

Abstract
Environmental factors such as diet are known to play important roles in inflammatory bowel disease (IBD). Epidemiological studies have indicated that a high-fat diet is a risk factor for IBD. In addition, the balance between effector T cells (Teff) and regulatory T cells (Treg) contributes to the pathogenesis of mucosal inflammation. The aim of this study was to understand the mechanisms by which a high-fat diet can regulate susceptibility to intestinal inflammation. Wild-type C57BL/6 mice were fed either a commercial high-fat diet or a normal diet, then exposed to dextran sulphate sodium (DSS) to induce colonic inflammation. Intraepithelial lymphocytes (IEL) were isolated from the colon, and their phenotype and cytokine profile were analysed by flow cytometry. Mice receiving the high-fat diet were more susceptible to DSS-induced colitis. They had higher numbers of non-CD1d-restricted natural killer (NK) T cells in the colonic IEL, when compared to mice fed a normal diet. These cells expressed tumour necrosis factor (TNF)-α and interferon (IFN)-γ, which are up-regulated by high-fat diets. Mice fed the high-fat diet also had decreased levels of colonic Treg. Depletion of colonic NK T cells or adoptive transfer of Treg reduced the DSS colitis in these mice, and reduced the colonic expression of TNF-α and IFN-γ. We conclude that a high-fat diet can increase non-CD1d-restricted NK T cells and decrease Treg in the colonic IEL population. This altered colonic IEL population leads to increased susceptibility to DSS-induced colitis. This effect may help to explain how environmental factors can increase the susceptibility to IBD.
Keywords: nutrition, inflammatory bowel disease, intraepithelial lymphocytes
Introduction
Our current understanding of the pathogenesis of inflammatory bowel disease (IBD) suggests that nutrition and dietary factors play a significant role in this condition [1–4]. There is strong epidemiological evidence linking dietary factors to IBD: for example, the incidence of IBD is high in populations that consume western-style diets, and the incidence of IBD has risen sharply among those from developing countries who have migrated to developed countries or who have adopted industrialized urban lifestyles with concomitant change in dietary habits (e.g. ingesting more fast food with high fat content) [5]. High-fat diets are also associated with an increased risk of developing ulcerative colitis (UC) [2,6]. Despite strong evidence linking dietary factors to IBD, however, the mechanisms involved are still poorly understood.
It is well known that nutrition and dietary factors can modulate immune function [7]. Dietary fatty acids such as n-3 polyunsaturated fatty acids can exert an anti-inflammatory effect that is related to their ability to reduce proinflammatory cytokine production [8,9]. On the other hand, high-fat diets can cause increased liver inflammation that is associated with depletion of NK T cells from the liver [10].
One theory regarding the pathogenesis of IBD suggests that this condition represents an inappropriate and exaggerated mucosal immune response to normal intestinal flora that can be attributed, at least in part, to an imbalance between effector T cells (Teff) and regulatory T cells (Treg) [11]. In this view, experimental inflammation occurs as a result of either excessive Teff function [12] or deficient Treg function [13–15]. However, little is known about whether dietary factors can influence intestinal lymphocytes, particularly Teff and Treg.
In the present study, we have asked whether a high-fat diet can increase the risk of intestinal inflammation in a mouse model of colitis. Furthermore, we have examined whether the effects of the high-fat diet are a result of altered Teff and Treg function in the colon. We discovered that mice fed a high-fat diet exhibited more severe dextran sulphate sodium (DSS)-induced colitis than did those fed a normal diet, and this increased severity was associated with an alteration in the intraepithelial lymphocyte population in the colon. These findings may explain how high-fat (i.e. westernized) diets can lead to an increased risk of IBD.
Materials and methods
Animal experiments
Adult (6–8-week-old) male wild-type C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). The mice were fed commercial diets containing either a high amount of fat (50% of the total kcal, F3282, BioServ, Inc., Frenchtown, NJ, USA) or normal amount of fat (11% of the total kcal) for 4–6 weeks. CD1d-deficient mice on a C57BL/6 background were kindly provided by Dr Albert Bendelac [16]. Natural killer (NK) cell-deficient LY49a-transgenic (NKD) mice on a C57BL/6 background were kindly provided by Dr Wayne Yokoyama [17] and maintained in our laboratory. Both CD1d knock-out and NK cell-deficient mice were crossed extensively into the C57BL/6 background (more than 10 generations). All mice were maintained in a temperature- and light-controlled facility and allowed to consume water and pellet chow ad libitum. All animal experiments fulfilled National Institutes of Health (NIH) and Johns Hopkins University (JHU) criteria for the humane treatment of laboratory animals.
DSS-induced colitis
After receiving a high-fat or control diet for 4–6 weeks, mice were exposed to 2·5–3% (w/v) DSS (MW 36–50 kDa; MP Biomedicals, Solon, OH, USA) in their drinking water. At the end of the treatment period the colonic histology was evaluated in a blinded fashion by a gastrointestinal pathologist (M. T.) for severity of inflammation according to the following criteria: 0 = no active inflammation, 1 = cryptitis, 2 = crypt abscesses, 3 = erosions and or ulcers. The worst degree of inflammation observed was recorded as the score. Because C57BL/6 mice are sensitive to the effects of DSS in inducing colonic inflammation, the concentrations of DSS in the diet experiments were titrated. A subpathogenic concentration (2·5% w/v) was used to achieve a little inflammation in the normal diet group and to help distinguish the degree of inflammation between the two groups. The mice were allowed to consume the water with DSS ad libitum. Daily water consumption was recorded.
NK and NK T cell depletion
Mice received a tail vein injection of anti-NK1·1 antibody (50 μg/mouse; Pharmingen, San Diego, CA, USA) on the day before (−1) and day +3 of DSS treatment.
Isolation of colonic lymphocytes and cell-surface labelling
Colonic intraepithelial lymphocytes (IEL) were isolated as described previously [18], with minor modification. The colonic mucosa was dissociated in RPMI-1640 containing 10% heat-inactivated fetal calf serum (FCS), 1 mM dithiothreitol (DTT) (Sigma-Aldrich, St Louis, MO, USA) and 1 mM ethylenediamine tetraacetic acid (EDTA) for 20 min at room temperature. The cell preparation was passed through a glass wool column to remove debris. The IEL fraction was isolated from the interface between the 80% and 40% fractions of a Percoll gradient (Amersham Pharmacia Biotech, Piscataway, NJ, USA) after centrifugation at 2000 g for 30 min. A previous study showed a high purity of IEL with little contamination of lamina propria cells using this method [19].
After isolation, the IELs were labelled with various anti-mouse antibodies against NK1·1, CD3, CD4, CD8, CD25 (Pharmingen) and CD1 tetramer loaded with a ligand (PBS-57, an analogue of α-GalCer; NIH tetramer facility). The IELs were then evaluated by flow cytometry (Becton Dickinson, Palo Alto, CA, USA). Data were analysed using CellQuest software (Becton Dickinson).
Intracellular cytokines and forkhead box P3 (FoxP3) labelling
After isolation, the colonic IELs were incubated with lymphocyte activation cocktail, labelled with surface markers, permeabilized and labelled for intracellular cytokines with an intracellular staining kit (Pharmingen) according to the manufacturer's instructions. For intracellular FoxP3 staining, colonic IELs were permeabilized directly and stained using a FoxP3 staining kit (eBioscience, San Diego, CA, USA) according to the manufacturer's instructions.
Adoptive transfer of Treg cells
CD4+ CD25+ Treg were isolated from naive C57BL/6 mouse spleens using an MACS regulatory T cell isolation kit (Milltenyi Biotec, Auburn, CA, USA). A purity greater than 95% for the Treg separation was confirmed by CD4+ CD25+ FoxP3+ staining and flow cytometry. Before colitis induction, 1·5 × 106 CD4+ CD25+ Treg were injected into each recipient mouse via the tail vein; the same amount of CD4+ CD25– T cells was injected into each of the control mice.
Statistical analysis
All values are expressed as means ± standard deviation (s.d.). The group means were compared by Student's t-test using Microsoft Excel (Microsoft, Redmond, WA, USA). P-values of < 0·05 were considered statistically significant.
Results
A high-fat diet increases the susceptibility of mice to DSS-induced colitis
After 4–6 weeks, mice fed a high-fat diet gained more weight and developed more hyperlipidaemia, hyperglycaemia and insulin resistance than did the mice fed a normal diet, as described previously [10,20,21]. We saw no gross or histological differences between the colons of the mice after 4–6 weeks on the high-fat diet or normal diet (data not shown). However, when these mice were subjected to a DSS challenge, mice receiving the high-fat diet developed more severe colitis, as reflected by a significant colonic shortening (Fig. 1a), weight loss (Fig. 1b) and severe histological inflammation with crypt abscesses, mucosal erosions and/or ulcers when compared to those receiving the normal diet (Fig. 1c–e). The mice in both groups drank similar amounts of water (data not shown); therefore, it is unlikely that the increased inflammation in the mice receiving the high-fat diet was related to increased DSS ingestion. When mice on the high-fat diet were returned to a normal diet for 3–4 weeks, they lost the extra weight that they gained during the high-fat diet feeding, and were no longer susceptible to DSS (data not shown).
Fig. 1.
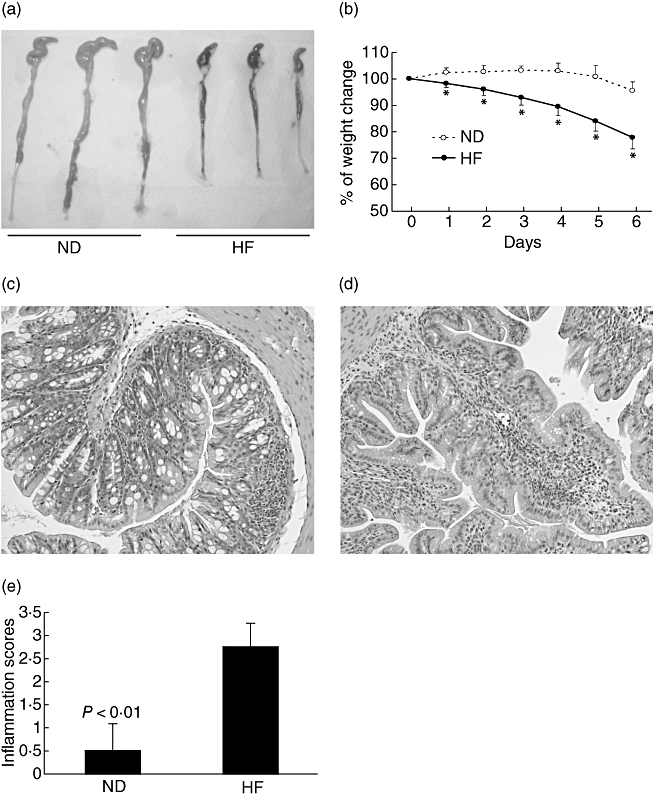
A high-fat diet increases the susceptibility of mice to dextran sulphate sodium (DSS)-induced colitis. Wild-type C57BL/6 mice were fed a normal diet (ND) or high-fat diet (HF) for 4–6 weeks and then exposed to subpathogenic DSS (2·5%, w/v) in drinking water for 6 days. (a) Gross anatomy of the mouse colon. Mice fed the high-fat diet have significant colonic shortening with visible colonic bleeding (black stool) in the lumen. (b) Mean [± standard deviation (s.d.)] change in body weight during the DSS treatment; *P < 0·01 (n = 10). (c, d) Haematoxylin and eosin staining of colonic sections (× 64) from mice fed a normal diet (c) or high-fat diet (d). (e) Mean (± s.d.) colonic inflammation scores for mice fed a normal diet or high-fat diet (n = 8).
A high-fat diet increases the number of non-classical NK T cells in the colon
We first evaluated the effect of a high-fat diet on colonic IEL populations. Unlike small intestinal IEL that are mainly T cells, the colonic IEL have a significant number of non-T cells, including NK cells (CD3– NK1·1+) and NK T cells (CD3+ NK1·1+) (Fig. 1). This may reflect a predominant innate immunity in response to high bacterial content of the colon. The high-fat diet increased the population of CD3+ NK1·1+ (NK T) cells significantly in the colonic IEL (Fig. 2a–b). The high-fat diet also increased the population of CD3– NK1·1+ (NK) cells in the colonic IEL (Fig. 2a–b). However, it was unlikely that colonic NK cells contributed to the increased susceptibility of high-fat fed mice to DSS-induced colitis, as depletion of NK cells in transgenic mice does not alter the susceptibility of these mice to DSS-induced colitis (see our data below). Unlike the classical CD1d-restricted NK T cells in the liver, the majority of the NK T cells up-regulated by the high-fat diet in the colon were not CD1d-restricted (Fig. 2c), based upon their inability to bind with the CD1 tetramer. In addition, the colonic NK T cells express both T cell receptor (TCR)α/β and TCRγ/δ (Fig. 2d) instead of invariant Vα14Jα18, as in classical CD1d-restricted NK T cells [22]. Classical CD1d-restricted NK T cells are either CD4+ or CD4– CD8– (Fig. 2e). The majority of colonic NK T cells were CD4–, but significant numbers of CD8+ cells were also present (Fig. 2f). Therefore, these cells represent a heterogeneous cell population that is distinguishable from the classical CD1d-restricted NK T cells, despite their expression of both the NK1·1 and TCR markers. A detailed description of different phenotypes of these cells is listed in Table 1.
Fig. 2.
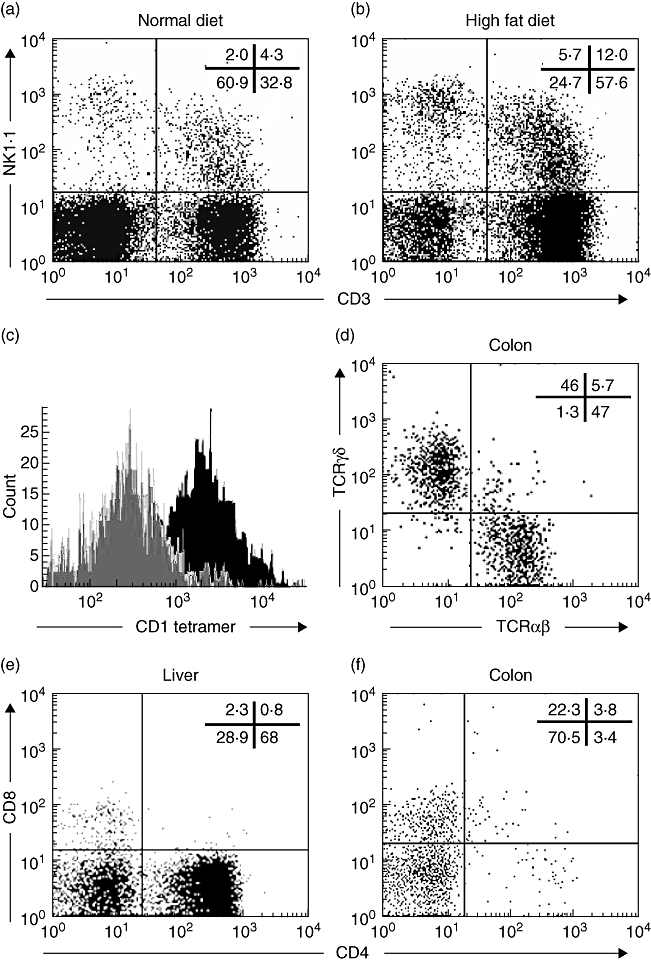
A high-fat diet increases the number of colonic non-classical natural killer (NK) T cells. Wild-type C57BL/6 mice were fed a normal diet or high-fat diet. After 4–6 weeks, colonic intraepithelial lymphocytes (IEL) and liver total mononuclear cells were isolated and analysed by flow cytometry. Numbers of total colonic IEL were similar for mice fed the high-fat and normal diets; therefore, the percentage of NK T cells reflects the number of NK T cells in the colon. The experiments were repeated five times (n = 10). (a, b) Representative dot plots of the CD3 and NK1·1 staining of colonic IEL with the percentage of each quadrant listed. P < 0·05 in NK T cells (CD3+, NK1·1+) from normal diet versus high-fat diet. (c) Histogram of the CD1d tetramer staining of colonic NK T cells (grey) and hepatic NK T cells (black) as gated CD3+ NK1·1+ cells. (d–f) Representative dot plots of hepatic (e) and colonic (d, f) NK T cells (gated CD3+ NK1·1+ cells) with the percentage of each quadrant listed.
Table 1.
Phenotypes of the isolated cells.
| Name | CD1d tetramer | NK 1·1 | CD3 | CD4 | CD8 | TCRαβ | TCRγδ | CD25 | FoxP3 |
|---|---|---|---|---|---|---|---|---|---|
| Colonic non-classical NK T cells | – | + | + | – | ± | ± | ± | n.a. | n.a. |
| Hepatic classical NK T cells | + | + | + | ± | – | + | – | n.a. | n.a. |
| Tregs | n.a. | n.a. | + | + | n.a. | n.a. | n.a. | + | + |
| Teffs | n.a. | n.a. | + | + | n.a. | n.a. | n.a. | – | – |
+: Positive; –: negative; ±, partially positive; n.a.: not tested; NK: natural killer; TCR: T cell receptor; FoxP3: forkhead box P3.
Colonic non-classical NK T cells contribute to increased DSS-induced colitis
In order to confirm that these non-classical NK T cells in colonic IEL actually contribute to the increased DSS-induced colitis in mice receiving the high-fat diet, we performed cell-depletion experiments. Because there is no reagent that targets specifically non-classical NK T cells, we accomplished the depletion by injecting the mice with anti-NK1·1 antibody. After depletion with anti-NK1·1 antibody, mice receiving the high-fat diet became more resistant to DSS-induced colitis. They had less weight loss and a markedly lower degree of inflammation in the colon (Fig. 3a,b). However, systematic administration of NK1·1 antibody also depletes NK cells and classical CD1d-restricted NK T cells. Therefore, in order to demonstrate that the protective effect of the NK1·1 antibody was not due to depletion of NK cells and/or classical NK T cells, we utilized transgenic mice that had been depleted selectively of either NK cells (NKD mice) [17] or classical NK T cells (CD1d knock-out mice). NKD and NKDXCD1dko mice had a similar degree of weight loss and colonic inflammation to the wild-type mice after they were fed with a high-fat diet and challenged with DSS. However, when NKDXCD1dko mice were given anti-NK1·1 antibody to eliminate the only non-classical NK T cells remaining in these mice, they had significantly less weight loss and colonic inflammation (Fig. 3a,b). Thus, the protective effect of the anti-NK1·1 antibody that we observed is probably the result of depletion of non-classical NK T cells rather than NK cells or classical NK T cells. Similar experiments were repeated in mice fed a normal diet to avoid any additional effect of the high-fat diet on non-classical NK T cells. These normal diet-fed mice were also exposed to a standard pathogenic concentration (3%) of DSS for a longer exposure time (9 days) to induce adequate colonic inflammation. The NKD mice, CD1dko and NKDXCD1dko mice all demonstrated a susceptibility to DSS-induced colitis that was similar to that of wild-type mice (Fig. 3c,d). When NKDXCD1dko mice were treated with anti-NK1·1 antibody, they showed significant improvement in weight loss and colonic inflammation (Fig. 3c,d). These results indicated that it was non-classical NK T cells, but not NK cells or classical CD1d-restricted NK T cells, that contributed to high-fat diet-induced increased susceptibility of colitis.
Fig. 3.
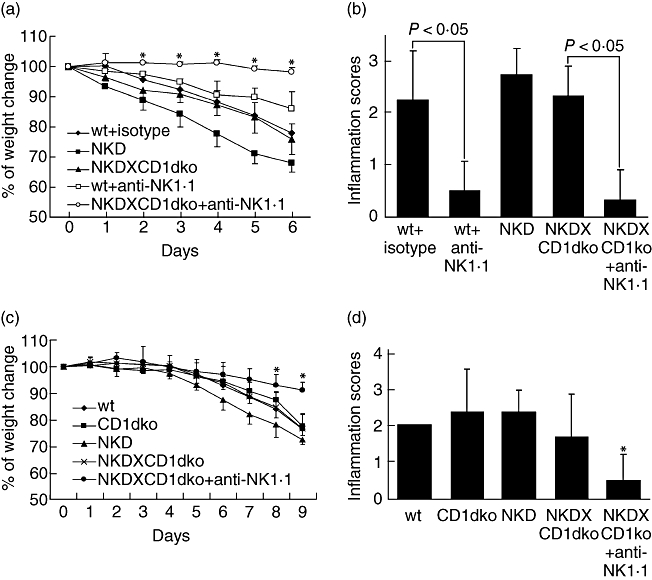
Colonic non-classical NK T cells contribute to increased dextran sulphate sodium (DSS)-induced colitis. Wild-type mice, or transgenic mice that had been selectively depleted of either natural killer (NK) cells (NKD), or NK cells and classical NK T cells (NKDXCD1dko) were fed either a high-fat diet (a, b) or normal diet (c, d). These mice were then exposed to a standard concentration (c, d) or a low concentration of DSS (a, b) in drinking water. Some of the mice were also received anti-NK1·1 antibody or isotype Ig control injections before they were exposed to DSS. (a) Mean [± standard deviation (s.d.)] change in weight during the low concentration of DSS treatment after being fed a high-fat diet (n = 18). (b) Mean (± s.d.) colonic inflammation scores for mice injected with anti-NK1 or isotype Ig control (n = 18). (c) Mean (± s.d.) change in weight of mice treated with a standard concentration of DSS and normal diet (n = 17). (d) Mean (± s.d.) change in colonic inflammation score during the standard concentration of DSS treatment with normal diet (n = 17). *P < 0·05 versus wt.
A high-fat diet also reduces the number of colonic Treg
Because intestinal inflammation can result not only from excessive Teff function [12] but also from deficient Treg function [13–15], we evaluated the Treg population in the colonic IEL of mice receiving the high-fat diet. Compared to those on the normal diet, the mice receiving the high-fat diet had a significantly reduced number of CD4+ CD25+ T cells in the colonic IEL (Fig. 4a,b,d). The majority of these cells were also positive for intracellular FoxP3 (Fig. 4c), indicating that they were Treg.
Fig. 4.
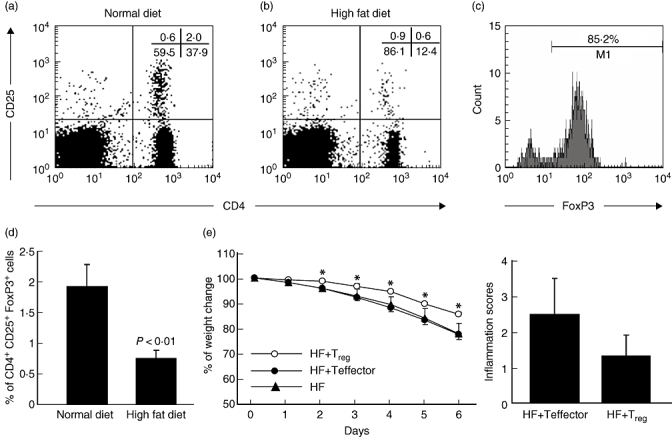
A high-fat diet (HF) reduces the number of colonic regulatory T cells. Wild-type C57BL/6 mice were fed a normal diet or high-fat diet, and colonic intraepithelial lymphocytes (IEL) were isolated and analysed by flow cytometry. The total numbers of colonic IEL were similar in both groups; therefore, the percentage of T regulatory cells (Treg) reflects the number of Treg in the colon. (a, b) Representative flow cytometry data are shown; the experiments were repeated five times (n = 10). (c) Intracellular forkhead box P3 (FoxP3) staining of CD4+ CD25+ cells from normal diet-fed mouse colonic IEL. Intracellular FoxP3 staining of CD4+ CD25+ cells from high-fat diet-fed mice became technically infeasible due to the rarity of colonic CD4+ CD25+ cells. (d) Mean [± standard deviation (s.d.)]% of colonic Treg (CD4+ CD25+ FoxP3+) cells from three experiments are graphed (n = 10). In separate experiments (e, f), either CD4+ CD25+ cell (HF + Treg) or CD4+ CD25– cells (HF + Teffector) were transferred adoptively into mice that had received a high-fat diet and were then exposed to 2·5% dextran sulphate sodium (DSS) (w/v) in drinking water. (e) Mean (± s.d.) change in weight during the DSS treatment; *P < 0·01 (n = 7). (f) Mean (± s.d.) colonic inflammation scores from the same mice (n = 7).
To confirm further the effect of reducing colonic regulatory T cells on increased susceptibility to colitis, we performed adoptive transfer experiments. CD4+ CD25+ Treg were isolated from the spleens of mice fed the normal diet and transferred adoptively to the mice fed the high-fat diet before the recipient mice were subjected to DSS challenge. Control mice received CD4+ CD25– cells. As expected, adoptive transfer of Treg reduced significantly the degree of weight loss associated with the colitis (Fig. 4e). It also lessened the degree of histological inflammation, although the difference between the control and experimental groups was not statistically significant (Fig. 4f). Adoptive transfer of regulatory T cells did not reduce the number of non-classical NK T cells in the colon of the mice fed high-fat diets (data not shown). However, it did reduce the production of inflammatory cytokines by non-classical NK T cells (see below).
A high-fat diet increases the colonic expression of inflammatory cytokines
To characterize further the mechanisms governing the increased susceptibility to colitis in mice fed a high-fat diet, we analysed the intracellular cytokine production by colonic IEL. Mice fed a high-fat diet displayed a significantly higher expression of inflammatory cytokine-producing cells, i.e. interferon (IFN)-γ and tumour necrosis factor (TNF)-α, within their total colonic IEL (Fig. 5b,d) and colonic non-classical NK T cells (Fig. 5c,e). Adoptive transfer of Treg reduced significantly the expression of these inflammatory cytokines (Fig. 5a–e).
Fig. 5.
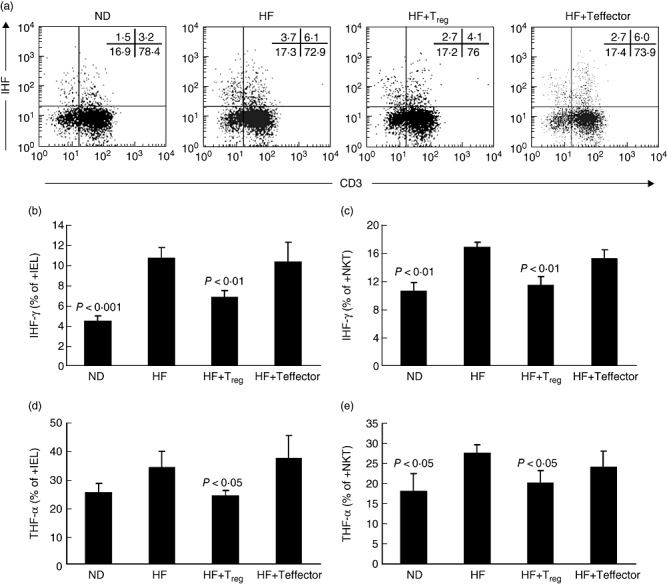
A high-fat diet (HF) increases the colonic expression of inflammatory cytokines. Wild-type C57BL/6 mice were fed either a normal diet (ND) or high-fat diet. Some of the mice fed the high-fat diet also received either CD4+ CD25+ cells [HF + T regulatory (Treg] or CD4+ CD25– cells (HF + Teffector) by adoptive transfer. The mice were then exposed to dextran sulphate sodium (DSS), and the intracellular cytokines tumour necrosis factor (TNF)-α (d, e) and interferon (IFN)-γ (b, c) from total colonic intraepithelial lymphocytes (IEL) (b, d) and colonic natural killer (NK) T cells (c, e, gated as CD3+ NK1·1+) cells were measured by flow cytometry. (a) Representative flow cytometry data for intracellular IFN-γ staining of cells from mice fed a normal diet or high-fat diet, with or without adoptive transfer. (b–e) Mean (± standard deviation) levels of intracellular TNF-α or IFN-γ in the four groups of mice (n = 12). P-values are ND or HF + Treg versus HF.
Discussion
The pathogenesis of IBD is believed to involve a deregulation of intestinal immune responses that may be genetic and/or environmental in nature. High-fat diets are an important environmental risk factor that contribute to the high incidence of IBD in western, industrialized countries [2,23]. Despite the strong epidemiological evidence linking dietary factors to IBD, little is known about the mechanisms underlying this relationship. Although there is no perfect animal model to mimic the condition of human IBD, the mouse model of DSS-induced colitis has been used widely to study intestinal inflammation. In order to analyse the effects of a high-fat diet on susceptibility to DSS-induced colitis, we used a mouse model with many characteristics of the human metabolic syndromes induced by high-fat diets. In the present study, we have shown that a high-fat diet can alter the nature of the lymphocyte population in the colon, increasing the number of non-classical NK T cells and decreasing the number of Treg in the colonic IEL. This alteration in colonic IEL leads in turn to greater production of inflammatory cytokines. It is known that the balance between Teff and Treg plays a significant role in regulating mucosal inflammation and that experimental inflammation occurs as a result of either excessive effector T cell function [12] or deficient regulatory T cell function [13–15]. The percentage of CD4+ cells in the colonic IEL is higher than those in the small intestinal IEL, which is consistent with previous findings [24]. However, most previous studies have described that CD4+ CD25+ FoxP3+ cells are located in the lamina propria [25]. To our knowledge, this is the first report to identify the colonic intraepithelial regulatory T cells that may contribute to the pathogenesis of colitis. It also provides a potential novel mechanism by which high-fat diets can increase the risk of intestinal inflammation. This is also the first time that non-classical NK T cells have been shown to play the role as Teff in the intestinal mucosa of a murine model of experiment colitis. A previous study has shown that non-classical NK T cells contribute the pathogenesis of UC in humans. Human colonic non-classical NK T cells have atypical T helper 2 (Th2) response and cytotoxic potential [26]. In addition, dietary factors are known to increase the risk of UC in humans [6]. Therefore, it is possible that, in humans, dietary factors also alter colonic non-classical NK T cells that contribute to the pathogenesis of UC. However, definitive study in humans is still lacking. Our murine model may provide a useful animal model to study these mechanisms.
NK T cells express both T cell surface markers (e.g. CD3) and NK cell-surface markers (i.e. NK1·1). They are a heterogeneous population that can be divided into classical (CD1-dependent) and non-classical (CD1-independent) subsets [27,28]. In mice, classical NK T cells express predominantly a conserved αβ TCR with invariant Vα14Jα18 and are referred to as Vα14iNK T, or iNK T cells. They represent 30–40% of liver αβ T cells and are present in lower numbers in other lymphoid organs [27,29]. iNK T cells recognize glycolipid antigens bound to the major histocompatibility complex (MHC) class I-like molecule CD1d, which is expressed with β2 microglobulin on various antigen-presenting cells [22]. Upon activation, these iNK T cells produce both Th2 cytokines (e.g. IL-4) and Th1 cytokines (e.g. IFN-γ) [30]. In contrast to these cells, relatively little is known about the non-classical T cells, which also express NK markers but are not CD1d-restricted. These cells are abundant in the colon, as shown in the current study and demonstrated previously by other investigators [31]. Unlike classical NK T cells, these cells produce a large amount of IFN-γ but little IL-4 [31]. Earlier studies also have shown that CD8+ T cells can be induced to express the NK1·1 marker [32–34]. It is possible that some of these cells are active CD8+ T cells with NK1·1 expression. However, a large number of these cells are both CD4– CD8–. We believe that the non-classical NK T cells in the colon represent a unique group of Teff that regulate intestinal inflammation, and further studies are needed to characterize these cells.
T cells, including NK T cells, play an important role in the pathogenesis of intestinal inflammation [12,35–38]. Previous studies have generally indicated that NK T cells play a protective role in regulating intestinal inflammation. Activation of NK T cells has been shown to protect mice from DSS-induced colitis [36], adoptive transfer of NK T cells is able to alleviate the symptoms of colitis [37,39] and caloric restriction ameliorates experimental colitis by increasing the number of hepatic NK T cells [38]. However, all these studies have focused on CD1-dependent, classical NK T cells. In the current study, we have demonstrated that the levels of CD1-independent, non-classical NK T cells are modulated by dietary fat and that an increase in the number of these cells is associated directly with intestinal inflammation. At present, the relationship between the classical and non-classical NK T cells is largely unknown, and whether the classical NK T cells regulate the non-classical NK T cells, or vice versa, is still under investigation.
In summary, we have shown that a high-fat diet can alter the balance of Teff and Treg in the colon by increasing the number of non-classical NK T cells and decreasing the number of Treg in the colonic IEL. These changes lead to increased levels of inflammatory cytokines and an increased susceptibility to DSS-induced colitis. The detailed mechanisms responsible for the dietary regulation of the colonic non-classical NK T cells are still under investigation.
Acknowledgments
We would like thank Ms Alena Calm and the Hopkins Digestive Disease Basic Research Development Center (R24 DK064388-04) for providing technical support and Dr Deborah McClellan for editorial assistance. This work was supported partially by an AGA Fellowship/Faculty Transition Award (Z. L.), AGA Roche Research Scholar Award in Liver Diseases (Z. L.), Shanghai Leading Academic Discipline Project Grant (Y0205, X. M.) and an award from National Natural Science Foundation of China (no. 30571730, X. M.).
References
- 1.Sakamoto N, Kono S, Wakai K, et al. Dietary risk factors for inflammatory bowel disease: a multicenter case–control study in Japan. Inflamm Bowel Dis. 2005;11:154–63. doi: 10.1097/00054725-200502000-00009. [DOI] [PubMed] [Google Scholar]
- 2.Reif S, Klein I, Lubin F, Farbstein M, Hallak A, Gilat T. Pre-illness dietary factors in inflammatory bowel disease. Gut. 1997;40:754–60. doi: 10.1136/gut.40.6.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tragnone A, Valpiani D, Miglio F, et al. Dietary habits as risk factors for inflammatory bowel disease. Eur J Gastroenterol Hepatol. 1995;7:47–51. [PubMed] [Google Scholar]
- 4.Russel MG, Engels LG, Muris JW, et al. ‘Modern life’ in the epidemiology of inflammatory bowel disease: a case–control studywith special emphasis on nutritional factors. Eur J Gastroenterol Hepatol. 1998;10:243–9. doi: 10.1097/00042737-199803000-00010. [DOI] [PubMed] [Google Scholar]
- 5.Loftus EV., Jr Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology. 2004;126:1504–17. doi: 10.1053/j.gastro.2004.01.063. [DOI] [PubMed] [Google Scholar]
- 6.Geerling BJ, Dagnelie PC, Badart-Smook A, Russel MG, Stockbrugger RW, Brummer RJ. Diet as a risk factor for the development of ulcerative colitis. Am J Gastroenterol. 2000;95:1008–13. doi: 10.1111/j.1572-0241.2000.01942.x. [DOI] [PubMed] [Google Scholar]
- 7.Grimble RF. Nutritional modulation of immune function. Proc Nutr Soc. 2001;60:389–97. doi: 10.1079/pns2001102. [DOI] [PubMed] [Google Scholar]
- 8.Grimble RF. Dietary lipids and the inflammatory response. Proc Nutr Soc. 1998;57:535–42. doi: 10.1079/pns19980078. [DOI] [PubMed] [Google Scholar]
- 9.Endres S, Ghorbani R, Kelley VE, et al. The effect of dietary supplementation with n-3 polyunsaturated fatty acids on the synthesis of interleukin-1 and tumor necrosis factor by mononuclear cells. N Engl J Med. 1989;320:265–71. doi: 10.1056/NEJM198902023200501. [DOI] [PubMed] [Google Scholar]
- 10.Li Z, Soloski MJ, Diehl AM. Dietary factors alter hepatic innate immune system in mice with nonalcoholic fatty liver disease. Hepatology. 2005;42:880–5. doi: 10.1002/hep.20826. [DOI] [PubMed] [Google Scholar]
- 11.Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu Rev Immunol. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
- 12.Wirtz S, Finotto S, Kanzler S, et al. Cutting edge: chronic intestinal inflammation in STAT-4 transgenic mice: characterization of disease and adoptive transfer by TNF- plus IFN-gamma-producing CD4+ T cells that respond to bacterial antigens. J Immunol. 1999;162:1884–8. [PubMed] [Google Scholar]
- 13.Faubion WA, De Jong YP, Molina AA, et al. Colitis is associated with thymic destruction attenuating CD4+25+ regulatory T cells in the periphery. Gastroenterology. 2004;126:1759–70. doi: 10.1053/j.gastro.2004.03.015. [DOI] [PubMed] [Google Scholar]
- 14.Mottet C, Uhlig HH, Powrie F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J Immunol. 2003;170:3939–43. doi: 10.4049/jimmunol.170.8.3939. [DOI] [PubMed] [Google Scholar]
- 15.Liu H, Hu B, Xu D, Liew FY. CD4+CD25+ regulatory T cells cure murine colitis: the role of IL-10, TGF-{beta}, and CTLA4. J Immunol. 2003;171:5012–17. doi: 10.4049/jimmunol.171.10.5012. [DOI] [PubMed] [Google Scholar]
- 16.Park S-H, Guy-Grand D, Lemonnier FA, Wang C-R, Bendelac A, Jabri B. Selection and expansion of CD8{alpha}/{alpha}1 T cell receptor {alpha}/{beta}1 intestinal intraepithelial lymphocytes in the absence of both classical major histocompatibility complex class I and nonclassical CD1 molecules. J Exp Med. 1999;190:885–90. doi: 10.1084/jem.190.6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim S, Iizuka K, Aguila HL, Weissman IL, Yokoyama WM. In vivo natural killer cell activities revealed by natural killer cell-deficient mice. Proc Natl Acad Sci USA. 2000;97:2731–6. doi: 10.1073/pnas.050588297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davies A, Lopez-Briones S, Ong H, et al. Infection-induced expansion of a MHC Class Ib-dependent intestinal intraepithelial gammadelta T cell subset. J Immunol. 2004;172:6828–37. doi: 10.4049/jimmunol.172.11.6828. [DOI] [PubMed] [Google Scholar]
- 19.Davies MD, Parrott DM. Preparation and purification of lymphocytes from the epithelium and lamina propria of murine small intestine. Gut. 1981;22:481–8. doi: 10.1136/gut.22.6.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harte RA, Kirk EA, Rosenfeld ME, LeBoeuf RC. Initiation of hyperinsulinemia and hyperleptinemia is diet dependent in C57BL/6 mice. Horm Metab Res. 1999;31:570–5. doi: 10.1055/s-2007-978797. [DOI] [PubMed] [Google Scholar]
- 21.Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6 mice. Diabetes. 1988;37:1163–7. doi: 10.2337/diab.37.9.1163. [DOI] [PubMed] [Google Scholar]
- 22.Lantz O, Bendelac A. An invariant T cell receptor alpha chain is used by a unique subset of major histocompatibility complex class I-specific CD4+ and CD4-8- T cells in mice and humans. J Exp Med. 1994;180:1097–106. doi: 10.1084/jem.180.3.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hunter JO. Nutritional factors in inflammatory bowel disease. Eur J Gastroenterol Hepatol. 1998;10:235–7. doi: 10.1097/00042737-199803000-00008. [DOI] [PubMed] [Google Scholar]
- 24.Camerini V, Panwala C, Kronenberg M. Regional specialization of the mucosal immune system. Intraepithelial lymphocytes of the large intestine have a different phenotype and function than those of the small intestine. J Immunol. 1993;151:1765–76. [PubMed] [Google Scholar]
- 25.Makita S, Kanai T, Nemoto Y, et al. Intestinal lamina propria retaining CD4+CD25+ regulatory T cells is a suppressive site of intestinal inflammation. J Immunol. 2007;178:4937–46. doi: 10.4049/jimmunol.178.8.4937. [DOI] [PubMed] [Google Scholar]
- 26.Fuss IJ, Heller F, Boirivant M, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–7. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Godfrey DI, MacDonald HR, Kronenberg M, Smyth MJ, Van Kaer L. NKT cells: what's in a name? Nat Rev Immunol. 2004;4:231–7. doi: 10.1038/nri1309. [DOI] [PubMed] [Google Scholar]
- 28.Eberl G, Lees R, Smiley ST, Taniguchi M, Grusby MJ, MacDonald HR. Tissue-specific segregation of CD1d-dependent and CD1d-independent NK T cells. J Immunol. 1999;162:6410–19. [PubMed] [Google Scholar]
- 29.Klugewitz K, Adams DH, Emoto M, Eulenburg K, Hamann A. The composition of intrahepatic lymphocytes: shaped by selective recruitment? Trends Immunol. 2004;25:590–4. doi: 10.1016/j.it.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 30.Benlagha K, Kyin T, Beavis A, Teyton L, Bendelac A. A thymic precursor to the NK T cell lineage. Science. 2002;296:553–5. doi: 10.1126/science.1069017. [DOI] [PubMed] [Google Scholar]
- 31.Bannai M, Kawamura T, Naito T, et al. Abundance of unconventional CD8(+) natural killer T cells in the large intestine. Eur J Immunol. 2001;31:3361–9. doi: 10.1002/1521-4141(200111)31:11<3361::aid-immu3361>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 32.Kambayashi T, Assarsson E, Michaelsson J, et al. Emergence of CD8+ T cells expressing NK cell receptors in influenza A virus-infected mice. J Immunol. 2000;165:4964–9. doi: 10.4049/jimmunol.165.9.4964. [DOI] [PubMed] [Google Scholar]
- 33.Assarsson E, Kambayashi T, Sandberg JK, et al. CD8+ T cells rapidly acquire NK1.1 and NK cell-associated molecules upon stimulation in vitro and in vivo. J Immunol. 2000;165:3673–9. doi: 10.4049/jimmunol.165.7.3673. [DOI] [PubMed] [Google Scholar]
- 34.Slifka MK, Pagarigan RR, Whitton JL. NK markers are expressed on a high percentage of virus-specific CD8+ and CD4+ T cells. J Immunol. 2000;164:2009–15. doi: 10.4049/jimmunol.164.4.2009. [DOI] [PubMed] [Google Scholar]
- 35.Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C.B-17 SCID mice. Int Immunol. 1993;5:1461–71. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- 36.Saubermann LJ, Beck P, De Jong YP, et al. Activation of natural killer T cells by alpha-galactosylceramide in the presence of CD1d provides protection against colititis in mice. Gastroenterology. 2000;119:119–128. doi: 10.1053/gast.2000.9114. [DOI] [PubMed] [Google Scholar]
- 37.Shibolet O, Kalish Y, Klein A, et al. Adoptive transfer of ex vivo immune-programmed NKT lymphocytes alleviates immune-mediated colitis. J Leukoc Biol. 2004;75:76–86. doi: 10.1189/jlb.0703351. [DOI] [PubMed] [Google Scholar]
- 38.Shibolet O, Alper R, Avraham Y, Berry EM, Ilan Y. Immunomodulation of experimental colitis via caloric restriction: role of Nk1.1+ T cells. Clin Immunol. 2002;105:48–56. doi: 10.1006/clim.2002.5260. [DOI] [PubMed] [Google Scholar]
- 39.Menachem Y, Trop S, Kolker O, et al. Adoptive transfer of NK 1.1+ lymphocytes in immune-mediated colitis: a pro-inflammatory or a tolerizing subgroup of cells? Microbes Infect. 2005;7:825–35. doi: 10.1016/j.micinf.2005.03.019. [DOI] [PubMed] [Google Scholar]