

Abstract
Angiotensin IV (ANG IV), an active ANG II fragment, has been shown to induce systemic and renal cortical effects by binding to ANG IV (AT4) receptors and activating unique signaling transductions unrelated to classical type 1 (AT1) or type 2 (AT2) receptors. We tested whether ANG IV exerts systemic and renal cortical effects on blood pressure, renal microvascular smooth muscle cells (VSMCs), and glomerular mesangial cells (MC) and, if so, whether AT1 receptor-activated signaling is involved. In anesthetized rats, systemic infusion of ANG II, ANG III, or ANG IV (0.01, 0.1, and 1.0 nmol·kg−1 ·min−1 iv) caused dose-dependent increases in mean arterial pressure (MAP) and decreases in renal cortical blood flow (CBF; P < 0.01). ANG II also induced dose-dependent reductions in renal medullary blood flow (P < 0.01), whereas ANG IV did not. ANG IV-induced pressor and renal cortical vasoconstriction were completely abolished by AT1 receptor blockade with losartan (5 mg/kg iv; P < 0.05). When ANG IV (1 nmol·kg−1 ·min−1) was infused directly in the renal artery, CBF was reduced by > 30%, and the response was also blocked by losartan (P < 0.01). In the renal cortex, unlabeled ANG IV displaced 125I-labeled [Sar1,Ile8]ANG II binding, whereas unlabeled ANG II (10 μM) inhibited 125I-labeled Nle1-ANG IV (AT4) binding in a concentration-dependent manner (P < 0.01). In freshly isolated renal VSMCs, ANG IV (100 nM) increased intracellular Ca2+ concentration, and the effect was blocked by losartan and U-73122, a selective inhibitor of phospholipase C/inositol trisphosphate/Ca2+ signaling (1 μM). In cultured rat MCs, ANG IV (10 nM) induced mitogen-activated protein kinase extracellular/signal-regulated kinase 1/2 phosphorylation via AT1 receptor- and phospholipase C-activated signaling. These results suggest that, at nanomolar concentrations, ANG IV can increase MAP and induce renal cortical effects by interacting with AT1 receptor-activated signaling.
Keywords: angiotensin II, angiotensin IV, angiotensin type 1 receptor, angiotensin type 4 receptor, renal cortical blood flow, mitogen-activated protein kinases
High-affinity receptor binding sites and the physiological role of bioactive angiotensin fragments are gaining increasing attention after molecular cloning of type 1 (AT1) and type 2 (AT2) receptors for the octapeptide ANG II. It is now established that AT1 receptors mediate most (if not all) classic effects of ANG II, including potent vasoconstriction, aldosterone synthesis, cell growth, and body fluid and electrolyte homeostasis, whereas AT2 receptors oppose most (if not all) AT1 receptor-mediated effects in cardiovascular and renal cells (4, 10, 16, 39, 40). By contrast, neither receptor pharmacology nor the physiological or pathological role of other ANG II fragments is fully understood, with the possible exception of ANG III (des-Asp1-ANG II), which also activates AT1 receptors in most tissues or cells (3, 5, 12, 13, 16). ANG IV, which is formed by removing the first NH2-terminal amino acid (Arg2) from ANG III with aminopeptidase N and/or aminopeptidase B (3, 12–14, 16), was initially thought to be biologically inert but has recently been shown to have various effects in different tissues or cells by binding to high-affinity angiotensin type 4 receptors (AT4) or insulin-regulated aminopeptidase (IRAP; see Refs. 2, 13, 17, 23, 35, 37).
It is unclear whether ANG IV acts as an exclusive agonist for the putative AT4 receptor alone or as a partial but active agonist for the AT1 receptor, mediating its widely reported cardiovascular and renal effects (1, 17, 28–32, 34). ANG IV has been reported to cause both vasodilatation and vasoconstriction (1, 15, 18, 19, 35). For example, infusion of ANG IV directly in cerebral or renal arteries increases cerebral blood flow and renal cortical blood flow (CBF) via a mechanism that appears to be mediated by the AT4 receptor and nitric oxide (NO; see Refs. 15, 22, 37). In contrast, systemic or intrarenal arterial administration of ANG IV reportedly caused systemic and renal vasoconstriction that was completely prevented by pretreatment with losartan, suggesting an AT1 receptor-mediated response (11, 18, 19). However, it is not clear whether the reported different responses to ANG IV are only secondary to a systemic effect or the result of a direct intrarenal action, because previous studies dealt primarily with larger regional arterial or whole kidney blood flow responses to ANG IV and none of them has directly compared systemic and intrarenal effects of ANG IV.
To resolve the differences between renal cortical vasoconstrictor and vasodilator effects of ANG IV, it is important to study whether classical AT1 receptor-activated signaling pathways are involved at the cellular levels. The present study was therefore performed to determine 1) concentration-dependent systemic arterial pressure and renal CBF responses to systemic infusion of ANG IV, ANG III, and ANG II; 2) whether AT1 receptors are involved in systemic ANG IV-induced responses; 3) whether direct intrarenal arterial infusion of ANG IV induces renal cortical vasoconstriction by activating AT1 receptors; 4) whether ANG IV competes for AT1 receptor binding in the rat kidney; and 5) whether ANG IV activates classical AT1 receptor-mediated signaling in two well-described ANG II-targeted renal cells [microvascular smooth muscle cells (VSMCs) and mesangial cells (MCs)]. Our results support the view that, at subnanomolar to nanomolar concentrations, ANG IV is an active AT1 receptor agonist in both systemic and intrarenal microvasculature and glomerular MCs.
MATERIALS AND METHODS
Effects of Systemic and Intrarenal Arterial Infusion of ANG IV on Mean Arterial Pressure and Renal Cortical Blood Perfusion
Animals and surgical preparation
Twenty-four adult male Sprague-Dawley (SD) rats (250 g; Charles River Laboratories) were used for in vivo studies of systemic blood pressure [mean arterial pressure (MAP)] and renal cortical (CBF) and medullary blood flow (MBF) responses to systemic ANG IV, ANG III, or ANG II. Eight additional rats were used for studying the effects of intrarenal arterial infusion of ANG IV on renal CBF and MAP. Animals were maintained on a normal rat diet and allowed free access to tap water. On the day of the experiment, rats were anesthetized with inactin (100 mg/kg ip) and prepared as described previously (39, 40). Briefly, the right jugular vein and right carotid artery were cannulated with catheters (SP-50) for systemic infusion of saline and drugs and measurement of intra-arterial blood pressure, respectively. To maintain intrarenal arterial infusion of ANG IV, a catheter (SP-10) was inserted in the lower abdominal aorta and slowly advanced in the left renal artery. The left kidney was exposed through a flank incision and placed in a micropuncture cup to prevent movement. Laser-Doppler probes were inserted in the cortex (~2 mm deep) and the inner stripe of the outer medulla (~5 mm deep) to monitor CBF and MBF responses using a two-channel laser-Doppler flowmeter (floLAB; Moor Instruments; see Ref. 39). MAP, renal CBF, and MBF were recorded continuously using an eight-channel PowerLab data acquisition system interfaced with a Microsoft Work Station 4.0 (ADInstruments, Sydney, Australia). Hemodynamic variables were analyzed using the Chart for Windows Data Analysis System (39). These experiments were approved by the Animal Experimental Ethics Committee of Howard Florey Institute of Experimental Physiology and Medicine and the Henry Ford Health System Institutional Animal Care and Use Committee.
Dose-Dependent MAP and Renal CBF and MBF Responses to Equimolar ANG IV, III, and II
Upon completion of surgery, saline was infused (n = 8) at a rate of 37.5 μl/min to maintain constant blood pressure and renal hemodynamics, as described previously (39, 40). A minimal 30-min equilibration period was allowed to stabilize MAP and renal CBF and MBF before infusion of equimolar angiotensin peptides (0, 0.01, 0.1, and 1.0 nmol·kg−1 ·min−1 iv) in the following order: ANG IV, ANG III, and ANG II (Peninsula Laboratories). For each peptide, we started with the lowest dose for the first 30 min, allowing MAP and renal CBF responses to return to baseline before infusing subsequent doses, which usually occurred within 5 min after halting the infusion. The doses we used were based on previous studies in which ANG IV was shown to cause either vasodilatation or vasoconstriction in vivo in rats (11, 15, 18, 19). MAP and renal CBF and MBF responses were recorded throughout the experiment and analyzed using Chart Data Analysis (ADInstruments).
Effects of AT1 and AT2 Receptor Blockade on Systemic ANG IV-Induced Responses of MAP, Renal CBF, and MBF
Because the above protocol showed that ANG IV caused dose-dependent increases in MAP and decreases in renal CBF at 0.1 nmol·kg−1 ·min−1 and above, we next examined whether systemic and renal vasoconstrictor effects of ANG IV could be blocked by an AT1 or AT2 receptor antagonist. Rats (n = 8) were surgically prepared as described above. After a 30-min equilibration period, ANG IV was infused intravenously at a rate of 1 nmol·kg−1 ·min−1 throughout the experiment. The AT2 receptor antagonist PD-123319 (5 mg/kg bolus followed by 50 μg·kg−1 ·min−1 iv; Pfizer) was added for 30 min, followed by a 30-min infusion of the AT1 receptor antagonist losartan (5 mg/kg bolus followed by 50 μg·kg−1 ·min−1 iv; Du Pont Merck Pharmaceuticals). We have previously shown that these doses of PD-123319 and losartan completely abolish AT2 and AT1 receptor binding in the rat kidney and adrenal gland (39, 41), respectively, and also inhibit ANG II-induced pressor and renal hemodynamic responses after intravenous administration (39, 40). To explore the possibility that the vasodilatory effects of ANG IV may be uncovered in the absence of AT1 receptors, losartan was administered in an additional group of rats (n = 8) for the first 30 min before infusing ANG IV for an additional 30 min. MAP and renal CBF responses were recorded throughout the experiment.
Effects of AT1 Receptor Blockade on Intrarenal Arterial Administration of ANG IV on Renal CBF
To exclude the possibility that the observed renal cortical vasoconstriction induced by systemic infusion of ANG IV may be secondary to a systemic pressor response, ANG IV (1 nmol·kg−1 ·min−1 in 30 μl) was infused directly in the left renal artery of an additional group of rats (n = 8). In this protocol, 30 min equilibration was allowed after surgery before saline (30 μl/min) was infused in the left renal artery, and baseline MAP and CBF were recorded for 30 min. ANG IV was then infused, and MAP and the renal CBF response to ANG IV was measured for 30 min. Finally, ANG IV and losartan (5 mg/kg iv bolus followed by 50 μg·kg−1 ·min−1 ia) were coadministered and MAP, and renal CBF was recorded for an additional 30 min.
Effects of ANG IV on Renal Cortical AT1 Receptor Binding and Effects of ANG II or Losartan on Renal Cortical AT4 Receptor Binding as Visualized by Quantitative In Vitro Autoradiography
To study whether ANG IV induces renal cortical vasoconstriction by binding to AT1 receptors in the renal cortex, we performed competitive inhibition binding studies to determine whether ANG IV competes for AT1 receptor binding sites in the rat kidney using quantitative in vitro autoradiography (39, 42, 43). Renal AT1 receptors were mapped using the radioligand 125I-labeled [Sar1,Ile8]ANG II (Biochem). Frozen kidney sections (20 μm thick) from SD rats were preincubated in 10 mmol/l sodium phosphate buffer (pH 7.4) for 15 min to remove endogenously bound angiotensin peptides, which bind to their respective receptors. The sections were then incubated for 1 h in fresh buffer containing ~100 pmol/l of 125I-[Sar1,Ile8]ANG II at 22°C. Nonspecific binding was determined in parallel incubations containing an excess (10 μmol/l) of unlabeled ANG II or ANG IV (Peninsula). To determine the competitive potency of ANG II, ANG IV, and losartan in displacing specific AT1 receptor binding, 0.1 nM to 100 μmol/l unlabeled peptides or compounds were added to the incubation buffer to compete for AT1 receptor binding. To explore whether ANG II and losartan can displace renal cortical AT4 receptor binding sites, kidney sections were incubated with ~100 pmol/l of ANG IV receptor radioligand, 125I-labeled Nle1-ANG IV at 22°C for 60 min (kindly provided by Dr. Robert Speth, University of Mississippi Peptide Radioiodination Service Center). Unlabeled ANG IV, the ANG IV receptor antagonist divalinal-ANG IV, ANG II, or losartan (10 μM each) was added in parallel incubations to compete for 125I-Nle1-ANG IV binding. After incubation and subsequent washes, the sections were exposed to X-ray film (Agfa-Gevaert) together with a set of 125I-radioactivity standards for 7 days. The films were developed, and the levels of AT1 and AT4 receptor binding in the renal cortex were analyzed by computerized densitometry (MCID, Imaging Research, Ontario, Canada), as previously described (39, 41, 42, 43). Binding competition data were analyzed using GraphPad Prism 4.0 (GraphPad Software).
Effects of ANG IV on Intracellular Ca2+ Concentration Levels in Renal VSMCs
Increased intracellular Ca2+ concentration ([Ca2+]i) is the fore-most classical signaling for AT1 receptor-mediated vasoconstriction by ANG II (4, 12, 16, 25, 33). To determine whether ANG IV induces renal cortical vasoconstriction by increasing [Ca2+]i in renal VSMCs, SD rats were anesthetized, and renal VSMCs were isolated as described previously for Ca2+ imaging experiments (25, 33). Freshly isolated renal VSMCs were plated on cover slips and loaded with the Ca2+-sensitive fluorescent dye fura 2 (Molecular Probes) at 2 μM for 30 min at 37°C. After washes, cover slips were mounted on a perfusion chamber maintained at 37°C, which in turn was mounted on a Nikon Eclipse TE2000-U inverted fluorescence microscope coupled with a Lambda DG4 illumination system (Sutter Instruments). Ratio-metric Ca2+ measurements (340/380 ratio) in response to ANG IV were made continuously at 3-s intervals for up to 10 min using a MetaFluor Fluorescence Imaging System (Universal Imaging). To enable calculation of the average magnitude of peak [Ca2+]i responses to ANG IV, the imaging system was first calibrated using a fura 2 Calcium Imaging Calibration Kit with Ca2+ concentrations ranging from 0 to 10 mM (Molecular Probes). Ca2+ responses to ANG IV were examined further in cells pretreated with losartan (10 μM) or U-73122 (1 μM) for 30 min, a selective inhibitor for phospholipase C (PLC)-activated Ca2+ signaling.
Effects of ANG IV on Mitogen-activated Protein Kinase Extracellular/Signal-Regulated Kinase 1/2 Phosphorylation in Rat Glomerular MCs
Activation of mitogen-activated protein kinase extracellular/signal-regulated kinase (ERK) 1/2 phosphorylation is another classical signaling pathway for AT1 receptor-mediated effects of ANG II (4, 16, 20, 21). If ANG IV interacts with AT1 receptor-activated signaling, we expect that ANG IV would also induce ERK1/2 phosphorylation in a manner similar to ANG II. Cultured rat MCs, a well-described target for AT1 receptor-mediated effects of ANG II, were obtained from ATCC and subcultured to 80% confluence in six-well plates containing RPMI-1640 medium supplemented with 12% FBS. MCs were first starved for 24 h in serum-free medium before stimulation by ANG IV (10 nM) for 5 min. The effects of ANG IV on ERK1/2 phosphorylation were examined further in the presence of losartan (10 μM) or U-73122 (1 μM) to determine the role of AT1 receptor-activated PLC/inositol trisphosphate (IP3)/[Ca2+]i signaling. After stimulation, MCs were washed with ice-cold PBS and lysed with a modified RIPA buffer, and protein samples were extracted. Protein concentrations were determined using a BCA protein assay kit (Pierce), and total and phosphorylated ERK1/2 were measured by Western blot using selective antibodies targeted to total (SC-93; 1:5,000) or phosphorylated ERK1/2 (SC-7383; 1:200; Santa Cruz).
Data Analysis and Statistics
Data are presented as means ± SE. Differences between experimental periods within each group were compared using one-way ANOVA with repeated comparisons (Tukey’s test). Differences between ANG IV and ANG II at the same concentration(s) were analyzed by unpaired t-test. The competing effects of unlabeled ANG IV for AT1 receptor binding or unlabeled ANG II and losartan for AT4 receptor binding were analyzed using an unpaired Student’s t-test. P < 0.05 was considered significant.
RESULTS
Concentration-Dependent Responses of MAP, Renal CBF, and MBF to Systemic Administration of ANG IV, ANG III, or ANG II
At the lowest dose (0.01 nmol·kg−1 ·min−1 iv), ANG IV did not alter MAP, whereas ANG II increased it by 30 ± 5 mmHg (Fig. 1A). However, ANG IV increased MAP significantly at 0.1 (~20%, P < 0.05) and 1 (~33%, P < 0.05) nmol·kg−1 ·min−1 in a dose-dependent manner. By contrast, ANG II induced dose-dependent pressor effects at all concentrations examined (P < 0.05, Fig. 1A). ANG IV at the lowest dose had no effect on renal CBF; however, at higher concentrations it decreased CBF in a dose-dependent manner (Fig. 1B, P < 0.05). Again, ANG II induced more potent renal cortical vasoconstriction in a concentration-dependent fashion (Fig. 1B, P < 0.05). ANG IV had no effect on MBF at all concentrations examined [Fig. 1C, not significant (NS)]. By contrast, ANG II was potent at the two higher doses (Fig. 1C, P < 0.05). The effects of ANG III on MAP, renal CBF, and MBF were between those of ANG IV and ANG II.
Fig. 1.
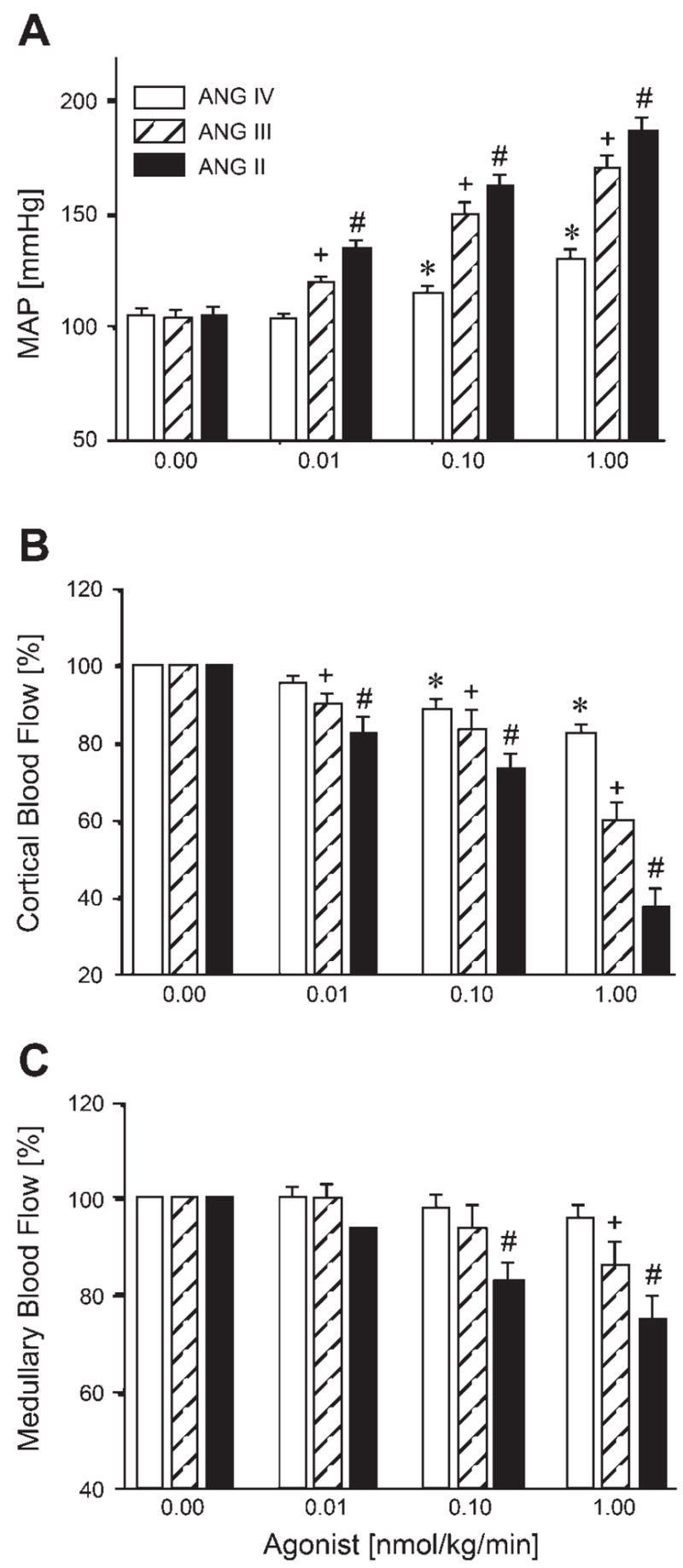
Dose-dependent pressor and renal vasoconstrictor effects of iv infusion of increasing concentrations of ANG II, ANG III, and ANG IV. All three ANG peptides increased mean arterial pressure (MAP) and decreased renal cortical blood flow (CBF) in a dose-dependent fashion, exhibiting potency in the order: ANG II > ANG III > ANG IV. ANG II and ANG III also decreased renal medullary blood flow (MBF) at higher doses, whereas ANG IV had no effect. P < 0.05 vs. baseline ANG IV response (*), vs. baseline ANG III response (+), and vs. baseline ANG II response (#).
Effects of AT1 and AT2 Receptor Blockade on Systemic ANG IV-Induced Pressor and Renal CBF Responses
Because ANG IV produced pressor and renal vasoconstrictor effects at 0.1 and 1 nmol·kg−1 ·min−1, we next tested whether these effects were mediated by interaction with AT1 or AT2 receptors. As shown in Fig. 2, AT2 receptor blockade with PD-123319 had no effect on ANG IV-induced MAP (top) and renal CBF responses (middle), suggesting that the AT2 receptor is not involved. By contrast, blockage of the AT1 receptor with losartan abolished the pressor effect of ANG IV (Fig. 2, top, P < 0.05). Losartan also reversed the ANG IV-induced decrease in CBF to a level significantly above control (Fig. 2, middle) and increased MBF (Fig. 2, bottom), indicating a tonic influence of endogenous ANG II via AT1 receptors on the renal cortical and medullary microcirculation (P < 0.05). In a reverse protocol designed to clarify whether ANG IV can induce systemic or renal vasodilatation after AT1 receptor blockade, pretreatment with losartan alone before ANG IV infusion did not decrease MAP significantly but did prevent ANG IV-induced increases in MAP (data not shown).
Fig. 2.
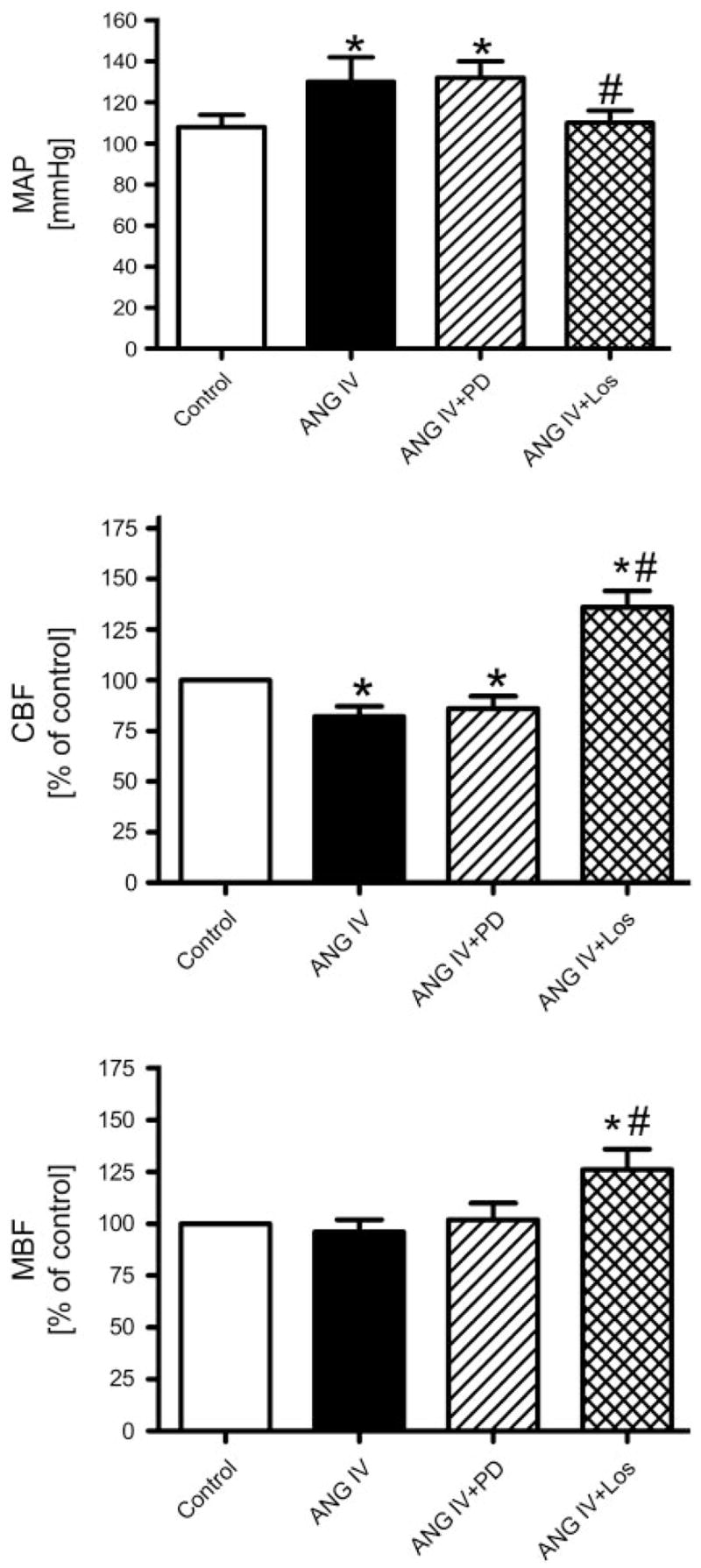
Effects of angiotensin type 2 (AT2) and/or type 1 (AT1) receptor blockade on MAP and CBF and MBF responses to iv infusion of ANG IV (1 nmol ·kg−1 ·min−1). ANG IV increased MAP and decreased CBF. These responses were not altered by the AT2 receptor antagonist PD-123319 (PD) but were reversed by the AT1 receptor antagonist losartan (Los). P < 0.05 vs. control (*) and vs. ANG IV response (#).
Effects of Direct Intrarenal Arterial Infusion of ANG IV on MAP and Renal CBF
Figure 3 shows that intrarenal arterial infusion of ANG IV had no effect on MAP (top) but significantly decreased renal CBF by 30% (70.4 ± 3.2% of control, P < 0.01; Fig. 3, bottom), suggesting that ANG IV directly induced renal cortical vasoconstriction. Coadministration of ANG IV with losartan restored renal CBF to a level not significantly different from control (96 ± 5.3% of control, NS).
Fig. 3.
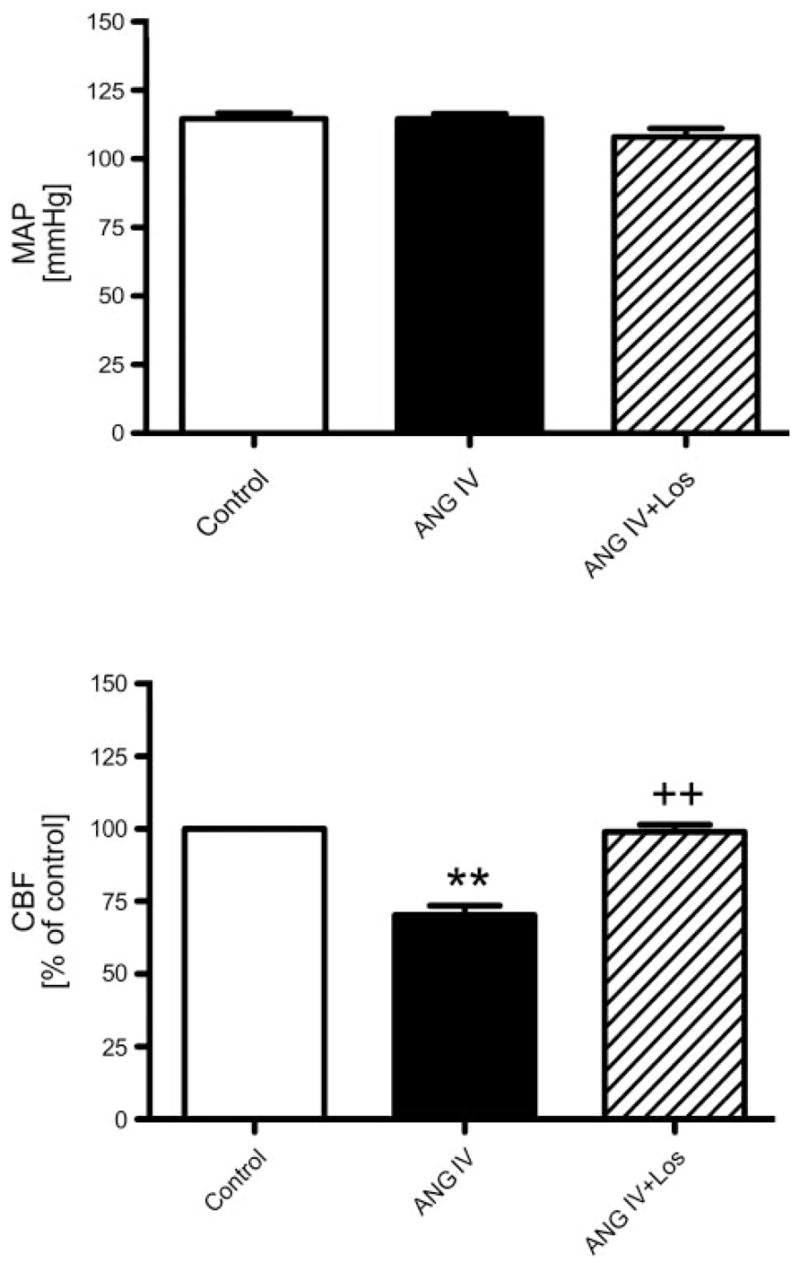
Effects of intrarenal arterial infusion of ANG IV (1 nmol ·kg−1 ·min−1) on MAP and renal CBF in anesthetized rats. Intrarenal infusion of ANG IV did not affect MAP but reduced renal CBF. Coadministration of ANG IV with losartan blocked intrarenal ANG IV-induced renal cortical vasoconstriction. P < 0.01 vs. control (**) and vs. ANG IV (++).
Effects of ANG IV on Renal Cortical AT1 Receptor Binding as Visualized by Quantitative In Vitro Autoradiography
Quantitative in vitro autoradiography was performed to examine whether ANG IV competes for AT1 receptor binding in the renal cortex. As expected, ANG II receptors in the renal cortex were predominantly the AT1 subtype (Fig. 4). AT1 receptors are located primarily in the cortex and the inner stripe of the outer medulla (39, 41–43). Renal AT1 receptor binding was completely inhibited by 10 μM unlabeled ANG II (Fig. 4B) and losartan (Fig. 4D) and also partially displaced by unlabeled ANG IV (10 μM; Fig. 4C). As shown in Fig. 4D, unlabeled ANG II, ANG IV, and losartan competed for specific AT1 receptor binding in a concentration-dependent manner. The inhibitory potency (IC50) on AT1 receptor binding for ANG II, ANG IV, and losartan was 3.8 ± 0.3, 300 ± 15, and 10.2 ± 0.5 nM, respectively (Fig. 4D).
Fig. 4.
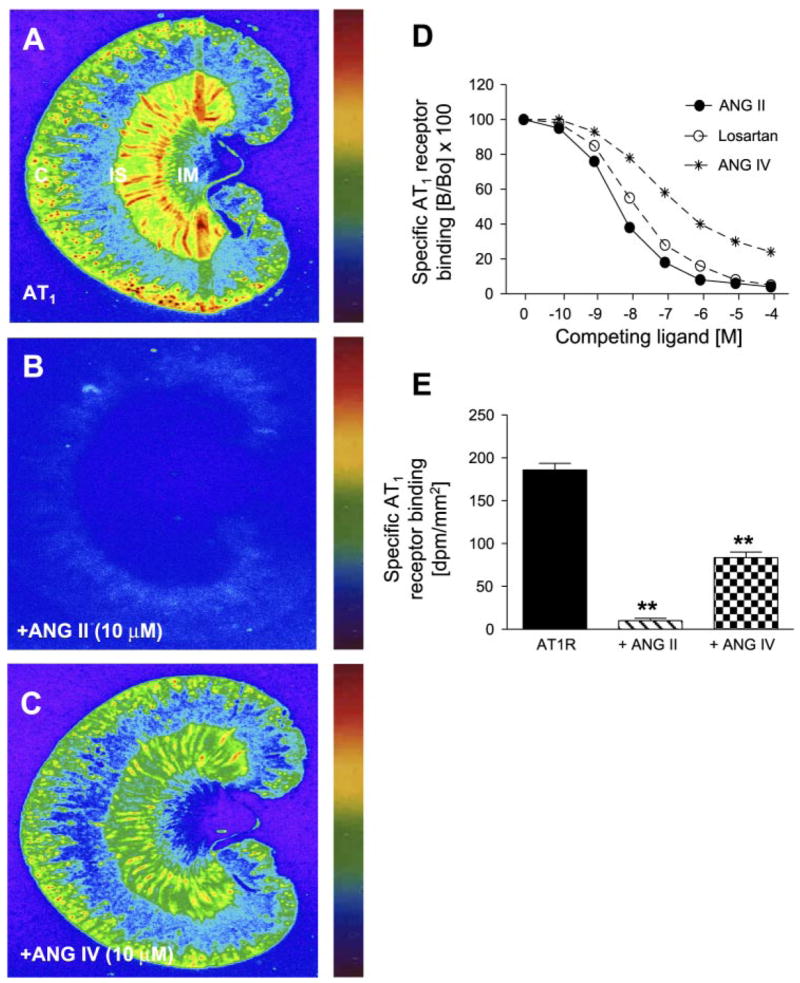
Autoradiographs showing the effects of unlabeled ANG II or IV on specific AT1 receptor binding in the rat kidney. A: AT1 receptor binding. B: AT1 receptor binding was completely displaced by unlabeled ANG II (10 μM). C: AT1 receptor binding was partially inhibited by unlabeled ANG IV (10 μM). D: concentration-dependent inhibition of AT1 receptor binding by increasing concentrations of unlabeled ANG II, ANG IV, and losartan (10−10 to 10−4 M). E: quantitative levels of AT1 receptor (R) binding in the absence and presence of unlabeled ANG II or ANG IV. Color bars: red represents the highest level of binding, whereas blue shows the background level. **P < 0.01 vs. total AT1 receptor binding in the absence of unlabeled ANG II or ANG IV (10 μM). C, cortex; IS, inner stripe of the outer medulla; IM, inner medulla.
Effects of ANG II or Losartan on Renal Cortical AT4 Receptor Binding as Visualized by Quantitative In Vitro Autoradiography
Figure 5 shows renal cortical AT4 receptor binding using a radiolabeled, specific ANG IV agonist (125I-Nle1-ANG IV) and the effects of unlabeled ANG IV, divalinal ANG IV (an ANG IV receptor-selective antagonist), ANG II, losartan (AT1 receptor-selective antagonist), and PD-123319 (AT2 receptor-selective antagonist) on 125I-Nle1-ANG IV receptor binding. Specific AT4 receptor binding predominated in the inner cortex with a moderate level in the superficial cortex (Fig. 5A). Both unlabeled ANG IV (Fig. 5B) and divalinal ANG IV (Fig. 5C) displaced 80–90% of ANG IV receptor binding, whereas unlabeled ANG II (Fig. 5D) and losartan (Fig. 5E) inhibited ANG IV receptor binding by between 30 and 50%. However, PD-123319 had no effect on ANG IV receptor binding (Fig. 5F).
Fig. 5.
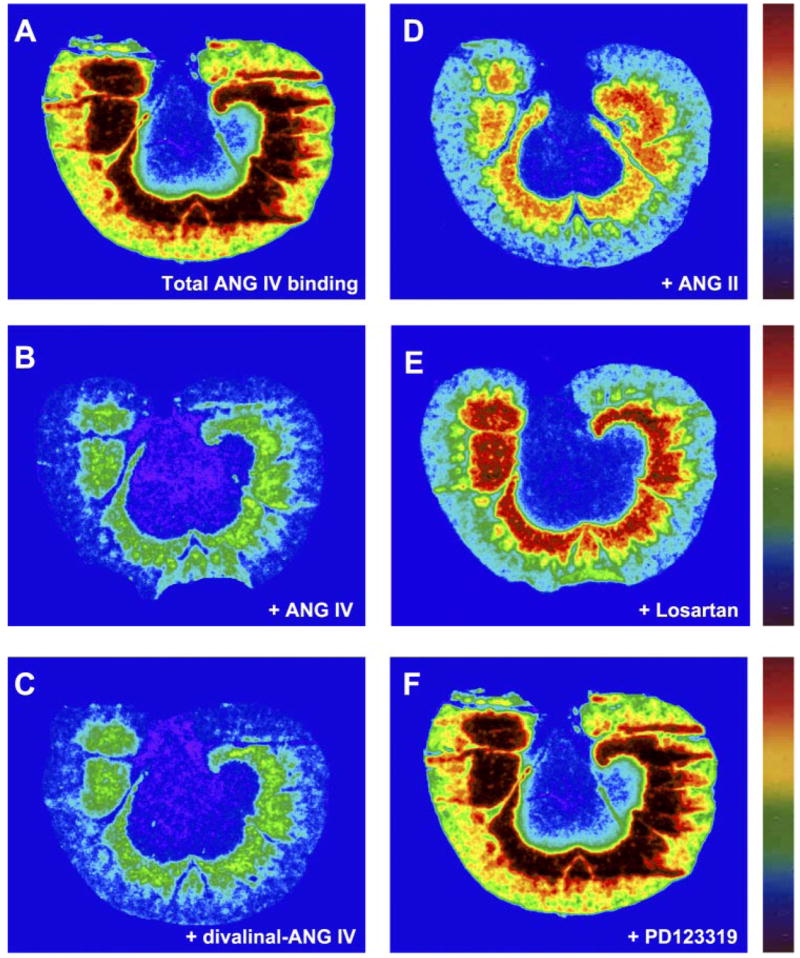
Autoradiographs showing the effects of unlabeled ANG II (10 μM) and losartan (10 μM) on specific AT4 receptor binding in the rat kidney. AT4 receptors were labeled by 125I-Nor-Leu-ANG IV, a selective agonist for ANG IV. Note that unlabeled native ANG IV (B, 10 μM) and the antagonist divalinal-ANG IV (C, 10 μM) displaced most AT4 receptor binding. AT4 receptor binding was also displaced to a significant extent by unlabeled ANG II (D) or losartan (E, 10 μM) but not by PD-123319 (F, 10 μM).
Effects of ANG IV on [Ca2+]i Levels in Renal VSMCs
Whether ANG IV can increase [Ca2+]i in renal VSMCs has not been studied to our knowledge; however, Chansel et al. (12) showed that at 100 nM to 1 μM, ANG IV induced [Ca2+]i responses in rat glomerular MCs via activation of AT1 receptors. Figure 6 shows that ANG IV (100 nM) induced a sustained increase in [Ca2+]i in two representative renal VSMCs (top). Basal [Ca2+]i in renal VSMCs averaged 86 ± 18 nM, which was increased to 262 ± 25 nM during ANG IV stimulation (Fig. 6, bottom). ANG IV-induced increases in [Ca2+]i levels were prevented by pretreating the cells with losartan (10 μM, P < 0.01).
Fig. 6.
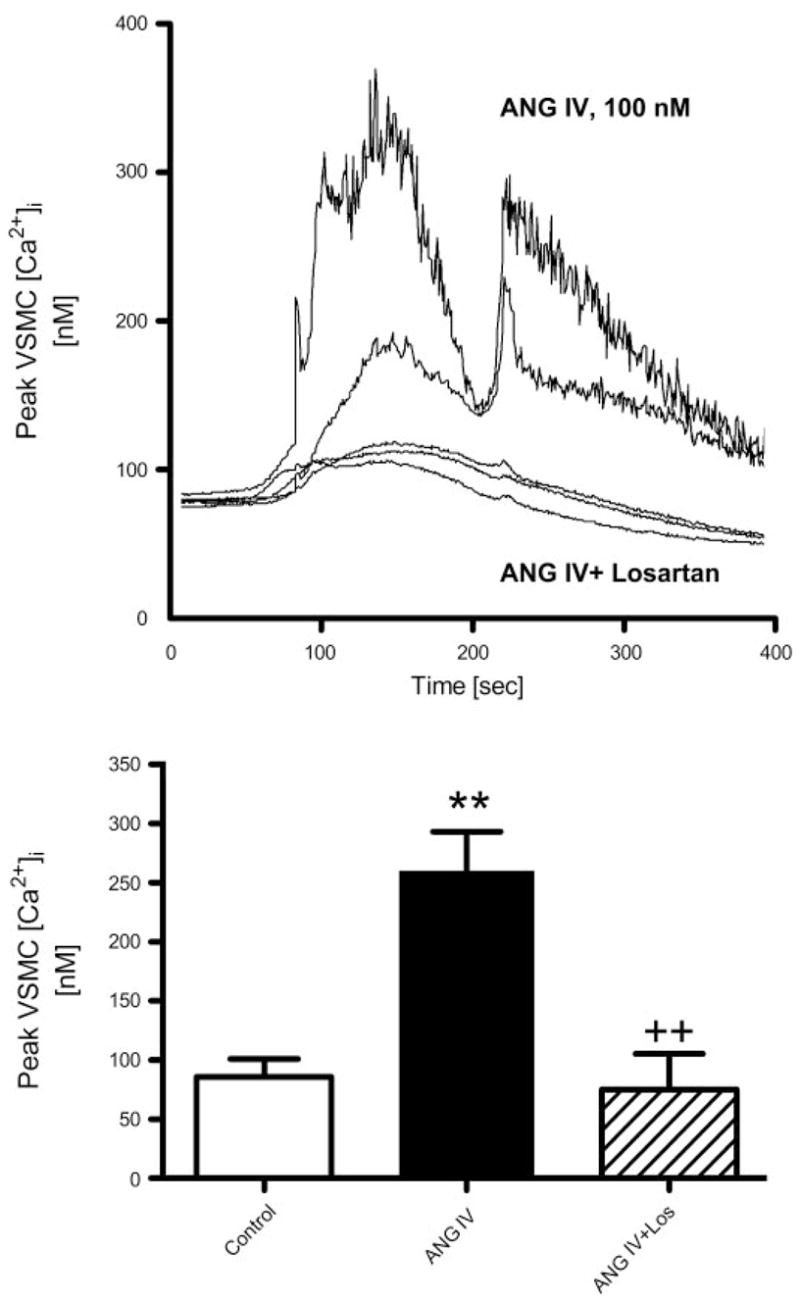
Effects of ANG IV (100 nM) on intracellular Ca2+ concentration ([Ca2+]i) in renal microvascular smooth muscle cells (VSMCs) determined using fura 2 ratiometric Ca2+ imaging (340/380). Top: time-dependent [Ca2+]i responses to ANG IV stimulation in two representative VSMCs and three other cells pretreated with losartan (10 μM) before stimulation by ANG IV. Bottom: averaged peak [Ca2+]i responses in renal VSMCs treated with perfusate only (time control), ANG IV, or ANG IV in the presence of losartan. Ratiometric [Ca2+]i imaging (340/380) was recorded continuously at 3-s intervals for up to 10 min (n = 8–10 cells/group). P < 0.01 vs. control (**) and vs. ANG IV (++).
Effects of ANG IV on Mitogen-Activated Protein Kinase ERK1/2 Phosphorylation in Rat Glomerular MCs
Glomerular MCs are another well-described target for ANG II, acting via AT1 receptors in the renal cortex. ANG II has been shown to activate ERK1/2 phosphorylation via AT1 receptors in MCs (20, 21). As an active agonist of ANG II, ANG IV would be expected to activate AT1 receptor-mediated phosphorylation of mitogen-activated protein kinase ERK1/2, inducing a downstream AT1 receptor signaling in MCs (4, 16, 20, 21). As expected, ANG II (1 nM) induced a twofold increase in ERK1/2 phosphorylation via activation of AT1 receptors (data not shown). ANG IV (10 nM) also more than doubled ERK1/2 phosphorylation in MCs (Fig. 7). Pretreatment with losartan (10 μM) for 30 min significantly inhibited ANG IV-induced ERK1/2 phosphorylation, whereas losartan alone had no effect (Fig. 7). U-73122, a selective inhibitor of PLC/IP3/[Ca2+]i signaling, also significantly attenuated ANG IV-induced ERK1/2 signaling (Fig. 8).
Fig. 7.
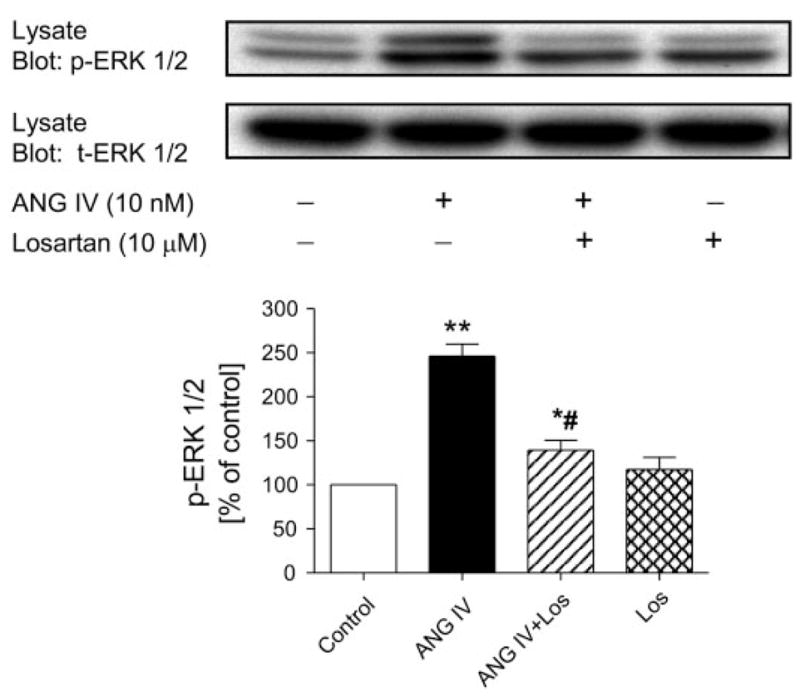
Effects of ANG IV (10 nM) on mitogen-activated protein kinase extracellular/signal-regulated kinase (ERK) 1/2 phosphorylation in rat mesangial cells. Top: representative Western blots of phosphorylated (p-ERK1/2) and total (t-ERK1/2) ERK1/2. Bottom: semiquantitative levels of p-ERK1/2. ANG IV significantly increased ERK1/2 phosphorylation, and the effect was blocked by losartan (10 μM; n = 6 each). *P < 0.05, **P < 0.01 vs. control. #P < 0.05 vs. ANG IV.
Fig. 8.
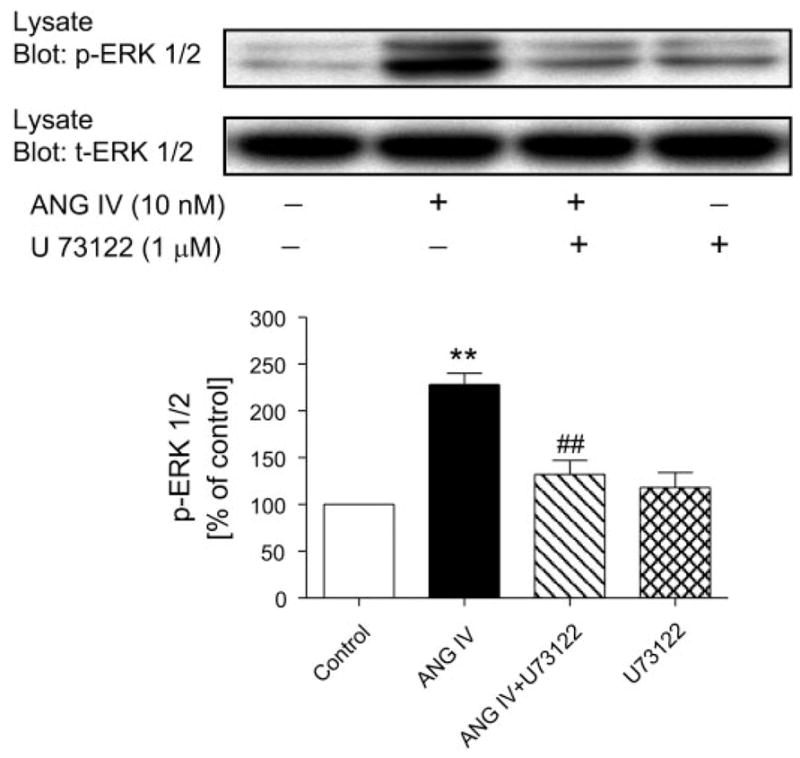
Effects of inhibition of phospholipase C with U-73122 (1 μM) on ANG IV-induced ERK1/2 phosphorylation in rat mesangial cells. Top: Western blots of phosphorylated and total ERK1/2. Bottom: semiquantitative levels of p-ERK1/2. ANG IV significantly increased ERK1/2 phosphorylation, and the effect was blocked by U-73122, suggesting involvement of phospholipase C (n = 6 each). **P < 0.01 vs. control. ##P < 0.01 vs. ANG IV.
DISCUSSION
The present study demonstrates the following five key findings: 1) in anesthetized rats, systemic infusion of ANG IV increased MAP and decreased renal CBF in a dose-dependent manner without affecting MBF in the inner stripe of the outer medulla; 2) ANG IV-induced systemic and renal cortical vasoconstriction was abolished by the AT1 receptor antagonist losartan but not by the AT2 receptor antagonist PD-123319; 3) direct intrarenal infusion of ANG IV also induced renal cortical vasoconstriction, and the response was blocked by losartan; 4) unlabeled ANG IV inhibited AT1 receptor binding, and conversely unlabeled ANG II, and losartan inhibited AT4 receptor binding to some extent in the rat kidney; and 5) ANG IV activated AT1 receptors to increase [Ca2+]i in rat renal VSMCs and induced mitogen-activated protein kinase ERK1/2 phosphorylation in MCs. Our results are consistent with the concept that, at subnanomolar to nanomolar concentrations, ANG IV behaves as an active agonist for the AT1 receptor (5, 11, 12, 18, 19, 28) and may play a physiological role in the regulation of blood pressure and intrarenal microcirculation.
Currently, there are conflicting reports on the systemic and renal hemodynamic effects of ANG IV. Both systemic and/or renal vasoconstrictor (11, 18, 19, 30, 36) or vasodilator responses to ANG IV (15, 22, 35, 37) have been observed. Coleman et al. (15) were the first to describe the renal vasodilator response to intrarenally administered ANG IV using laser-Doppler flowmetry in the rat superficial renal cortex. Because the renal vasodilator effects of ANG IV were not affected by an AT1 (losartan) or AT2 receptor blocker (PD-123319), but were abolished by divalinal-ANG IV, an ANG IV receptor antagonist, or by blocking NO release with NG-nitro-L-arginine methyl ester, they suggested that activation of intrarenal AT4 receptors mediates intrarenal vasodilatation via an NO-dependent mechanism (15). However, divalinal-ANG IV and ANG IV have been shown to substantially increase [Ca2+]i levels in human proximal tubule cells, a characteristic signal for AT1 receptor activation (24). Subsequent in vivo and in vitro studies using other approaches in various vascular beds failed to uncover a vasodilator effect for the hexapeptide (11, 18, 19, 36). Using chronically implanted pulsed Doppler flow probes in conscious rats, Gardiner et al. found that, at doses up to 125 pmol/kg, ANG IV did not alter blood pressure, renal and mesenteric blood flow, or vascular conductance; however, at higher doses it increased blood pressure and significantly reduced renal and mesenteric blood flow in a dose-dependent manner. Pressor and renal vasoconstrictor responses to ANG IV were abolished by pretreatment with losartan but were not altered by L-arginine, suggesting an AT1 receptor-mediated event independent of NO (19). Furthermore, Fitzgerald et al. (18) monitored the whole kidney blood flow response to increasing doses of ANG IV (10–1,000 pmol/min) infused directly in the renal artery of anesthetized rats and observed dose-related reductions in total renal blood flow using transit-time flow probes. As before, pretreatment with losartan abolished the vasoconstrictor response to ANG IV (18). Finally, van Rodijnen et al. (36) recently described the AT1 receptor-mediated vasoconstrictor effects of the ANG II fragments ANG IV and ANG (I-VII) in rat renal interlobular arteries and in afferent and efferent arterioles using the isolated perfused hydronephrotic kidney. Thus most (if not all) studies suggest that ANG IV exerts an AT1 receptor-mediated vasoconstrictor effect on systemic blood pressure and large renal blood vessels.
However, it can be argued that renal vasoconstrictor effects of ANG IV may be secondary to a systemic pressor response because none of these studies have directly compared the renal cortical hemodynamic responses to intrarenal vs. systemic ANG IV administration, or may only apply to large renal arteries or total renal blood flow (18, 19, 36) because those approaches may not uncover intrarenal microvascular vasodilatation induced by ANG IV (15, 22). Based on our findings, we believe this is unlikely. Like Coleman et al. (15), we also used laser-Doppler flowmetry to monitor renal CBF responses to ANG IV and ANG II in the renal cortex and MBF responses in the inner stripe of the outer medulla in anesthetized rats. Although this technique cannot measure absolute regional blood flow, it has been widely used to monitor changes in microvascular perfusion in the renal cortex and medulla in response to a given peptide or drug. Although our experimental design differed from earlier studies in several aspects (see below), a similar conclusion can be drawn, namely, that ANG IV might activate the AT1 receptor to induce both systemic pressor responses and intrarenal vasoconstrictor effects.
In the present study, we tested the hypothesis that AT1 receptor-activated signaling mediates ANG IV-induced renal cortical vasoconstrictor responses using complementary in vivo and in vitro approaches. First, we examined the pressor and renal cortical vasoconstrictor effects of ANG IV and compared them with equimolar concentrations of its more potent precursors ANG II and ANG III in the same animal and experimental settings; moreover, the concentrations we used (0.01, 0.1, and 1 nmol·kg−1 ·min−1) were comparable with those previously associated with either vasoconstriction or vasodilatation in vivo or in vitro (11, 15, 18, 19, 36). Second, pressor and renal cortical vasoconstrictor responses to ANG IV were monitored during a constant infusion rather than a single bolus injection before and after blockade of AT2 and/or AT1 receptors. We did not observe any vasodilatation response throughout ANG IV infusion before PD-123319 or losartan was administered. Instead, we observed systemic and renal cortical vasoconstriction at two higher concentrations of ANG IV (0.1 and 1.0 nmol·kg−1 ·min−1), which increased MAP and reduced renal CBF (Fig. 1). The increase in renal CBF observed after losartan administration can be attributed to AT1 receptor blockade and a tonic influence of endogenous ANG II acting via AT1 receptors on the intrarenal microvasculature (Fig. 2). Further experiments with reverse protocols again confirmed that losartan blocked ANG IV-induced increases in MAP and reductions in renal CBF, whereas PD-123319 did not. Thus our results exclude the possibility that blockage of AT1 receptors with losartan before administration of ANG IV may uncover additional systemic and renal vasodilator effects of ANG IV. Third, to exclude the influence of systemic factors on ANG IV-induced renal CBF responses, we infused the hexapeptide directly in the renal artery. Although intrarenal infusion of ANG IV did not alter MAP, it reduced CBF by 30%, and again the response was completely blocked by losartan (Fig. 3). Fourth, we found that unlabeled ANG IV was able to displace renal AT1 receptor binding in a concentration-dependent manner (Fig. 4), and conversely unlabeled ANG II and losartan inhibited AT4 receptor binding to some extent (Fig. 5). Our results suggest that, even though ANG IV has less affinity for AT1 receptors than ANG II and losartan, at higher concentrations it can still compete for (or interact with) AT1 receptors to induce systemic and intrarenal effects. Indeed, a recent study shows that ANG IV is a potent agonist for AT1 receptors in CHO-K1 cells, which express mutant human AT1 receptors (28).
AT1 receptor-mediated increases in [Ca2+]i in response to ANG II stimulation constitute one of the most important signaling pathways in cardiovascular and renal cells (4, 12, 16, 25, 38). Previous studies have shown that ANG II increases [Ca2+]i via AT1 receptor-dependent mechanisms in preglomerular VSMCs (25, 38), but it is not known whether ANG IV induces renal cortical microvascular vasoconstriction by similar intracellular mechanisms. ANG IV and its antagonist divalinal-ANG IV have been reported to activate mutant human AT1 receptors to increase intracellular IP3 accumulation in CHO-K1 cells (28). Increased intracellular IP3 accumulation would be expected to induce [Ca2+]i responses. Handa (24) reported that both ANG IV and divalinal-ANG IV markedly increase [Ca2+]i in human proximal tubule cells, but he did not clarify whether these responses were mediated by activation of AT1 receptors. In rat glomerular MCs, Chansel et al. (12) showed that at 100 nM, ANG IV does stimulate [Ca2+]i, and this stimulation was completely inhibited by losartan or candesartan, suggesting that AT1 receptors are involved. The concentrations of ANG IV they used to elicit [Ca2+]i responses in MCs were 10-fold higher than ANG II, but pharmacologically it behaved identically to ANG II. In the present study, we observed similar [Ca2+]i responses to ANG IV stimulation in rat renal VSMCs at the concentrations used by Chansel et al. (12). Pretreatment of renal VSMCs with losartan or a PLC-selective inhibitor (U-73122) effectively abolished the effects of ANG IV on [Ca2+]i. Thus these results indicate that ANG IV induces renal microvascular vasoconstriction by stimulating AT1 receptors to increase [Ca2+]i.
In addition to inducing renal cortical microvascular constriction by increasing [Ca2+]i, ANG IV also appears to mimic ANG II in activating another important AT1 receptor-mediated signal, phosphorylation of mitogen-activated protein kinase ERK1/2 in renal cortical cells (14, 16, 20, 21). We used rat glomerular MCs for the following two reasons: the difficulty of obtaining sufficient amounts of protein samples from renal VSMCs for Western blot of ERK1/2 phosphorylation and the fact that renal VSMCs and MCs both express abundant AT1 receptors and respond in a similar manner to ANG II and ANG IV (12, 16, 25, 38). ANG II is well known to induce ERK1/2 phosphorylation in VSMCs via AT1 receptor activation, but it is not clear whether ANG IV has similar effects on ERK1/2 phosphorylation. Our results show that ANG IV (10 nM) was able to induce ERK1/2 phosphorylation in rat MCs, whereas pretreatment with losartan significantly inhibited ANG IV-induced ERK1/2 phosphorylation. These actions of ANG IV are identical to ANG II (1 nM). Thus ANG IV behaves as an active agonist of ANG II by acting on AT1 receptors in rat glomerular MCs (12). The increases in mitogen-activated protein kinase ERK1/2 phosphorylation induced by ANG IV suggests that the hexapeptide may be involved in AT1 receptor-mediated effects of angiotensin peptides, including cell growth and proliferation in addition to intrarenal microvascular responses (29).
In summary, the present study demonstrates that, at nano-molar concentrations, ANG IV can act as an active agonist of ANG II by activating AT1 receptor signaling in blood pressure regulation, renal VSMCs, and glomerular MCs. Early structure-activity studies suggest that the three NH2-terminal amino acids (Asp1-Arg2-Val3) are important for pressor activity of ANG II and its active fragments ANG III and ANG IV (2, 5, 26). Complete removal of these three amino acids abolishes the biological activities of ANG II, suggesting that ANG IV may be a minimal requirement for pressor or vasoconstrictor effects of angiotensin peptides (26, 28). Thus it is not surprising that ANG IV can compete for AT1 receptor binding sites and interact with AT1 receptors to increase blood pressure and induce constriction in various vascular beds (11, 12, 18, 19, 36). However, it should be emphasized that the pressor and renal cortical vasoconstrictor effects of ANG IV were achieved only at subnanomolar to nanomolar concentrations, which were often 10- to 100-fold higher than those of ANG II. Therefore, for ANG IV to exert physiological or pathophysiological effects on blood pressure control and intrarenal microvascular regulation, nanomolar levels of ANG IV may be required. We have previously reported femtomolar ANG II levels in rat plasma and kidn*ey under physiological conditions (7–9), which increases by severalfold during ANG II-induced hypertension (39, 44). Because ANG IV is mainly derived from the metabolism of its precursors ANG II and ANG III, its levels in the circulation and kidney unlikely reach nanomolar concentrations (6–9). Indeed, we found that the levels of ANG III and ANG IV are much lower than those of ANG II in normal rat and human plasma (7–9), although they may be increased significantly after treatment with eprosartan in hypertensive humans (9). Taken together, our results suggest that ANG IV most likely plays a relatively minor role in physiological regulation of arterial blood pressure and intrarenal hemodynamics by angiotensin peptides. However, because radioreceptor binding studies have shown separate AT1 and AT4 receptors in the central nervous system and other peripheral tissues (16), ANG IV may have effects that are mediated by AT4 receptors or IRAP (2, 10, 14, 16, 17, 24, 37).
Acknowledgments
GRANTS
This work was supported, in part, by National Institute of Diabetes and Digestive and Kidney Diseases Grant RO1DK-067299, American Heart Association Greater Midwest Affiliate Grant-in-Aid 0355551Z, and a National Kidney Foundation of Michigan Grant-in-Aid to J. L. Zhuo. A portion of the work was supported by the National Health and Medical Research Council of Australia while J. L. Zhuo worked at the Howard Florey Institute of Experimental Physiology and Medicine, University of Melbourne, Australia (Grant No. 983001). D. J. Campbell is a recipient of Career Development Fellowship Award CR02M 0829 from the National Heart Foundation of Australia. M. Ohishi was supported by an International Research Fellowship Award from the High Blood Pressure Research Council of Australia.
References
- 1.Abrahamsen CT, Pullen MA, Schnackenberg CG, Grygielko ET, Edwards RM, Laping NJ, Brooks DP. Effects of angiotensin II and IV on blood pressure, renal function, and PAI-1 expression in the heart and kidney of the rat. Pharmacology. 2002;66:26–30. doi: 10.1159/000063252. [DOI] [PubMed] [Google Scholar]
- 2.Albiston AL, McDowall SG, Matsacos D, Sim P, Clune E, Mustafa T, Lee J, Mendelsohn FA, Simpson RJ, Connolly LM, Chai SY. Evidence that the angiotensin IV [AT(4)] receptor is the enzyme insulin-regulated aminopeptidase. J Biol Chem. 2001;276:48623–48626. doi: 10.1074/jbc.C100512200. [DOI] [PubMed] [Google Scholar]
- 3.Ardaillou R, Chansel D. Synthesis and effects of active fragments of angiotensin II. Kidney Int. 1997;52:1458–1468. doi: 10.1038/ki.1997.476. [DOI] [PubMed] [Google Scholar]
- 4.Berk BC, Corson MA. Angiotensin II signal transduction in vascular smooth muscle cells: role of tyrosine kinase. Circ Res. 1997;80:607–616. doi: 10.1161/01.res.80.5.607. [DOI] [PubMed] [Google Scholar]
- 5.Blair-West JR, Coghlan JP, Denton DA, Funder JW, Scoggins BA. The effect of the heptapeptide (2–8) and hexapeptide (3–8) fragments of angiotensin II on aldosterone secretion. J Clin Invest. 1971;32:575–578. doi: 10.1210/jcem-32-4-575. [DOI] [PubMed] [Google Scholar]
- 6.Cain MD, Catt KJ, Coghlan JP. Immunoreactive fragments of angiotensin II in blood. Nature. 1969;223:617–618. doi: 10.1038/223617a0. [DOI] [PubMed] [Google Scholar]
- 7.Campbell DJ, Kladis A. Simultaneous radioimmunoassay of six angiotensin peptides in arterial and venous plasma of man. J Hypertens. 1990;8:165–172. doi: 10.1097/00004872-199002000-00011. [DOI] [PubMed] [Google Scholar]
- 8.Campbell DJ, Lawrence AC, Towrie A, Kladis A, Valentijin AJ. Differential regulation of angiotensin peptide levels in plasma and kidney of the rat. Hypertension. 1991;18:763–773. doi: 10.1161/01.hyp.18.6.763. [DOI] [PubMed] [Google Scholar]
- 9.Campbell DJ, Krum H, Murray DS. Losartan increases bradykinin levels in hypertensive humans. Circulation. 2005;111:315–320. doi: 10.1161/01.CIR.0000153269.07762.3B. [DOI] [PubMed] [Google Scholar]
- 10.Carey RM, Siragy HM. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev. 2003;24:261–271. doi: 10.1210/er.2003-0001. [DOI] [PubMed] [Google Scholar]
- 11.Champion HC, Czapla MA, Kadowitz PJ. Responses to angiotensin peptides are mediated by AT1 receptors in the rat. Am J Physiol Endocrinol Metab. 1998;274:E115–E123. doi: 10.1152/ajpendo.1998.274.1.E115. [DOI] [PubMed] [Google Scholar]
- 12.Chansel D, Vandermeersch S, Oko A, Curat C, Ardaillou R. Effects of angiotensin IV and angiotensin-(1–7) on basal and angiotensin II-stimulated cytosolic Ca2+ in mesangial cells. Eur J Pharmacol. 2001;414:165–175. doi: 10.1016/s0014-2999(01)00791-9. [DOI] [PubMed] [Google Scholar]
- 13.Chansel D, Czekalski S, Vandermeersch S, Ruffet E, Fournie-Zaluski M, Ardaillou R. Characterization of angiotensin IV-degrating enzymes and receptors on rat mesangial cells. Am J Physiol Renal Physiol. 1998;275:F535–F542. doi: 10.1152/ajprenal.1998.275.4.F535. [DOI] [PubMed] [Google Scholar]
- 14.Chen JK, Zimpelmann J, Harris RC, Burns KD. Angiotensin IV induces tyrosine phosphorylation of focal adhesion kinase and paxillin in proximal tubule cells. Am J Physiol Renal Physiol. 2001;280:F980–F988. doi: 10.1152/ajprenal.2001.280.6.F980. [DOI] [PubMed] [Google Scholar]
- 15.Coleman JK, Krebs LT, Hamilton TA, Ong B, Lawrence KA, Sardinia MF, Harding JW, Wright JW. Autoradiographic identification of kidney angiotensin IV binding sites and angiotensin IV-induced renal cortical blood flow changes in rats. Peptides. 1998;19:269–277. doi: 10.1016/s0196-9781(97)00291-x. [DOI] [PubMed] [Google Scholar]
- 16.de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International Union of Pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52:415–472. [PubMed] [Google Scholar]
- 17.Dulin N, Madhum ZT, Chang CH, Berti-Mattera L, Dickens D, Douglas JG. Angiotensin IV receptors and signaling in opossum kidney cells. Am J Physiol Renal Fluid Electrolyte Physiol. 1995;268:F644–F652. doi: 10.1152/ajprenal.1995.269.5.F644. [DOI] [PubMed] [Google Scholar]
- 18.Fitzgerald SM, Evans RG, Bergstrom G, Anderson WP. Renal hemodynamic responses to intrarenal infusion of ligands for the putative angiotensin IV receptor in anesthetized rats. J Cardiovasc Pharmacol. 1999;34:206–211. doi: 10.1097/00005344-199908000-00005. [DOI] [PubMed] [Google Scholar]
- 19.Gardiner SM, Kemp PA, March JE, Bennett T. Regional haemodynamic effects of angiotensin II (3–8) in conscious rats. Br J Pharmacol. 1993;110:159–162. doi: 10.1111/j.1476-5381.1993.tb13786.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Geiger MUF, Herrero H, Zeuzem M, Piiper A. Involvement of the platelet-derived growth factor receptor in angiotensin II-induced activation of extracellular regulated kinases 1 and 2 in human glomerular mesangial cells. FEBS Lett. 2000;472:129–132. doi: 10.1016/s0014-5793(00)01433-2. [DOI] [PubMed] [Google Scholar]
- 21.Gorin Y, Ricono JM, Wagner B, Kim NH, Bhandari B, Choudhury GG, Abboud HE. Angiotensin II-induced ERK1/ERK2 activation and protein synthesis are redox-dependent in glomerular mesangial cells. Biochem J. 2004;381:231–239. doi: 10.1042/BJ20031614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haberl RL, Decker PJ, Einhaupl KM. Angiotensin degradation products mediate endothelium-dependent dilation of rabbit brain arterioles. Circ Res. 1991;68:1621–1627. doi: 10.1161/01.res.68.6.1621. [DOI] [PubMed] [Google Scholar]
- 23.Handa RK, Handa SE, Elgemark MK. Autoradiographic analysis and regulation of angiotensin receptor subtypes AT4, AT1, and AT1–7 in the kidney. Am J Physiol Renal Physiol. 2001;281:F936–F947. doi: 10.1152/ajprenal.2001.281.5.F936. [DOI] [PubMed] [Google Scholar]
- 24.Handa RK. Characterization and signaling of the AT4 receptor in human proximal tubule epithelial (HK-2) cells. J Am Soc Nephrol. 2001;12:440–449. doi: 10.1681/ASN.V123440. [DOI] [PubMed] [Google Scholar]
- 25.Inscho EW, Mason MJ, Schroeder AC, Deichmann PC, Stiegler KD, Imig JD. Agonist-induced calcium regulation in freshly isolated renal microvascular smooth muscle cells. J Am Soc Nephrol. 1997;8:569–579. doi: 10.1681/ASN.V84569. [DOI] [PubMed] [Google Scholar]
- 26.Khosla MC, Smeby RR, Bumpus FM. Structure-activity relationship in angiotensin analogs. In: Page IH, Bumpus FM, editors. Handbook of Experimental Pharmacology. XXXVII. Angiotensin. Berlin: Springer-Verlag; 1974. pp. 126–161. [Google Scholar]
- 27.Kono T, Ikeda F, Oseko F, Ohmori Y, Nakano R, Muranaka H, Taniguchi A, Imura H, Khosla MC, Bumpus FM. Biological activity of des-asp1-des-arg2-angiotensin II in man. Acta Endocrinol (Copenh) 1982;99:577–584. doi: 10.1530/acta.0.0990577. [DOI] [PubMed] [Google Scholar]
- 28.Le MT, Vanderheyden PM, Szaszak M, Hunyady L, Vauquelin G. Angiotensin IV is a potent agonist for constitutive active human AT1 receptors. Distinct roles of the N- and C-terminal residues of angiotensin II during AT1 receptor activation. J Biol Chem. 2002;277:23107–23110. doi: 10.1074/jbc.C200201200. [DOI] [PubMed] [Google Scholar]
- 29.Li YD, Block ER, Patel JM. Activation of multiple signaling modules is critical in angiotensin IV-induced lung endothelial cell proliferation. Am J Physiol Lung Cell Mol Physiol. 2002;283:L707–L716. doi: 10.1152/ajplung.00024.2002. [DOI] [PubMed] [Google Scholar]
- 30.Lochard N, Thibault G, Silversides DW, Touyz RM, Reudelhuber TL. Chronic production of angiotensin IV in the brain leads to hypertension that is reversible with an angiotensin II AT1 receptor antagonist. Circ Res. 2004;94:1451–1457. doi: 10.1161/01.RES.0000130654.56599.40. [DOI] [PubMed] [Google Scholar]
- 31.Loufrani L, Henrion D, Chansel D, Ardaillou R, Levy BI. Functional evidence for an angiotensin IV receptor in rat resistance arteries. J Pharmacol Exp Ther. 1999;291:583–588. [PubMed] [Google Scholar]
- 32.Petrescu G, Costuleanu M, Slatineanu SM, Costuleanu N, Foia L, Costuleanu A. Contractile effects of angiotensin peptides in rat aorta are differentially dependent on tyrosine kinase activity. J Renin Angiotensin Aldosterone Syst. 2001;2:180–187. doi: 10.3317/jraas.2001.025. [DOI] [PubMed] [Google Scholar]
- 33.Ruan X, Arendshort WJ. Calcium entry and mobilization signaling pathways in Ang II-induced renal vasoconstriction in vivo. Am J Physiol Renal Fluid Electrolyte Physiol. 1996;270:F398–F405. doi: 10.1152/ajprenal.1996.270.3.F398. [DOI] [PubMed] [Google Scholar]
- 34.Skurk T, Lee YM, Rohrig K, Hauner H. Effect of angiotensin peptides on PAI-1 expression and production in human adipocytes. Horm Metab Res. 2001;33:196–200. doi: 10.1055/s-2001-14948. [DOI] [PubMed] [Google Scholar]
- 35.Swanson GN, Hanesworth JM, Sardinia MF, Coleman JK, Wright JW, Hall KL, Miller-Wing AV, Stobb JW, Cook VI, Harding JW. Discovery of a distinct binding site for angiotensin II (3–8), a putative angiotensin IV receptor. Regul Pept. 1992;40:409–419. doi: 10.1016/0167-0115(92)90527-2. [DOI] [PubMed] [Google Scholar]
- 36.van Rodijnen WF, van Lambalgen TA, van Wijhe MH, Tangelder GJ, Ter Wee PM. Renal microvascular actions of angiotensin II fragments. Am J Physiol Renal Physiol. 2002;283:F86–F92. doi: 10.1152/ajprenal.00121.2001. [DOI] [PubMed] [Google Scholar]
- 37.Wright JW, Krebs LT, Stobb JW, Harding JW. The angiotensin IV system: functional implications. Front Neuroendocrinol. 1995;16:23–52. doi: 10.1006/frne.1995.1002. [DOI] [PubMed] [Google Scholar]
- 38.Zhu Z, Arendshort WJ. Angiotensin II-receptor stimulation of cytosolic calcium concentration in cultured renal resistence arterioles. Am J Physiol Renal Fluid Electrolyte Physiol. 1996;271:F1239–F1247. doi: 10.1152/ajprenal.1996.271.6.F1239. [DOI] [PubMed] [Google Scholar]
- 39.Zhuo JL, Ohishi M, Mendelsohn FA. Roles of AT1 and AT2 receptors in the hypertensive Ren-2 gene transgenic rat kidney. Hypertension. 1999;33:347–353. doi: 10.1161/01.hyp.33.1.347. [DOI] [PubMed] [Google Scholar]
- 40.Zhuo JL, Thomas D, Harris PJ, Skinner SL. The role of endogenous angiotensin II in the regulation of renal haemodynamics and proximal fluid reabsorption in the rat. J Physiol. 1992;453:1–13. doi: 10.1113/jphysiol.1992.sp019214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhuo JL, Song K, Abdelrahman A, Mendelsohn FA. Blockade by intravenous losartan of AT1 angiotensin II receptors in rat brain, kidney and adrenals demonstrated by in vitro autoradiography. Clin Exp Pharmacol Physiol. 1994;21:557–567. doi: 10.1111/j.1440-1681.1994.tb02555.x. [DOI] [PubMed] [Google Scholar]
- 42.Zhuo JL, Song K, Harris PJ, Mendelsohn FA. In vitro autoradiography reveals predominantly AT1 angiotensin II receptors in rat kidney. Renal Physiol Biochem. 1992;15:231–239. doi: 10.1159/000173458. [DOI] [PubMed] [Google Scholar]
- 43.Zhuo JL, Moeller I, Jenkins T, Chai SY, Allen AM, Ohishi M, Mendelsohn FAO. Mapping tissue angiotensin-converting enzyme and angiotensin AT1, AT2 and AT4 receptors. J Hypertens. 1997;16:2027–2037. doi: 10.1097/00004872-199816121-00026. [DOI] [PubMed] [Google Scholar]
- 44.Zhuo JL, Imig JD, Hammond TG, Orengo S, Benes E, Navar LG. Ang II accumulation in rat renal endosomes during Ang II-induced hypertension: role of AT1 receptor. Hypertension. 2002;39:116–121. doi: 10.1161/hy0102.100780. [DOI] [PubMed] [Google Scholar]