protein Bax, as measured by Western blotting analysis (Supplementary Fig. 3c). Inhibition of the cAMP/PKA signaling pathway by H-89 pretreatment blocked/suppressed forskolin- or lithium-induced increase in levels of pCREBSer133, pGSK-3βSer9 and Bcl-2, and also blocked forskolin- or lithium-elicited decrease in Bax protein levels in cultured cortical neurons (Supplementary Fig. 3d). These results demonstrate the proficiency of PKA signaling in activating CRE-dependent genes, and suggest that cAMP/PKA signaling is involved in the modulation of lithium’s regulation on CRE-dependent transactivation.
Figure 4.
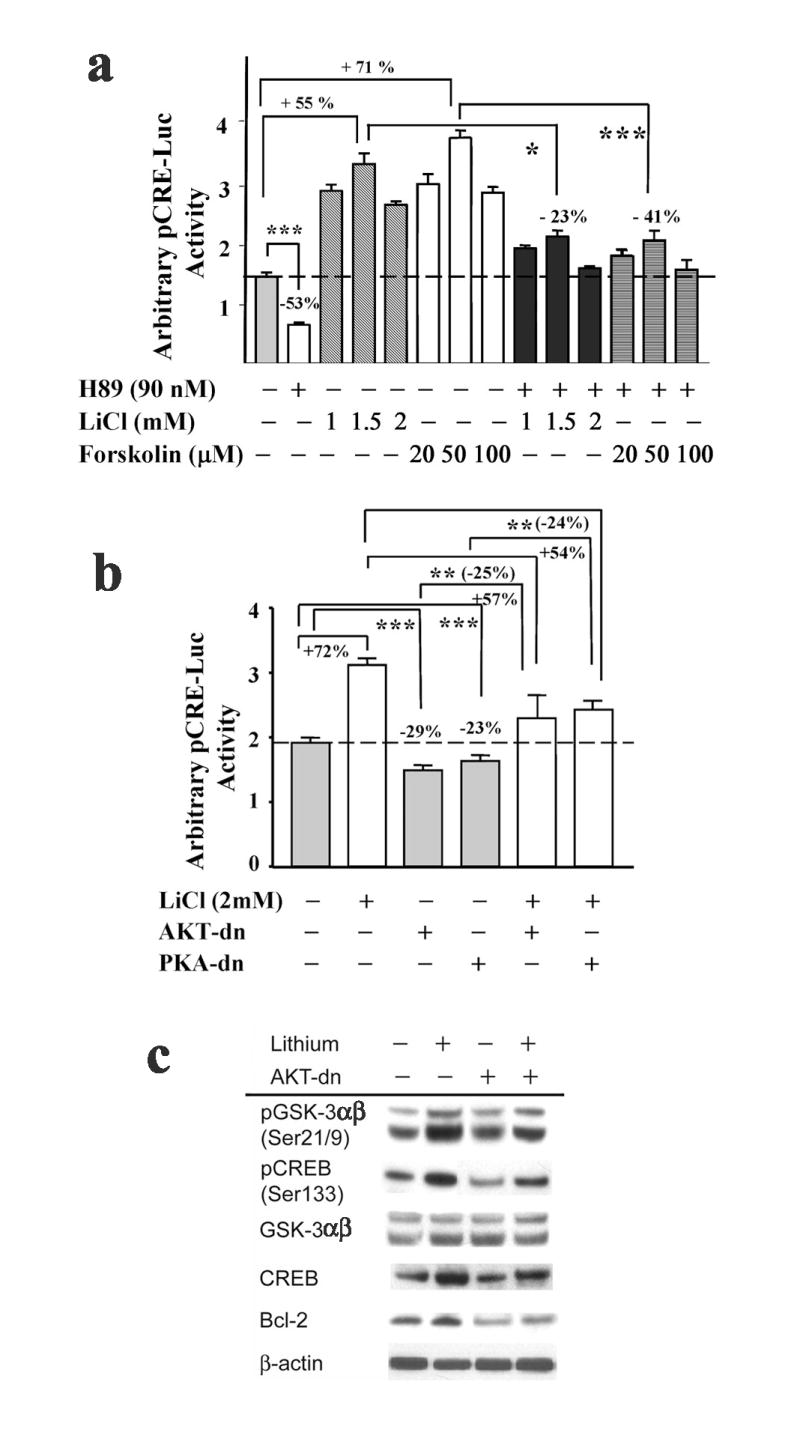
Lithium-induced CRE-dependent responses are partially blocked by the inhibition of AKT and PKA signaling. (a) Cortical neurons were transfected with pCRE-Luc reporter construct at the time of plating and pretreated with either the PKA specific inhibitor, H-89, or the cAMP-elevating agent forskolin at the indicated concentrations for 4 h, followed by an incubation with or without the indicated concentrations of lithium on 9-DIV. Incubations were continued for 48 h, after which luciferase activities were determined on 11-DIV. (b) Cortical neurons were transfected with pCRE-Luc reporter construct with or without dominant negative mutant forms of AKT or PKA (AKT-dn, PKA-dn, respectively) at the time of plating and treated with or without 2 mM lithium on 9-DIV. Incubations were continued for 48 h, after which luciferase activities were determined on 11-DIV. Representative results shown as arbitrary luciferase activities are means ± SEM of triplicates of one set of data from three independent experiments. * p<0.05; ** p< 0.01; *** p<0.001, compared to the respective control using one-way ANOVA. (c) Representative Western blotting analysis of protein levels of pGSK-3αβ (Ser21/9), pCREB (Ser133), GSK-3αβ, CREB, and Bcl-2 from three experiments. Cortical neurons were transfected with a dominant negative form of AKT at the time of plating with or without treatment of 1.5 mM lithium on 9-DIV for 48 h. Cell lysates were collected on 11-DIV for Western blotting analysis. β-actin was used as a loading control.
Our results suggest that lithium activates pCREBSer133 phosphorylation and CRE-dependent transactivaton partially through AKT/GSK-3β and cAMP/PKA signaling pathways. We next examined whether cAMP/PKA signaling predominantly regulates lithium’s upregulation in CRE-dependent transactivation, because PKA can induce CREB serine133 phosphorylation through two mechanisms: directly by phosphorylation of CREB and indirectly through the inhibition of GSK-3β. As shown in Fig. 4b, overexpression of both AKT-dn and PKA-dn mutants attenuated CRE-dependent transactivation to a similar degree (29% vs 23%). Lithium-induced increase in CRE-dependent transcriptional activity was reduced by overexpression of either AKT-dn or PKA-dn mutants. Overexpression of AKT-dn largely suppressed lithium-induced increase in protein levels of pGSK-3αβSer21/9, pCREBSer133 and Bcl-2 (Fig. 4c), although it was noted that AKT-dn alone also reduced the levels of pCREBSer133 and Bcl-2. Collectively, our results demonstrate the critical roles of both PKA and AKT/GSK-3β signaling pathways in lithium’s modulation on CRE-dependent transactivation and its downstream target gene expression.
Inhibitory effects of lithium on Smad3/4 transactivation are partially mediated through CREB
We next investigated the role of CREB, a downstream effector of both AKT and PKA signaling, in PKA-activated CRE-dependent transcription. Overexpression of the catalytic subunit of PKA caused a 3-fold increase in CRE-dependent transcriptional activation (Fig. 5a). PKA-induced CRE-dependent transcriptional upregulation was completely blocked by inactivating endogenous CREB with exogenously expressed CREB dominant negative mutants, dn-CREB133 (mutated PKA-specific phosphorylation site at serine 133, thus preventing transcription) and dn-KCREB (mutated in DNA-binding domain, thus blocking CREB’s ability to bind CRE) (Fig. 5a). These data were further supported by a reduction in protein levels of several PKA-induced anti-apoptotic proteins including pCREBSer133, Bcl-2, and Bcl-xL in neurons overexpressing both dominant negative mutant forms of CREB (Fig. 5b). These results indicate that the PKA-CREB signaling cascade regulates CRE-dependent transactivation and its downstream gene expression. Moreover, PKA activation by overexpression of the PKA catalytic subunit caused an increase in pGSK-3αβSer21/9 phosphorylation levels, which was partially blocked by dn-CREB133 overexpression and completely blocked by dn-KCREB overexpression (Fig. 5b) raising the possibility of feedback regulation by CREB on PKA activity or GSK-3 serine phosphorylation. Further, the overexpression of cotransfected-wild-type CREB (wt-CREB) and the catalytic subunit of PKA resulted in an increase in protein levels of pCREBSer133, pGSK-3αβSer21/9, Bcl-2, and Bcl-xL, similar to the effects of PKA overexpression.
Figure 5.

The inhibitory effects of lithium, PKA signaling and AKT signaling on Smad3/4-dependent transcription are modulated through CREB. (a) Cortical neurons were transfected with pCRE-Luc reporter construct together with or without expression vectors for the catalytic subunit of PKA and/or either one of two mutant forms of CREB, dn-CREB133 and dn-KCREB, at the time of plating. Luciferase activities were determined on 11-DIV. Representative results shown as arbitrary luciferase activities are means ± SEM of triplicates of one set of data from three independent experiments. *** p<0.001, compared to the PKA alone using one-way ANOVA. (b) Representative Western blotting analysis of protein levels of pCREB (Ser133), pGSK-3αβ (Ser21/9), Bcl-2, Bcl-xL, GSK-3αβ, and CREB from three experiments. β-actin was used as a loading control. Cortical neurons were transfected with the expression vectors for the catalytic subunit of PKA and wild type (wt-CREB) or mutant form of CREB (dn-CREB133 and dn-KCREB) at the time of plating. Cell lysates were collected on 11-DIV for Western blotting analysis. (c) Cortical neurons were transfected with pSmad3/4-Luc reporter construct together with the expression vectors for either the catalytic subunit of PKA and wild type (wt-CREB) or mutant CREB (dn-CREB133 and dn-KCREB) at the time of plating. Alternatively, the cultures were transfected with pSmad3/4-Luc reporter construct together with the expression vectors for either wild type (wt-CREB) or mutant CREB (dn-CREB133 and dn-KCREB) at the time of plating followed by lithium (2 mM) treatment on 9-DIV for 48 h. (d) Cortical neurons were transfected with pSmad3/4-Luc reporter construct together with the expression vectors for the constitutively active form of AKT and either the wild-type (wt-CREB) or mutant form of CREB (dn-CREB133 and dn-KCREB) at the time of plating. Luciferase activities were determined on 11-DIV. Representative results shown as arbitrary luciferase activities are means ± SEM of triplicates of one set of data from three independent experiments. ** p<0.01;***p<0.001, compared to the respective control using one-way ANOVA.
We subsequently determined whether CREB was able to interfere directly with Smad3/4-dependent transcription. For this purpose, expression vectors for the catalytic subunit of PKA or the wild-type CREB (wt-CREB) transcription factor were cotransfected together with Smad3/4-Luc reporter vector into cultured neurons. As shown in Fig. 5c, overexpression of either wt-CREB or PKA robustly suppressed Smad3/4-dependent transcriptional activation, even to a greater extent than lithium’s effects. However, cotransfection of PKA and wt-CREB did not produce additional inhibitory effects, suggesting a convergent action. Overexpression of dominant negative mutant dn-CREB133 or dn-KCREB largely blocked PKA-induced suppression of Smad3/4-dependent transcription. The blocking action of dn-KCREB also suggests that the effect of CREB on Smad3/4 transactivation was only partially dependent on PKA activity. Similarly, CREB’s negative regulation in Smad3/4-dependent transcriptional activation was observed in neurons treated with lithium (Fig. 5c) or overexpressing AKT-CA constitutively active mutant (Fig. 5d). Moreover, dn-CREB133 or dn-KCREB overexpression blocked AKT-CA-induced downregulation of Smad3/4-dependent transactivation. Thus, our results indicate the interference of the PKA-CREB and AKT-CREB cascades with the Smad3/4 pathway. This cross-talk demonstrates a critical role of CREB and its upstream signalings in the regulation of Smad3/4-dependent transcriptional activation.
Transcriptional coactivator, p300, recruited by activated CREB is critical to the interference of PKA signaling and regulation of Smad 3/4-dependent TGF-β gene promoters, 3TP and p21
It has been documented that CBP and p300 function as essential transcriptional coactivators for Smad-dependent gene expression and that competition for CBP/p300 could be a mechanism to mediate signal-induced transcriptional repression (Hottiger and Nabel, 1998; Schiller et al., 2003). CBP and p300 are also known to be coactivators of CREB (Chrivia et al., 1993). We thus hypothesized that compensation of transcriptional coactivators by overexpressing either CBP or p300 might reverse lithium’s inhibitory effects against TGF-β/Smad3/4-dependent transactivation resulting from pCREB-CRE interaction. Accordingly, the effects of recruitment of transcriptional coactivator CBP or p300 in the transcriptional regulation of TGF-β/Smad-dependent gene targets were examined.
By using p3TP-Luc, a PAI-1 promoter reporter construct, we first demonstrated that lithium suppressed 3TP promoter activity of the PAI-1 gene (Fig. 1c, and 6a and b). We found that overexpression of both CBP and p300 increased basal PAI-1 promoter activities (Fig. 6a and b), an observation suggesting limited availability of CBP/p300 in the cell nucleus and the role of CBP/p300 as a Smad transcriptional coactivator. However, only histone acetyltransferase (HAT)-deficient p300 (Fig. 6b), but not HAT-deficient CBP (Fig. 6a), inhibited basal PAI-1 promoter activity, suggesting that p300 is a primary transcriptional coactivator recruited by transcription factors to the PAI-1 gene promoter. Of interest, overexpression of p300 (Fig. 6b), but not CBP (Fig. 6a), completely antagonized lithium-induced reduction of PAI-1 promoter activity. Importantly, lithium treatment increased pCREB-p300 complex formation but decreased levels of pSmad2/3 (Ser433/435) in cortical cultures (Fig. 6c). This data strongly suggests that immuno-precipitated p300 (IP-p300) bound to pCREBSer133 is in a more abundant amount than that bound to pSmad2/3Ser433/435 in cells treated with lithium. However, overexpression of wild type p300 expression vector reversed the effects of lithium in suppressing binding activity of pSmad3 with p300 coactivator (Fig. 6c). The overexpressed p300 thus restores normal levels of p300 in the transcriptional complex bound to Smad3/4-dependent gene promoters and salvages the potentially suppressed Smad3/4 transcription from the lack of sufficient amounts of p300 in the neuronal nuclei. Together, our data suggest that the inhibitory effects of lithium in Smad3/4 signaling are due to complex formation of activated CREB and p300, resulting in limited interactions of p300 with the transcription factors/Smad complexes, and thus preventing efficient Smad3/4-dependent transcription of TGF-β-responsive target genes.
Figure 6.

Overexpression of transcriptional coactivator p300 abolishes lithium’s inhibitory regulation on the promoter activities of Smad3/4-dependent TGF-β-responsive downstream gene, 3TP. (a) Cultured cortical neurons were transfected with p3TP-Luc reporter construct together with expression vectors for wild-type CBP (CBP) or HAT-deficient CBP mutant (CBP-ΔHAT) at the time of plating. Lithium (1.5 mM) was added starting at 9-DIV and incubations were continued for 48 h before luciferase activity was determined on 11-DIV. (b) Cortical neurons were transfected with p3TP-Luc reporter construct together with expression vectors for wild-type p300 (p300) or HAT-deficient p300 mutant (p300-ΔHAT) at the time of plating. Lithium treatment and luciferasea activity assay were carried out as described in Fig. 5c. Representative results shown as arbitrary luciferase activities are means ± SEM of triplicates of one set of data from three independent experiments. *** p<0.001, compared to the respective controls. n.s,, not significant from the comparative control using one-way ANOVA. (c) Representative IP-Western blotting (WB) analysis of protein levels of pCREB(Ser133) and pSmad2/3(Ser433/435) from three experiments. β-actin was used as an internal control. Cortical neurons were transfected with or without p300 wild-type (WT) expression vector at the time of plating. Lithium treatment was initiated on 9-DIV for 48 h. Cell lysates were collected on 11-DIV for anti-β-actin antibody immunoprecipitation (control and lithium) or anti-p300 and anti-β-actin antibodies co-immunoprecipitation (p300-WT and p300-WT plus lithium) and Western blotting analysis. (d) Representative Western blotting analysis of protein levels of pCREB (Ser133), CREB, Bcl-2, and β-catenin from three experiments. β-actin was used as a loading control. Cortical neurons were transfected with expression vectors for either wild-type or HAT-deficient CBP/p300 at the time of plating. Lithium treatment was carried out on 9-DIV for 48 h. Cell lysates were collected on 11-DIV for Western blotting analysis.
Downregulation of Smad3/4-dependent gene transcription by the formation of the CREB-p300 complex was further studied with another TGF-β-responsive gene promoter construct, p21P-Luc. Lithium modestly inhibited p21 promoter activity in cultured cortical neurons (Supplementary Fig. 4a and b). Intriguingly, overexpression of CBP wild-type and CBP-ΔHAT mutant did not significantly affect the basal state or lithium’s effect on p21 promoter activity, indicating that CBP may not be involved in p21 transcriptional regulation (Supplementary Fig. 4a). On the other hand, p300 overexpression activated p21 promoter transcription, and completely blocked lithium’s negative regulation of p21 gene transcription (Supplementary Fig. 4b), a result similar to the effects on PAI-1 gene transcription (Fig. 6b). Conversely, overexpression of p300-ΔHAT markedly decreased p21 promoter activity in the absence or presence of lithium treatment (Supplementary Fig. 4b). The biochemical effects of overexpressed wild-type CBP and p300 transcriptional coactivators in cultured cortical neurons were profiled by immunoblotting analysis (Fig. 6d). As expected, overexpression of both wild-type transcriptional coactivators increased the protein levels of pCREBSer133 and Bcl-2, effects similar to those of lithium (Fig. 6d). The protein levels of β-catenin, a downstream transcriptional activator of Wnt signaling, were also increased in neurons transfected with wild-type CBP or p300 (Fig. 6d), suggesting the inhibition of GSK-3 activity. On the other hand, overexpression of HAT-deficient mutants of both coactivators caused a decrease in protein levels of pCREB(Ser133), Bcl-2, and β-catenin.
Thus far, our results suggest the involvement of p300, in mediating lithium’s negative effects on 3TP and p21 promoter activity (Fig. 6 and Supplementary Fig. 4). We next performed a CHIP assay to examine whether lithium-induced decrease in 3TP and p21 gene promoter activity was primarily associated with p300, but not CBP. Cultured cortical neurons treated with lithium were cross-linked with formaldehyde and subjected to nuclei isolation and sonication. The sheared chromatins were then immunoprecipitated with antibodies against either p300 or CBP. The immunoprecipitated DNA was next purified and amplified by real-time fluorescent PCR. The results showed a stronger amplification (16-fold resulting from 4 cycle differences) of DNA immunoprecipitated with anti-p300 (30 cycles on log fluorescence for p300-IP samples) than with anti-CBP (at least 34 cycles on linear log fluorescence for CBP-IP samples) in the PAI-1/Serp1 promoter (Supplementary Fig. 5a). These results are consistent with the notion that p300 is bound with a greater amount than CBP to the PAI-1/Serp1 promoter to regulate its transcription. Interestingly, no detectable signal was observed in PCR amplification of the p21/cdkn1a gene promoter when anti-CBP antibody was used in immunoprecipitation, even though robust amplification was detected with anti-p300 antibody (Supplementary Fig. 5b). Gel electrophoresis also showed more PCR amplification of the PAI-1/Serp1 and p21/cdkn1a gene promoters in DNA from neurons that were immunoprecipitated with p300 compared with DNA immunoprecipitated with anti-CBP (Supplementary Fig. 5c). Together, these results further confirm the results in Supplementary Fig. 4, and demonstrate the involvement of p300, but not CBP, in the coactivator-transcription factor complex bound to the p21/cdkn1a gene promoter.
Discussion
In this study, we used the methods of pharmacological treatments, gene silencing, and gene delivery to provide the first evidence that lithium suppresses Smad3/4-dependent TGF-β-responsive gene transactivation through cross-talk of CRE-dependent, cAMP/PKA, and PI3-K/AKT/GSK-3β signaling pathways in rat cortical cultures. These results suggest that lithium suppresses Smad3/4-dependent gene transcription via an increased sequestration of p300 by pCREBSer133 and a disruption of the Smad-p300 complex. A schematic illustration showing the proposed sites of actions of lithium and interactions among these effectors/pathways in regulating Smad3/4 transcription is shown in Fig. 7. These actions of lithium likely involve the activation of CRE-dependent gene transcription via increased CREB Ser133 phosphorylation and subsequent pCREBSer133-p300 complex formation (Jope, 1999a). Our results strongly suggest that the sequestration of p300 by lithium-induced activated pCREBSer133 results in the reduction of Smad3-p300 interaction in the promoter of two natural Smad3/4-dependent TGF-β-responsive target genes, PAI-1 and p21. We also showed that these effects of lithium can be overcome by the presence of a sufficient amount of p300, but not CBP, in the cell nucleus. Lithium also increases the protein level of tPA (data not shown), a direct target of PAI-1 (Wind et al., 2002). Multiple roles of the tPA-plasmin system in the central nervous system have been suggested, ranging from neural organization to brain disorders (reviewed in Samson and Medcalf, 2006; Vivien and Ali, 2006). Our study shows that lithium regulates the PAI-1-tPA-plasmin system through cross-talk among TGF-β/Smad and lithium-targeted signaling pathways, and thus provides critical information regarding lithium’s novel molecular and cellular actions.
Figure 7.

Schematic illustration of lithium’s negative regulation on Smad/TGF-β signaling mediated through the interactions of three signaling pathways: Smad/TGF-β, cAMP-PKA and PI3-K/AKT/GSK-3β. Three signaling pathways, namely, Smad/TGF-β, cAMP-PKA and PI3-K/AKT/GSK-3β signalings can be triggered by stimulation with TGF-β and BDNF through their surface receptors, and the interactions of these pathways are shown. Lithium treatment causes Smad3/4-dependent transcriptional suppression of two TGF-β-responsive genes, PAI-1 and p21. These effects of lithium are caused by the sequestration of transcriptional coactivator p300 due to an increase of phospho-CREBSer133 levels, which results in the increase of CRE-dependent transactivation and the expression of survival factors, BDNF and Bcl-2. The inhibitory regulation by lithium of Smad3/4 transactivation involves stimulation of cAMP-PKA and PI3-K-AKT signaling pathways, but inhibition of GSK-β activity.
 : Direct stimulatory modification,
: Direct stimulatory modification,
 : Direct inhibitory modification,
: Direct inhibitory modification,
 : Nuclear translocation. The stimulatory (broken line,
: Nuclear translocation. The stimulatory (broken line,
 ) and inhibitory (broken line,
) and inhibitory (broken line,
 ) effects of various treatments of cortical neurons with lithium, inhibitors/analogs specific to PKA and PI3-K or the exogenous overexpression of PKA and AKT mutants (constitutively active and dominant negative mutants) are shown.
) effects of various treatments of cortical neurons with lithium, inhibitors/analogs specific to PKA and PI3-K or the exogenous overexpression of PKA and AKT mutants (constitutively active and dominant negative mutants) are shown.
We have previously shown that Smad3/4-dependent transcriptional activation is regulated by GSK-3 α and β isofrms in opposite manners (Liang and Chuang, 2006). Thus, inactivation of GSK-3α and GSK-3β results in activation and inhibition of Smad3/4-dependent transactivation, respectively. Current data indicates an inhibitory effect of lithium in its modulation of Smad3/4 transactivation, an outcome similar to GSK-3β. Lithium is known to inhibit both GSK-3 isoforms by multiple mechanisms (Klein and Melton, 1996; Stambolic et al., 1996). Direct GSK-3 inhibition of lithium was identified to be through competition for magnesium in the active site of GSK-3 (Gurvich and Klein, 2002; Ryves and Harwood, 2001). Original measures of lithium’s inhibitory potency found 50% inhibition at concentrations of 1–2 mM (Klein and Melton, 1996; Stambolic et al., 1996). This moderate direct inhibition by lithium is thought to be amplified through an increase in serine21/9 phosphorylation. Proposed mechanisms for lithium’s indirect GSK-3 inhibition include GSK-3-regulated decreased activity of protein phosphatase 1 (Zhang et al, 2003), increased activity of PKC (Kirshenboim et al, 2004), or increased Akt activity (Chalecka-Franaszek and Chuang, 1999). We thus propose that lithium-induced GSK-3β inhibition causes increased serine phosphorylation of both isoforms, which in turn induces CREB phosphorylation and downstream Smad3/4 transcriptional suppression.
It has been reported that lithium activates PI3-K/AKT signaling and subsequently inhibits GSK-3β in order to induce binding activity of pCREBSer133 and the CREB responsive element of various downstream anti-apoptotic genes (Jope, 1999a; Grimes and Jope, 2001a; Rowe and Chuang, 2004). We thus hypothesized that the increased interaction of lithium-induced pCREBSer133 with the transcriptional coactivators CBP/p300 concurrently suppresses Smad3/4-dependent gene transcription due to the sequestration of CBP/p300 by activated pCREBSer133. Recent studies demonstrate that AKT can directly interact with non-phosphorylated Smad3, promoting the sequestration of Smad3 in the cytoplasm and thereby impairing Smad3-dependent transcriptional activity (Conery et al., 2004; Remy et al., 2004). Although Smad3 phosphorylation is not affected by lithium treatment in cultured cortical neurons (unpublished data), we have not investigated whether lithium treatment alters AKT-Smad3 interaction or Smad3 nuclear translocation. The possibility of Smad3/4 transcriptional suppression caused by lithium-induced AKT-Smad3 interaction thus cannot be ruled out. However, the fact that inhibition of GSK-3β with isoform-specific siRNAs or a dominant negative mutant causes Smad3/4-dependent transcriptional suppression (Liang and Chuang, 2006), suggests that the inhibitory effect of lithium on Smad3/4 transactivation is partially mediated through the AKT-GSK-3β-CREB cascade.
It has been reported that lithium at supra-therapeutic levels inhibits TGF-β-induced epithelial mesenchymal transition (Stump et al., 2006). It has also been reported that lithium causes the upregulation of CRE-dependent gene transcription resulting in the expression of the neuronal survival factors BDNF and Bcl-2 (reviewed in Jope, 1999a; Rowe and Chuang, 2004). Our results not only confirm lithium’s neuroprotective action, but also provide insight into how this drug’s negative regulation of Smad3/4-TGF-β signaling is mediated through upregulation of CRE-dependent transactivation. Specifically, we show that lithium-induced suppression of two Smad3/4-regulated genes, PAI-1 and p21, is mediated by CREB and p300 at their promoters. This effect is most likely due to the squelching effects of CREB in p300/CREB complexes, resulting in the blockade of Smad-dependent transcription. This notion is supported by a previous report that, in TGF-β-stimulated human HaCaT keratinocytes, the inhibition of TGF-β-induced Smad3/4-dependent transcription of the PAI-1 gene by cAMP-elevating agents is mediated through PKA activation and disruption of the Smad-CBP/p300 complex (Schiller et al., 2003). Our results thus confirm CREB’s inhibitory effects and further demonstrate that lithium’s inhibitory effect on PAI-1 promoter transactivation is partially mediated through a PKA-dependent mechanism in an unstimulated state of cortical neurons. Our study also suggests that both AKT/GSK-3β and PKA signaling are involved in the regulation of lithium-induced TGF-β transcriptional suppression. Additionally, we show that transcriptional coactivator p300 is primarily responsible for the inhibition of Smad3/4-dependent PAI-1 gene transcription. The difference between the results of our and their studies likely lies at the differences in cell types (primary cortical cultures vs keratinocytes, respectively) used.
Similar to the transcriptional regulation for the PAI-1 gene, we found that only transcriptional coactivator p300 is involved in the lithium-induced down-regulation of p21 promoter transcription. This observation is supported by a report that retinoic acid–induced cell differentiation and transcriptional upregulation of p21Cip1 requires normal levels of p300, but not CBP, in F9 cells (Kawasaki et al., 1998). CBP and p300 share a high degree of structural homology and both bind the transcription factor CREB (Chan and La Thangue, 2001; Vo and Goodman, 2001). CBP/p300 degradation was reported to lead to an irreversible decrease in CREB-dependent transcription in apoptotic conditions, thus compromising neuroprotection (Rouaux et al., 2003). However, fundamental differences between CBP and p300 involving regulation and function have been reported. For example, the activation of CREB-dependent transcription by CBP depends on a secondary regulatory mechanism, namely, CBP phosphorylation at Ser301, an event that does not occur for p300 (Impey et al., 2002). Several cell-based and in vivo studies on CBP and p300 (reviewed in Kalkhoven, 2004) indicate that they have unique functions in addition to their common roles. These functional differences could be due to differential association with other proteins, such as gene-specific transcription factors, and variations in substrate specificity between these two histone acetyltransferases. The latter case is demonstrated in an earlier study indicating that Smad7, a TGF-β response gene that negatively regulates TGF-β signaling, is acetylated by p300 and becomes stabilized and protected from proteasome (Smurf-1)-mediated degradation (Gronroos et al., 2002). Conversely, histone deacetylases (HDAC) interact with Smad7 and promotes the degradation of Smad7 protein (Simonsson et al., 2005). The reduction of Smad7 protein expression could further lead to the upregulation of Smad3/4-dependent TGF-β signaling and cause an increase in transcription of downstream genes, such as PAI-1 and p21.
A cross-talk between TGF-β and Wnt signaling pathways through physical association of β-catenin and Smad7 has been reported (Edlund et al., 2005). This cross-talk was found to be important for TGF-β-induced apoptosis. β-catenin acts as a downstream transcriptional activator of Wnt signaling by accumulating in the nucleus and forming a complex with DNA-binding proteins, such as lymphoid enhancer factor (LEF-1) and T-cell factor (TCF) to regulate TCF-dependent transactivation (reviewed in Grimes and Jope, 2001a). Both protein phosphorylation and protein–protein interactions have been implicated in the mechanism of Wnt-mediated inactivation of GSK-3β. Phosphorylation of β-catenin by GSK-3β results in rapid degradation of β-catenin (Rubinfeld et al., 1996; Yost et al., 1996). It has also been shown that GSK-3β activity is regulated by protein complex formation mediated by GSK-3β-binding proteins, such as GSK-3 binding protein (GBP) (Yost et al., 1998). Complex formation between GSK-3β and proteins in the GBP family, which include Frat 1 and 2, is an important mechanism for the inhibitory control of GSK-3β activity. This mechanism was shown to allow local inhibition of GSK-3β, leading to stabilization of β-catenin due to reduced GSK-3β-facilitated degradation of β-catenin. The accumulation of β-catenin caused by AKT activation and lithium treatment (Supplementary Fig. 3a) thus implies the inhibition of GSK-3 activity.
Our results indicate that lithium treatment causes a decrease in gene transcription and protein expression of PAI-1, a target gene of Smad signaling. PAI-1 is a serine protease inhibitor known to bind and inactivate both tissue-type and urokinase-type plasminogen activators (tPA and uPA), respectively (Melchor and Strickland, 2005). PAI-1 is found in low levels in the adult brain (Sawdey and Loskutoff, 1991), but its expression can be upregulated in astrocytes by stimulation with TGF-β1 and plasminogen (Datto et al., 1995; Buisson et al., 1998). Intravenous tPA has been used to treat acute stroke because of its thrombolytic activity and its ability to restore circulation to the brain (Hacke et al., 1995). Controversial results suggest that the tPA-plasmin system can be either protective or harmful in response to neurological insults (Tsirka , 2002; Maddika et al., 2007). For example, sufficient release of PAI-1 from astrocytes into the extracellular environments of neurons prevents neuronal apoptosis (Soeda et al., 2001; Docagne et al., 2002). PAI-1 was also reported to rescue neurons from kainate-induced death in both wild-type and tPA-/- mice (Tsirka et al., 1996). Additionally, tPA has also been shown to exacerbate neuronal damage in tPA-deficient mice after excitotoxic insult (Tsirka et al., 1995) or focal cerebral ischemia in mice (Wang et al., 1998). Of interest, a recent report demonstrates that tPA in the amygdala is critical for stress-induced neuronal remodeling and the development of anxiety-like behavior, and is subsequently inhibited by PAI-1 (Pawlak et al., 2003). The tPA-plasminogen proteolytic cascade accelerates the clearance of fibrin and protects the brain from damage in stroke models or when the blood-brain barrier breaks down (Tabrizi et al., 1999; Akassoglou et al, 2003), and contributes to Aβ degradation and pathology (Melchor et al., 2003), which suggest tPA’s protective effect against the neurodegenerative progression of Alzheimer’s disease. Because the tPA-plasminogen proteolytic cascade contributes to Aβ degradation, it may suggest the utility of tPA in blocking Aβ-induced cell death in the brain of Alzheimer’s disease (reviewed in Maddika et al., 2007).
Many reports indicate an involvement of the tPA-plasmin system in synaptic plasticity and remodeling (Baranes et al., 1998; Fiumelli et al., 1999; Neuhoff et al., 1999), control of the direction of BDNF’s functions (Martinowich et al., 2007), regulation of long-term potentiation (Pang et al., 2004), pathophysiology of mood disorders (Eskandari et al., 2005; Tsai, 2006) and stress-induced anxiety (Pawlak et al., 2003). The neurotrophic effects of lithium in stress-induced hippocampal atrophy and antidepressant effects of this drug in rodent models (Wood et al., 2004; O’Brien et al., 2004) suggest a critical role of BDNF in these lithium-induced actions (reviewed in Chuang, 2004). Our results of lithium-induced suppression of PAI-1 transcription thus imply that PAI-1 may function as a target of lithium’s neuroprotective and mood stabilizing effects through the PAI-1-tPA-plasmin pathway.
In conclusion, our results provide strong evidence that lithium suppresses Smad3/4-depedendent gene transcription by a unique mechanism depending on increased CREB activation and subsequent pCREBSer133-p300 complex formation, and these events are mediated through CRE-dependent, cAMP-PKA and PI3-K-AKT-GSK-3β signaling pathways. These effects of lithium likely result from the fine tuning and cross-talk of four pathways: Smad/TGF-β, CRE-dependent, cAMP-PKA and PI3-K-AKT-GSK-3β signalings. The TGF-β signaling pathway has been implicated as a therapeutic target in neurodegeneration (Wyss-Coray, 2006), and the PAI-1-tPA-plasmin system has also been suggested to play a critical role in the pathogenesis of mood disorders (Eskandari et al., 2005; Higgins, 2006; Tsai, 2006). Thus, further elucidation of the molecular details involved in lithium-induced regulation of Smad/TGF-β signaling will advance our understanding of the neuroprotective and therapeutic mechanisms of this drug.
Experimental Methods
Rat primary cerebral cortical culture
Cerebral cortical neurons were prepared from 18-day-old embryonic rats and cultured as described previously with slight modifications (Liang and Chuang, 2006). Briefly, cells from dissected brain cortex were separated by enzymatic digestion with trypsin (Invtrogen, Carlsbad, CA) and DNase I (Sigma), followed by centrifugation at 80 g for 5 min at 4 °C. Separated cortical cells were seeded at a density of 1 × 106 cells/cm2 on poly-D-lysine-coated culture plates and maintained in DMEM supplemented with 10% fetal bovine serum (Invitrogen) for 18 h. Cortical cultures were then maintained in Neurobasal media (Invitrogen) with B27 supplement and L-glutamine (Invitrogen). Cortical neurons were used after 9 days in vitro (DIV), unless otherwise indicated. On 10-DIV, non-neuronal cells represented less than 10% of the population, as revealed by FACS (fluorescence-activated cell sorter) analysis. NIH animal care guidelines were followed in this study.
Biochemical reagents
Lithium salt (LiCl) was obtained from Sigma-Aldrich (Saint Louis, MO). Three PKA inhibitors (KT5720, H-89 and PKI), two cAMP elevating agents (8-Br-cAMP and forskolin), LY-294002 and Wortmannin were obtained from Calbiochem (San Diego, CA).
Plasmids
The reporter plasmid pSmad3/4-Luc was purchased from Panomics (Redwood City, CA). The pRL-TK reporter plasmid was purchased from Promega (Madison, WI). The reporter construct pCRE-Luc (Mercury Pathway Profiling Luciferase System; Clontech, Mountain View, CA) was used to determine CRE-driven transcription. The vector expressing the catalytic subunit of PKA, wild-type pCMV-CREB and two mutant expressing vectors, named pCMV-CREB133 and pCMV-KCRE, were obtained from Clontech. The expression vector PKA-dn, a dominant negative mutant of PKA, was a gift from Dr. Jeffery J.Y. Yen (IBMS, Academia Sinica in Taiwan). The promoter constructs, p21P-Luc and p15-Luc (Datto et al., 1995), were gifts from Dr. Xiao-Fan Wang (Duke University, Durham, NC). The promoter construct p3TP-Lux and expression vectors Flag-p300 (WT) and Flag-p300 (HAT-defective) were gifts from Drs. Takeshi Imamura and Kohei Miyazono (The JFCR Cancer Institute, Tokyo, Japan). Expression vectors AKT-CA (constitutively active mutant) and AKT-dn (dominant negative mutant) were gifts from Dr. Benoit Dérijard (Université de Nice Sophia Antipolis, France) with the permission of Dr. Anne Brunet (Stanford University, Palo Alto, CA). The expression vector of wild-type CBP with HA tag was a gift from Drs. Richard H. Goodman and Ryan Cleland (Oregon Health & Science University, Portland, OR). Expression vector HAT-defective point mutant CBP (F1541A) was a gift from Dr. Tony Kouzarides (The Welcome Trust/Cancer Research UK Gurdon Institute, University of Cambridge, UK).
Transfection assays
Transfection of expression vectors into primary cortical neurons was established using an Amaxa Nucleofector (Amaxa, Cologne, Germany) and a rat neuron nucleofector kit (Amaxa) formulated for primary rat cortical neurons as described previously (Liang and Chuang, 2006). The transfection efficiency was about 58 ± 7 %.
Non-radioactive PKA assay
Cultured cortical neurons (1 × 107) treated with LiCl at various concentrations were harvested on 11-DIV and washed with cold PBS. The PKA assay was carried out using the PepTag® assay for non-radioactive detection of PKA (Promega) according to the manufacturer’s protocol using PKA-specific PepTag® A1 peptide substrate. The activity was detected by the amount of phosphorylated substrate migrating toward the anode. The intensity of fluorescence of phosphorylated peptides, which reflects PKA activity, was quantified using spectrofluorometry at an excitation wavelength of 560 nm and an emission wavelength of 590 nm.
cAMP measurement analysis
Intracellular cAMP levels of cultured cortical neurons harvested on 11-DIV (2000 cells/well in a 96-well poly-D-lysine coated culture plate) were measured using cAMP Biotrak™ enzyme immunoassay system (Amersham Biosciences, Little Chalfont, UK) in conjunction with a microtiter plate reader at 450 nm according to the manufacturer’s suggestion in protocol 3 of the cAMP immunoassay protocol (Amersham Biosciences).
Immunoblotting analysis
Cultured cortical neurons were washed twice with 5 ml of phosphate-buffered saline (PBS) and harvested by scraping into cell lysis buffer (Cell Signaling Technology, Beverly, MA) supplemented with protease inhibitor cocktail (Roche Diagnostics GmbH, Basel, Switzerland). Protein concentration measurement and Western blotting analysis were carried out as described previously (Liang and Chuang, 2006). Antibodies against PAI-1, tPA, GSK-3α, GSK -3β, β-actin, β-catenin, pSmad2/3(Ser433/435), and Bax were purchased from Santa Cruz (Santa Cruz, CA). Antibody against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was obtained from Upstate (Charlottesville, VA). Antibodies against pGSK3αSer21, pGSK3βSer9, pGSK-3αβTyr279/216, pCREBSer133, phospho-extracellular receptor kinase (pERK (p44/42), pAKTSer476, Bcl-2, Bcl-xL, CREB, ERK, and AKT were products of Cell Signaling Technology. ECL Plus Western blotting detection system (Amersham Biosciences) was used to obtain Western blot signals. Autoradiographs were scanned, and the intensity of immunoblot signals was analyzed using Scion Image software (Scion Image for Windows, NIH).
Dual luciferase reporter assay
Dual luciferase assaying was performed using the dual luciferase reporter assay system (Promega) as described previously (Liang and Chuang, 2006). Briefly, dual reporter-transfected cultured cortical neurons were lysed in lysis buffer (Promega) and subjected to the detection of firefly luciferase activity and subsequently, Renilla luciferase activity, which serves as an internal control. Luciferase activities were determined by using a plate-reading luminometer (Packard Bioscience Company, Downers Grove, IL), and the activities of the experimental reporter were normalized to the activities of the Renilla luciferase.
Chromatin immunoprecipitation–real-time quantitative PCR assay (CHIP-qPCR)
CHIP assays were performed using the CHIP-IT assay kit protocol as recommended by the manufacturer (Active Motif, Carlsbad, CA) with slight modifications. Briefly, 5 × 106 cultured cortical neurons were treated with LiCl (1.5 mM) starting on 7-DIV for 96 h. Cortical cells were then cross-linked with 1% formaldehyde at 37 °C for 10 min, followed by repeated washings with ice-cold PBS. A total of 4 × 107 cross-linked cortical cells from eight individual treatments were pooled together and lysed in lysis buffer (Active Motive). Nuclear pellets were then resuspended in shearing buffer and sonicated for 20 pulses of 15 sec each, using a sonicator (Sonic and Materials, Newtown, CT) to shear DNA into 500-1000 bp fragments. All of the samples were precleared with salmon sperm DNA/protein G agarose. Twenty five percent of the mixture of protein/DNA complex was taken for “input DNA” analysis. An equal amount of the protein/DNA complex was then incubated with an antibody against either p300 (anti-p300, Santa Cruz) or CBP (anti-CBP, Santa Cruz) at 4°C overnight, followed by incubation with salmon sperm DNA/protein G agarose for 2 h. Immunoprecipitated DNA was then eluted from protein G agarose. The cross-linking was reversed and the DNA was purified. The PAI-1/Serp1 promoter region (GenBank accession number: NM_012620.1) was PCR amplified using forward primer 5’-aactctgcagtccgcccta-3’ (position -926 relative to translation initiation (ATG) codon of the gene) and reverse primer 5’-ggaaacgcagtttgaccatc-3’ (position -857). The p21/cdkn1a promoter region (GenBank accession number: NM_080782.3) was PCR amplified using forward primer 5’-aaggtccagggcacttttg-3’ (position -503) and reverse primer 5’-ccctatcctgccgtcctt-3’ (position -443). Primers and probes were designed using the Universal Probe Library Assay Design Center from Roche Diagnostics (www.universalprobelibrary.com). Primers were chemically synthesized by Operon (Huntsville, AL). The PCR amplification was performed in a total volume of 20 μl reaction mixture including 3 μl of chromatin immunoprecipitated DNA, 200 nM primers, 100 nM FAM-labeled probes (Universal Probe Library, probe number 114 for Serpine1 and probe number 4 for cdkn1a assay obtained from Roche Applied Science), and Platinum Quantitative PCR SuperMix-UDG (Invitrogen). All samples were run in triplicate, including a no-template control, which consistently showed no amplification. Reactions were performed in white hard-shell PCR plates (Bio-Rad, Hercules, CA) in an MJ Chromo4 (Bio-Rad) continuous fluorescence detector connected to a PC running Opticon Monitor software version 3.1. Thermocycling consisted of 2 min at 50 °C (UDG activation), 2 min at 95 °C (initial denaturation and Taq polymerase activation) and 45 cycles of denaturation at 95 °C for 15 sec, combined annealing and extension at 56 °C for 1 min and fluorescence acquisition. Baseline correction was performed using “global minimum” subtraction. After amplification, triplicates were pooled and electrophoresed on a 3.5% agarose gel (Invitrogen), stained with ethidium bromide (Sigma-Aldrich) and visualized by UV illumination.
Statistical analysis
All results presented are means ± SEM from three to four independent experiments, with three or four replicates for each data point. The results were analyzed for statistical significance in GraphPad InStat (GraphPad, San Diego, CA) by one-way ANOVA. In ANOVA, a Q-Q plot was adopted for normal distribution tests, and Tukey’s post hoc test was adopted if the variances between groups were similar.
Supplementary Material
Inhibitory effects of lithium on Smad3/4-regulated promoter activity of down-stream genes. (a) Cultured cortical neurons were transfected with pSmad3/4-Luc reporter construct at the time of plating and treated with or without lithium (1.5 mM) for various times starting at 6-DIV. (b) Cultured cortical neurons were transfected with pSmad3/4-Luc reporter construct at the time of plating and treated with or without lithium at various concentrations for various times starting at 10-DIV. (c) Cultured cortical neurons were transfected with p21P-Luc reporter construct at the time of plating and treated with or without lithium on 9-DIV at various concentrations for 48 h. (d) Cultured cortical neurons were transfected with p15-Luc reporter construct at the time of plating and treated with or without lithium on 9-DIV at various concentrations for 48 h. Luciferase activities were determined on 11-DIV. Results represented as arbitrary luciferase activities are means ± SEM of three independent experiments. ** p<0.01; *** p<0.001, compared to the untreated control using one-way ANOVA.
Negative regulation by lithium on PKA signaling. (a) Cultured cortical neurons were transfected with pSmad3/4-Luc reporter construct at the time of plating and treated with or without cAMP-elevating agents, 8-Br-cAMP and forskolin, at the indicated concentrations on 10-DIV for 24 h, or treated with 2 mM lithium chloride on 9-DIV for 48 h. (b) Cultured cortical neurons were transfected with pSmad3/4-Luc reporter construct at the time of plating and treated with or without PKA specific inhibitors, KT5720, H-89 and PKI, at the indicated concentrations on 10-DIV for 24 h, or treated with 2 mM LiCl at 9-DIV for 48 h. Luciferase activities were determined on 11-DIV. Results represented as arbitrary luciferase activities are means ± SEM of three independent experiments. * p<0.05; ** p<0.01; *** p<0.001, compared to the untreated control using one-way ANOVA. (c) Quantification data for Western blot from Fig. 2c. Data points of PAI-, pCREB(Ser133), and CREB were first normalized to that of β-actin. Fold changes of normalized data from experiment groups (lithium, H-89, and H-89 plus lithium) were obtained by comparing to that of the control (Ctl).
Effects of AKT-CA and lithium on Smad signaling and protein expression levels. (a) At the time of plating, cortical neurons were either co-transfected with 4 μg of pSmad3/4-Luc reporter construct and the expression vector of AKT constitutively active mutant or transfected with pSmad3/4-Luc reporter construct followed by treatment with 100 μM forskolin or 8-Br-cAMP treatment for 24 h on 10-DIV. The luciferase activities of cell lysates were determined on 11-DIV. Representative results shown as arbitrary luciferase activities are means ± SEM of triplicates of one set of data from three independent experiments. (b) Representative Western blotting analysis of levels of PAI-1, pAKT (Thr308), AKT, and β-catenin from three experiments. β-actin was used as a loading control. Cultured cortical neurons were transfected with the expression vector for the constitutively active form of AKT at the time of plating, and followed by lithium treatment for 48 h on 9-DIV. Cell lysates were harvested on 11-DIV for immunoblotting analysis. (c) Representative Western blotting analysis of protein levels of pCREB (Ser133), CREB, Bcl-2, Bcl-xL and Bax from three independent experiments. β-actin was used as a loading control. Cortical neurons were treated with lithium at the indicated concentrations on 9-DIV for 48 h. Cell lysates were collected on 11-DIV for immunoblotting analysis. (d) Representative Western blotting analysis of protein levels of pCREB (Ser133), pGSK-3β (Ser9), CREB, GSK-3β, Bcl-2 and Bax from three experiments. β-actin was used as a loading control. Cortical neurons were pretreated with 90 nM H-89 for 4 h on 9-DIV followed by treatment with either 1.5mM LiCl or 100 μM forskolin for 48 h. Cell lysates were collected on 11-DIV for Western blotting analysis.
Overexpression of transcriptional coactivator p300 abolishes lithium’s inhibitory regulation on the promoter activity of Smad3/4-dependent TGF-β-responsive downstream gene, p21. (a) Cortical neurons were transfected with p21P-Luc reporter construct together with expression vectors for wild-type CBP (CBP) or HAT-deficient CBP mutant (CBP-ΔHAT) at the time of plating. Lithium treatment and luciferase activity assay were carried out as described in Fig. 6a. (b) Cortical neurons were transfected with p21P-Luc reporter construct together with expression vectors for wild-type p300 (p300) or HAT-deficient p300 mutant (p300-ΔHAT) at the time of plating. Lithium treatment and luciferase activity assay were carried out as described in Fig. 6b. Results represented as arbitrary luciferase activities are means ± SEM of three independent experiments. * p<0.05; *** p<0.001, compared to the respective controls. n.s, not significant from the comparative control using one-way ANOVA.
The involvement of p300 in gene promoters of PAI-1/Serp1 and p21/cdkn1a determined by CHIP-qPCR assay. Cortical neurons treated with 1.5 mM LiCl for 96 h were cross-linked with 1% formaldehyde on 11-DIV, and the chromatins were prepared and sheared by sonication as described in Materials and the Methods. The protein/DNA complex was then incubated with anti-p300 antibody (p300-AB), anti-CBP antibody (CBP-AB) or without antibody (No AB, Input) for CHIP analysis. The immunoprecipitated DNA was then purified and used for qPCR amplification of PAI-1/Serp1 gene promoter (a) or p21/cdkn1a (b). Representative log fluorescence graphs are shown for the comparison of qPCR amplification of gene promoters among different treatments. The circle denotes the differences in the PCR cycles to reach the same level of log fluorescence (broken line) for PAI-1/Serp1 using DNA immunoprecipitated with anti-p300 or anti-CBP. NTC: no template control. (c) Representative gel electrophoresis of PCR amplification of gene promoter of PAI-1/Serp1 and p21/cdkn1a.
Acknowledgments
This work was supported by the Intramural Research Program of the NIMH, NIH (Project number: Z01MH002468-18).
We thank Charlotte Wiest and Dr. Peixiong Yuan for their technical assistance, and Peter Leeds of the Section for his critical reading of this manuscript. The superb editorial assistance from the NIH Fellows Editorial Board is also greatly appreciated.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akassoglou K, Akpinar P, Murray S, Strickland S. Fibrin is a regulator of Schwann cell migration after sciatic nerve injury in mice. Neurosci Lett. 2003;338:185–188. doi: 10.1016/s0304-3940(02)01387-3. [DOI] [PubMed] [Google Scholar]
- Baranes D, Lederfein D, Huang YY, Chen M, Bailey CH, Kandel ER. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron. 1998;21:813–825. doi: 10.1016/s0896-6273(00)80597-8. [DOI] [PubMed] [Google Scholar]
- Buisson A, Nicole O, Docagne F, Sartelet H, Mackenzie ET, Vivien D. Upregulation of a serine protease inhibitor in astrocytes mediates the neuroprotective activity of transforming growth factor beta1. FASEB J. 1998;12:1683–1691. [PubMed] [Google Scholar]
- Chalecka-Franaszek E, Chuang D-M. Lithium activates the serine/threonine kinase Akt-1 and suppresses glutamate-induced inhibition of Akt-1 activity in neurons. Proc Natl Acad Sci USA. 1999;96:8745–8750. doi: 10.1073/pnas.96.15.8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan HM, La Thangue NB. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. J Cell Sci. 2001;114:2363–2373. doi: 10.1242/jcs.114.13.2363. [DOI] [PubMed] [Google Scholar]
- Chang A, Li PP, Warsh JJ. Altered cAMP-dependent protein kinase subunit immunolabeling in post-mortem brain from patients with bipolar affective disorder. J Neurochem. 2003;84:781–791. doi: 10.1046/j.1471-4159.2003.01605.x. [DOI] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Chuang D-M. Neuroprotective and neurotrophic actions of the mood stabilizer lithium: can it be used to treat neurodegenerative diseases? Crit Rev Neurobiol. 2004;16:83–90. doi: 10.1615/critrevneurobiol.v16.i12.90. [DOI] [PubMed] [Google Scholar]
- Chuang D-M, Priller J. Potential use of lithium in neurodegenerative disorders. In: Bauer M, Grof P, Muller-Oerlinghausen B, editors. Lithium in neuropsychiatry. The comprehensive guide. Taylor & Francis Books Ltd; London, UK: 2006. pp. 381–398. [Google Scholar]
- Conery AR, Cao YE, Thompson A, Townsend CM, Jr, Ko TC, Luo K. Akt interacts directly with Smad3 to regulate the sensitivity to TGF-beta induced apoptosis. Nat Cell Biol. 2004;6:366–372. doi: 10.1038/ncb1117. [DOI] [PubMed] [Google Scholar]
- Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF. Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci USA. 1995;92:5545–5549. doi: 10.1073/pnas.92.12.5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signaling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- Docagne F, Nicole O, Gabriel C, Fernandez-Monreal M, Lesne S, Ali C, et al. Smad3-dependent induction of plasminogen activator inhibitor-1 in astrocytes mediates neuroprotective activity of transforming growth factor-beta 1 against NMDA-induced necrosis. Mol Cell Neurosci. 2002;21:634–644. doi: 10.1006/mcne.2002.1206. [DOI] [PubMed] [Google Scholar]
- Drevets WC. Neuroimaging and neuropathological studies of depression: implications for the cognitive-emotional features of mood disorders. Cur Opin Neurobiol. 2001;11:240–249. doi: 10.1016/s0959-4388(00)00203-8. [DOI] [PubMed] [Google Scholar]
- Edlund S, Lee SY, Grimsby S, Zhang S, Aspenstrom P, Heldin CH, et al. Interaction between Smad7 and beta-catenin: importance for transforming growth factor beta-induced apoptosis. Mol Cell Biol. 2005;25:1475–1488. doi: 10.1128/MCB.25.4.1475-1488.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskandari F, Mistry S, Martinez PE, Torvik S, Kotila C, Sebring N, et al. Younger, premenopausal women with major depressive disorder have more abdominal fat and increased serum levels of prothrombotic factors: implications for greater cardiovascular risk. Metabolism. 2005;54:918–924. doi: 10.1016/j.metabol.2005.02.006. [DOI] [PubMed] [Google Scholar]
- Fiumelli H, Jabaudon D, Magistretti PJ, Martin JL. BDNF stimulates expression, activity and release of tissue-type plasminogen activator in mouse cortical neurons. Eur J Neurosci. 1999;11:1639–1646. doi: 10.1046/j.1460-9568.1999.00580.x. [DOI] [PubMed] [Google Scholar]
- Gomes FC, Sousa Vde O, Romao L. Emerging roles for TGF-β1 in nervous system development. Int J Dev Neurosci. 2005;23:413–424. doi: 10.1016/j.ijdevneu.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Gould TD, Quiroz JA, Singh J, Zarate CA, Manji HK. Emerging experimental therapeutics for bipolar disorder: insights from the molecular and cellular actions of current mood stabilizers. Mol Psychiatry. 2004;9:734–755. doi: 10.1038/sj.mp.4001518. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog Neurobiol. 2001a;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Grimes CA, Jope RS. CREB DNA binding activity is inhibited by glycogen synthase kinase-3β and facilitated by lithium. J Neurochem. 2001b;78:1219–1232. doi: 10.1046/j.1471-4159.2001.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronroos E, Hellman U, Heldin CH, Ericsson J. Control of Smad7 stability by competition between acetylation and ubiquitination. Mol Cell. 2002;10:483–493. doi: 10.1016/s1097-2765(02)00639-1. [DOI] [PubMed] [Google Scholar]
- Gurvich N, Klein PS. Lithium and valproic acid: parallels and contrasts in diverse signaling contexts. Pharmacol Ther. 2002;96:45–66. doi: 10.1016/s0163-7258(02)00299-1. [DOI] [PubMed] [Google Scholar]
- Hacke W, Kaste M, Fieschi C, Toni D, Lesaffre E, von Kummer R, et al. Intravenous thrombolysis with recombinant tissue plasminogen activator for acute hemispheric stroke. The European Cooperative Acute Stroke Study (ECASS) JAMA. 1995;274:1017–1125. [PubMed] [Google Scholar]
- Higgins PJ. The TGF-β1/upstream stimulatory factor-regulated PAI-1 gene: potential involvement and a therapeutic target in Alzheimer’s disease. J Biomed Biotechnol. 2006;2006:15792–15797. doi: 10.1155/JBB/2006/15792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hottiger MO, Nabel GJ. Interaction of human immunodeficiency virus type 1 Tat with the transcriptional coactivators p300 and CREB binding protein. J Virol. 1998;72:8252–8256. doi: 10.1128/jvi.72.10.8252-8256.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, Fong AL, Wang Y, Cardinaux JR, Fass DM, Obrietan K, et al. Phosphorylation of CBP mediates transcriptional activation by neural activity and CaM kinase IV. Neuron. 2002;34:235–244. doi: 10.1016/s0896-6273(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Jope RS. Anti-bipolar therapy: mechanism of action of lithium. Mol Psychiatry. 1999a;4:117–128. doi: 10.1038/sj.mp.4000494. [DOI] [PubMed] [Google Scholar]
- Jope RS. A bimodal model of the mechanism of action of lithium. Mol Psychiatry. 1999b;4:21–25. doi: 10.1038/sj.mp.4000444. [DOI] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Milman A, Weizman A, Pick CG, Eldar-Finkelman H. Rapid antidepressive-like activity of specific glycogen synthase kinase-3 inhibitor and its effect on β-catenin in mouse hippocampus. Biol Psychiatry. 2004;55:781–784. doi: 10.1016/j.biopsych.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Kalkhoven E. CBP and p300: HATs for different occasions. Biochem Pharmacol. 2004;68:1145–1155. doi: 10.1016/j.bcp.2004.03.045. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Eckner R, Yao TP, Taira K, Chiu R, Livingston DM, et al. Distinct roles of the co-activators p300 and CBP in retinoic-acid-induced F9-cell differentiation. Nature. 1998;393:284–289. doi: 10.1038/30538. [DOI] [PubMed] [Google Scholar]
- Kirshenboim N, Plotkin B, Shlomo SB, Kaidanovich-Beilin O, Eldar-Finkelman H. Lithium-mediated phosphorylation of glycogen synthase kinase-3beta involves PI3 kinase-dependent activation of protein kinase C-alpha. J Mol Neurosci. 2004;24:237–245. doi: 10.1385/JMN:24:2:237. [DOI] [PubMed] [Google Scholar]
- Klein PS, Melton DA. Molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystosek A, Seeds NW. Plasminogen Activator Secretion by Granule Neurons in Cultures of Developing Cerebellum. Proc Natl Acad Sci USA. 1981;78:7810–7814. doi: 10.1073/pnas.78.12.7810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Gururaj AE, Barnes CJ. p21-activated kinases in cancer. Nat Rev Cancer. 2006;6:459–471. doi: 10.1038/nrc1892. [DOI] [PubMed] [Google Scholar]
- Liang M-H, Chuang D-M. Differential roles of glycogen synthase kinase-3 isoforms in the regulation of transcriptional activation. J Biol Chem. 2006;281:30479–30484. doi: 10.1074/jbc.M607468200. [DOI] [PubMed] [Google Scholar]
- Liang M-H, Chuang D-M. Regulation and function of glycogen synthase kinase-3 isoforms in neuronal survival. J Biol Chem. 2007;282:3904–3917. doi: 10.1074/jbc.M605178200. [DOI] [PubMed] [Google Scholar]
- Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A, et al. Cell survival, cell death and cell cycle pathways are interconnected: Implications for cancer therapy. Drug Resist Updat. 2007;10:13–29. doi: 10.1016/j.drup.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Manji HK, Zarate CA. Molecular and cellular mechanisms underlying mood stabilization in bipolar disorder: implications for the development of improved therapeutics. Mol Psychiatry. 2002;7(Suppl 1):S1–S7. doi: 10.1038/sj.mp.4001068. [DOI] [PubMed] [Google Scholar]
- Martinowich K, Manji H, Lu B. New insights into BDNF function in depression and anxiety. Nature Neurosci. 2007;10:1089–1093. doi: 10.1038/nn1971. [DOI] [PubMed] [Google Scholar]
- Melchor JP, Pawlak R, Strickland S. The tissue plasminogen activator-plasminogen proteolyc cascade accelerates amyloid-beta (Abeta) degradation and inhibits Abeta-induced neurodegeneration. J Neurosci. 2003;23:8867–8871. doi: 10.1523/JNEUROSCI.23-26-08867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchor JP, Strickland S. Tissue plasminogen activator in central nervous system physiology and pathology. Thromb Haemost. 2005;93:655–660. doi: 10.1160/TH04-12-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montezinho LP, Duarte B, Fonseca CP, Glinka Y, Layden B, Mota de Freitas D, et al. Intracellular lithium and cyclic AMP levels are mutually regulated in neuronal cells. J Neurochem. 2004;90:920–930. doi: 10.1111/j.1471-4159.2004.02551.x. [DOI] [PubMed] [Google Scholar]
- Moonen G, Grau-Wagemans MP, Selak I. Plasminogen activator-plasmin system and neuronal migration. Nature (London) 1982;298:753–755. doi: 10.1038/298753a0. [DOI] [PubMed] [Google Scholar]
- Moore GJ, Bebchuk JM, Hasanat K, Chen G, Seraji-Bozorgzad N, Wilds IB, et al. Lithium increases N-acetyl-aspartate in the human brain: in vivo evidence in support of bcl-2’s neurotrophic effects? Biol Psychiatry. 2000a;48:1–8. doi: 10.1016/s0006-3223(00)00252-3. [DOI] [PubMed] [Google Scholar]
- Moore GJ, Bebchuk JM, Wilds IB, Chen G, Manji HK. Lithium-induced increase in human brain grey matter. Lancet. 2000b;356:1241–1242. doi: 10.1016/s0140-6736(00)02793-8. [DOI] [PubMed] [Google Scholar]
- Neuhoff H, Roeper J, Schweizer M. Activity-dependent formation of perforated synapses in cultured hippocampal neurons. Eur J Neurosci. 1999;11:4241–4250. doi: 10.1046/j.1460-9568.1999.00856.x. [DOI] [PubMed] [Google Scholar]
- O’Brien WT, Harper AD, Jove F, Woodgett JR, Maretto S, Piccolo S et al. Glycogen synthase kinase-3β haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24:6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang PT, Teng HK, Zaitsev E, Woo NT, Sakata K, Zhen S, Teng KK, Yung WH, Hempstead BL, Lu B. Cleavage of proBDNF by tPA/plasmin is essential for long-term hippocampal plasticity. Science. 2004;306:487–91. doi: 10.1126/science.1100135. [DOI] [PubMed] [Google Scholar]
- Pawlak R, Magarinos AM, Melchor J, McEwen B, Strickland S. Tissue plasminogen activator in the amygdala is critical for stress-induced anxiety-like behavior. Nat Neurosci. 2003;6:168–174. doi: 10.1038/nn998. [DOI] [PubMed] [Google Scholar]
- Pawlak R, Rao BS, Melchor JP, Chattarji S, McEwen B, Strickland S. Tissue plasminogen activator and plasminogen mediate stress-induced decline of neuronal and cognitive functions in the mouse hippocampus. Proc Natl Acad Sci USA. 2005;102:18201–18206. doi: 10.1073/pnas.0509232102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Z, Gilbert M, Colicos M, Kandel E, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling and long-term potentiation. Nature (London) 1993;361:453–456. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-β signaling through a direct interaction with Smad3. Nat Cell Biol. 2004;6:358–365. doi: 10.1038/ncb1113. [DOI] [PubMed] [Google Scholar]
- Rouaux C, Jokic N, Mbebi C, Boutillier S, Loeffler JP, Boutillier AL. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003;22:6537–6549. doi: 10.1093/emboj/cdg615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe MK, Chuang D-M. Lithium neuroprotection: molecular mechanisms and clinical implications. Expert Rev Mol Med. 2004;6:1–18. doi: 10.1017/S1462399404008385. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri R, Fiol C, Munemitsu S, Polakis P. Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- Ryves WJ, Harwood AJ. Lithium inhibits glycogen synthase kinase-3 by competition for magnesium. Biochem Biophys Res Commun. 2001;280:720–725. doi: 10.1006/bbrc.2000.4169. [DOI] [PubMed] [Google Scholar]
- Samson AL, Medcalf RL. Tissue-type plasminogen activator: a multifaceted modulator of neurotransmission and synaptic plasticity. Neuron. 2006;50:673–678. doi: 10.1016/j.neuron.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Sanyal S, Kim SM, Ramaswami M. Retrograde regulation in the CNS; neuron-specific interpretations of TGF-β signaling. Neuron. 2004;41:845–848. doi: 10.1016/s0896-6273(04)00152-7. [DOI] [PubMed] [Google Scholar]
- Sawdey MS, Loskutoff DJ. Regulation of murine type 1 plasminogen activator inhibitor gene expression in vivo. Tissue specificity and induction by lipopolysaccharide, tumor necrosis factor-alpha and transforming growth factor-β. J Clin Invest. 1991;88:1346–1353. doi: 10.1172/JCI115440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller M, Verrecchia F, Mauviel A. Cyclic adenosine 3’,5’-monophosphate-elevating agents inhibit transforming growth factor-beta-induced SMAD3/4-dependent transcription via a protein kinase A-dependent mechanism. Oncogene. 2003;22:8881–8890. doi: 10.1038/sj.onc.1206871. [DOI] [PubMed] [Google Scholar]
- Seeds NW, Basham ME, Haffke SP. Neuronal migration is retarded in mice lacking the tissue plasminogen activator gene. Proc Natl Acad Sci USA. 1999;96:14118–14123. doi: 10.1073/pnas.96.24.14118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeds NW, Williams BL, Bickford PC. Tissue plasminogen activator induction in Purkinje neurons after cerebellar motor learning. Science. 1995;270:1992–1994. doi: 10.1126/science.270.5244.1992. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Simonsson M, Heldin CH, Ericsson J, Gronroos E. The balance between acetylation and deacetylation controls Smad7 stability. J Biol Chem. 2005;280:21797–21803. doi: 10.1074/jbc.M503134200. [DOI] [PubMed] [Google Scholar]
- Soeda S, Oda M, Ochiai T, Shimeno H. Deficient release of plasminogen activator inhibitor-1 from astrocytes triggers apoptosis in neuronal cells. Brain Res Mol Brain Res. 2001;91:96–103. doi: 10.1016/s0169-328x(01)00133-4. [DOI] [PubMed] [Google Scholar]
- Song K, Cornelius SC, Reiss M, Danielpour D. Insulin-like growth factor-I inhibits transcriptional responses of transforming growth factor-β by phosphatidylinositol 3-kinase/Akt-dependent suppression of the activation of Smad3 but not Smad2. J Biol Chem. 2003;278:38342–38351. doi: 10.1074/jbc.M304583200. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics Wingless signalling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- Stump RJ, Lovicu FJ, Ang SL, Pandey SK, McAvoy JW. Lithium stabilizes the polarized lens epithelial phenotype and inhibits proliferation, migration, and epithelial mesenchymal transition. J Pathol. 2006;210:249–257. doi: 10.1002/path.2049. [DOI] [PubMed] [Google Scholar]
- Tabrizi P, Wang L, Seeds N, McComb JG, Yamada S, Griffin JH, Carmeliet P, Weiss MH, Zlokovic BV. Tissue plasminogen activator (tPA) deficiency exacerbates cerebrovascular fibrin deposition and brain injury in a murine stroke model: studies in tPA-deficient mice and wild-type mice on a matched genetic background. Arterioscler Thromb Vasc Biol. 1999;19:2801–2806. doi: 10.1161/01.atv.19.11.2801. [DOI] [PubMed] [Google Scholar]
- Tsai SJ. The possible role of tissue-type plasminogen activator and the plasminogen system in the pathogenesis of major depression. Med Hypotheses. 2006;66:319–322. doi: 10.1016/j.mehy.2005.10.009. [DOI] [PubMed] [Google Scholar]
- Tsirka SE. Tissue plasminogen activator as a modulator of neuronal survival and function. Biochem Soc Trans. 2002;30:222–225. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- Tsirka SE, Gualandris A, Amaral DG, Strickland S. Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature. 1995;377:340–344. doi: 10.1038/377340a0. [DOI] [PubMed] [Google Scholar]
- Tsirka SE, Rogove AD, Strickland S. Neuronal cell death and tPA. Nature. 1996;384:123–124. doi: 10.1038/384123b0. [DOI] [PubMed] [Google Scholar]
- Vivien D, Ali C. Transforming growth factor-β signaling in brain disorders. Cytokine Growth Factor Rev. 2006;17:121–128. doi: 10.1016/j.cytogfr.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Vo N, Goodman RH. CREB-binding protein and p300 in transcriptional regulation. J Biol Chem. 2001;276:13505–13508. doi: 10.1074/jbc.R000025200. [DOI] [PubMed] [Google Scholar]
- Wang YF, Tsirka SE, Strickland S, Stieg PE, Soriano SG, Lipton SA. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- Wind T, Hansen M, Jensen JK, Andreasen PA. The molecular basis for anti-proteolytic and non-proteolytic functions of plasminogen activator inhibitor type-1: roles of the reactive centre loop, the shutter region, the flexible joint region and the small serpin fragment. J Biol Chem. 2002;383:21–36. doi: 10.1515/BC.2002.003. [DOI] [PubMed] [Google Scholar]
- Wood GE, Young LT, Reagan LP, Chen B, McEwen BS. Stress-induced structural remodeling in hippocampus: prevention by lithium treatment. Proc Natl Acad Sci USA. 2004;101:3973–3978. doi: 10.1073/pnas.0400208101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T. TGF-β pathway as a potential target in neurodegeneration and Alzheimer’s. Curr Alzheimer Res. 2006;3:191–195. doi: 10.2174/156720506777632916. [DOI] [PubMed] [Google Scholar]
- Yost C, Torres M, Miller JR, Huang E, Kimelman D, Moon RT. The axis-inducing activity, stability, and subcellular distribution of β-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev. 1996;10:1443–1454. doi: 10.1101/gad.10.12.1443. [DOI] [PubMed] [Google Scholar]
- Yost C, Farr GH, Pierce SB, Ferkey DM, Chen MM, Kimelman D. GBP, an inhibitor of GSK-3, is implicated in Xenopus development and oncogenesis. Cell. 1998;93:1031–1041. doi: 10.1016/s0092-8674(00)81208-8. [DOI] [PubMed] [Google Scholar]
- Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem. 2003;278:33067–33077. doi: 10.1074/jbc.M212635200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Inhibitory effects of lithium on Smad3/4-regulated promoter activity of down-stream genes. (a) Cultured cortical neurons were transfected with pSmad3/4-Luc reporter construct at the time of plating and treated with or without lithium (1.5 mM) for various times starting at 6-DIV. (b) Cultured cortical neurons were transfected with pSmad3/4-Luc reporter construct at the time of plating and treated with or without lithium at various concentrations for various times starting at 10-DIV. (c) Cultured cortical neurons were transfected with p21P-Luc reporter construct at the time of plating and treated with or without lithium on 9-DIV at various concentrations for 48 h. (d) Cultured cortical neurons were transfected with p15-Luc reporter construct at the time of plating and treated with or without lithium on 9-DIV at various concentrations for 48 h. Luciferase activities were determined on 11-DIV. Results represented as arbitrary luciferase activities are means ± SEM of three independent experiments. ** p<0.01; *** p<0.001, compared to the untreated control using one-way ANOVA.
Negative regulation by lithium on PKA signaling. (a) Cultured cortical neurons were transfected with pSmad3/4-Luc reporter construct at the time of plating and treated with or without cAMP-elevating agents, 8-Br-cAMP and forskolin, at the indicated concentrations on 10-DIV for 24 h, or treated with 2 mM lithium chloride on 9-DIV for 48 h. (b) Cultured cortical neurons were transfected with pSmad3/4-Luc reporter construct at the time of plating and treated with or without PKA specific inhibitors, KT5720, H-89 and PKI, at the indicated concentrations on 10-DIV for 24 h, or treated with 2 mM LiCl at 9-DIV for 48 h. Luciferase activities were determined on 11-DIV. Results represented as arbitrary luciferase activities are means ± SEM of three independent experiments. * p<0.05; ** p<0.01; *** p<0.001, compared to the untreated control using one-way ANOVA. (c) Quantification data for Western blot from Fig. 2c. Data points of PAI-, pCREB(Ser133), and CREB were first normalized to that of β-actin. Fold changes of normalized data from experiment groups (lithium, H-89, and H-89 plus lithium) were obtained by comparing to that of the control (Ctl).
Effects of AKT-CA and lithium on Smad signaling and protein expression levels. (a) At the time of plating, cortical neurons were either co-transfected with 4 μg of pSmad3/4-Luc reporter construct and the expression vector of AKT constitutively active mutant or transfected with pSmad3/4-Luc reporter construct followed by treatment with 100 μM forskolin or 8-Br-cAMP treatment for 24 h on 10-DIV. The luciferase activities of cell lysates were determined on 11-DIV. Representative results shown as arbitrary luciferase activities are means ± SEM of triplicates of one set of data from three independent experiments. (b) Representative Western blotting analysis of levels of PAI-1, pAKT (Thr308), AKT, and β-catenin from three experiments. β-actin was used as a loading control. Cultured cortical neurons were transfected with the expression vector for the constitutively active form of AKT at the time of plating, and followed by lithium treatment for 48 h on 9-DIV. Cell lysates were harvested on 11-DIV for immunoblotting analysis. (c) Representative Western blotting analysis of protein levels of pCREB (Ser133), CREB, Bcl-2, Bcl-xL and Bax from three independent experiments. β-actin was used as a loading control. Cortical neurons were treated with lithium at the indicated concentrations on 9-DIV for 48 h. Cell lysates were collected on 11-DIV for immunoblotting analysis. (d) Representative Western blotting analysis of protein levels of pCREB (Ser133), pGSK-3β (Ser9), CREB, GSK-3β, Bcl-2 and Bax from three experiments. β-actin was used as a loading control. Cortical neurons were pretreated with 90 nM H-89 for 4 h on 9-DIV followed by treatment with either 1.5mM LiCl or 100 μM forskolin for 48 h. Cell lysates were collected on 11-DIV for Western blotting analysis.
Overexpression of transcriptional coactivator p300 abolishes lithium’s inhibitory regulation on the promoter activity of Smad3/4-dependent TGF-β-responsive downstream gene, p21. (a) Cortical neurons were transfected with p21P-Luc reporter construct together with expression vectors for wild-type CBP (CBP) or HAT-deficient CBP mutant (CBP-ΔHAT) at the time of plating. Lithium treatment and luciferase activity assay were carried out as described in Fig. 6a. (b) Cortical neurons were transfected with p21P-Luc reporter construct together with expression vectors for wild-type p300 (p300) or HAT-deficient p300 mutant (p300-ΔHAT) at the time of plating. Lithium treatment and luciferase activity assay were carried out as described in Fig. 6b. Results represented as arbitrary luciferase activities are means ± SEM of three independent experiments. * p<0.05; *** p<0.001, compared to the respective controls. n.s, not significant from the comparative control using one-way ANOVA.
The involvement of p300 in gene promoters of PAI-1/Serp1 and p21/cdkn1a determined by CHIP-qPCR assay. Cortical neurons treated with 1.5 mM LiCl for 96 h were cross-linked with 1% formaldehyde on 11-DIV, and the chromatins were prepared and sheared by sonication as described in Materials and the Methods. The protein/DNA complex was then incubated with anti-p300 antibody (p300-AB), anti-CBP antibody (CBP-AB) or without antibody (No AB, Input) for CHIP analysis. The immunoprecipitated DNA was then purified and used for qPCR amplification of PAI-1/Serp1 gene promoter (a) or p21/cdkn1a (b). Representative log fluorescence graphs are shown for the comparison of qPCR amplification of gene promoters among different treatments. The circle denotes the differences in the PCR cycles to reach the same level of log fluorescence (broken line) for PAI-1/Serp1 using DNA immunoprecipitated with anti-p300 or anti-CBP. NTC: no template control. (c) Representative gel electrophoresis of PCR amplification of gene promoter of PAI-1/Serp1 and p21/cdkn1a.