

Abstract
We have shown that the tetrapeptide N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) inhibited endothelin-1 (ET-1)-induced cell proliferation and collagen synthesis in cultured rat cardiac fibroblasts (CFs) and reduced left ventricle collagen deposition in rats with aldosterone (salt)- and ANG II-induced hypertension. However, it is not known whether these effects are mediated by receptor binding sites specific for Ac-SDKP. We hypothesized that Ac-SDKP exerts antifibrotic effects by binding to specific receptor sites in cultured rat CFs, which mediate the inhibitory effects of Ac-SDKP on ET-1-stimulated collagen synthesis. Ac-SDKP binding sites in rat CFs and hearts were characterized by a specific radioligand, 125I-labeled 3-(p-hydroxy-phenyl)-propionic acid (or desaminotyrosine) (Hpp)-Aca-SDKP, a biologically active analog of Ac-SDKP. 125I-labeled Hpp-Aca-SDKP bound to rat CFs and fractionated membranes with similar affinities and specificity in a concentration- and time-dependent fashion. Scat-chard plot analyses revealed a single class of high-affinity Hpp-Aca-SDKP binding sites (maximal binding: 1,704 ± 198 fmol/mg protein; dissociation constant: 3.3 ± 0.6 nM). 125I-labeled Hpp-Aca-SDKP binding in CFs was displaced by unlabeled native peptide Ac-SDKP (inhibition constant: 0.69 ± 0.15 nM) and the analog Hpp-Aca-SDKP (inhibition constant: 10.4 ± 0.2 nM) but not the unrelated peptide ANG II or ET-1 (10 μM). In vitro, both Ac-SDKP and Hpp-Aca-SDKP inhibited ET-1-stimulated collagen synthesis in CFs in a dose-dependent fashion, reaching a maximal effect at 1 nM (control: 7.5 ± 0.4, ET-1: 19.9 ± 1.2, ET-1+SDKP: 7.7 ± 0.4, ET-1+Hpp-Aca-SDKP: 9.7 ± 0.1 μg/mg protein; P < 0.001). Ac-SDKP also significantly attenuated ET-1-induced increases in intracellular calcium and MAPK ERK1/2 phosphor-ylation in CFs. In the rat heart, in vitro autoradiography revealed specific 125I-labeled Hpp-Aca-SDKP binding throughout the myocardium, primarily interstitially. We believe that these results demonstrate for the first time that Hpp-Aca-SDKP is a functional ligand specific for Ac-SDKP receptor binding sites and that both Ac-SDKP and Hpp-Aca-SDKP exert antifibrotic effects by binding to Ac-SDKP receptors in rat CFs.
Keywords: collagen synthesis, heart, autoradiography, receptor
N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) is a naturally occurring inhibitor of pluripotent hematopoietic stem cell entry into the S phase of the cell cycle and hepatocyte proliferation (4, 15). Ac-SDKP is normally present in nanomolar concentrations in human plasma and circulating mononuclear cells (24). This tetrapeptide is released primarily from its precursor thymosin-β4 and hydrolyzed exclusively by angiotensin I-converting enzyme (ACE) (2). Because chronic ACE inhibition in humans is associated with a four- to fivefold increase in serum Ac-SDKP levels, it has been suggested that changes in serum Ac-SDKP levels could be used as a marker of effective ACE inhibition in humans (2, 3). However, we and others have recently shown that Ac-SDKP exerts antifibrotic effects in cultured rat cardiac fibroblasts (CFs) in vitro and in rat hearts in vivo, thus uncovering a new role for Ac-SDKP (22, 25–27). In vitro, Ac-SDKP inhibits DNA and collagen synthesis by rat CFs when stimulated with FBS or endothelin (26). In rats with renovascular or mineralocorticoid hypertension, Ac-SDKP inhibits collagen deposition and cell proliferation in the heart (fibroblasts) and kidney (22, 27). In addition, our group (25) showed that, after myocardial infarction, Ac-SDKP both prevented and regressed collagen synthesis and deposition in the interstitial and perivascular spaces of the rat heart and inhibited infiltration of monocytes and macrophages in the left ventricle. Together, these studies suggest that Ac-SDKP plays an important role in the regulation of cardiac remodeling and function beyond its involvement as a biological marker of ACE inhibition.
Although the antifibrotic effects of Ac-SDKP are well documented, the cellular mechanisms involved are not fully understood. Ac-SDKP does not alter systemic blood pressure (22, 25, 27) and has no effect on cardiomyocyte hypertrophy after myocardial infarction, nor does it significantly affect cardiac function (25, 30). However, its antifibrotic actions appear to involve inhibition of macrophage infiltration, transforming growth factor-β expression (14), and MAPK activity (26, 23), which are important mechanisms of cytokine-induced tissue fibrosis. However, at present, we do not know whether the effects of Ac-SDKP are mediated by specific receptor binding sites present in CFs, since no receptors (or binding sites) for Ac-SDKP have been identified or localized in any cardiovascular cells or tissues because of the lack of a specific radioligand. In the present study, we tested the hypothesis that Ac-SDKP-induced antiproliferation and anticollagen deposition in rat CFs are mediated by specific Ac-SDKP receptor binding sites. To characterize these binding sites, we developed a novel radioligand called 3-(p-hydroxyphenyl)-propionic acid (or desaminotyrosine) (Hpp)-Aca-SDKP, an 125I-labeled analog of Ac-SDKP, for radioreceptor assays in cultured rat CFs and cardiac tissue. We believe our results demonstrate for the first time that Hpp-Aca-SDKP is a functional ligand specific for Ac-SDKP receptor binding sites and that both Ac-SDKP and Hpp-Aca-SDKP exert antifibrotic effects by binding to Ac-SDKP receptors in CFs.
MATERIALS AND METHODS
Primary culture of rat CFs
CFs from adult Sprague-Dawley rats were isolated according to our own protocols and those described by Eghbali et al. (10, 26). Briefly, two or three hearts from adult Sprague-Dawley rats weighing 200–250 g (~8 wk old) were rapidly excised under pentobarbital sodium anesthesia (50 mg/kg ip), and atrial tissue was removed. All protocols or procedures involving use of rats were approved by the Institutional Animal Care and Use Committee of Henry Ford Health System. Ventricles were minced with scissors and placed in tubes containing 5 ml calcium bicarbonate-free Hanks’ solution with HEPES (CBFHH) buffer with 0.067% collagenase B (Boehringer Mannheim) along with a small magnetic bar. Tubes were kept on Magnetic Stir 1 (setting 2) for 15 min; 5 ml of CBFHH buffer were added to each tube, and minced tissue was pipetted up and down 30 times. Cell suspensions from three digestion periods were pooled together in a 50-ml tube containing 5 ml FCS on ice and pelleted at 1,000 rpm for 5 min. The pellet was washed twice with 10% FCS-DMEM. Suspensions from six sequential digestions were combined, centrifuged, and resuspended in 8 ml 10% FCS-DMEM and then passed through a 60 mesh screen (Sigma) into a 100-mm tissue culture dish. Cells were incubated for 45 min at 37°C in 5% CO2. Those that remained unattached were discarded, whereas attached cells were washed twice with 10% FCS-DMEM and allowed to grow to confluence before passaging. Cell passage was performed with a trypsin-based solution in 1:3 dilutions. All cells used in these experiments were from passages 3–4 (10, 23, 26).
Synthesis of Hpp-Aca-Ser-Asp-Lys-Pro-OH
In preliminary binding experiments using [3H]Ac-SDK*P as a radioligand, we found very high nonspecific binding (~50%), which could not be easily displaced with unlabeled Ac-SDKP (10 μM) and made it difficult to construct saturation binding curves and perform Scatchard analyses. To develop a functional ligand for Ac-SDKP, we synthesized an Ac-SDKP analog by adding desaminotyrosine to the sequence of the tetrapeptide (Hpp-Aca-SDKP), which can be labeled with 125I (20, 21). Briefly, Hpp-Aca-Ser-Asp-Lys-Pro-OH was synthesized on solid phase support with a peptide synthesizer (Applied Biosystems 430A), using a Merrifield resin (substitution 0.885 meq/g grain size, 100–200 mesh, 1% divinylbenzene) with the first amino acid (L-proline) attached (Chem-Impex International). N-t-Boc-protected amino acid [Boc-Lys(2-ClZ)-OH, Boc-Asp(OBzl)-OH, and Boc-Ser(Bzl)-OH; Chem-Impex], used for coupling, were activated by DCC/HOBT in 1-methyl-2-pyrro-lidinone as solvent. Couplings of Boc-Aca-OH (N-t-butyloxycarbonyl-6-amino caproic acid; Aldrich) and Hpp (Sigma) were achieved with TBTU/HOBT [O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (Chem-Impex) and 1-hydroxybenzotriazole (Peptides International)], using N-methyl morpholine as a base. The final peptide was cleaved from the resin with anhydrous hydrogen fluoride, using anisol as a scavenger (20, 21). The resulting crude peptide was purified by reverse-phase (C18) chromatography, and its identity was confirmed by mass spectrometry, yielding a mass of 707 Da. The full chemical structure of Hpp-Aca-SDKP is shown in Fig. 1.
Fig. 1.
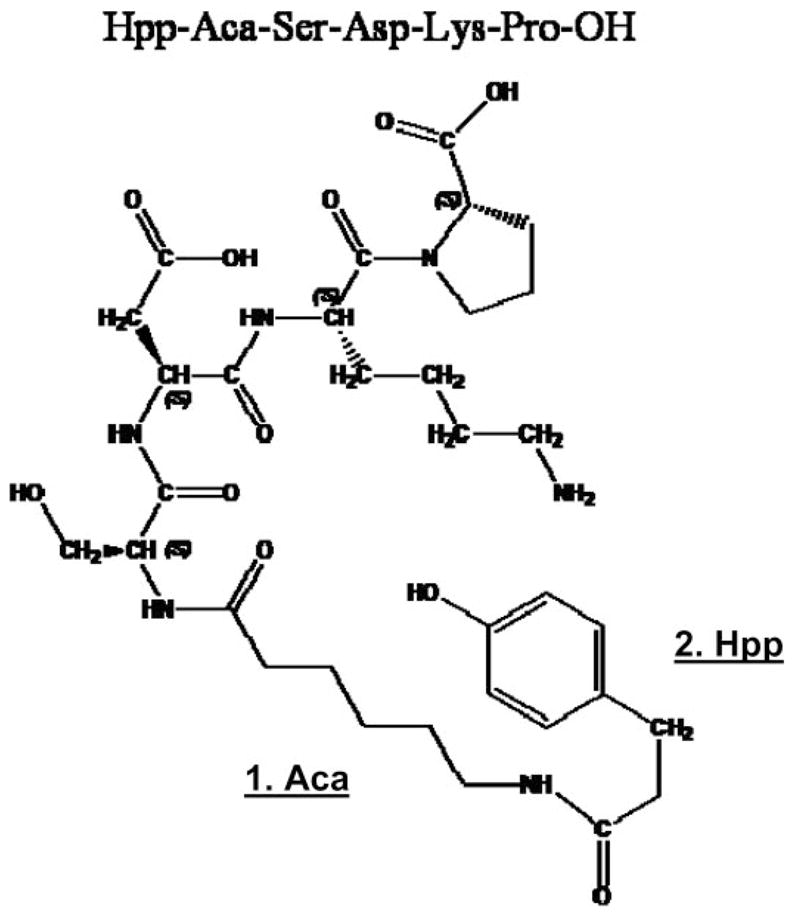
Chemical structure of the synthesized Ac-SDKP analogue Hpp-Aca-SDKP was confirmed by mass spectrometry, which yielded a mass of 707 Da. Aca-OH, N-t-butyloxycarbonyl-6-amino caproic acid (Aldrich); Hpp, 3-(p-hydroxyphenyl)-propionic acid or desaminotyrosine; SDKP, N-acetyl-seryl-aspartyl-lysyl-proline. The added desaminotyrosine can be labeled with 125I for Ac-SDKP radioreceptor assays in cultured cells and receptor localization in tissues.
Biological activity of Hpp-Aca-SDKP
We next tested whether the synthesized analog, Hpp-Aca-SDKP, retains the biological activity of its natural tetrapeptide, Ac-SDKP, in cultured rat CFs, examining whether Hpp-Aca-SDKP inhibits endothelin-1 (ET-1)-induced collagen synthesis in cultured rat CFs similarly to what we reported for Ac-SDKP (26). To compare Hpp-Aca-SDKP with Ac-SDKP (1 nmol/l), Hpp-Aca-SDKP (0–1,000 nmol/l) was added to CFs in the presence of captopril (1 μmol/l) and ET-1 (10 nmol/l) for 48 h at 37°C. Captopril was added to inhibit ACE and prevent Ac-SDKP degradation (7, 8, 11, 12). At the end of the experiment, the medium was examined for collagen synthesis by hydroxyproline assay (26). Cells were harvested, and protein content was determined. Collagen synthesis was expressed as micrograms collagen per milligram protein (26).
125I-labeled Hpp-Aca-SDKP receptor binding in cultured rat CFs
Fibroblasts were split and subcultured in six-well plates in DMEM + 10% FCS until 80% subconfluent (3–5 days culture: ~5 × 105 cells) (23, 26). Subconfluent cells were washed twice with 2 ml PBS, pH 7.4, and then incubated with the radioligand for 60 min at 37°C in 2 ml of 10 mM sodium phosphate buffer, pH 7.4, containing 0.3 μCi/ml (~1 nM) 125I-labeled Hpp-Aca-SDKP (125I-Hpp-Aca-SDKP, specificity of 4,565 Ci/mmol; Bachem), 150 mM NaCl, 5 mM Na2EDTA, 0.2% NaN3, 0.2% BSA, 0.5 mg/ml bacitracin, 10 μM captopril, and 1 μM pepstatin A. After incubation, cells were washed twice with 2 ml of ice-cold PBS and scraped with a cell lifter. Cell-bound and unbound radioligands were separated by rapid filtration with a 48-well filtration device (Brandel) followed by four washes with 1 ml of ice-cold buffer (31, 34, 36). Bound radioactivity on the filter disks was counted. To exclude the possibility that 125I-Hpp-Aca-SDKP binding in rat CFs may reflect receptor uptake or internalization, parallel binding assays were separately performed in membrane fractions prepared from fractionated rat CFs as described (31, 36). Total binding was determined in the presence of 125I-Hpp-Aca-SDKP (~1 nM) without competition by unlabeled compounds. Nonspecific binding was determined in the presence of 10 μM unlabeled Hpp-Aca-SDKP. Specific binding was taken as the difference between total and nonspecific binding as described (31–33).
Saturation 125I-Hpp-Aca-SDKP receptor binding and Scatchard plot analyses
Saturation radioligand binding experiments measure specific radioligand binding at equilibrium at various concentrations, allowing us to determine maximal binding sites (Bmax) and dissociation constant (Kd). To carry out these experiments, cultured rat CFs were subcultured and assayed with the binding buffer and conditions described above. Specific 125I-Hpp-Aca-SDKP binding was determined in a six-well plate containing increasing concentrations of 125I-Hpp-Aca-SDKP (0, 0.03, 0.1, 0.3, 1.0, 3.0, and 10 nM) in the incubation buffer for 60 min at 37°C. In separate experiments, a constant concentration (10 μM) of unlabeled Ac-SDKP was added into incubations to determine nonspecific binding. Cells were washed and scraped, and bound and unbound radioligands were separated by rapid filtration. Binding data were analyzed with GraphPad Prism 4 as described (31–33, 35, 36).
Competition for 125I-Hpp-Aca-SDKP binding by unlabeled SDKP analogs and unrelated peptides
To determine the specificity of 125I-Hpp-Aca-SDKP binding sites in cultured rat CFs, binding experiments were performed with increasing concentrations of unlabeled Hpp-Aca-SDK and Ac-SDKP (10−10 to 10−4 M) or 10 μM each of Hpp-Aca-SDKP, Ac-SDKP, SDKP (dehydro), or tripeptide SDK at 37°C for 60 min. Additionally, we also tested the effects of unrelated peptides ANG II (10 μM) or ET-1 (10 μM) on 125I-Hpp-Aca-SDKP binding in CFs to determine whether these peptides compete for Ac-SDKP binding. After incubation, the cells were washed and scraped, and bound and free radioligands were separated by rapid filtration. Radioactivity of each sample was counted as described (31–33, 35, 36).
Effects of Ac-SDKP on ET-1-induced intracellular calcium responses in rat CFs
Our group (25–27) has previously shown that Ac-SDKP significantly attenuated ET-1-induced cell proliferation and collage synthesis in rat CFs. However, we do not know the cellular mechanisms involved in these effects. ET-1 is a potent vasoconstrictor and growth factor, and increased intracellular calcium concentration ([Ca2+]i) induced by ET-1 is one of the major signaling pathways (1, 9, 18). To determine whether Ac-SDKP attenuates ET-1-stimulated collagen synthesis in part by inhibiting [Ca2+]i responses to ET-1, we pretreated subconfluent CFs plated on glass coverslips with serum-free medium, Ac-SDKP (10 nM), or thapsigargin (1 μM), an inhibitor of sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA), to deplete intracellular calcium stores and loaded these with the calcium indicator fura 2 (2 μM) at 37°C for 30 min as described previously (16, 17, 33). After two washes with PBS, coverslips were placed inside a perfusion chamber maintained at 37°C, mounted on a Nikon Eclipse TE 2000-U fluorescence microscope coupled with a Lambda DG4 illumination system (Sutter Instruments). Fura 2-loaded CFs were alternately excited at 340 and 380 nM every 3 s. Basal ratiometric calcium imaging was first recorded for 5 min with serum-free medium alone. CFs were then stimulated with ET-1 (1 nM), and 340/380 ratiometric calcium images were continuously recorded at 3-s intervals for up to 10 min. At the end of experiment, CFs were tested on whether they responded to the calcium ionophore A-23187 (10 μM). The effects of Ac-SDKP on ET-1-increased [Ca2+]i were determined by calculating the average magnitude of the peak [Ca2+]i responses during the entire ET-1 stimulation (200 s) (16, 17, 33).
Effects of Ac-SDKP on ET-1-induced activation of MAPK ERK1/2
Growth factors or inflammatory cytokines such as ET-1 or ANG II play an important role in inducing cell growth and proliferation, partly through mobilization of [Ca2+]i. ET-1 has been shown to induce activation of MAPKs in cultured vascular smooth muscle cells (13) and rat mesangial cells (29). Conversely, we have previously shown that the MAPK inhibitor PD-98059 was able to block ET-1-stimulated collagen synthesis, similar to the effect of Ac-SDKP (26). To determine whether Ac-SDKP attenuates ET-1-stimulated collagen synthesis in rat CFs in part by inhibiting ET-1-induced MAPK ERK1/2 phosphorylation, subconfluent CFs were treated with ET-1 (1 nM) in the absence or presence of Ac-SDKP (10 nM). The effects of Ac-SDKP on ET-1-induced ERK1/2 phosphorylation were further compared in CFs pretreated with a PLC and/or the inositol 1,4,5-trisphos-phate-specific inhibitor U-73122 (1 μM) or with thapsigargin (1 μM), a potent inhibitor of SERCA ATPase to deplete intracellular calcium stores (16, 17, 33). After treatment, the medium was removed and CFs were washed twice with ice-cold PBS and lysed with a modified RIPA buffer containing an inhibitor cocktail (Roche). Protein was measured with the use of a bicinchoninic acid kit (Pierce). Protein (10 μg) from each sample was separated on 8%–16% SDS-PAGE and transferred semi-dry onto an Immobilon-P membrane (Millipore). Total and phosphorylated ERK1/2 were detected by immunoblotting with a rabbit ERK1/2 monoclonal antibody (Cell Signaling Technology) or a mouse antiphosphorylated ERK1/2 monoclonal antibody against an amino acid sequence containing phosphorylated Tyr-204 of human ERK1/2 (Santa Cruz) (16, 17). Immunoblots were visualized by enhanced chemiluminescence using horseradish peroxidase-conjugated secondary antibodies (Santa Cruz), scanned, and analyzed with a microcomputer imaging device (MCID, Imaging Research, Ontario, Canada) (16, 17).
125I-Hpp-Aca-SDKP receptor binding in rat heart sections visualized by quantitative in vitro autoradiography
Because we have demonstrated specific 125I-Hpp-Aca-SDKP binding sites in cultured rat CFs in vitro, we next wanted to determine whether specific 125I-Hpp-Aca-SDKP binding sites are present in the rat heart in vivo. Quantitative in vitro autoradiography was employed to visualize the anatomic distribution of Ac-SDKP binding sites, as described for localization of ACE and ANG II receptor subtypes in rat and human blood vessels and hearts (31, 32, 35, 36). Rats were decapitated, and the hearts were removed quickly. The cardiac chambers were filled with tissue medium and snap frozen in isopentane on ice. Heart sections (10 μm thick) were cut on a Cryostat and mounted on glass slides, which were dried overnight under reduced pressure. Tissue sections were first preincubated for 15 min with 10 mM sodium phosphate buffer to clear the endogenous ligand and then incubated with 125I-Hpp-Aca-SDKP (~1 nM) for 60 min at 37°C in 10 ml of 10 mM sodium phosphate buffer, pH 7.4, containing 150 mM NaCl, 5 mM Na2EDTA, 0.2% NaN3, 0.2% BSA, 0.5 mg/ml bacitracin, 10 μM captopril, and 10 μM pepstatin A. After incubation, sections were washed four times with fresh buffer without BSA (1 min each) and air dried. Sections were then exposed to X-ray film in light-tight cassettes with a set of radioactivity standards for up to 30 days. To enable cellular localization, some sections were coated with LM-1 liquid emulsion (Amersham), dried, and exposed in light-tight cassettes for 60 days as described previously (31–33, 34, 36). After exposure, the films and emulsion-coated sections were developed, and autoradiographs were analyzed by a microcomputer imaging device (Image Research). To determine whether Ac-SDKP binding is localized to cardiomyocytes or the cardiac interstitium, adjacent sections were stained with fluorescein-labeled peanut lectin (Vector Laboratories) to identify the myocyte border as described previously (22, 25, 27, 30).
Statistical analysis
Results are expressed as means ± SE. Ac-SDKP receptor binding constant (Kd) and Bmax in cultured CFs were analyzed by GraphPad Prism 4. Total and nonspecific binding were compared by unpaired t-test.
RESULTS
Effects of Hpp-Aca-SDKP on ET-1-stimulated collagen synthesis in cultured rat CFs
To determine whether the synthesized analog Hpp-Aca-SDKP is a functional agonist of its native tetrapeptide Ac-SDKP, we compared their inhibitory effects on ET-1-stimulated collagen synthesis in cultured rat CFs. As shown in Fig. 2, Hpp-Aca-SDKP inhibited ET-1-induced collagen synthesis in a dose-dependent fashion, reaching maximum inhibition at a concentration of 1 nM. However, Hpp-Aca-SDKP lost its inhibitory effect on ET-1-stimulated collagen synthesis at 1 μM. As our group reported previously (26), Ac-SDKP (1 nmol/l) completely blocked ET-1-induced collagen synthesis (Fig. 2) but also lost its inhibitory activity at 1 μM (26). These data suggest that Hpp-Aca-SDKP exhibits pharmacological properties similar to Ac-SDKP and can be used as a functional ligand for pharmacological characterization of Ac-SDKP receptor binding sites.
Fig. 2.
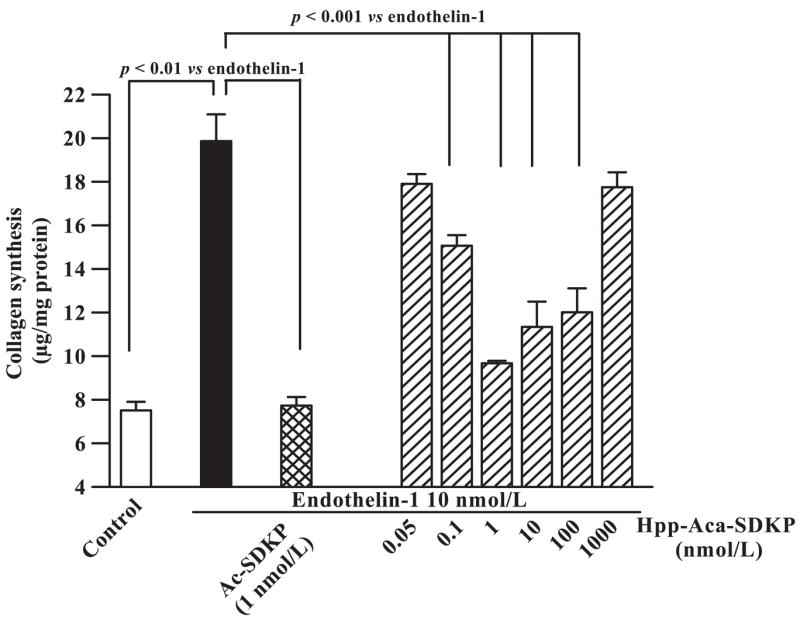
Effects of Ac-SDKP and its analog Hpp-Aca-SDKP on endothelin-1 (ET-1)-stimulated collagen synthesis in cultured rat cardiac fibroblasts (CFs). Note that ET-1 (10 nM) markedly increased CF collagen synthesis (solid bar) compared with control (open bar). Hpp-Aca-SDKP (hatched bars) had no effect on ET-1-induced collagen synthesis at 0.05 nM but inhibited the response at 0.1 and 1 nM in a concentration-dependent manner. However, Hpp-Aca-SDKP lost its inhibitory activity at 1 μM. Ac-SDKP (1 nM) also potently inhibited ET-1-induced collagen synthesis (cross-hatched bar), as described previously (26).
Specific Ac-SDKP receptor binding in cultured rat CFs or fractionated membranes with 125I-Hpp-Aca-SDKP as radioligand
CFs or fractionated CF membranes were incubated with 1 nM 125I-Hpp-Aca-SDKP (Fig. 3). Total binding was determined without adding any competing ligand, whereas nonspe-cific binding was determined in the presence of an excess concentration of unlabeled Hpp-Aca-SDKP (10 μM), which displaces 125I-Hpp-Aca-SDKP binding sites. Under equilibrium conditions, both intact CFs and isolated membranes displayed highly specific 125I-Hpp-Aca-SDKP binding (>85% of total binding), whereas nonspecific binding represented only ~15% of the total (Fig. 3A). Thus the level of nonspecific binding is consistent with those found in ACE and AT1-receptor binding using 125I-labeled radioligands (31–36).
Fig. 3.
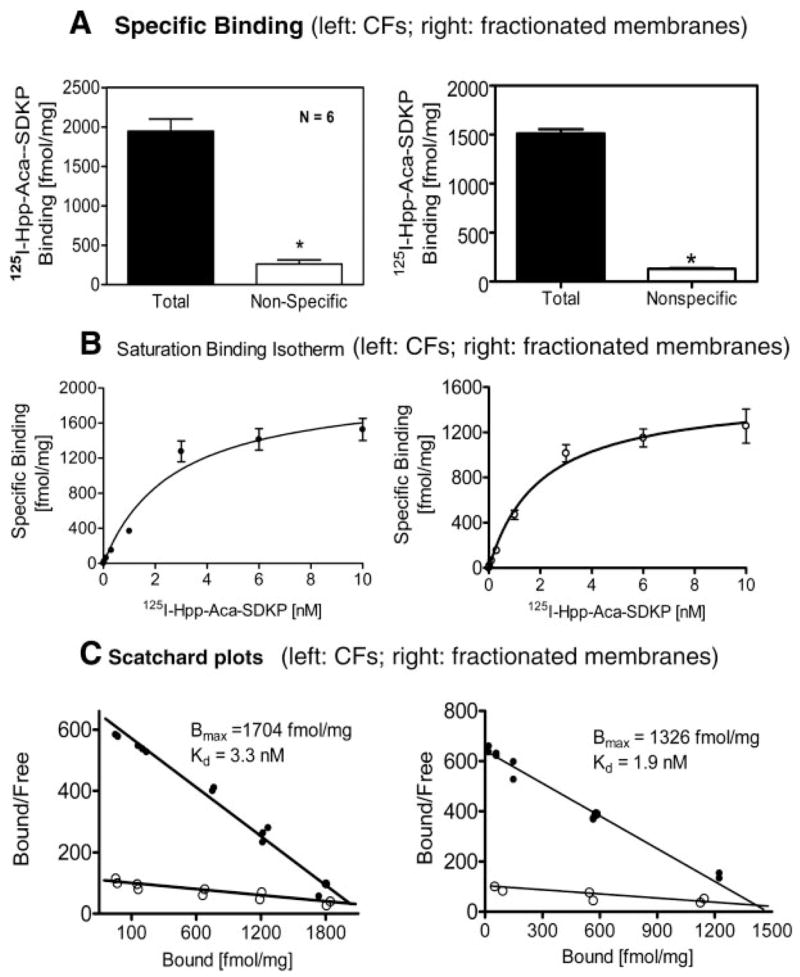
Total and nonspecific binding in the presence of excess unlabeled Hpp-Aca-SDKP (10 μM) (A), saturation binding isotherms with increasing concentrations of 125I-labeled Hpp-Aca-SDKP (125I-Hpp-Aca-SDKP, 0–10 nM) (B), and Scatchard plot analysis (C) of 125I-Hpp-Aca-SDKP receptor binding sites in intact CFs (left) or fractionated cell membranes (right) (n = 6 from 3 experiments). Note that both intact CFs and fractionated CF membranes bound 125I-Hpp-Aca-SDKP with similar maximal binding site (Bmax) and dissociation constant (Kd) results.
Saturation binding experiments and Scatchard plot analyses of 125I-Hpp-Aca-SDKP binding in cultured rat CFs and fractionated membranes
Figure 3B shows one of three representative saturation binding isotherms with 125I-Hpp-Aca-SDKP in intact rat CFs or fractionated membranes. Specific Hpp-Aca-SDKP binding was increased in a concentration-dependent manner, reaching a plateau at 10 nM. Analysis of the binding data with nonlinear regression (curve fit) and one-site binding (hyperbola) showed a single class of binding sites with high affinity for 125I-Hpp-Aca-SDKP in both intact CFs and fractionated membranes. As shown in Fig. 3C, the level (Bmax) of 125I-Hpp-Aca-SDKP binding was slightly lower in CF membranes, but it was not significantly different from that of intact CFs. In either CFs or fractionated membranes, we were unable to curve fit the two-site binding data (hyperbola). Best-fit values for Bmax and Kd are shown in the inserted Scatchard plots (Fig. 3C).
Competition for 125I-Hpp-Aca-SDKP receptor binding by unlabeled Hpp-Aca-SDKP and other Ac-SDKP analogs and in cultured rat CFs
To confirm the specificity of 125I-Hpp-Aca-SDKP binding in CFs, we compared the inhibitory potencies of Hpp-Aca-SDKP, the native peptide Ac-SDKP, and other SDKP analogs (Fig. 4). Both Hpp-Aca-SDKP (Fig. 4A) and Ac-SDKP (Fig. 4B) displaced 125I-Hpp-Aca-SDKP binding in CFs in a concentration-dependent manner, but the native peptide [inhibition constant (Ki) = 0.69 ± 0.2 nM] was more potent than the synthesized analog (Ki = 10.4 ± 0.2 nM). Other analogs such as SDKP (dehydro) and the tripeptide SDK were less potent in displacing 125I-Hpp-Aca-SDKP binding than Ac-SDKP or Hpp-Aca-SDKP, whereas the nonrelated peptides ET-1 or ANG II had no effect on 125I-Hpp-Aca-SDKP binding even at 10 μM.
Fig. 4.
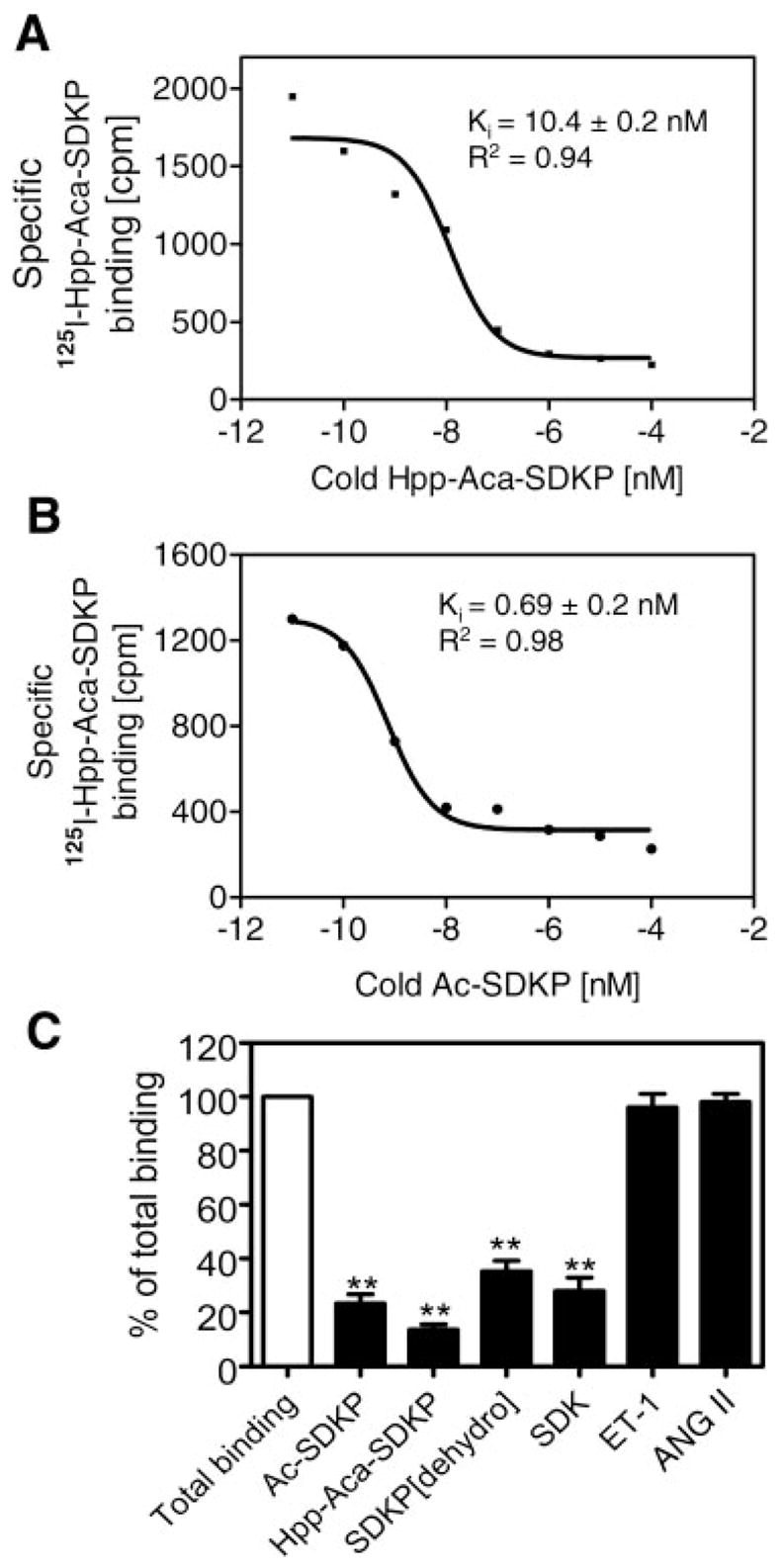
Effects of increasing concentrations of unlabeled competing ligands, Hpp-Aca-SDKP (A) and Ac-SDKP (B), or excess concentrations (10 μM) of Ac-SDKP, Hpp-Aca-SDKP, SDKP (dehydro), SDK, ET-1, and ANG II (C) on specific 125I-Hpp-Aca-SDKP receptor binding in cultured rat CFs, representing 4 different experiments. Ki, inhibition constant. Note that the inhibitory potencies of these compounds on 125I-Hpp-Aca-SDKP receptor binding were in the order of Hpp-Aca-SDKP ≥ Ac-SDKP > SDK > SDKP (dehydro) and that unrelated peptide ET-1 and ANG II did not displace 125I-Hpp-Aca-SDKP receptor binding in CFs. **P < 0.01 vs. total binding.
Effects of Ac-SDKP on ET-1-induced [Ca2+]i mobilization in rat CFs
ET-1 binds to its cell surface receptors to induce a variety of cellular responses, with increased [Ca2+]i being one of the most important signaling pathways (1, 9, 18). We have shown that Ac-SDKP inhibited ET-1-stimulated collagen synthesis in rat CFs (26), but the cellular mechanisms by which Ac-SDKP exerts its effect are not known. In the present study, Ac-SDKP alone did not significantly alter basal [Ca2+]i (basal: 93 ± 26 nM vs. Ac-SDKP: 116 ± 33 nM; not significant) (Fig. 5A). However, ET-1 induced a rapid increase in [Ca2+]i from basal levels (328 ± 52 nM; P < 0.01) (Fig. 5B), which was significantly attenuated by pretreatment of CFs with Ac-SDKP (10 nM) (156 ± 38 nM, P < 0.01 vs. ET-1) (Fig. 5C). Pretreatment of CFs with thapsigargin to deplete [Ca2+]i stores and Ac-SDKP also abolished [Ca2+]i responses to ET-1 (88 ± 22 nM; P < 0.01 vs. ET-1) (Fig. 5D).
Fig. 5.
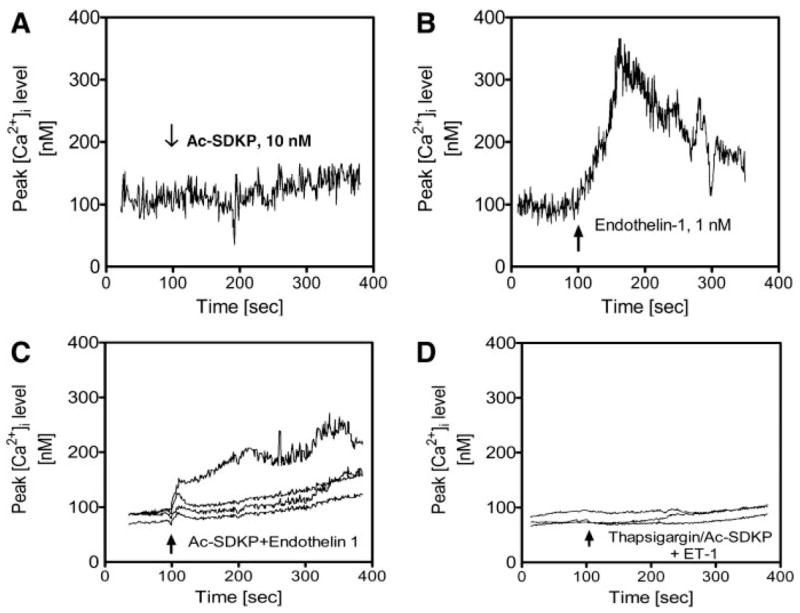
Effects of Ac-SDKP on basal and ET-1-induced intracellular Ca2+concentration ([Ca2+]i) responses in rat CFs. CFs were loaded with the Ca2+ indicator fura 2 (2 μM), and changes in ratiometric calcium ratio (340/380 nm) in response to Ac-SDKP and/or ET-1 were recorded by MetaFlor fluorescence imaging system (Universal Imaging). A: Ac-SDKP (10 nM) did not alter [Ca2+]i significantly from baseline. B: ET-1 markedly increased [Ca2+]i, as expected. C: pretreatment of CFs with Ac-SDKP significantly attenuated [Ca2+]i responses to ET-1 stimulation (1 nM). D: pretreatment of CFs with thapsigargin (1 μM) and Ac-SDKP (10 nM) completely abolished [Ca2+]i responses to ET-1 stimulation in 4 different CFs (1 nM). Note that the experiments were repeated 3–5 times with >12 CFs studied in each protocol.
Effects of Ac-SDKP on ET-1-induced activation of MAPK ERK1/2 in rat CFs
Activation or phosphorylation of MAPK ERK1/2 by growth-promoting and vasoactive factors is well recognized as an important intracellular signaling pathway in cell proliferation, protein synthesis, and transformation (5, 13, 16, 17, 19, 29). Figure 6 shows that ET-1 (1 nM) induced more than threefold increases in phosphorylation of ERK1/2 (control: 100% vs. ET-1: 320 ± 39%; P < 0.01), as expected. Pretreatment of CFs with Ac-SDKP (10 nM) for 30 min inhibited ET-1-induced ERK1/2 phosphorylation by almost 50% (205 ± 29%; P < 0.01 vs. ET-1). However, Ac-SDKP alone appeared to have a small effect on basal phosphorylated ERK1/2 (132 ± 16%; P < 0.05 vs. ET-1). Depletion of intracellular calcium stores with thapsigargin attenuated ET-1-stimulated ERK1/2 phosphorylation (ET-1: 282 ± 13% vs. ET-1 + thapsigargin: 158 ± 38%; P < 0.01), whereas inhibition of PLC with U-73122 had no effect on ERK1/2 response to ET-1 stimulation (252 ± 10% vs. ET-1; not significant) (Fig. 7).
Fig. 6.
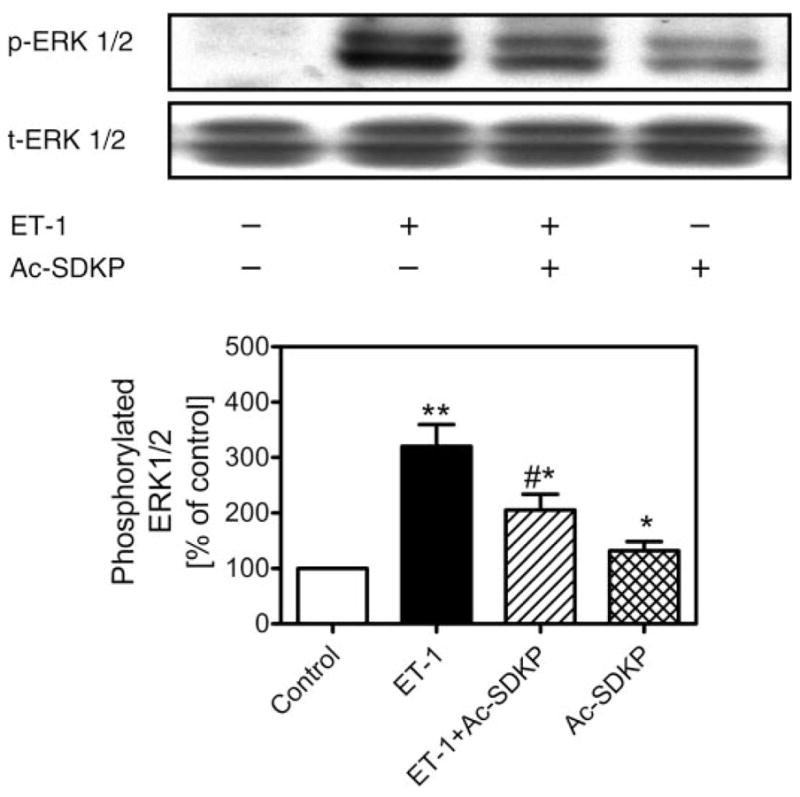
Effects of Ac-SDKP on ET-1-induced MAPK ERK1/2 phosphorylation in rat CFs. CFs were stimulated with ET-1 (1 nM) in the absence or presence of Ac-SDKP (10 nM) at 37°C for 5 min. Top: Western blots of phosphorylated and total ERK1/2. Bottom: semi-quantitative data as % of control from triplicate blots for each protocol. Note that ET-1 markedly increased ERK1/2 phosphorylation, which was attenuated by pretreatment of cells with Ac-SDKP (10 nM). Total ERK1/2 was not changed by any treatment and also served to confirm equal protein loading. Note that, in double bands, bottom represents ERK2 (p42) and top band represents ERK1 (p44). p, Phosphorylated ERK1/2; t, total ERK1/2. *P < 0.05 and **P < 0.01 vs. control; #P < 0.05 vs. ET-1.
Fig. 7.
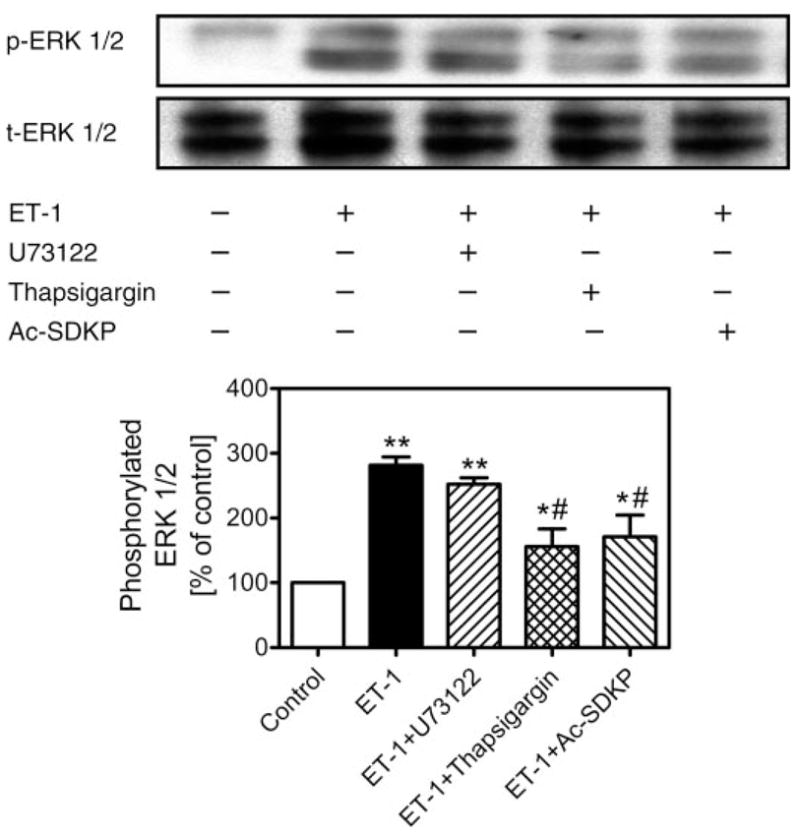
Effects of inhibition of PLC with U-73122 (1 μM) or intracellular sarco(endo)plasmic reticulum Ca2+-ATPase with thapsigargin (1 μM) on ET-1-induced ERK1/2 phosphorylation in rat CFs. Top: Western blots of phosphorylated (p-ERK1/2) and total (t-ERK1/2) ERK1/2. Bottom: semi-quantitative data as % of control from triplicate blots for each protocol. Note that ET-1 markedly increased ERK1/2 phosphorylation, which was attenuated by pretreatment of cells with thapsigargin or Ac-SDKP (10 nM) but not by U-73122 (1 μM). Total ERK1/2 was not changed by any treatment and also served to confirm equal protein loading. *P < 0.05 and **P < 0.01 vs. control; #P < 0.05 vs. ET-1.
Autoradiographic and cellular localization of 125I-Hpp-Aca-SDKP binding sites in the rat heart
Figure 8 shows anatomic localization of 125I-Hpp-Aca-SDKP binding in the rat heart. Specific binding occurred throughout the heart, including atria and ventricles, septum, and blood vessels, with relatively higher levels in the left ventricle and septum (Fig. 8A). Emulsion autoradiographs revealed that silver grains clearly outlined cardiomyocytes (Fig. 8C, arrows) and overlapped with fluorescein-labeled peanut lectin staining, primarily in the interstitium (Fig. 8E). Nonspecific binding was only ~14% of total binding, suggesting that specific 125I-Hpp-Aca-SDKP binding could be displaced by an excess of unlabeled Hpp-Aca-SDKP (10 μM) (Fig. 8, B and D). Relative quantitative levels of 125I-Hpp-Aca-SDKP in the rat heart are shown in Table 1 (n = 6).
Fig. 8.
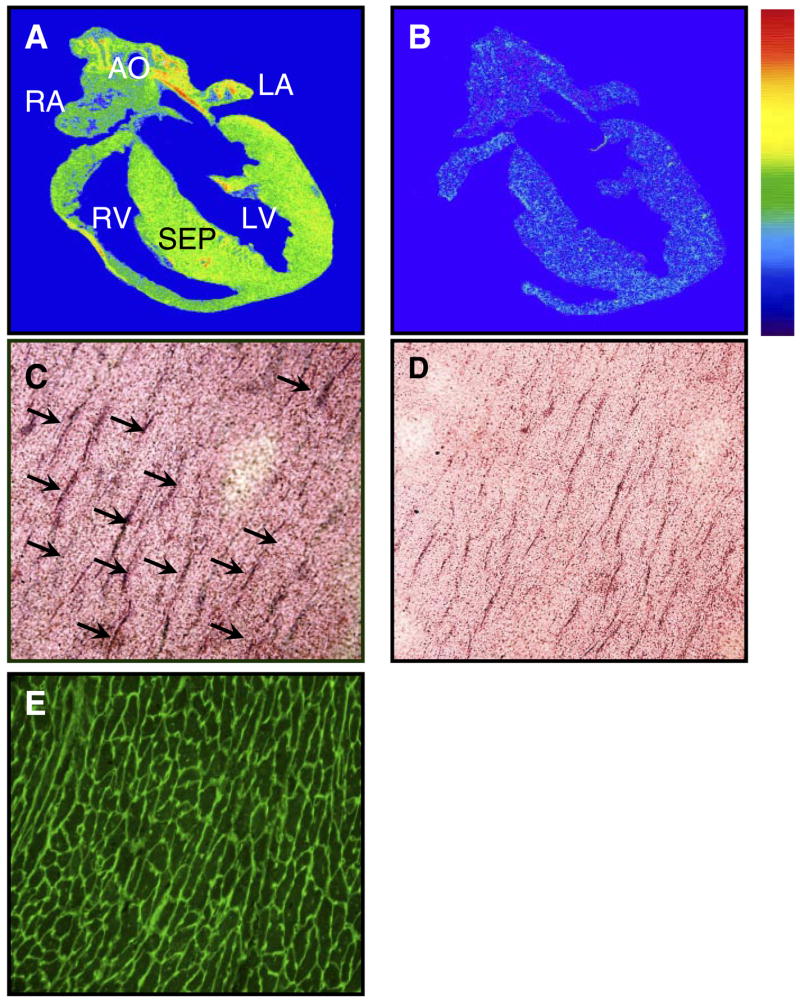
Anatomic and cellular localization of 125I-Hpp-Aca-SDKP receptor binding in a representative rat heart (n = 5). A: total 125I-Hpp-Aca-SDKP receptor binding. B: nonspecific binding in the presence of excess competing unlabeled Hpp-Aca-SDKP (10 μM). 125I-Hpp-Aca-SDKP receptor binding is indicated by the color bar, with red being the highest and blue being the lowest (background). C: emulsion autoradiograph showing 125I-Hpp-Aca-SDKP receptor binding (silver grains indicated by arrows) surrounding cardiomyocytes. D: adjacent section of C, which was preincubated with an excess of unlabeled Hpp-Aca-SDKP (10 μM) before being labeled by 125I-Hpp-Aca-SDKP. E: also an adjacent section of C, which was stained with fluorescein-labeled peanut lectin to identify the myocyte border, and suggests interstitial localization of 125I-Hpp-Aca-SDKP receptor binding in the rat heart. AO, aorta; LA, left atrium; RA, right atrium; LV, left ventricle; RV, right ventricle; SEP, septum.
Table 1.
Cardiac distribution of specific 125I-Hpp-Aca-SDKP receptor binding in rats as visualized by quantitative in vitro autoradiography
| Left Atrium | Right Atrium | Left Ventricle | Right Ventricle | Septum | |
|---|---|---|---|---|---|
| Specific binding, dpm/mm2 | 25.8 ± 3.5 | 23.6 ± 1.6 | 34.7 ± 2.6 | 29.6 ± 3.9 | 29.3 ± 2.2 |
Values are means ± SE. Specific binding was determined as the difference between total binding and binding in the presence of excess unlabeled Hpp-Aca-SDKP (10 μM). Hpp, 3-(p-hydroxyphenyl)-propionic acid; SDKP, N-acetyl-seryl-aspartyl-lysyl-proline. There were no significant differences in specific 125I-labeled Hpp-Aca-SDKP receptor binding between different cardiac structures (one-way ANOVA).
DISCUSSION
Our group has previously shown that Ac-SDKP potently inhibits DNA and collagen synthesis in cultured rat CFs in vitro (25, 26) and attenuates the fibrotic effects of aldosterone (salt)- and ANG II-induced hypertension in rats in vivo (22, 25, 27). This tetrapeptide also prevents and reverses cardiac fibrosis in rats with chronic heart failure after myocardial infarction (30). The antifibrotic effects of Ac-SDKP appear to involve multiple signaling pathways, including inhibition of transforming growth factor-β and connective tissue growth factor expression in CFs mediated via p42/p44 MAPK (14, 23, 26) and inhibition of plasminogen activator-1 expression in human mesangial cells (mediated via Smads) (14). However, we do not know whether these effects involve specific Ac-SDKP receptor binding sites. Using a new radioligand, 125I-Hpp-Aca-SDKP, we demonstrated for the first time that there is a single class of high-affinity receptor binding sites for Hpp-Aca-SDKP in cultured CFs. Because 125I-Hpp-Aca-SDKP receptor binding was potently displaced by unlabeled Hpp-Aca-SDKP and the native peptide Ac-SDKP and because Hpp-Aca-SDKP, like Ac-SDKP, also potently inhibits ET-1-stimulated collagen synthesis in CFs, our results suggest that Hpp-Aca-SDKP is a functional ligand for Ac-SDKP receptor binding sites in CFs and tissues.
Although the effects of Ac-SDKP have been extensively studied, its specific receptor binding sites have not been characterized to our knowledge, nor has the location of Ac-SDKP receptor binding in cardiac tissues been determined. Ac-SDKP is a small peptide containing only four amino acids (4, 15). It does not possess tyrosine, which is generally required for iodination with 125I. The only radioligand available for characterization of Ac-SDKP receptor binding is [3H]SDK*P (Amersham), an analog of Ac-SDK-(tetrahydroproline). However, we found no reports of Ac-SDKP receptor binding using this radioligand in the literature. In a preliminary study, we attempted to characterize and localize Ac-SDKP receptor binding in isolated rat CFs and rat hearts using [3H]SDK*P. Although we performed binding assays using different buffers and conditions, we were unable to reduce nonspecific binding to an acceptable level, and it remained relatively high (~50%) despite the excess concentration of unlabeled Ac-SDKP (10 μM) used to displace [3H]SDK*P binding. Similar high levels of nonspecific binding were also observed in rat cardiac sections as visualized by quantitative in vitro autoradiography. Because we added a high concentration of captopril (10 μM) to the binding buffer to inhibit ACE, it is unlikely that the persistent high levels of nonspecific binding were due to degradation of the radioligand by ACE, which almost exclusively breaks down Ac-SDKP both in vivo and in vitro (6, 8, 11, 12). These results suggest that [3H]SDK*P is not an ideal radioligand for Ac-SDKP receptor binding, probably due to the chemical properties of the 3H-labeled compound.
To develop a functional ligand for pharmacological characterization of Ac-SDKP receptor binding sites, we synthesized an Ac-SDKP analog for the first time by adding desaminotyrosine to the sequence of the tetrapeptide, which can be labeled with 125I (20, 21). To exclude the possibility that the analog may lose its biological activity when desaminotyrosine is added, we first examined whether it would exert effects similar to its native tetrapeptide on ET-1-stimulated collagen synthesis in cultured rat CFs. At 1 nM, both Ac-SDKP and Hpp-Aca-SDKP potently attenuated ET-1-induced collagen synthesis (34). However, their inhibitory effects on ET-1-stimulated collagen synthesis were lost at micromolar concentrations (Fig. 2) (26). The cellular mechanisms or signaling pathways underlying these biphasic effects of Ac-SDKP and Hpp-Aca-SDKP are not fully understood at present, but it may relate to the physiological levels (nM) of endogenous Ac-SDKP in the circulation and tissues (6, 24, 25, 27). Furthermore, persistent exposure of receptor binding sites to these ligands may lead to full occupancy and subsequent desensitization of the receptors. Nevertheless, our data confirm that Hpp-Aca-SDKP is pharmacologically similar to Ac-SDKP and can be used as a functional ligand for Ac-SDKP receptor binding in cells and tissues. Indeed, in radioreceptor binding assays using 125I-labeled Hpp-Aca-SDKP, we were able to reduce nonspecific binding substantially, from ~50% with [3H]Ac-SDK*P to <15% with 125I-labeled Hpp-Aca-SDKP (Fig. 3A). Levels of specific 125I-labeled Hpp-Aca-SDKP receptor binding were comparable to those reported for other peptide receptors such as ANG II, ET-1, or bradykinin (31–33, 35, 36). In addition to increasing the level of specific binding, this radioligand made it possible to perform saturation studies to determine the binding characteristics of Ac-SDKP receptors, including Bmax and Kd (Fig. 3). Saturation binding and Scatchard plot analyses revealed only a single class of receptor binding sites, since the best curve fit was with the one-site rather than the two-site model. Specificity of 125I-Hpp-Aca-SDKP binding was examined further with different Ac-SDKP analogs and other unrelated peptides. Again, Ac-SDKP and Hpp-Aca-SDKP potently displaced 125I-Hpp-Aca-SDKP binding, with a Ki of ~0.69 and 10.4 nM, respectively (Fig. 4). The Ki values of the tetrapeptide and its analog Hpp-Aca-SDK are close to the concentrations at which they exert their maximal inhibitory effects on ET-1-induced collagen synthesis (23, 26, 34). Interestingly, the tripeptide analog SDK also significantly blocked 125I-Hpp-Aca-SDKP binding, probably due to its similar chemical structure to Ac-SDKP (Fig. 4). In fact, Thierry et al. (28) showed that SDK was even more potent than Ac-SDKP in biological activity. We also found that SDK exhibited inhibitory effects on ET-1-stimulated collagen synthesis in CFs (Peng H, Carretero OA, and Rhaleb NE, unpublished observations). By contrast, the nonrelated peptides ET-1 and ANG II had no effect on 125I-labeled Hpp-Aca-SDKP binding in CFs. Thus we believe that our results confirm for the first time the presence of a single class of high-affinity and high-specificity Ac-SDKP receptor binding sites in rat CFs.
Although the use of 125I-Hpp-Aca-SDKP enabled us to characterize Ac-SDKP receptor binding sites in cultured rat CFs, it is important to determine whether there are specific Ac-SDKP receptor binding sites in the heart and localize them at the cellular level. We have shown that Ac-SDKP prevents and reverses cardiac fibrosis in rats with aldosterone (salt)- and ANG II-induced hypertension, as well as rats with myocardial infarction (22, 25, 27, 30). These antifibrotic effects of Ac-SDKP in vivo may be mediated via specific Ac-SDKP receptor binding sites, but such sites have not been reported in any tissues to our knowledge. Localization of Ac-SDKP receptors in tissues is important to study the cellular mechanisms by which this tetrapeptide exerts beneficial effects on cardiac tissue fibrosis or target organ damage (25, 27, 30). To achieve this goal, we employed quantitative in vitro autoradiography to localize 125I-labeled Hpp-Aca-SDKP receptor binding sites in the rat heart, which is a powerful tool to localize peptide receptors at the tissue and cellular levels. Indeed, we have used this technique to extensively study vasoactive peptide receptor distribution in the rat heart, blood vessels, and kidneys (31–36). As shown in Fig. 8, 125I-Hpp-Aca-SDKP receptor binding occurred at moderate levels throughout the rat heart, including the atria, septum, and ventricles, with maximal binding in the left ventricle and septum. The pattern of 125I-Hpp-Aca-SDKP binding in the rat heart indicates a homogeneous distribution, but this does not permit cellular localization. Further studies with emulsion light microscopic autoradiography revealed binding at the border of cardiomyocytes, illustrating a pattern consistent with the cardiac interstitium. The presence of specific 125I-Hpp-Aca-SDKP binding in the rat cardiac interstitium is therefore consistent with our findings with cultured rat CFs, further supporting our hypothesis that there are high-affinity and high-specificity binding sites for Ac-SDKP in CFs.
The significance of the finding that specific Ac-SDKP receptor binding sites are present in rat CFs remains to be determined. At present, we still do not know the precise size or structure of the receptor protein that binds Ac-SDKP, nor do we know which class of peptide receptor it belongs to. However, we did find that Ac-SDKP consistently inhibited ET-1-stimulated collagen synthesis and cell proliferation in rat CFs in vitro and prevented and reversed cardiac fibrosis induced by aldosterone (salt)- or ANG II-induced hypertension in vivo (22, 25–27, 30). There are two important mechanisms or signaling pathways that mediate the vasoconstrictor and growth effects of ET-1 (1, 9, 18). One is receptor-mediated mobilization of [Ca2+]i, and the other is activation of various MAPKs such as ERK1/2 or JNK (1, 13, 18, 29). Increased [Ca2+]i is also associated with MAPK activation in renal microvascular cells and mesangial cells (16, 17). In the present study, Ac-SDKP did not alter basal [Ca2+]i, but it significantly attenuated [Ca2+]i responses to ET-1, an effect comparable to thapsigargin, an inhibitor of SERCA ATPase (Fig. 5). This suggests that the effects of Ac-SDKP may involve modulation of Ca2+ release from intracellular calcium stores, although the precise mechanisms are not known. Indeed, we further found that ET-1-induced ERK1/2 phosphorylation, as expected (13, 29), and this effect of ET-1 was significantly attenuated by Ac-SDKP (Fig. 6). Thapsigargin, a potent inhibitor of SERCA ATPase, attenuated ET-1-induced ERK1/2 phosphorylation, whereas U-73122, an inhibitor of PLC, did not (Fig. 7). One plausible explanation for this finding would be that the inhibitory effects of Ac-SDKP on ET-1-increased [Ca2+]i and activation of MAPK ERK1/2 were probably mediated in part by intracellular mechanisms other than PLC-activated signaling. However, more work is required to study how Ac-SDKP binds and activates its receptor binding sites to inhibit cardiac fibroblast proliferation and collagen synthesis. Only when the receptor is cloned or specific receptor antagonists are developed will we be able to fully elucidate the cellular mechanisms (including signaling pathways) involved in the antifibrotic effects of Ac-SDKP in CFs in vitro, as well as the role of Ac-SDKP in cardiac remodeling during target organ damage due to hypertension and myocardial infarction. Nevertheless, our work on specific Ac-SDKP receptor binding sites in rat CFs will likely open up a new field in studying interactions between this novel peptide, ACE inhibition, and cardiac remodeling during hypertension and chronic heart failure.
Acknowledgments
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-28982 (to O. A. Carretero) and HL-71806 to N.-E. Rhaleb. J. L. Zhuo is supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant 5RO1 DK-067299, American Heart Association Greater Midwest Affiliate Grant 0355551Z, and the National Kidney Foundation of Michigan.
Footnotes
Portions of this work were presented at the 58th Annual Conference and Scientific Sessions of the Council for High Blood Pressure Research in association with the Council on the Kidney in Cardiovascular Disease in October 2004 and are published as an abstract (Hypertension 44: 498, 2004).
References
- 1.Ansari HR, Kaddour-Djebbar I, Abdel-Latif AA. Involvement of Ca2+ in endothelin-a-induced MAP kinase phosphorylation, myosin light chain phosphorylation and contraction in rabbit iris sphincter smooth muscle. Cell Signal. 2004;16:609–619. doi: 10.1016/j.cellsig.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Azizi M, Rousseau A, Ezan E, Guyene TT, Michelet S, Grognet JM, Lenfant M, Corvol P, Ménard J. Acute angiotensin-converting enzyme inhibition increases the plasma level of the natural stem cell regulator N-acetyl-seryl-aspartyl-lysyl-proline. J Clin Invest. 1996;97:839–844. doi: 10.1172/JCI118484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Azizi M, Ezan E, Nicolet L, Grognet JM, Menard J. High plasma level of N-acetyl-seryl-aspartyl-lysyl-proline: a new marker of chronic angiotensin-converting enzyme inhibition. Hypertension. 1997;30:1015–1019. doi: 10.1161/01.hyp.30.5.1015. [DOI] [PubMed] [Google Scholar]
- 4.Bonnet D, Lemoine FM, Pontvert-Delucq S, Baillou C, Najman A, Guigon M. Direct and reversible inhibitory effect of the tetrapeptide acetyl-N-Ser-Asp-Lys-Pro (Seraspenide) on the growth of human CD34+ subpopulations in response to growth factors. Blood. 1993;82:3307–3314. [PubMed] [Google Scholar]
- 5.Campbell SE, Katwa LC. Angiotensin II stimulated expression of transforming growth factor-β1 in cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol. 1997;29:1947–1958. doi: 10.1006/jmcc.1997.0435. [DOI] [PubMed] [Google Scholar]
- 6.Cavasin MA, Rhaleb NE, Yang XP, Carretero OA. Prolyl oligopeptidase is involved in release of the antifibrotic peptide Ac-SDKP. Hypertension. 2004;43:1140–1145. doi: 10.1161/01.HYP.0000126172.01673.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chisi JE, Wdzieczak-Bakala J, Riches AC. Inhibitory action of the peptide AcSDKP on the proliferative state of hematopoietic stem cells in the presence of captopril but not lisinopril. Stem Cells. 1997;15:455–460. doi: 10.1002/stem.150455. [DOI] [PubMed] [Google Scholar]
- 8.Comte L, Lorgeot V, Volkov L, Allegraud A, Aldigier JC, Praloran V. Effects of the angiotensin-converting enzyme inhibitor enalapril on blood haematopoietic progenitors and acetyl-N-Ser-Asp-Lys-Pro concentrations. Eur J Clin Invest. 1997;27:788–790. doi: 10.1046/j.1365-2362.1997.1980737.x. [DOI] [PubMed] [Google Scholar]
- 9.Davenport AP. International Union of Pharmacology. XXIX. Update on endothelin receptor nomenclature. Pharmacol Rev. 2002;54:219–226. doi: 10.1124/pr.54.2.219. [DOI] [PubMed] [Google Scholar]
- 10.Eghbali M, Tomek R, Woods C, Bhambi B. Cardiac fibroblasts are predisposed to convert into myocyte phenotype: specific effect of transforming growth factor β. Proc Natl Acad Sci USA. 1991;88:795–799. doi: 10.1073/pnas.88.3.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gaudron S, Grillon C, Thierry J, Riches A, Wierenga PK, Wdzieczak-Bakala J. In vitro effect of acetyl-N-Ser-Asp-Lys-Pro (AcSDKP) analogs resistant to angiotensin I-converting enzyme on hematopoietic stem cell and progenitor cell proliferation. Stem Cells. 1999;17:100–106. doi: 10.1002/stem.170100. [DOI] [PubMed] [Google Scholar]
- 12.Grillon C, Lenfant M, Wdzieczak-Bakala J. Optimization of cell culture conditions for the evaluation of the biological activities of the tetrapeptide N-acetyl-ser-asp-lys-pro, a natural hemoregulatory factor. Growth Factors. 1993;9:133–138. doi: 10.3109/08977199309010828. [DOI] [PubMed] [Google Scholar]
- 13.Iwasaki H, Eguchi S, Ueno H, Marumo F, Hirata Y. Endothelin-mediated vascular growth requires p42/p44 mitogen-activated protein kinase and p70 S6 kinase cascades via transactivation of epidermal growth factor receptor. Endocrinology. 1999;140:4659–4668. doi: 10.1210/endo.140.10.7023. [DOI] [PubMed] [Google Scholar]
- 14.Kanasaki K, Koya D, Sugimoto T, Isono M, Kashiwagi A, Haneda M. N-acetyl-seryl-aspartyl-lysyl-proline inhibits TGF-β-mediated plasminogen activator inhibitor-1 expression via inhibition of Smad pathway in human mesangial cells. J Am Soc Nephrol. 2003;14:863–872. doi: 10.1097/01.asn.0000057544.95569.ec. [DOI] [PubMed] [Google Scholar]
- 15.Lenfant M, Wdzieczak-Bakala J, Guittet E, Prome JC, Sotty D, Frindel E. Inhibitor of hematopoietic pluripotent stem cell proliferation: purification and determination of its structure. Proc Natl Acad Sci USA. 1989;86:779–782. doi: 10.1073/pnas.86.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li XC, Campbell DJ, Ohishi M, Yuan S, Zhuo JL. AT1 receptor-activated signaling mediates angiotensin IV-induced renal cortical vasoconstriction in rats. Am J Physiol Renal Physiol. 2006;290:F1024–F1033. doi: 10.1152/ajprenal.00221.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li XC, Carretero OA, Shao Y, Zhuo JL. Glucagon receptor-mediated extracellular signal-regulated kinase 1/2 phosphorylation in rat mesangial cells: role of protein kinase A and phospholipase C. Hypertension. 2006;47:580–585. doi: 10.1161/01.HYP.0000197946.81754.0a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Masaki T, Vane JR, Vanhoutte PM. International Union of Pharmacology Nomenclature of endothelin receptors. Pharmacol Rev. 1994;6:137–142. [PubMed] [Google Scholar]
- 19.Moriguchi Y, Matsubara H, Mori Y, Murasawa S, Masaki H, Maruyama K, Tsutsumi Y, Shibasaki Y, Tanaka Y, Nakajima T, Oda K, Iwasaka T. Angiotensin II-induced transactivation of epidermal growth factor receptor regulates fibronectin and transforming growth factor-β synthesis via transcriptional and posttranscriptional mechanisms. Circ Res. 1999;84:1073–1084. doi: 10.1161/01.res.84.9.1073. [DOI] [PubMed] [Google Scholar]
- 20.Neugebauer W, Gratton JP, Ihara M, Bkaily G, D’Orléans-Juste P. Solid phase synthesis of head-to-tail cyclic peptide, ETA antagonist. In: Ramage R, Epton R, editors. Peptides 1996; Proceedings of the Twenty-Fourth European Peptide Symposium; September 8–13, 1996; Edinburgh, Scotland. European Peptide Society; 1998. pp. 677–678. [Google Scholar]
- 21.Neugebauer W, Elliott P, Cuello AC, Escher E. Synthesis and immunological evaluation of N-terminal, noncrossreactive tachykinin antigens. J Med Chem. 1988;31:1907–1910. doi: 10.1021/jm00118a007. [DOI] [PubMed] [Google Scholar]
- 22.Peng H, Carretero OA, Raij L, Yang F, Kapke A, Rhaleb NE. Antifibrotic effects of N-acetyl-seryl-aspartyl-lysyl-proline on the heart and kidney in aldosterone-salt hypertensive rats. Hypertension. 2001;37:794–800. doi: 10.1161/01.hyp.37.2.794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pokharel S, Rasoul S, Roks AJM, van Leeuwen REW, van Luyn MJA, Deelman LE, Smits JF, Carretero O, van Gilst WH, Pinto YM. N-acetyl-Ser-Asp-Lys-Pro inhibits phosphorylation of Smad2 in cardiac fibroblasts. Hypertension. 2002;40:155–161. doi: 10.1161/01.hyp.0000025880.56816.fa. [DOI] [PubMed] [Google Scholar]
- 24.Pradelles P, Frobert Y, Créminon C, Liozon E, Massé A, Frindel E. Negative regulator of pluripotent hematopoietic stem cell proliferation in human white blood cells and plasma as analysed by enzyme immunoassay. Biochem Biophys Res Commun. 1990;170:986–993. doi: 10.1016/0006-291x(90)90489-a. [DOI] [PubMed] [Google Scholar]
- 25.Rasoul S, Carretero OA, Peng H, Cavasin MA, Zhuo J, Sanchez-Mendoza A, Brigstock DR, Rhaleb NE. Antifibrotic effect of Ac-SDKP and angiotensin-converting enzyme inhibition in hypertension. J Hypertens. 2004;22:593–603. doi: 10.1097/00004872-200403000-00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhaleb NE, Peng H, Harding P, Tayeh M, LaPointe MC, Carretero OA. Effect of N-acetyl-seryl-aspartyl-lysyl-proline on DNA and collagen synthesis in rat cardiac fibroblasts. Hypertension. 2001;37:827–832. doi: 10.1161/01.hyp.37.3.827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhaleb NE, Peng H, Yang XP, Liu YH, Mehta D, Ezan E, Carretero OA. Long-term effect of N-acetyl-seryl-aspartyl-lysyl-proline on left ventricular collagen deposition in rats with 2-kidney, 1-clip hypertension. Circulation. 2001;103:3136–3141. doi: 10.1161/01.cir.103.25.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thierry J, Papet MP, Saez-Servent Plissonneau-Haumont J, Potier P, Lenfant M. Synthesis and activity of NAcSerAspLysPro analogues on cellular interactions between T-cell and erythrocytes in Rosette formation. J Med Chem. 1990;33:2122–2127. doi: 10.1021/jm00170a012. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Simonson MS, Pouyssegur J, Dunn MJ. Endothelin rapidly stimulates mitogen-activated protein kinase activity in rat mesangial cells. Biochem J. 1992;287:589–594. doi: 10.1042/bj2870589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang F, Yang XP, Liu YH, Xu J, Cingolani O, Rhaleb NE, Carretero OA. Ac-SDKP reverses inflammation and fibrosis in rats with heart failure after myocardial infarction. Hypertension. 2004;43:229–236. doi: 10.1161/01.HYP.0000107777.91185.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhuo JL, Yamada H, Allen AM, Sun Y, Mendelsohn FAO. Localization and properties of angiotensin converting enzyme and angiotensin II receptors in the heart. In: Lindpaintner K, Ganten D, editors. The Cardiac Renin-Angiotensin System. New York: Futura Scientific; 1994. pp. 63–88. [Google Scholar]
- 32.Zhuo JL, Mendelsohn FAO, Ohishi M. Perindopril alters vascular angiotensin-converting enzyme, AT1 receptor, and nitric oxide synthase expression in patients with coronary heart disease. Hypertension. 2002;39:634–638. doi: 10.1161/hy0202.103417. [DOI] [PubMed] [Google Scholar]
- 33.Zhuo JL, MacGregor DP, Mendelsohn FAO. Presence of angiotensin II AT-2 receptors in the adventitia of human kidney vasculature. Clin Exp Pharmacol Physiol. 1996;23:S147–S153. [PubMed] [Google Scholar]
- 34.Zhuo JL, Carretero OA, Peng HM, Regoli D, Neugebauer W, Rhaleb NE. Effects of Ac-SDKP on collagen synthesis are mediated by specific Ac-SDKP receptor binding sites in rat cardiac fibroblasts (Abstract) Hypertension. 2004;44:498. [Google Scholar]
- 35.Zhuo JL, Allen AM, Alcorn D, MacGregor D, Aldred Mendelsohn FAOGP. The distribution of angiotensin II receptors. In: Laragh JH, Brenner BM, editors. Hypertension: Pathology, Diagnosis, and Management. 2. New York: Raven; 1995. pp. 1739–1762. [Google Scholar]
- 36.Zhuo JL, Dean R, Maric C, Aldred PG, Harris P, Alcorn D, Mendelsohn FA. Localization and interactions of vasoactive peptide receptors in renomedullary interstitial cells of the kidney. Kidney Int. 1998;67:S22–S28. doi: 10.1046/j.1523-1755.1998.06705.x. [DOI] [PubMed] [Google Scholar]