

Abstract
Purpose of review
Diabetic nephropathy is one of the most common complications in diabetes mellitus. Multiple pathogenic mechanisms are now believed to contribute to this disease, including inflammatory cytokines, autacoids and oxidative stress. Numerous studies have shown that the kallikrein–kinin system may be involved in these mechanisms. This review focuses on recent research advance on the potential role of the kallikrein–kinin system in the development of diabetic nephropathy, and its clinical relevance.
Recent findings
A collection of recent studies has shown that angiotensin-converting enzyme inhibitors, which inhibit angiotensin II formation and degradation of bradykinin, and vasopeptidase inhibitors attenuated the development of diabetic nephropathy in experimental animals and clinical settings. The role of the kallikrein–kinin system in diabetes is further supported by findings that diabetic nephropathy is worsened in diabetic mice lacking bradykinin B2 receptors. Although long-acting bradykinin B2 receptor agonists have been shown to have renal protective effects, their therapeutic benefits have not been well studied.
Summary
Current experimental investigations demonstrated that pharmacological intervention of the kallikrein–kinin system improved renal conditions in diabetes mellitus. These findings suggest that the kallikrein–kinin system may be a therapeutic target in preventing and treating diabetic nephropathy.
Keywords: bradykinin, diabetic nephropathy, kallikrein-kinin system
Introduction
The kallikrein–kinin system (KKS) is a complex multi-enzyme system that can be divided into a circulating and a tissue/renal KKS. The circulating KKS belongs to the coagulation system, whereas the renal/tissue KKS acts in a paracrine or autocrine fashion involving the local synthesis and release of kinins, such as bradykinin and kallidin, mainly from kininogens by the kininogenase, kallikrein (KLK) [1]. Kinins exert their biological effects by selective stimulation of two distinct G protein-coupled receptors termed bradykinin B1-receptor (B1R) and B2-receptor (B2R) [2]. Both receptors are primarily linked to phospholipase C activation, which induces intracellular calcium mobilization through inositol 1,4,5-triphosphate and additional intracellular effects [3]. Although B1R and B2R activate similar transduction pathways, both receptors differ with regard to their translational and transcriptional regulation [4]. The B1R is expressed at low levels in different tissues. Its expression is induced in response to inflammatory stimuli such as lipopolysaccharides, endotoxins, and cytokines (IL-1β and TNF-α) [5–8]. In diabetes mellitus, activation of both B1R and B2R signaling has been observed [7,9,10]. Oxidative stress, pro-inflammatory cytokines [7], such as IL-1β or tumor necrosis factor (TNF)-α, as well as stimuli from other vasoactive peptides, such as the renin–angiotensin system (RAS) [11–14], are implicated in the regulation of both receptors. Kinins are rapidly inactivated by several peptidases, the kininases, including carboxypeptidases N and M, angiotensin-converting enzyme (ACE), aminopeptidase P and neutral endopeptidase 24–11 (NEP) in the circulation and at tissue sites [15].
All components of the KKS are also expressed in the kidney and are important regulators for renal hemodynamic and tubular function [16,17]. The kidney produces local kinins, like bradykinin, at much higher levels than those present in blood [16]. Major effects of activation of the renal KKS include diuresis and natriuresis. Hence, it is believed to play a pivotal role in the regulation of fluid and electrolyte balance, mostly through its renal actions [18]. Accumulating evidence supports the emerging role for oxidative stress and inflammatory cytokines in the development of diabetic complications, including diabetic nephropathy. This review focuses on the role of the KKS in the development of diabetic nephropathy and underlying mechanisms involved.
The implication of the kallikrein–kinin system in diabetic nephropathy
It has been shown that renal KLK production can be reduced in patients with mild renal disease and more markedly in patients with severe renal failure [19,20]. Moreover, KLK reversed salt-induced renal fibrosis and glomerular hypertrophy, and reduced recruitment and accumulation of inflammatory cells in the tubulo-interstitium and vasculature of hypertensive rats [21]. Furthermore, kinins restored renal nitric oxide production and significantly inhibited salt-induced NADH oxidase activity, superoxide formation and expression of leucocyte adhesion molecules. These results indicate a novel role of the KKS in the protection against salt-induced and hypertension-induced renal injury by inhibiting oxidative stress and inflammatory responses.
There is increasing evidence that supports the relevance of the KKS and renal injury under diabetic conditions [22]. Streptozotocin (STZ)-induced diabetic rats with moderate hyperglycemia show increased renal and urinary excretion of active KLK kinins, in conjunction with reduced renal vascular resistance and increased glomerular filtration rate (GFR) and renal plasma flow (RPF) [23]. Acute treatment of these hyperfiltrating diabetic rats with aprotinin (a kallikrein inhibitor) or with a B2R-receptor antagonist, nearly normalized GFR and RPF [23,24••]. By contrast, in severe STZ-induced noninsulin-treated hypofiltrating rats, KLK activity and expression have been found to be downregulated, indicating a direct correlation of the activity of KLK and the degree of GFR and RPF [25]. In addition to their hemodynamic actions, kinins may have important anti-inflammatory and antiproliferative properties under diabetic conditions. In this respect, the renal KKS may belong to an important system that can provide important renal protection. In particular, bradykinin has been shown to have nitric oxide-releasing properties, induce endothelium-dependent vasodilation, and exert antifibrotic and antihypertrophic actions, as well as stimulating glucose uptake [2]. The last effect has been suggested to be beneficial under diabetic conditions [2].
Increased glomerular basement membrane thickness, diffused mesangial sclerosis, and hyaline arteriosclerosis are hallmarks of diabetes mellitus-induced glomerulosclerosis [26]. Furthermore, tubular and interstitial lesions are also present in diabetic nephropathy [27,28]. In a review article regarding the mechanisms of diabetic complications in the kidney, Brownlee et al. [27] showed that high glucose concentrations (hyperglycemia) first induce proliferation of mesangial cells, followed by the development of hypertrophy, which eventually progresses to glomerulosclerosis. In this regard, remaining renal cells may play a pivotal role in regulating renal structure and function under diabetic conditions. These cells can be stimulated directly or indirectly by hyperglycemia leading to release of a variety of pro-inflammatory cytokines and/or growth factors. Further, stimulation of these resident cells can stimulate mesangial cell growth and promote ECM production in an autocrine or paracrine manner [28,29]. The transforming growth factor (TGF)-β serves as a key mediator for mesangial cell expansion and matrix deposition [30]. Recent evidence indicates that the profibrotic signals initiated by TGF-β are mediated via activation of connective tissue growth factor (CTGF) [31]. While the beneficial influence of the KKS on mesangial cells is well documented under basal conditions, the manner of regulation by KKS remains controversial. Bradykinin can induce proliferation of quiescent renal cells via B2R activation [32–34]. In contrast, under proliferating conditions, bradykinin is antiproliferative in a variety of different cell types, including mesangial cells [35]. In addition, Alric et al. [35] demonstrated that bradykinin reduced mesangial cell proliferation under diabetic conditions. A possible association between the KKS and diabetes-induced mesangial cell proliferation may involve key regulators in diabetes such as insulin-like growth factor-I (IGF-I) and, to a lesser extent, insulin. Both IGF-1 and insulin induce mesangial cell proliferation and collagen secretion, most likely via activation of the IGF-I receptor. Insulin thus may be involved in the early stages of diabetic nephropathy [25]. IGF-I is a growth factor synthesized by many cells and tissues including mesangial cells. An increase in renal IGF-I levels has been implicated during the development of diabetic glomerulosclerosis by promoting cell proliferation and collagen secretion [36]. It has been shown that the effect of bradykinin on cell proliferation may be also dependent upon growth factor receptor activation [35]. Intracellular mitogen-activated kinases like ERK 1/2 could be identified as a relevant mediator for IGF-1 actions in mesangial cells [35]. Bradykinin is known to be able to reduce IGF-I-induced ERK 1/2 activation in mesangial cells. Finally, it has been shown that bradykinin can reduce mesangial cell proliferation induced by IGF-I, supporting a crosstalk between the renal KKS and IGF-1 [35], indicating important antiproliferative effects of the renal KKS. Thus, these conflicting findings of both proliferative and antiproliferative effects of bradykinin suggest that differences in experimental settings may play an important role in the regulation of the renal KKS under diabetic conditions [24••,37].
Although blockade of the B2R in STZ-induced diabetic rats by the B2R antagonist icatibant (HOE 140) did not greatly improve diabetic nephropathy [38], recent studies in mice lacking the B2R implicated the role of the KKS in the development of diabetic nephropathy [39]. In diabetic mice lacking the B2R, the condition of the diabetic nephropathy worsens, which was associated with a further increase in glomerular mesangial sclerosis. Although these mice also showed a further rise in renal B1R expression, it is obvious that this increase did not fully compensate for the loss of B2R in this model. The role of the renal B2R under diabetic conditions is further supported by the finding that a loss of the B2R also leads to an induction of an aging of renal tubule cells and to an increase of oxidative stress in diabetic mice [39]. These data therefore suggest that the B2R may be a potential therapeutic target in diabetic nephropathy.
Interactions between inhibition of the renin–angiotensin system and activation of the renal kallikrein–kinin system
The investigation of the therapeutic actions of angiotensin-converting enzyme inhibitors (ACEI) and angiotensin type 1 (AT1) receptor antagonists revealed complex interactions between the RAS and KKS [11]. Such interactions are supported by the following findings. ACE efficiently degrades kinins or bradykinin. Angiotensin fragments such as ANG-(1–7) exert kinin-like effects. KLK probably serves as a prorenin-activating enzyme. Several studies [40–42] have demonstrated experimentally that the protective effects of ACEI are at least partly mediated by a direct potentiation of kinin receptor response to bradykinin stimulation. We [38] previously showed that kinins are partly involved in the antiproteinuric action of ACEI in experimental diabetic nephropathy. Furthermore, studies on AT1 antagonists, which do not directly influence kinin degradation, and studies on angiotensin-receptor transgenic mice, have revealed additional interactions between the RAS and the KKS. There is mounting evidence to suggest that an autocrine cascade including kinins, nitric oxide, prostaglandins, and cyclic GMP is involved in at least some of the angiotensin type 2 receptor effects [43]. In addition, Siragy et al. [44] showed that Ang II tonically stimulates renal kinin peptide production due to AT2 receptor stimulation. Although the mechanisms have not been well understood, the interaction of the KKS and RAS is further supported by our findings, showing that the renal expression of ACE in transgenic rats expressing the human KLK1 gene is reduced, whereas it is increased in kidneys of kininogen-deficient rats (Fig. 1) [45]. The clinical relevance of the newly elucidated mechanisms of ACE inhibitor inducing a cross-talk between ACE and B2 receptors, the role of ANG-(1–7)-dependent bradykinin-accumulation and the importance of AT2 receptor-dependent KKS stimulation during AT1 antagonism still remain to be determined, however.
Figure 1. Basal renal ACE binding of Sprague Dawley control rats, transgenic rats harbouring the human kallikrein gene [TGR(KLK1)], known to be characterized by increased kinin levels, and kininogen-deficient Brown Norway (BN-K) rats.
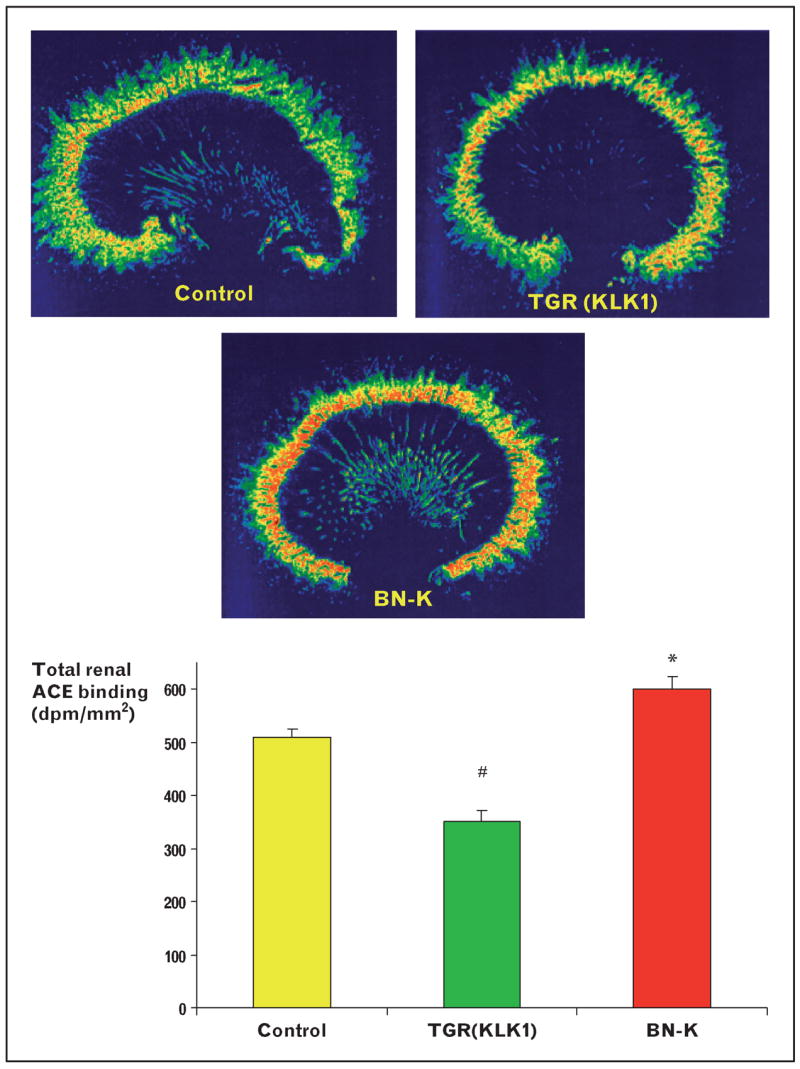
Total renal ACE was mapped by 125I-labeled 351A, a tyrosyl derivative of the ACE inhibitor lisinopril (Merck). The binding properties of this radioligand and its validity for quantitative in-vitro autoradiography of tissue ACE have been established [12]. ACE binding was reduced in TGR(KLK1) compared with controls and BN-K rats. TGR(KLK1) rats are characterized by increased basal bradykinin levels [45]. Kininogen deficiency led to a significant increase in renal ACE binding, suggesting an interaction between the renal kallikrein–kinin system and the renin–angiotensin system. #P < 0.05 compared with control and BN-K; *P < 0.05 compared with controls and TGR(KLK1), n = 6 per group. Experiments were performed by C.T. and J.L.Z. at the Howard Florey Institute, Australia.
Targeting the renal kallikrein–kinin system by vasopeptidase inhibition
A vasopeptidase inhibitor (AVE7688) has been shown to prevent the development of nephropathy or slow its progress in Zucker diabetic rats [46]. Vasopeptidase inhibition markedly reduced both glomerular and tubulo-interstitial damage, and this protective effect was partially prevented by combination with the B2R antagonist, icatibant. The vasopeptidase inhibitor omapatrilat has been tested clinically in hypertension and heart failure but not in diabetes. As the clinical safety of omapatrilat has been established, and animal studies have shown that this inhibitor may be superior to ACEI as a nephroprotective agent, it would be very interesting to test its clinical relevance in human kidney diseases with and without diabetes.
Targeting the renal kallikrein–kinin system by bradykinin receptor agonists
There are no selective, nonpeptide bradykinin receptor agonists currently available. Peptide bradykinin mimetics that are longer-lasting than bradykinin have been developed, including Sar-[D-Phe8]des-Arg9-bradykinin, which is resistant to ACE, and labradimil, which is resistant to aminopeptidases and neutral endopeptidases [47]. These agents and the selective B2R antagonists (FR 173657 and LF 16.0687) may be useful in further determining the role of bradykinin and B2R in nephroprotection. Selective nonpeptide B2 receptor agonists would be useful in determining the role of this receptor in nephroprotection, and may have therapeutic potential in nephropathology. Stimulation of B2R is a target in treating diabetic nephropathy. Presently, in the absence of selective nonpeptide B2 receptor agonists, the best way to do this is probably with vasopeptidase inhibitors that will increase the levels of bradykinin [48]. Vasopeptidase inhibitors also reduce the levels of Ang II, which is also known to be nephroprotective. ACEI and AT1 antagonists, however, known to interact with the activation of the renal KKS, remain the gold standard in the treatment of diabetic nephropathy clinically.
Conclusions
Despite intensified metabolic control and antihypertensive treatment of diabetic patients, the development of diabetic nephropathy continues to be a serious clinical problem. Growing evidence suggests that diabetic kidney diseases are multifactorial diseases and therefore require different therapeutic approaches. Recent investigations suggest that the KKS plays an important role in the development and progression of diabetic nephropathy. The precise mechanisms by which the KKS is involved in the development of diabetic nephropathy remain to be further investigated. Future studies using novel therapeutic strategies will likely play an important role in demonstrating the significance of the KKS as a target in preventing and treating diabetic nephropathy.
Acknowledgments
This work was supported by grants from the Deutsche Forschungsge-sellschaft (DFG; GRK 865 and 64-1/2) to C.T. Part of this work was carried out at the Howard Florey Institute of Experimental Physiology and Medicine, University of Melbourne, in collaboration with J.L.Z. J.L.Z. is supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant (RO1DK067299) and American Heart Association Greater Midwest Affiliate Grant-in-Aid (0355551Z).
Abbreviations
- ACE
angiotensin-converting enzyme
- ACEI
angiotensin-converting enzyme inhibitor
- AT1
angiotensin type 1 receptor
- GFR
glomerular filtration rate
- IGF-I
insulin-like growth factor-I
- KKS
kallikrein–kinin system
- KLK
kallikrein
- RAS
renin–angiotensin system
- RPF
renal plasma flow
- STZ
streptozotocin
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
• of special interest
•• of outstanding interest Additional references related to this topic can also be found in the Current World Literature section in this issue (p. 52).
- 1.Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- 2.Spillmann F, Van Linthout S, Schultheiss HP, Tschope C. Cardioprotective mechanisms of the kallikrein–kinin system in diabetic cardiopathy. Curr Opin Nephrol Hypertens. 2006;15:22–29. doi: 10.1097/01.mnh.0000199009.56799.2b. [DOI] [PubMed] [Google Scholar]
- 3.Marceau F, Regoli D. Bradykinin receptor ligands: therapeutic perspectives. Nat Rev Drug Discov. 2004;3:845–852. doi: 10.1038/nrd1522. [DOI] [PubMed] [Google Scholar]
- 4.De Vriese AS, Verbeuren TJ, Van de Voorde J, et al. Endothelial dysfunction in diabetes. Br J Pharmacol. 2000;130:963–974. doi: 10.1038/sj.bjp.0703393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Couture R, Harrisson M, Vianna RM, Cloutier F. Kinin receptors in pain and inflammation. Eur J Pharmacol. 2001;429:161–176. doi: 10.1016/s0014-2999(01)01318-8. [DOI] [PubMed] [Google Scholar]
- 6.Mombouli JV, Vanhoutte PM. Kinins and endothelial control of vascular smooth muscle. Annu Rev Pharmacol Toxicol. 1995;35:679–705. doi: 10.1146/annurev.pa.35.040195.003335. [DOI] [PubMed] [Google Scholar]
- 7.Phagoo SB, Poole S, Leeb-Lundberg LM. Autoregulation of bradykinin receptors: agonists in the presence of interleukin-1beta shift the repertoire of receptor subtypes from B2 to B1 in human lung fibroblasts. Mol Pharmacol. 1999;56:325–333. doi: 10.1124/mol.56.2.325. [DOI] [PubMed] [Google Scholar]
- 8.Zhou X, Prado GN, Taylor L, et al. Regulation of inducible bradykinin B1 receptor gene expression through absence of internalization and resensitization. J Cell Biochem. 2000;78:351–362. doi: 10.1002/1097-4644(20000901)78:3<351::aid-jcb1>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 9.Duka I, Kintsurashvili E, Gavras I, et al. Vasoactive potential of the b(1) bradykinin receptor in normotension and hypertension. Circ Res. 2001;88:275–281. doi: 10.1161/01.res.88.3.275. [DOI] [PubMed] [Google Scholar]
- 10.Ignjatovic T, Tan F, Brovkovych V, et al. Activation of bradykinin B1 receptor by ACE inhibitors. Int Immunopharmacol. 2002;2:1787–1793. doi: 10.1016/s1567-5769(02)00146-7. [DOI] [PubMed] [Google Scholar]
- 11.Tschope C, Schultheiss HP, Walther T. Multiple interactions between the renin-angiotensin and the kallikrein-kinin systems: role of ACE inhibition and AT1 receptor blockade. J Cardiovasc Pharmacol. 2002;39:478–487. doi: 10.1097/00005344-200204000-00003. [DOI] [PubMed] [Google Scholar]
- 12.Dean R, Murone C, Lew RA, et al. Localization of bradykinin B2 binding sites in rat kidney following chronic ACE inhibitor treatment. Kidney Int. 1997;52:1261–1270. doi: 10.1038/ki.1997.451. [DOI] [PubMed] [Google Scholar]
- 13.Marin-Castano ME, Schanstra JP, Neau E, et al. Induction of functional bradykinin b(1)-receptors in normotensive rats and mice under chronic angiotensin-converting enzyme inhibitor treatment. Circulation. 2002;105:627–632. doi: 10.1161/hc0502.102965. [DOI] [PubMed] [Google Scholar]
- 14.Sabourin T, Morissette G, Bouthillier J, et al. Expression of kinin B(1) receptor in fresh or cultured rabbit aortic smooth muscle: role of NF-kappa B. Am J Physiol Heart Circ Physiol. 2002;283:H227–H237. doi: 10.1152/ajpheart.00978.2001. [DOI] [PubMed] [Google Scholar]
- 15.Cyr M, Lepage Y, Blais C, Jr, et al. Bradykinin and des-Arg(9)-bradykinin metabolic pathways and kinetics of activation of human plasma. Am J Physiol Heart Circ Physiol. 2001;281:H275–H283. doi: 10.1152/ajpheart.2001.281.1.H275. [DOI] [PubMed] [Google Scholar]
- 16.Campbell DJ. Towards understanding the kallikrein-kinin system: insights from measurement of kinin peptides. Braz J Med Biol Res. 2000;33:665–677. doi: 10.1590/s0100-879x2000000600008. [DOI] [PubMed] [Google Scholar]
- 17.Carretero OA, Scicli AG. The renal kallikrein-kinin system. Am J Physiol. 1980;238:F247–F255. doi: 10.1152/ajprenal.1980.238.4.F247. [DOI] [PubMed] [Google Scholar]
- 18.Katori M, Majima M. Pivotal role of renal kallikrein-kinin system in the development of hypertension and approaches to new drugs based on this relationship. Jpn J Pharmacol. 1996;70:95–128. doi: 10.1254/jjp.70.95. [DOI] [PubMed] [Google Scholar]
- 19.Price RG. Urinary enzymes, nephrotoxicity and renal disease. Toxicology. 1982;23:99–134. doi: 10.1016/0300-483x(82)90092-0. [DOI] [PubMed] [Google Scholar]
- 20.Naicker S, Naidoo S, Ramsaroop R, et al. Tissue kallikrein and kinins in renal disease. Immunopharmacology. 1999;44:183–192. doi: 10.1016/s0162-3109(99)00089-2. [DOI] [PubMed] [Google Scholar]
- 21.Zhang JC, Claffey K, Sakthivel R, et al. Two-chain high molecular weight kininogen induces endothelial cell apoptosis and inhibits angiogenesis: partial activity within domain 5. FASEB J. 2000;14:2589–2600. doi: 10.1096/fj.99-1025com. [DOI] [PubMed] [Google Scholar]
- 22.Harvey JN, Edmundson AW, Jaffa AA, et al. Renal excretion of kallikrein and eicosanoids in patients with type 1 (insulin-dependent) diabetes mellitus: relationship to glomerular and tubular function. Diabetologia. 1992;35:857–862. doi: 10.1007/BF00399932. [DOI] [PubMed] [Google Scholar]
- 23.Harvey JN, Jaffa AA, Margolius HS, Mayfield RK. Renal kallikrein and hemodynamic abnormalities of diabetic kidney. Diabetes. 1990;39:299–304. doi: 10.2337/diab.39.3.299. [DOI] [PubMed] [Google Scholar]
- 24••.Tan Y, Wang B, Keum JS, Jaffa AA. Mechanisms through which bradykinin promotes glomerular injury in diabetes. Am J Physiol Renal Physiol. 2005;288:F483–F492. doi: 10.1152/ajprenal.00165.2004. Latest review describing interactions between bradykinin and glomerula injury. [DOI] [PubMed] [Google Scholar]
- 25.Tschope C, Reinecke A, Seidl U, et al. Functional, biochemical, and molecular investigations of renal kallikrein-kinin system in diabetic rats. Am J Physiol. 1999;277(6 Pt 2):H2333–H2340. doi: 10.1152/ajpheart.1999.277.6.H2333. [DOI] [PubMed] [Google Scholar]
- 26.Mauer SM, Steffes MW, Brown DM. The kidney in diabetes. Am J Med. 1981;70:603–612. doi: 10.1016/0002-9343(81)90582-9. [DOI] [PubMed] [Google Scholar]
- 27.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 28.Derubertis FR, Craven PA. Activation of protein kinase C in glomerular cells in diabetes: mechanisms and potential links to the pathogenesis of diabetic glomerulopathy. Diabetes. 1994;43:1–8. doi: 10.2337/diab.43.1.1. [DOI] [PubMed] [Google Scholar]
- 29.Tsutsumi Y, Matsubara H, Masaki H, et al. Angiotensin II type 2 receptor overexpression activates the vascular kinin system and causes vasodilation. J Clin Invest. 1999;104:925–935. doi: 10.1172/JCI7886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma K, Ziyadeh FN. The emerging role of transforming growth factor-beta in kidney diseases. Am J Physiol. 1994;266(6 Pt 2):F829–F842. doi: 10.1152/ajprenal.1994.266.6.F829. [DOI] [PubMed] [Google Scholar]
- 31.Duncan MR, Frazier KS, Abramson S, et al. Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: down-regulation by camp. FASEB J. 1999;13:1774–1786. [PubMed] [Google Scholar]
- 32.Goldstein RH, Wall M. Activation of protein formation and cell division by bradykinin and des-Arg9-bradykinin. J Biol Chem. 1984;259:9263–9268. [PubMed] [Google Scholar]
- 33.Owen NE, Villereal ML. Lys-bradykinin stimulates Na+ influx and DNA synthesis in cultured human fibroblasts. Cell. 1983;32:979–985. doi: 10.1016/0092-8674(83)90082-x. [DOI] [PubMed] [Google Scholar]
- 34.Godin C, Smith AD, Riley PA. Bradykinin stimulates DNA synthesis in competent Balb/c 3T3 cells and enhances inositol phosphate formation induced by platelet-derived growth factor. Biochem Pharmacol. 1991;42:117–122. doi: 10.1016/0006-2952(91)90689-3. [DOI] [PubMed] [Google Scholar]
- 35.Alric C, Pecher C, Cellier E, et al. Inhibition of IGF-I-induced Erk 1 and 2 activation and mitogenesis in mesangial cells by bradykinin. Kidney Int. 2002;62:412–421. doi: 10.1046/j.1523-1755.2002.00475.x. [DOI] [PubMed] [Google Scholar]
- 36.Schreiber BD, Hughes ML, Groggel GC. Insulin-like growth factor-1 stimulates production of mesangial cell matrix components. Clin Nephrol. 1995;43:368–374. [PubMed] [Google Scholar]
- 37.El-Dahr SS, Dipp S, Baricos WH. Bradykinin stimulates the ERKright-arrowElk-1right-arrowFos/AP-1 pathway in mesangial cells. Am J Physiol Renal Physiol. 1998;275:F343–F352. doi: 10.1152/ajprenal.1998.275.3.F343. [DOI] [PubMed] [Google Scholar]
- 38.Tschope C, Seidl U, Reinecke A, et al. Kinins are involved in the antiproteinuric effect of angiotensin-converting enzyme inhibition in experimental diabetic nephropathy. Int Immunopharmacol. 2003;3:335–344. doi: 10.1016/S1567-5769(02)00273-4. [DOI] [PubMed] [Google Scholar]
- 39.Kakoki M, Kizer CM, Yi X, et al. Senescence-associated phenotypes in Akita diabetic mice are enhanced by absence of bradykinin B2 receptors. J Clin Invest. 2006;116:1302–1309. doi: 10.1172/JCI26958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Auch-Schwelk W, Kuchenbuch C, Claus M, et al. Local regulation of vascular tone by bradykinin and angiotensin converting enzyme inhibitors. Eur Heart J. 1993;14(Suppl I):154–160. [PubMed] [Google Scholar]
- 41.Hecker M, Blaukat A, Bara AT, et al. ACE inhibitor potentiation of bradykinin-induced venoconstriction. Br J Pharmacol. 1997;121:1475–1481. doi: 10.1038/sj.bjp.0701281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hecker M, Porsti I, Bara AT, Busse R. Potentiation by ACE inhibitors of the dilator response to bradykinin in the coronary microcirculation: interaction at the receptor level. Br J Pharmacol. 1994;111:238–244. doi: 10.1111/j.1476-5381.1994.tb14050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Katada J, Majima M. AT(2) receptor-dependent vasodilation is mediated by activation of vascular kinin generation under flow conditions. Br J Pharmacol. 2002;136:484–491. doi: 10.1038/sj.bjp.0704731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siragy HM, Jaffa AA, Margolius HS, Carey RM. Renin-angiotensin system modulates renal bradykinin production. Am J Physiol. 1996;271(4 Pt 2):R1090–R1095. doi: 10.1152/ajpregu.1996.271.4.R1090. [DOI] [PubMed] [Google Scholar]
- 45.Koch M, Wendorf M, Dendorfer A, et al. Cardiac kinin level in experimental diabetes mellitus: role of kininases. Am J Physiol Heart Circ Physiol. 2003;285:H418–H423. doi: 10.1152/ajpheart.00677.2002. [DOI] [PubMed] [Google Scholar]
- 46.Schafer S, Linz W, Bube A, et al. Vasopeptidase inhibition prevents nephropathy in Zucker diabetic fatty rats. Cardiovasc Res. 2003;60:447–454. doi: 10.1016/s0008-6363(03)00544-3. [DOI] [PubMed] [Google Scholar]
- 47.Emerich DF, Dean RL, Osborn C, Bartus RT. The development of the bradykinin agonist labradimil as a means to increase the permeability of the blood-brain barrier: from concept to clinical evaluation. Clin Pharmacokinet. 2001;40:105–123. doi: 10.2165/00003088-200140020-00003. [DOI] [PubMed] [Google Scholar]
- 48.Doggrell SA. Bradykinin B2 receptors: a target in diabetic nephropathy. Expert Opin Ther Targets. 2005;9:411–414. doi: 10.1517/14728222.9.2.411. [DOI] [PubMed] [Google Scholar]