

Abstract
Asthma is a heterogeneous disease that has been increasing in incidence throughout western societies and cytokines, including proinflammatory tumour necrosis factor alpha (TNF-α), have been implicated in the pathogenesis of asthma. Anti-TNF-α therapies have been established successfully in the clinic for diseases such as rheumatoid arthritis and Crohn's disease. TNF-α-blocking strategies are now being trialled in asthma; however, their mode of action is poorly understood. Based on the observation that TNF-α induces lymph node hypertrophy we have attempted to investigate this as a mechanism of action of TNF-α in airway inflammation by employing two models of murine airway inflammation, that we have termed short and long models, representing severe and mild/moderate asthma, respectively. The models differ by their immunization schedules. In the short model, characterized by eosinophilic and neutrophilic airway inflammation the effect of TNF-α blockade was a reduction in draining lymph node (DLN) hypertrophy, eosinophilia, interleukin (IL)-5 production and immunoglobulin E (IgE) production. In the long model, characterized by eosinophilic inflammation, TNF-α blockade produced a reduction in DLN hypertrophy and IL-5 production but had limited effects on eosinophilia and IgE production. These results indicate that anti-TNF-α can suppress DLN hypertrophy and decrease airway inflammation. Further investigations showed that anti-TNF-α-induced inhibition of DLN hypertrophy cannot be explained by preventing l-selectin-dependent capture of lymphocytes into the DLN. Given that overall TNF blockade was able to suppress the short model (severe) more effectively than the long model (mild/moderate), the results suggest that TNF-α blocking therapies may be more effective in the treatment of severe asthma.
Keywords: airway hypersensitivity, airway inflammation, IgE, T cells, TNF-α
Introduction
Asthma is a major global disease, whose pathophysiology remains poorly understood, despite its high prevalence. Over the last two decades its prevalence has doubled in Western countries and the associated health-care costs are escalating, emphasizing the need for effective treatment of the disease [1]. Asthma is a T helper 2 (Th2)-mediated disease, characterized by increased airway eosinophilia, goblet cell hyperplasia with associated mucus production, neutrophilia and increased numbers of lung macrophages and activated mast cells [2].
Cytokines play an important role in airway inflammation in asthma by promoting the development, differentiation, recruitment, activation and survival of inflammatory cells. Although Th2 cytokines including interleukin (IL)-4, IL-5 and IL-13 are important in asthma [3], tumour necrosis factor (TNF)-α, a cytokine usually associated with Th1 responses, has also been implicated in the inflammatory response seen in asthma [4]. TNF-α is a potent proinflammatory cytokine with immunoregulatory activities, and is produced by many cell types including monocytes, macrophages, lymphocytes, neutrophils, eosinophils and mast cells [5–7]. In the lung, TNF-α is synthesized and stored mainly in mast cells and alveolar macrophages [8]. It functions as a chemoattractant for neutrophils and monocytes, increasing microvascular permeability, and activating T cells, eosinophils and mast cells.
Furthermore, there is increased expression of TNF-α in the airway of asthmatic patients compared to normal subjects [8,9]. Mast cells are unique in being the only tissue-resident cells with granules containing preformed TNF-α, and consequently during the early stages of inflammation or infection they may be the sole source of this cytokine [10]. TNF-α is able to stimulate the production of IL-8, regulated upon activation normal T cell expressed and secreted (RANTES) and granulocyte–macrophage colony-stimulating factor (GM-CSF) by airway epithelial cells, which increase the expression of vascular adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1), which are involved in the recruitment of inflammatory cells to the airways [11]. TNF-α is also thought to be involved in airway remodelling through its effects on fibroblasts and by inducing eosinophils to release matrix metalloproteinase-9 [12]. These data have initiated a number of small trials with anti-TNF-α therapy in human asthmatics. Two recent trials looking at severe asthmatics showed improvement in lung function [13,14], while others have shown no benefit [13,15,16]. These have been small studies and large-scale double-blind randomized controlled trials are awaited.
Despite the apparent efficacy observed, the mechanism of action of anti-TNF-α therapy remains unclear. McLachlan and colleagues have postulated an association between mast cells, TNF-α and the regulation of inflammation and immunity [17,18]. They demonstrated in a murine model that injection of Escherichia coli into the footpad led to hypertrophy of the draining popliteal lymph node. This was attributed to an influx of T and B lymphocytes, and associated with mast cell degranulation in the footpad. Mast cell-deficient mice (W/W v) mice showed significantly reduced lymph node swelling which was reversed by reconstitution of the mice with wild-type mast cells, but not with TNF-deficient mast cells. As lymph nodes are the central inductive site of the adaptive immune response, facilitating the T cell–antigen-presenting cell (APC) synapse, such effects could be significant in a wide range of conditions including infection, autoimmunity and allergies where there is immune activation.
This study set out to investigate the mechanism of action of TNF-α blockade in airway inflammation. We investigated whether the mechanism of action of TNF-α blockade could be explained by modulating lymphocyte trafficking in the draining lymph nodes using murine models of airway inflammation in which ovalbumin (OVA)-specific T cell receptor (TCR) Tg (transgenic) T cells (KJ1·26 + T cells) have been transferred adoptively into naive mice, allowing us to track the movement of antigen-specific T cells. Two models have been employed: a short model, with both eosinophils and neutrophils driving the inflammatory pathology that is considered representative of the pathology seen in severe asthma; and a long model, where eosinophils are associated with the inflammatory response, that is considered more representative of mild to moderate asthma.
Materials and methods
Animals
BALB/c (H-2d/d) mice were purchased from Harlan-Olac (Oxon, Bicester, UK). Mice homozygous for the cOVA peptide323−339/I-Ad specific DO11·10 TCR transgenes [detected using the clonotypic monoclonal antibody (mAb) KJ1·26] on the BALB/c background [19] were used as donors. All animals were specified pathogen-free and were maintained under standard animal holding with water and chow ad libitum at the University of Glasgow Central Research Facilities in accordance with local and UK Home Office regulations.
Preparation of cell suspensions for adoptive transfer
Peripheral lymph nodes (PLN) (axillary, brachial, inguinal, cervical), mesenteric lymph nodes and spleens from DO11·10 BALB/c mice were pooled and prepared as single cell suspensions, by passing through a Nitex sieve (Cadisch Precision Meshes, London, UK) using a syringe plunger, and washed in sterile RPMI-1640 (Invitrogen Life Technologies, Paisley, UK). The percentages of KJ1·26+ CD4+ DO11·10 T cells were determined by flow cytometric analysis as described below and made up to required cell volume in phosphate-buffered saline (PBS).
OVA model of airway inflammation
Tg T cells, 3 × 106, in 200 μl were injected intravenously (i.v.) into age-matched naive BALB/c recipients on day −1. The mice were then immunized with an intraperitoneal (i.p.) injection of 100 μg chicken OVA (OVA, Fraction V; Sigma-Aldrich, Poole, UK) in a 1% alum suspension (Brenntag Biosector, Frederikssund, Denmark) made up to a volume of 200 μl. The short model was injected on day 0 only while the long model was injected on days 0, 7 and 14. Mice were anaesthetized i.p. with 250 μl avertin (1 : 1 w/v solution of 2,2,2-tribromoethanol in tert-amyl alcohol; Sigma-Aldrich) and challenged intranasally (i.n.) in 30 μl PBS with 50 μg OVA plus 2·5 μg lipopolysaccharide (LPS) or 50 μg OVA alone on days 10 or 21 for the short and long models, respectively. Mice were euthanased at various time-points by administration of a lethal dose of avertin.
Anti-TNF-α treatment
Mice were treated with 0·5 mg/kg recombinant human TNF receptor p80 Fc fusion protein (sTNFR:Fc) [20] (Etanercept, Wyeth Pharmaceuticals, Taplow, UK), as described in Results. Control mice were given 0·5 mg/kg human immunoglobulin (IgG).
Bronchoalveolar lavage (BAL) and blood sampling
Immediately after termination with avertin, blood was collected by cardiac puncture. BAL was harvested in two 0·8 ml aliquots of fluorescence activated cell sorter (FACS) buffer [2% fetal calf serum (FCS), 0·05% sodium azide in PBS]. Individual BAL samples were kept on ice before processing by centrifugation at 400 g for 5 min. The supernatants were collected and volumes measured before storage at −70°C until assayed for cytokines. The cell pellets were resuspended in 1 ml of PBS and counted in a haemocytometer. Cytospin preparations were prepared in a Cytospin (Thermo Shandon, Runcorn, UK) and were stained with Rapi-diff II (Triangle Biomedical Sciences, Durham, NC, USA). Blinded differential cell counting was performed using standard morphological criteria.
Lung histology
After BAL sampling, lungs were removed and inflated with 1 ml of 10% neutral-buffered formalin, fixed in 10% neutral-buffered formalin for 72 h and sections (8 μm) were stained with haematoxylin and eosin (H&E) and examined under × 20–100 magnification. Peribronchial and perivascular inflammation was assessed.
Lung scoring
Lung scoring was based on a system developed by Howie and colleagues [21]. H&E-stained sections of whole lungs were scored over 10 consecutive fields at × 200 magnification. In order to be included, each field had to contain a complete transection of at least one bronchiole less than half a field width in diameter, a blood vessel and an alveolar airway. Inflammatory cell infiltrate, i.e. the number of immune cells present, was evaluated around the blood vessel walls (perivascular compartment), the bronchiolar epithelium and the peri-bronchiolar alveolar tissue. Inflammation was also scored on the basis of increased alveolar wall thickness.
Flow cytometry
PLN, draining lymph node (DLN) and BAL fluid were harvested the day before airway challenge and between days 1 and 7 after airway challenge. Cell suspensions were prepared as described above and analysed by flow cytometry for CD4, and antigen-specific T cells were detected using the monoclonal antibody KJ1·26 as described previously [22]. In some experiments the transgenic T cells were stained for l-selectin. Where lungs were analysed, the thoracic cage was opened and the lungs were separated carefully from surrounding tissue by blunt dissection and removed en bloc with the heart. For flow cytometry, lungs were processed as described for lymph nodes.
Enzyme-linked immunosorbent assay (ELISAs)
To detect OVA-specific IgE in serum samples, Immulon 2 plates (Costar; Corning, NY, USA) were coated with OVA (20 μg/ml) in PBS at 4°C overnight. Plates were then washed at least three times with PBS/Tween 0·05% (Sigma-Aldrich) before being blocked with PBS–FCS 10% (v/v) for 1 h at 37°C. Plates were washed and incubated with diluted serum samples for 3 h at 37°C before further washing. IgE levels in serum were determined by incubation with biotinylated anti-IgE (1/8000; BD Pharmingen, Oxford, UK) for 1 h at 37°C. Plates were then washed and incubated with extravidin-horseradish peroxidase (HRP) (1/1000; Sigma-Aldrich) for 1 h at 37°C then washed again and tetramethylbenzidine (TMB) microwell peroxidase substrate (Kirkegaard & Perry Laboratories, Gaithersburg, MD, USA) was added to stop the reaction. Light absorbance was read on a plate reader at 630 nm.
Statistics
Results are expressed as mean ± standard error of the mean (s.e.m.). Significance was tested using Student's unpaired t-test. A value of P < 0·05 was regarded as significant.
Results
Effect of sTNFR:Fc in the short model of airways inflammation
The short adoptive transfer model consists of one immunization on day 1 and one intranasal airway challenge with OVA plus LPS on day 10, resulting in airway inflammation characterized by marked eosinophilia and an accompanying neutrophil influx (Fig. 1). The resulting inflammatory pathology is similar to that seen in severe asthmatic patients.
Fig. 1.
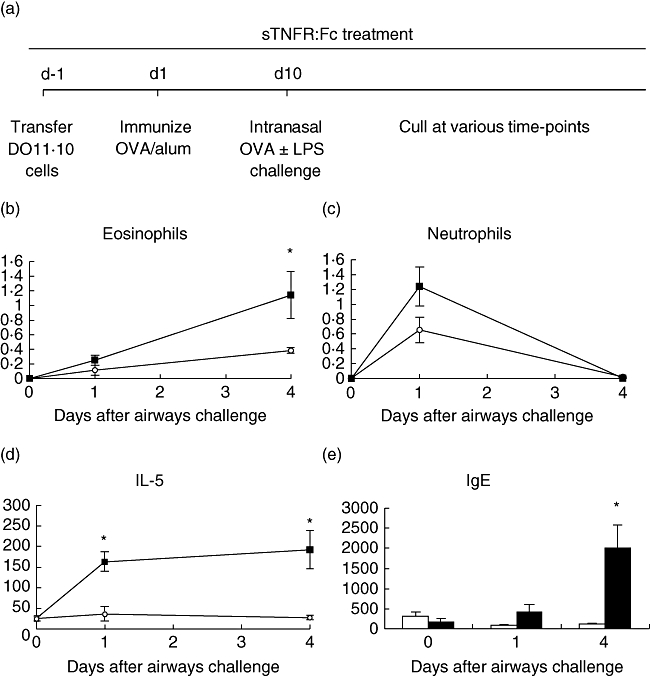
Ovalbumin (OVA)-specific (3 × 106) T cells were transferred adoptively into naive Balb/c recipients, which were then immunized with 100 μg OVA and 1% alum by subcutaneous (s.c.) injection. Ten days later, mice were challenged by intranasal administration of 50 μg OVA plus 2·5 μg lipopolysaccharide. Mice were treated throughout with 50 μg tumour necrosis factor (TNF) receptor p80 Fc fusion protein (sTNFR:Fc) (○) by s.c. injection every 2 days. Control mice were given 50 μg human IgG (▪) instead of sTNFR:Fc (a). Bronchoalveolar lavage (BAL) fluid was sampled and eosinophil (b) and neutrophil (c) content was analysed by cytospin preparation. BAL fluid was sampled and analysed for interleukin-5 production (d). Serum from the same mice was analysed for OVA-specific immunoglobulin E (e). Results are represented as mean ± standard error of the mean (*P < 0·05), n = 6 per group.
Administration of sTNFR:Fc (Etanercept), daily throughout the short model resulted in: a significant decrease in eosinophilia (Fig. 1b); a trend towards a decrease in neutrophil numbers (Fig. 1c); a significant reduction of IL-5 in BAL fluid (Fig. 1d) compared to no effect on IFN-γ (data not shown); and a significant reduction in OVA-specific anti-IgE production at day 4 post-airways challenge (Fig. 1e).
DLN weights in sTNFR:Fc-treated mice remained unchanged, while weight increase was observed in the control group (Fig. 2a). There was no difference observed in non-DLN weights between the two groups (Fig. 2b). The same trend was seen with cellularity of the DLN, where there were significantly more cells in the control group (Fig. 2c), and no difference was observed in the non-DLN (Fig. 2d). Antigen-specific T cell analysis by flow cytometry showed no significant difference between treated and control groups at day 4; however, subsequent data have shown that a transient peak of antigen-specific cells in control groups occurs between days 2 and 3 (data not shown).
Fig. 2.
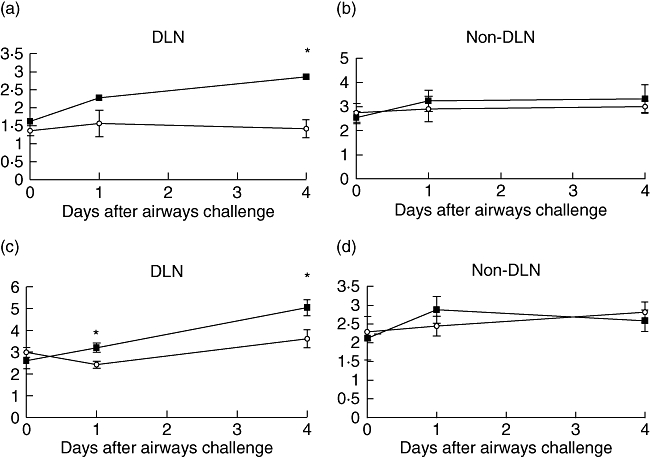
Ovalbumin (OVA)-specific (3 × 106) T cells were transferred adoptively into naive Balb/c recipients, which were then immunized with 100 μg OVA and 1% alum by intraperitoneal injection. Ten days later, mice were challenged by intranasal administration of 50 μg OVA plus 2·5 μg lipopolysaccharide. Mice were treated throughout with 50 μg tumour necrosis factor (TNF) receptor p80 Fc fusion protein (sTNFR:Fc) (○) by subcutaneous (s.c.) injection every 2 days. Control mice were given 50 μg human immunoglobulin (▪) instead of sTNFR:Fc. Draining and non-draining lymph nodes were removed and weighed (a, b), and cellularity was determined (c, d). Results are represented as mean ± standard error of the mean (*P < 0·05), n = 6 per group.
Effect of sTNFR:Fc in the long model of airways inflammation
The long adoptive transfer model consists of immunization on days 0, 7 and 14 and an intranasal challenge with OVA on day 21; this results in airways inflammation characterized by lymphocyte activation and eosinophilia (Fig. 3). This airway inflammation may be representative of that seen in mild to moderate asthmatic patients [23–25].
Fig. 3.
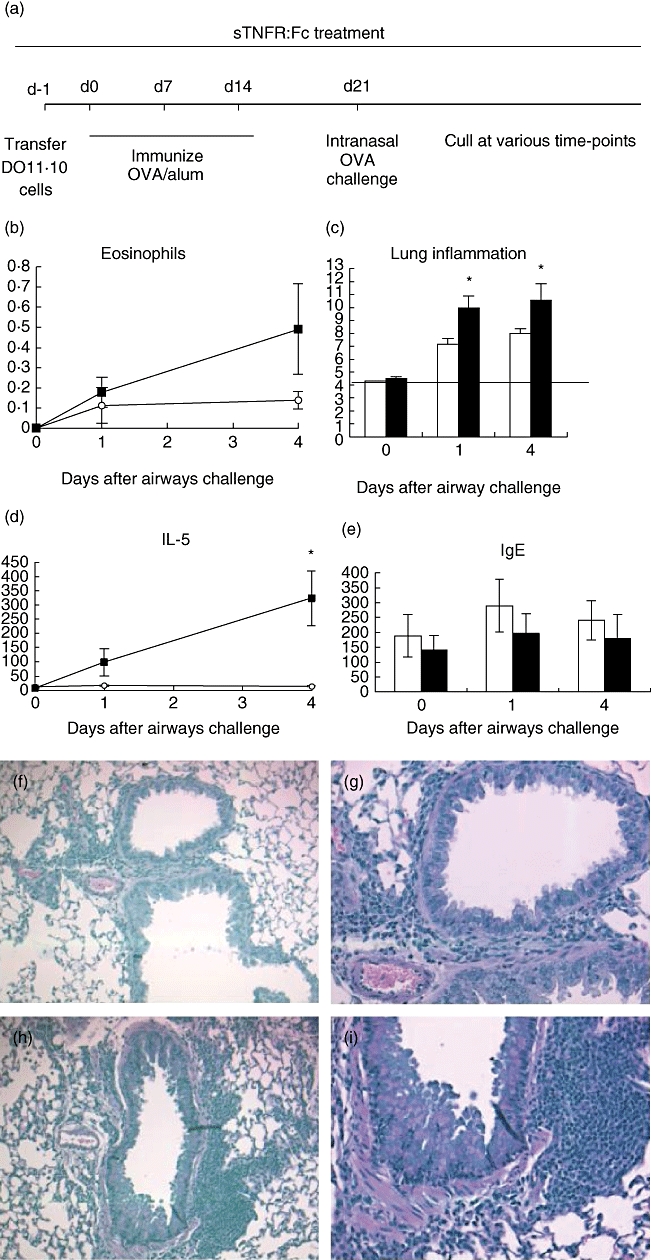
Ovalbumin (OVA)-specific (3 × 106) T cells were transferred adoptively into naive Balb/c recipients, which were then immunized with 100 μg OVA and 1% alum by subcutaneous (s.c.) injection on three occasions, each a week apart. One week after the final immunization, mice were challenged by intranasal administration of 50 μg OVA. Mice were treated throughout with 50 μg tumour necrosis factor (TNF) receptor p80 Fc fusion protein (sTNFR:Fc) (○ or white bars) by s.c. injection every 2 days. Control mice were given 50 μg human immunoglobulin (▪ or black bars) instead of sTNFR:Fc (a). Bronchoalveolar lavage (BAL) fluid was sampled and eosinophil content was analysed by cytospin preparation (b). Lung sections were scored for inflammation (c). Solid line represents baseline pathology. BAL fluid was sampled and analysed for interleukin-5 production (d). Serum from the same mice was analysed for OVA-specific immunoglobulin E (e). Lung sections were stained with haematoxylin and eosin. Sections show sTNFR:Fc- (f, g) and IgG (h, i)-treated mice at day 4 post-airways challenge at 10× and 20× magnification. Results are represented as mean ± standard error of the mean (*P < 0·05), n = 3–6 per group.
As TNF-α could play a role in the induction and/or effector mechanisms of asthma, we blocked its action during either or both of these phases. Our investigations show that TNF-α blockade during either the induction or effector phases caused no reduction in pathology associated with the disease (data not shown). However, blockade during the induction phase only prevented lymph node hypertrophy (data not shown).
Administration of TNF-α blockade daily throughout the long model resulted in a trend for reducing BAL eosinophilia (Fig. 3b); however, the reduction was not statistically significant and less marked than seen in the short model. Histological analysis of inflammation in lung sections showed that sTNFR:Fc treatment produced a significant reduction in lung pathology compared with the control group (Fig. 3c). IL-5 was reduced significantly in the BAL from sTNFR:Fc-treated mice (Fig. 3d), while IFN-γ was undetectable in either of the treated or control groups (data not shown). sTNFR:Fc treatment had no significant effect on serum OVA-specific IgE production (Fig. 3e). Decreased cell infiltrate and inflammation in sTNFR:Fc-treated mice can be seen in H&E-stained sections, compared to the control group (Fig. 3f–i).
The weight and cellularity of the DLNs in the sTNFR:Fc-treated group was decreased, while there was no difference in the non-DLNs. (Fig. 4). Antigen-specific T cell analysis by flow cytometry, showed no significant difference between treated and control groups at day 4 (data not shown); however, this may be the result of late sampling, as described above.
Fig. 4.
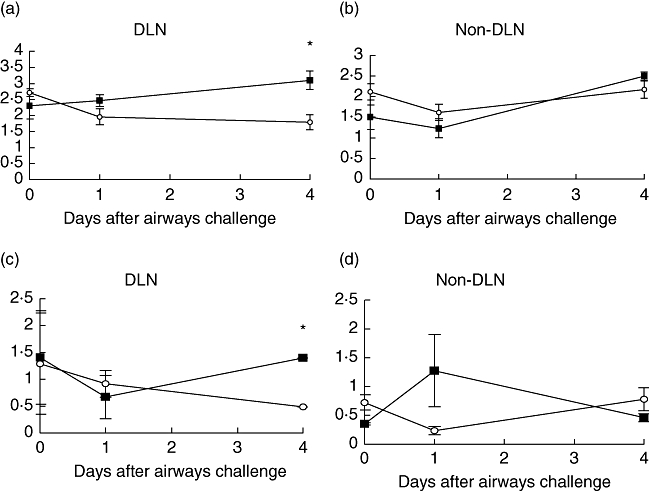
Ovalbumin (OVA)-specific (3 × 106) T cells were transferred adoptively into naive Balb/c recipients, which were then immunized with 100 μg OVA and 1% alum by subcutaneous (s.c.) injection on three occasions, each a week apart. One week after the final immunization, mice were challenged by intranasal administration of 50 μg OVA. Mice were treated throughout with 50 μg tumour necrosis factor (TNF) receptor p80 Fc fusion protein (sTNFR:Fc) (○) by s.c. injection every 2 days. Control mice were given 50 μg human IgG (▪) instead of sTNFR:Fc. Draining and non-draining lymph nodes were removed and weighed (a, b) and cellularity was determined (c, d). Results are represented as mean ± standard error of the mean (*P < 0·05), n = 6 per group.
l-Selectin expression on antigen-specific T cells
Antigen-specific T cells in DLN, non-DLN and lung tissue were assessed by flow cytometry for the expression of l-selectin low (effector/memory) and l-selectin high (naive) cells. There was an increase in the KJ1·26+, l-selectin low cells post-challenge in the DLN and lungs but not the non-DLN (Fig. 5). However, there was no effect of sTNFR:Fc treatment on KJ1·26+, l-selectin low cells. There was no effect post-challenge on the percentage of l-selectin high KJ1·26+ cells in any location treated with or without sTNFR:Fc (data not shown).
Fig. 5.
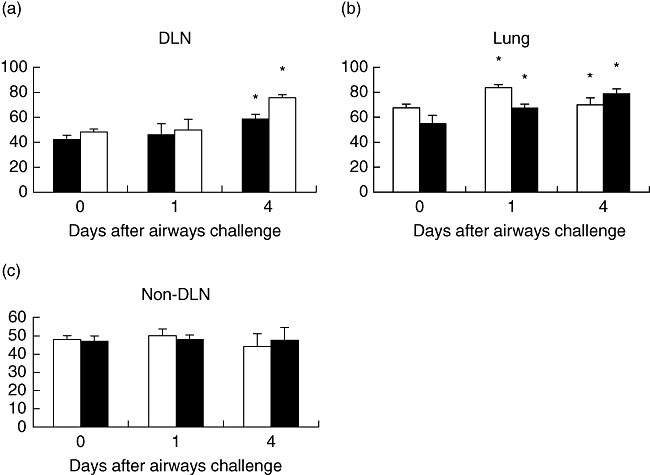
Ovalbumin (OVA)-specific (3 × 106) T cells were transferred adoptively into naive Balb/c recipients, which were then immunized with 100 μg OVA and 1% alum by subcutaneous (s.c.) injection. Ten days later, mice were challenged by intranasal administration of 50 μg OVA plus 2·5 μg lipopolysaccharide. Mice were treated throughout with 50 μg tumour necrosis factor (TNF) receptor p80 Fc fusion protein (sTNFR:Fc) (black) by s.c. injection every 2 days. Control mice were given 50 μg human immunoglobulin (white) instead of anti-TNF-α. Draining lymph nodes (a), lung tissue (b) and non-draining lymph nodes (c) were removed and l-selectin low expression (effector/memory) by OVA-specific T cell numbers were determined by flow cytometry. Results are represented as mean ± standard error of the mean (*P < 0·05), n = 3 per group.
Discussion
Based on preclinical studies, anti-TNF-α drugs are now being investigated as a therapy for asthma and recent clinical trials have shown beneficial effects in asthmatic patients [14,26]. In other trials, however, anti-TNF-α treatment did not show efficacy [15]. Although the reasons for the discrepancies are not established, the different types of anti-TNF-α inhibitors used and dosing regimens may be important contributory factors. Interestingly, clinical studies using differing strategies for TNF-α blockade have been shown to have differential effects in disease types; for example, anti-TNF-α antibody but not soluble receptor is beneficial in Crohn's disease [27,28].
Preclinical studies examining the role of TNF-α in airway inflammation models have mainly shown its deficiency or inhibition results in suppression of eosinophilia [29–31]; however, in one study using TNF-α receptor-deficient mice suppression was not observed [32]. The model used, the strategy of TNF-α inhibition or the dosing regimen employed may all contribute to discrepancies in results. Whereas many studies have used TNF receptor-deficient mice or anti-TNF-α antibodies, we have used Etanercept, which differs by binding to, and functionally inhibiting, soluble receptors [29–32].
In the short model employed in the studies reported here TNF-α blockade resulted in a significant reduction in BAL eosinophilia, IL-5 and antigen-specific IgE production. This was accompanied by a decrease in weight and cellularity of the draining lymph nodes. In the long model, representative of mild or moderate asthma, TNF-α blockade prevented lymph node hypertrophy accompanied by a decrease in IL-5 production, a reduction in lung inflammation, but a less marked reduction of BAL eosinophilia. This correlates with the recent studies suggesting that anti-TNF-α therapies are more beneficial in severe asthmatics compared to patients that exhibit mild or moderate symptoms [23–25].
These results indicate that TNF-α blockade suppresses immune airway inflammation but suppression of eosinophilic inflammation is more effective in a neutrophilic environment. Neutrophilic airway inflammation is found more often in severe asthma that provides linkage with our preclinical observations to the two clinical trials showing effects of TNF-α blockade in severe asthma [33,34]. The suppression of DLN hypertrophy in both the short and long models by TNF-α blockade is associated with decreased cellularity and we postulated that this a result of modulating lymphocyte trafficking into the draining lymph nodes.
Lymphocyte trafficking is not a random event, whereas naive T cells appear to circulate without preference for any one group of lymph nodes; the memory T cells return preferentially to the tissues associated with the lymph node groups in which they became sensitized [35]. l-Selectin mediates arteriolar and venular rolling on inflamed pulmonary endothelium [36–38], and is important in lymphocyte migration during an allergic inflammatory response [39]. Many studies demonstrate that in the absence of l-selectin there is reduced inflammation [40–42]. Some studies in murine models of asthma have shown that l-selectin is required for the development of airway hyperresponsiveness (AHR) but not airway inflammation [43]. In our studies we found no effect of TNF-α blockade on l-selectin expression.
Further studies will be required to establish if TNF-α blockade may modulate expression of other receptors involved in lymphocyte trafficking, such as sphingosine 1 phosphate receptors, important in lymphocyte egress from the lymph nodes [16].
In summary, TNF-α blockade in murine models of airway inflammation resulted in inhibition of DLN hypertrophy, reduction of DLN cellularity and an inhibition of airway inflammation. Inhibition of eosinophilic inflammation was more marked when the model was associated with a neutrophilic inflammation element, and this parallels observations found in the clinic where severe asthmatics with neutrophilic airway inflammation appear more responsive to anti-TNF-α therapy [14]. Preliminary studies investigating the mechanism of action of TNF-α blockade have shown that expression of l-selectin, a key receptor for lymphocyte capture into the lymph node, was not affected. An understanding of the mechanisms of TNF-α blockade will facilitate optimal therapeutic regimens in the clinic for allergic and autoimmune disease.
References
- 1.Schiller JS, Adams PF, Nelson ZC. Summary health statistics for the U.S. population: National Health Interview Survey, 2003. Vital Health Stat 10. 2005;224:1–104. [PubMed] [Google Scholar]
- 2.Hogg JC. Pathology of asthma. J Allergy Clin Immunol. 1993;92:1–5. doi: 10.1016/0091-6749(93)90029-f. [DOI] [PubMed] [Google Scholar]
- 3.Foster PS, Martinez-Moczygemba M, Huston DP, Corry DB. Interleukins-4, -5, and -13: emerging therapeutic targets in allergic disease. Pharmacol Ther. 2002;94:253–64. doi: 10.1016/s0163-7258(02)00220-6. [DOI] [PubMed] [Google Scholar]
- 4.Meiler F, Zimmermann M, Blaser K, Akdis CA, Akdis M. T-cell subsets in the pathogenesis of human asthma. Curr Allergy Asthma Rep. 2006;6:91–6. doi: 10.1007/s11882-006-0045-0. [DOI] [PubMed] [Google Scholar]
- 5.Gordon JR, Galli SJ. Release of both preformed and newly synthesized tumor necrosis factor alpha (TNF-alpha)/cachectin by mouse mast cells stimulated via the Fc epsilon RI. A mechanism for the sustained action of mast cell-derived TNF-alpha during IgE-dependent biological responses. J Exp Med. 1991;174:103–7. doi: 10.1084/jem.174.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Costa JJ, K Matossian K, Resnick MB, et al. Human eosinophils can express the cytokines tumor necrosis factor-alpha and macrophage inflammatory protein-1 alpha. J Clin Invest. 1993;91:2673–84. doi: 10.1172/JCI116506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bazzoni F, Beutler B. The tumor necrosis factor ligand and receptor families. N Engl J Med. 1996;334:1717–25. doi: 10.1056/NEJM199606273342607. [DOI] [PubMed] [Google Scholar]
- 8.Bradding P, Roberts JA, Britten KM, et al. Interleukin-4-, 5, and -6 and tumor necrosis factor-alpha in normal and asthmatic airways: evidence for the human mast cell as a source of these cytokines. Am J Respir Cell Mol Biol. 1994;10:471–80. doi: 10.1165/ajrcmb.10.5.8179909. [DOI] [PubMed] [Google Scholar]
- 9.Thomas PS. Tumour necrosis factor-alpha: the role of this multifunctional cytokine in asthma. Immunol Cell Biol. 2001;79:132–40. doi: 10.1046/j.1440-1711.2001.00980.x. [DOI] [PubMed] [Google Scholar]
- 10.Bischoff SC, Lorentz A, Schwengberg S, Weier G, Raab R, Manns MP. Mast cells are an important cellular source of tumour necrosis factor alpha in human intestinal tissue. Gut. 1999;44:643–52. doi: 10.1136/gut.44.5.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lassalle P, Gosset P, Delneste Y, et al. Modulation of adhesion molecule expression on endothelial cells during the late asthmatic reaction: role of macrophage-derived tumour necrosis factor-alpha. Clin Exp Immunol. 1993;94:105–10. doi: 10.1111/j.1365-2249.1993.tb05985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwingshackl A, Duszyk M, Brown N, Moqbel R. Human eosinophils release matrix metalloproteinase-9 on stimulation with TNF-alpha. J Allergy Clin Immunol. 1999;104:983–9. doi: 10.1016/s0091-6749(99)70079-5. [DOI] [PubMed] [Google Scholar]
- 13.Erin EM, Leaker BR, GC Nicholson GC, et al. The effects of a monoclonal antibody directed against tumor necrosis factor-alpha in asthma. Am J Respir Crit Care Med. 2006;174:753–62. doi: 10.1164/rccm.200601-072OC. [DOI] [PubMed] [Google Scholar]
- 14.Howarth PH, Babu KS, Arshad HS, et al. Tumour necrosis factor (TNFalpha) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax. 2005;60:1012–18. doi: 10.1136/thx.2005.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rouhani FN, Meitin CA, Kaler M, Miskinis-Hilligoss D, Stylianou M, Levine SJ. Effect of tumor necrosis factor antagonism on allergen-mediated asthmatic airway inflammation. Respir Med. 2005;99:1175–82. doi: 10.1016/j.rmed.2005.02.031. [DOI] [PubMed] [Google Scholar]
- 16.Idzko M, Hammad H, van Nimwegen M, et al. Local application of FTY720 to the lung abrogates experimental asthma by altering dendritic cell function. J Clin Invest. 2006;116:2935–44. doi: 10.1172/JCI28295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McLachlan JB, Hart JP, Pizzo SV, et al. Mast cell-derived tumor necrosis factor induces hypertrophy of draining lymph nodes during infection. Nat Immunol. 2003;4:1199–205. doi: 10.1038/ni1005. [DOI] [PubMed] [Google Scholar]
- 18.Choo-Kang BS, Hutchison S, Nickdel MB, et al. TNF-blocking therapies: an alternative mode of action? Trends Immunol. 2005;26:518–22. doi: 10.1016/j.it.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Murphy KM, Heimberger AB, Loh DY. Induction by antigen of intrathymic apoptosis of CD4+CD8+TCRlo thymocytes in vivo. Science. 1990;250:1720–3. doi: 10.1126/science.2125367. [DOI] [PubMed] [Google Scholar]
- 20.Mohler KM, Torrance DS, Smith CA, et al. Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. J Immunol. 1993;151:1548–61. [PubMed] [Google Scholar]
- 21.Fisher CE, Ahmad SA, Fitch PM, Lamb JR, Howie SE. FITC-induced murine pulmonary inflammation: CC10 up-regulation and concurrent Shh expression. Cell Biol Int. 2005;29:868–76. doi: 10.1016/j.cellbi.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 22.Smith KM, McAskill F, Garside P. Orally tolerized T cells are only able to enter B cell follicles following challenge with antigen in adjuvant, but they remain unable to provide B cell help. J Immunol. 2002;168:4318–25. doi: 10.4049/jimmunol.168.9.4318. [DOI] [PubMed] [Google Scholar]
- 23.Wenzel SE, Schwartz LB, Langmack EL, et al. Evidence that severe asthma can be divided pathologically into two inflammatory subtypes with distinct physiologic and clinical characteristics. Am J Respir Crit Care Med. 1999;160:1001–8. doi: 10.1164/ajrccm.160.3.9812110. [DOI] [PubMed] [Google Scholar]
- 24.Jatakanon A, Uasuf C, Maziak W, Lim S, Chung KF, Barnes PJ. Neutrophilic inflammation in severe persistent asthma. Am J Respir Crit Care Med. 1999;160:1532–9. doi: 10.1164/ajrccm.160.5.9806170. [DOI] [PubMed] [Google Scholar]
- 25.Ordonez CL, Shaughnessy TE, Matthay MA, Fahy JV. Increased neutrophil numbers and IL-8 levels in airway secretions in acute severe asthma: clinical and biologic significance. Am J Respir Crit Care Med. 2000;161:1185–90. doi: 10.1164/ajrccm.161.4.9812061. [DOI] [PubMed] [Google Scholar]
- 26.Berry MA, Hargadon B, Shelley M, Parker D, Shaw DE, Green RH. Evidence of a role of tumour necrosis factor in refractory asthma. N Engl J Med. 2006;381:77–80. doi: 10.1056/NEJMoa050580. [DOI] [PubMed] [Google Scholar]
- 27.Rutgeerts P, Feagan BG, Lichtenstein GR, et al. Comparison of scheduled and episodic treatment strategies of infliximab in Crohn's disease. Gastroenterology. 2004;126:402–13. doi: 10.1053/j.gastro.2003.11.014. [DOI] [PubMed] [Google Scholar]
- 28.Sandborn WJ, Feagan BG, Hanauer SB, et al. An engineered human antibody to TNF (CDP571) for active Crohn's disease: a randomized double-blind placebo-controlled trial. Gastroenterology. 2001;120:1330–8. doi: 10.1053/gast.2001.24042. [DOI] [PubMed] [Google Scholar]
- 29.Zuany-Amorim C. Induction of TNF-alpha autoantibody production by AutoVac TNF106: a novel therapeutic approach for the treatment of allergic diseases. Int Arch Allergy Immunol. 2004;133:154–63. doi: 10.1159/000076441. [DOI] [PubMed] [Google Scholar]
- 30.Choi IW, Sun K, Kim YS, et al. TNF-alpha induces the late-phase airway hyperresponsiveness and airway inflammation through cytosolic phospholipase A(2) activation. J Allergy Clin Immunol. 2005;116:537–43. doi: 10.1016/j.jaci.2005.05.034. [DOI] [PubMed] [Google Scholar]
- 31.Broide BH, Stachnick G, Castaneda D, Nayar J, Sriramarao P. Inhibition of eosinophilic inflammation in allergen-challenged TNF receptor p55/p75 − and TNF receptor p55-deficient mice. Am J Respir Cell Mol Biol. 2001;24:304–11. doi: 10.1165/ajrcmb.24.3.4071. [DOI] [PubMed] [Google Scholar]
- 32.Rudmann DG, Moore MW, Tepper JS, et al. Modulation of allergic inflammation in mice deficient in TNF receptors. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1047–57. doi: 10.1152/ajplung.2000.279.6.L1047. [DOI] [PubMed] [Google Scholar]
- 33.Kon OM, Sihra BS, Compton CH, Leonard TB, Kay AB, Barnes NC. Randomised, dose-ranging, placebo-controlled study of chimeric antibody to CD4 (keliximab) in chronic severe asthma. Lancet. 1998;352:1109–13. doi: 10.1016/S0140-6736(97)12261-9. [DOI] [PubMed] [Google Scholar]
- 34.Howarth PH, Babu K, Arshad HS, Lau L, Buckley M, McConnell W. Tumour necrosis factor (TNF alpha) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax. 2005;60:1012–18. doi: 10.1136/thx.2005.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wardlaw AJ, Guillen C, Morgan A. Mechanisms of T cell migration to the lung. Clin Exp Allergy. 2005;35:4–7. doi: 10.1111/j.1365-2222.2005.02139.x. [DOI] [PubMed] [Google Scholar]
- 36.Gebb SA, Graham JA, Hanger CC, et al. Sites of leukocyte sequestration in the pulmonary microcirculation. J Appl Physiol. 1995;79:493–7. doi: 10.1152/jappl.1995.79.2.493. [DOI] [PubMed] [Google Scholar]
- 37.Yamaguchi K, Nishio K, Sato N, et al. Leukocyte kinetics in the pulmonary microcirculation: observations using real-time confocal luminescence microscopy coupled with high-speed video analysis. Lab Invest. 1997;76:809–22. [PubMed] [Google Scholar]
- 38.Nishio K, Suzuki Y, Auki T, et al. Differential contribution of various adhesion molecules to leukocyte kinetics in pulmonary microvessels of hyperoxia-exposed rat lungs. Am J Respir Crit Care Med. 1998;157:599–609. doi: 10.1164/ajrccm.157.2.9704102. [DOI] [PubMed] [Google Scholar]
- 39.Keramidaris E, TD Merson TD, Steeber DA, Tedder TF, Tang ML. L-selectin and intercellular adhesion molecule 1 mediate lymphocyte migration to the inflamed airway/lung during an allergic inflammatory response in an animal model of asthma. J Allergy Clin Immunol. 2001;107:734–8. doi: 10.1067/mai.2001.114050. [DOI] [PubMed] [Google Scholar]
- 40.Tang ML, Hale LP, Steeber DA, Tedder TF. L-selectin is involved in lymphocyte migration to sites of inflammation in the skin: delayed rejection of allografts in L-selectin-deficient mice. J Immunol. 1997;158:5191–9. [PubMed] [Google Scholar]
- 41.Steeber DA, Tang ML, Zhang XQ, Muller W, Wagner N, Tedder TF. Efficient lymphocyte migration across high endothelial venules of mouse Peyer's patches requires overlapping expression of L-selectin and beta7 integrin. J Immunol. 1998;161:6638–47. [PubMed] [Google Scholar]
- 42.Spertini O, Luscinskas FW, Kansas GS, et al. Leukocyte adhesion molecule-1 (LAM-1, L-selectin) interacts with an inducible endothelial cell ligand to support leukocyte adhesion. J Immunol. 1991;147:2565–73. [PubMed] [Google Scholar]
- 43.Fiscus LC, Van Herpen J, Steeber DA, Tedder TF, Tang ML. L-Selectin is required for the development of airway hyperresponsiveness but not airway inflammation in a murine model of asthma. J Allergy Clin Immunol. 2001;107:1019–24. doi: 10.1067/mai.2001.114703. [DOI] [PubMed] [Google Scholar]