

Abstract
Frequent injections of the hormonal form of vitamin D3, 1,25 dihydroxyvitamin D3 (1,25D3) reportedly inhibits autoimmune type 1 diabetes (T1D) in non-obese diabetic (NOD) mice by correcting some of the abnormalities in antigen-presenting cells which contribute the development of pathogenic T cell responses. This route of administration greatly elevates the levels of these compounds in the bloodstream for hours after treatment, which requires mice to be fed diets formulated to contain much reduced levels of Ca to avoid the toxic effects of hypercalcaemia. In the current work, we demonstrate that feeding 1,25D3 or its synthetic precursor, 1alpha(OH) vitamin D3 (1alphaD3), as part of a T1D supportive chow diet containing normal levels of Ca, is an effective means of reducing the incidence of disease in NOD mice, but the doses required for protection elicited hypercalcaemia. However, T1D protection elicited by D3 analogue feeding appears, at least partially, to have an immunological basis, as splenic T cells from treated mice had a decreased capacity to adoptively transfer disease. Protection is associated with an increased proportion of T cells with CD4+ forkhead box P3+ regulatory phenotype within the islet infiltrate of treated animals. The 1alphaD3 precursor is converted rapidly to the active 1,25D3 isoform in vivo. However, feeding the 1alphaD3 analogue elicited stronger T1D protection than the 1,25D3 compound, but also induced more severe hypercalcaemia. In future, the dietary supplementation of novel low-calcaemic D3 analogues may enable their continuous delivery at levels that inhibit T1D development in susceptible humans consuming normal levels of Ca.
Keywords: 1,25-dihydroxyvitamin D3 analogues; calcium; dietary supplementation; NOD mouse; type 1 diabetes
Introduction
Prevention of type 1 diabetes (T1D) in humans, to date, has been unsuccessful. Compounds have been identified that exert T1D protective effects in animal models, yet most have little practical application in humans [1]. Among the few potential exceptions are various analogues of 1,25 dihydroxyvitamin D3 (1,25D3), the metabolically active form of vitamin D3, which inhibits T1D in non-obese diabetic (NOD) mice and may also represent a relatively benign method of altering the course of the disease in humans [2].
Vitamin D3 (D3) derivatives appear to compensate for some of the deficiencies of central and peripheral tolerance in NOD mice by functionally altering specific types of immune cells. Principal among these are dendritic cells (DC), which are the primary antigen-presenting cell (APC) subset involved in central [3] and peripheral tolerance induction [4–6]. The differentiation of tolerogenic APC, including DC, is impaired in NOD mice [6], biobreeding (BB) rats [7] and many T1D patients [8,9]. 1,25D3 has been proposed to inhibit T1D in NOD mice by reducing the capacity of DC to activate pathogenic T cells by down-regulating surface expression of major histocompatibility complex (MHC) class II and several co-stimulatory molecules including CD86, CD80 and CD83 [10]. 1,25D3 also inhibits the secretion of crucial APC cytokines, including interleukin (IL)-12, that are responsible for activation of T cells [10,11] and increases the immunosuppressive IL-10 cytokine [10,12,13].
Analogues of 1,25D3 that inhibit T1D in NOD mice are also reported to increase the frequency in pancreatic lymph nodes (PLN) of regulatory T cells (Tregs) which could suppress autoimmune responses [14]. It is thought that DC exposed to 1,25D3 partially induce tolerance by stimulating Tregs [15]. In addition to attenuating their activation by altering APC function, D3 analogues also affect T lymphocyes directly. Treatment of T cells with 1,25D3 in vitro reportedly inhibits antigen-stimulated proliferation, decreases secretion of key proinflammatory cytokines [interferon (IFN), IL-2] and arrests cell cycling [16–18]. This results in a highly skewed population of T cells with a T helper 2 (Th2) phenotype that putatively inhibits T1D in NOD mice [12,16–18]. At this point it is unclear to what extent the direct effects D3 analogues have on T cells versus APC contribute to the induction of T1D resistance in NOD mice.
The use of active forms of D3 can also elicit metabolic effects that must be taken into account when considering such compounds as potential T1D prevention agents. Fatal hypercalcaemia can occur once 1,25D3 exceeds physiological thresholds [19]. To circumvent this potential difficulty, previously published studies assessed the ability of 1,25D3 injection protocols to inhibit T1D in NOD mice fed diets containing very low levels of Ca [20,21]. It would be very difficult to achieve such low dietary levels of Ca in humans. It should also be considered that elevations in plasma Ca levels induced by D3 analogues could affect the insulin secretory capacity of pancreatic β cells [22,23].
Methods to reduce the hazards of D3 analogue treatment are being sought actively. D3 analogues have been identified that demonstrate much greater T1D protective effects compared to 1,25D3 [19] on a per molar basis, so that much less of these compounds is required for treatment. Another little-considered approach is the dietary administration of D3 analogues. Dietary supplementation may provide a useful means of ensuring that D3 analogues are supplied constantly to the bloodstream at safe levels while achieving the maximum desired immunological and/or metabolic effects. Oral administration would also be a preferential route for any potential future clinical use.
In the current work we examined the feasibility, and possible mechanisms involved, of reducing disease development in NOD mice by supplementing a normally T1D supportive chow diet with 1,25D3 or 1alpha hydroxyvitamin D3 (1alphaD3), also known as alfacalcidol. The rationale for utilizing the 1alphaD3 precursor is that hepatic enzyme activity will convert it immediately to the metabolically active 1,25D3 compound in vivo [24,25], but the former is far simpler to produce at a much lower cost [26], which ultimately could make it more desirable for clinical use. This study is unique, as it makes use of a Ca-adequate complete full-grain diet, analogous to a normal human diet, rather than the commonly used low-Ca diets or the inherently T1D protective purified diets [27,28].
Materials and methods
Mice and diet
NOD/Lt mice are maintained by brother–sister mating in a specific pathogen-free research colony at The Jackson Laboratory. T1D presently develops in 90% of females and 63% of males before a year of age. A NOD.scid stock deficient in B and T lymphocytes has been described previously [29] and is maintained at the N11 back-cross generation. A NOD stock transgenically expressing the rearranged T cell receptor (TCR)α (Vα 8)- and β (Vβ 2)-chains from the β cell autoreactive CD8-T cell clone AI4 (designated NOD.AI4) was developed previously [30] as well as a NOD.AI4 substock congenic for a functionally inactivated Rag1 gene (NOD.Rag1null.AI4) [31].
The basal diet purchased from Purina Test Diet (Land O'Lakes Purina Feed, LLC, Richmond, IN, USA) was a modified NIH rat and mouse 6% fat chow (5k52), with Ca and P levels reduced to meet the nutritional requirements of mice based on National Research Council estimates (0·5% Ca and 0·4% P) [32]. Vitamin D3 was included as part of the vitamin premix at a rate of 4·3 IU/g diet. To make the experimental diets, D3 analogues were first dissolved in ethanol (100 μg/ml and 500 μg/ml for 1,25D3 and 1alphaD3, respectively) and the appropriate amount of each was then added to propylene glycol to be mixed with the meal at 34 ml/kg diet, so that the final concentration of each D3 analogue was 40 nM/kg diet. The control diet was mixed identically using D3 analogue-free ethanol. Professor Hardy Edwards Jr, at the University of Georgia, kindly provided 1alphaD3 and 1,25D3 analogues and additional 1,25D3 was purchased from Sigma (D1530) (Sigma-Aldrich, St. Louis, MO, USA). Diets were caked with water and dried to make wafers that were fed to the mice.
Treatment regimens
In the initial incidence study, mice were divided into three groups of 15 animals each and fed either the 1,25D3, 1alphaD3 or control diet. All animals were fed the diets from shortly after weaning at 3–4 weeks of age until the termination of the experiment at 20 weeks of age or until they were diagnosed with disease. The mice were weighed between 16 and 18 weeks of age. For the adoptive transfer of T1D, total CD4 and CD8 T lymphocytes were purified, as indicated, from splenic leucocyte suspensions of female NOD mice fed the 1,25D3, 1alphaD3 or control diets for 7 weeks from weaning. Cells were then injected intravenously (i.v.) at a dose of 3 × 106 total T lymphocytes into 11–12-week-old NOD.scid recipients. T1D was assessed by weekly monitoring of glycosuric levels with Ames Diastrix (Diagnostics Division, Bayer Corporation, Elkhart, IN, USA), with disease onset defined by values of ≥ 3 on 2 consecutive days. Pancreata were taken from non-diabetic mice at the end of each experiment and sectioned and stained for the assessment of insulitis as described previously [33]. For animals injected with D3 analogues, ethanol stock solutions of 1,25D3 and 1alphaD3 were dissolved in phosphate-buffered saline (PBS) and infused intraperitoneally (i.p.) into 6–8-week-old NOD females. Treated animals received a single D3 analogue dose of 12 nM/kg body weight, while control animals received ethanol and PBS only (0·2 ml). For all experimental groups, two to four pools of plasma (100–500 μl per pool) were collected via retro-orbital bleeding from three to six mice per pool at 0·5, 1, 2, 3, 6, 8, 12 and 16 h time-points.
Glucose tolerance
Changes in blood glucose were determined after i.p. glucose challenge (2 g/kg) in fasted NOD mice fed either unsupplemented control diet or diets supplemented with 1alphaD3 or 1,25D3 (n = 4/group) for 23 weeks from weaning.
Analysis of plasma 1,25D3
Plasma and 1,25D3 standards and controls from Alpco Diagnostics (Salem, NH, USA) were applied to chromabond columns and incubated for 10 min. For sample volumes of less than 1000 μl, Tris-HCl buffer was used to wet the column before applying the plasma, so that the final volume in each column was 1000 μl. D3 analogues were extracted from the columns with 4 × 1 ml diisoproplyether (3 min for each elution). The eluate was allowed to drip into a dry silica cartridge after which the chromabond columns were removed. The silica cartridges were then washed with 5 × 2 ml isopropanol/hexane (4/96 vol/vol) followed by 3 × 2 ml isopropanol/hexane (6/94 vol/vol). 1,25D3 was then eluted from the silica cartridges into glass vials with 2 × 2 ml isopropanol/hexane (25/75 vol/vol). Eluates were evaporated using a vacuum centrifuge and redissolved in 20 μl of ethanol. A competitive enzyme immunoassay (EIA) using a selected monoclonal antibody recognizing 1,25D3 was performed subsequently according to the manufacturer's instructions (Alpco Diagnostics). Final 1,25D3 concentrations were adjusted for the initial volume of plasma applied to each chromabond column.
Plasma Ca and P analysis
Plasma Ca and P were determined using a Beckman Synchron CX5 Delta atomic absorption spectrometer (Beckman Coulter, Inc., Fullerton, CA, USA). For the Ca analyses, plasma was diluted with Arsenazo III that combines with available Ca to form Ca–Arsenazo, which can be monitored for change in absorbance at 650 nm. For the P analysis, plasma was diluted with a reagent containing ammonium molybdate that reacts with inorganic P in an acidic solution to form a coloured phosphomolybdate complex, which can be monitored for change in absorbance at 340 nm.
Histological analysis
Tissues from mice fed analogue or control diets for 31–33 weeks were fixed in Telly's fixative (100 ml EtOH 70%, 5 ml formalin, 5 ml glacial acetic acid), paraffin-embedded and sectioned at 5 μm. Sections were stained with haematoxylin and eosin (H&E).
Leucocyte purification
CD4 and CD8 T lymphocytes were purified from splenic leucocyte suspensions by removing B cells and myeloid cells labelled with biotin-conjugated anti-mouse polyvalent antibody (Sigma B-2016, Sigma-Aldrich Inc., St. Louis, MO, USA) and B220 (RA3-6B2) CD11b (M1/70) monoclonal antibodies (mAbs) from BD Pharmingen, San Jose, CA, USA using streptavidin-conjugated magnetic beads (Miltenyi Biotec, Bergisch Gladbach, Germany), as described previously [34]. Dendritic cells were purified from collagenase D (Roche, Basel, Switzerland) digested spleens by positive selection using CD11c-conjugated magnetic beads and LS columns following the manufacturer's directions (Miltenyi Biotec).
Flow cytometric analyses
Single cell suspensions were stained with the appropriate antibodies at 4°C for 30 min. Stained cells were washed and analysed on a fluorescence activated cell sorter (FACS) Calibur flow cytometer (BD Biosciences) using CellQuest software. Propidium iodide was used to gate out dead cells. For intracellular forkhead box P3 (FoxP3) staining, cells were surface stained using BD Pharmingen mAbs for CD4 (RM4-5) and/or CD25 (PC61) molecules before they were fixed and permeabilized with solutions contained within the eBioscience FoxP3 staining kit (eBioscience Inc., San Diego, CA, USA). The manufacturer's recommended staining protocol was followed using an anti-mouse/rat FoxP3 antibody (FJK-16s).
Analyses of cell types comprising insulitic lesions
Pancreatic islets from female NOD mice fed the experimental diets for 7 weeks from weaning were isolated and dispersed into single cell suspensions as previously described [35,36]. Three pools of islet cells were prepared from nine mice in each treatment group (three mice per pool). The proportion of total leucocytes within each islet cell pool was then assessed by flow cytometry with a CD45-specific BD Pharmingen mAb (30-F11). The proportions of different immune cells among CD45-positive leucocytes were assessed with T and B cell-specific antibodies. All analyses were restricted to cells that were shown to be viable by their exclusion of propidium iodide, with the exception of cells stained for FoxP3, as they were fixed and permeabilized for intracellular staining.
Results
T1D incidence is reduced in NOD mice fed 1,25D3 or 1alphaD3
Feeding NOD mice the 1alphaD3-supplemented diet significantly reduced the incidence of T1D compared to the control-fed group (Fig. 1a). However, the supplemented diet resulted in moderate to severe hypercalcaemia; analogue-fed mice weighed approximately 10% less than control-fed mice and their blood Ca levels were, on average, 30% higher (Table 1). In contrast, feeding the 1,25D3 compound did not reduce weight gain and only marginally increased plasma Ca levels (Table 1). Interestingly, this reduction in toxicity was associated with a decrease in disease protection and glucose tolerance compared to controls (Fig. 1a,b). 1,25D3 plasma levels, as a percentage of control-fed mice, were also elevated in 1alphaD3-fed mice, but not in 1,25D3-fed mice (Fig. 1c).
Fig. 1.
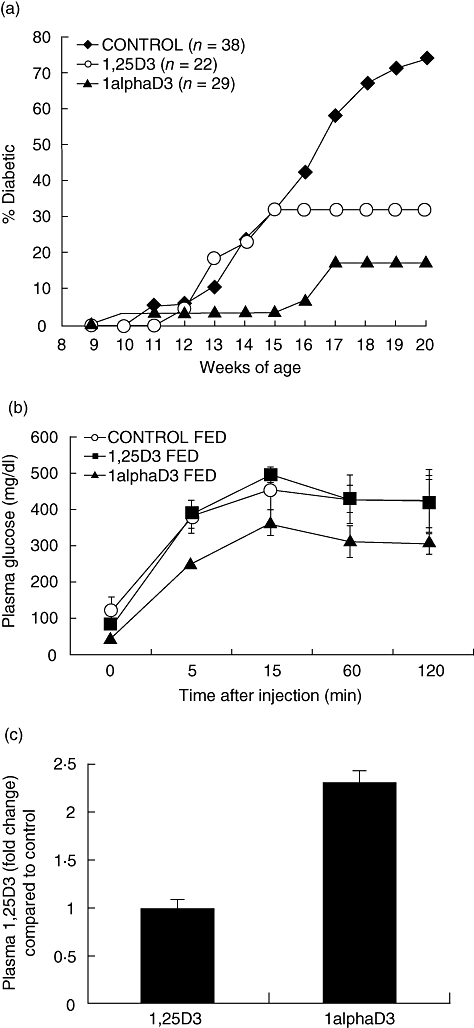
(a) Cumulative incidence of type 1 diabetes (T1D) in female non-obese diabetic mice (NOD) mice fed 1,25D3, 1alphaD3 or vehicle control supplemented chow diets from weaning until 20 weeks of age. Analogues were fed at a concentration of 40 nM/kg diet. T1D development was inhibited in 1,25D3- (P = 0·0126) and 1alphaD3 (P < 0·0001)-fed mice compared to control as determined via a Kaplan–Meier life table analysis. (b) Changes in blood glucose (mmol/l) after intraperitoneal (i.p.) glucose challenge (2 g/kg) in fasted NOD mice fed either unsupplemented control diet or diets supplemented with 1alphaD3 or 1,25D3 (n = 4/group) for 23 weeks from weaning. (c) Plasma 1,25D3 of 1,25D3- and 1alphaD3-fed mice as a ratio of mice fed the unsupplemented control diet. Three to four pools of plasma (500 μl per pool) were collected via retro-orbital bleed from three to four mice per pool for analysis by enzyme immunoassay (EIA). The plasma 1,25D3 ratio was no different from one (P = 0·763) for 1,25D3-fed mice versus control fed mice but significantly higher for 1alphaD3-fed mice (P = 0·0129) according to a comparison to one t-test.
Table 1.
Average plasma Ca and P concentrations and weights of female non-obese diabetic mice (NOD) mice fed 1,25D3, 1alphaD3 or vehicle control supplemented chow diets from weaning (Fig. 1a). Analogues were fed at a concentration of 40 nM/kg diet.
| Diet | n | Plasma Ca (mg/dl) | Plasma P (mg/dl) | n | 16–18-week weights (g) |
|---|---|---|---|---|---|
| Control | 14 | 11 ± 0·4c | 12 ± 0·7a | 6 | 22·7 ± 0·2a |
| 1,25D3 | 14 | 13 ± 0·6b | 13 ± 0·7a | 7 | 22·7 ± 0·6a |
| 1alphaD3 | 13 | 16 ± 0·7a | 13 ± 0·5a | 14 | 20·1 ± 0·5b |
| Analysis of variance | d.f. | P-values | d.f. | P-value | |
| Diet | 2 | < 0·0001 | 0·4719 | 2 | 0·0020 |
| Error | 38 | 24 | |||
Values within variables with different superscripts differ significantly (P < 0·05) when tested using Student's t-test. d.f.: degrees of freedom.
Values within variables with different superscripts differ significantly (P < 0·05) when tested using Student's t-test. d.f.: degrees of freedom.
Values within variables with different superscripts differ significantly (P < 0·05) when tested using Student's t-test. d.f.: degrees of freedom.
Tissue calcification is more severe in NOD mice fed 1,25D3 compared to 1alphaD3
Mice fed 1,25D3 had diffuse circumferential thickening of the cortices along the diaphysis and metaphysis of the long bones. Cortical expansion was characterized by a thin layer of woven bone overlying the pre-existing lamellar cortex (Fig. 2b). The cortices of the long bones were approximately twofold thicker than the control group.
Fig. 2.
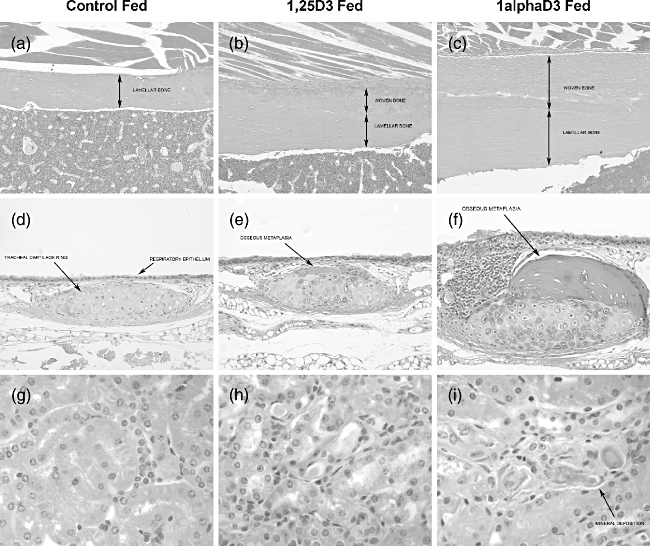
Tissues from mice fed analogue or control diets for 31–33 weeks were fixed in Telly's fixative, paraffin-embedded and sectioned at 5 μm. Sections were stained with haematoxylin and eosin (H&E). (a–c) Femur diaphysis × 100. Bone cortex was thickened by irregular deposition of woven bone. Compared to control-fed animals, the cortex was approximately double and triple the thickness for 1,25D3- and 1alphaD3-fed mice, respectively. (d–f) Tracheal ring cartilage × 200. Expansion of the cartilage ring by irregular nodules of well-differentiated osteoid. Compared to control-fed animals, cartilage rings were mildly and severely thickened and deformed by osteoid deposition in 1,25D3- and 1alphaD3-fed mice, respectively. (g–i) Renal tubules × 600. Mineral deposition along the basement membranes of distal tubules. Occasional mineral deposits were observed along the basement membranes of distal tubules in 1alphaD3-fed mice but rarely found in 1,25D3-fed mice.
Animals fed 1alphaD3 had a similar, although more prominent, thickening of the cortices of long bones as well as increased thickness of the bony trabeculae within the medullary cavity (Fig. 2c). Bone cortices thickness varied between two- to threefold the thickness of the control group. Bony trabeculae within the medullary cavity often coalesced and replaced haematopoietic tissue.
The hyaline cartilage in the tracheal rings of 1alphaD3-fed animals, compared to the control-fed group, was often mineralized and there was multi-focal osseous metaplasia of the cartilage along the trachea (Fig. 2d–f). This change was characterized by an irregular nodular thickening of the tracheal rings by well-differentiated, partially mineralized osteoid, containing osteocytes, and lined by plump osteoblasts.
Animals fed 1alphaD3 had multi-focal mineralization of basement membranes surrounding tubules in the kidney (Fig. 2i) as well as small foci of mineralization in the tracheal and upper respiratory submucosa (Fig. 2f). These foci were often surrounded by small numbers of macrophages. Submucosal mineralization was more abundant in animals fed 1alphaD3.
T1D is delayed in NOD.scid mice injected with T cells from 1alphaD3-fed NOD donors
Decreased T1D in 1alphaD3-fed mice could be due simply to the toxic metabolic effects of this compound. Hence, in order to determine whether feeding 1alphaD3 decreases the diabetogenicity of pathogenic lymphocytes, T cells were purified and adoptively transferred into immunodeficient NOD.scid recipients. Compared to those from 1,25D3- or control-fed animals (Fig. 3), T1D was delayed when T cells purified from mice fed the 1alphaD3 analogue from 3 to 7 weeks of age were transferred. Insulitis in surviving mice in all three treatments was severe, indicating that T cells from D3 analogue-fed mice retained their capacity to become effector cells (data not included).
Fig. 3.
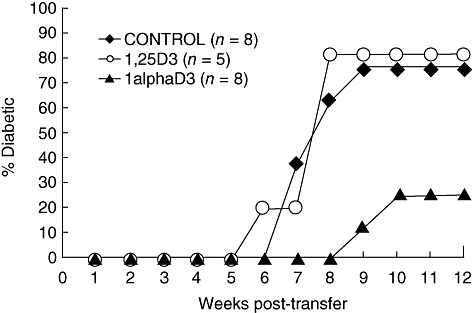
Cumulative incidence of type 1 diabetes (T1D) in female non-obese diabetic mice (NOD).scid mice that received 3 × 106 total CD4 and CD8 T cells from NOD mice fed 1,25D3, 1alphaD3 or vehicle control supplemented chow diets for 7 weeks from weaning. T1D development was inhibited in mice that received T cells from 1alphaD3-fed mice (P = 0·02) compared recipients of 1,25D3 or control T cells.
Injected 1alphaD3 is rapidly converted to 1,25D3 in vivo
Mice rapidly absorbed and degraded exogenous 1,25D3, with maximum circulating levels recorded 1 h after analogue injection (Fig. 4). Plasma 1,25D3 levels were markedly reduced 6 h after injection and no difference was found between treated and control mice from this time-point onwards. Mice injected with an equivalent molar amount of 1alphaD3 converted the synthetic analogue to 1,25D3 efficiently; however, plasma 1,25D3 levels increased more gradually compared to mice injected with 1,25D3, with the highest concentrations recorded 2 h after injection. Significantly (P < 0·05) more plasma 1,25D3 was detected in 1,25D3-injected mice compared to the 1alphaD3-injected mice at the 0·5, 1, 2 and 3 h time-points. In contrast to the diet studies described, neither compound increased plasma Ca levels after a single injection compared to the ethanol-treated controls (10 ± 0·8 mg/dl) at any time-point.
Fig. 4.
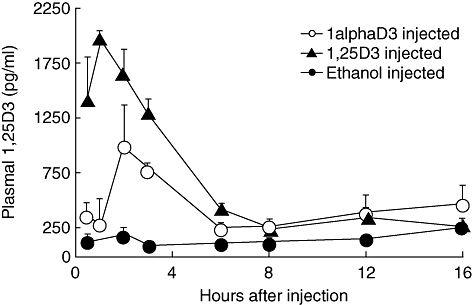
Plasma 1,25D3 concentrations from 6–8-week-old female non-obese diabetic mice (NOD) mice injected with a single dose of 12 nM/kg of 1,25D3, 1alphaD3 or vehicle control. Two to four pools of plasma (100–500 μl per pool) were collected via retro-orbital bleed from three to six mice per pool at 0·5, 1, 2, 3, 6, 8, 12 and 16 h time-points. Plasma 1,25D3 was significantly higher (P < 0·5) in the 1,25D3-fed mice at the 0·5, 1, 2 and 3 h time-points when compared using the Student's t-test.
NOD mice fed D3 analogues have an increased proportion of Tregs in their islet infiltrate
Treg cells were defined by co-expression of CD4 and FoxP3. We observed an increase in the proportion of CD4+ T cells that were FoxP3+ among leucocytes infiltrating the islets of prediabetic mice fed D3 analogues for 6 weeks after weaning (Fig. 5a). There was a corresponding decrease in the proportion of total islet-derived leucocytes that were CD4+FoxP3– in these same mice (Fig. 5b). At this point we can only conclude that feeding of either 1alphaD3 or 1,25D3 results in a numerical increase in cells with the CD4+FoxP3+ Treg phenotype in the pancreatic islets of NOD mice, but we do not know if the function of these potential immune regulators were differentially affected by the alternative D3 analogues tested. No difference was found in the proportion of other immune cells between treatments, including CD8+ T cells, B cells and macrophages (data not included).
Fig. 5.
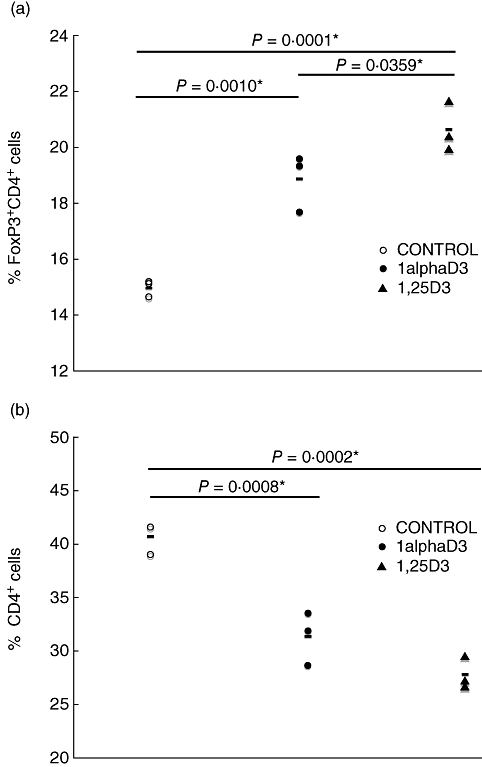
Conventional and regulatory T cells within islets of D3 analogue and control-fed mice. Pancreatic islets from female non-obese diabetic mice (NOD) mice fed the experimental diets for 6 weeks from weaning were isolated and dispersed into single cell suspensions. Three pools of islet cells were prepared from nine mice in each treatment group (three mice per pool). The proportion of total leucocytes within each islet cell pool was then assessed by flow cytometry with a CD45-specific monoclonal antibody. Differences between dietary treatments were established by one-way analysis of variance (P < 0·05). Differences between individual means were calculated according to Student's t-test. (a) Percentage of CD4+ T cells that were forkhead box P3+ (FoxP3+). (b) Percentage of leucocytes that were CD4+FoxP3–.
Discussion
In the current work we demonstrated that continuously supplementing the diet of NOD mice with 1,25D3 or 1alphaD3, an inexpensive synthetic precursor to 1,25D3, partially reduced the incidence of T1D. To our knowledge, only one other experiment has been published which demonstrates the T1D protective effects of dietary 1,25D3. However, in this previous study, even though control animals were kept in a vitamin D3-deficient state, the spontaneous disease incidence of the control mice (46% for females at 200 days) was unusually low, possibly because a purified diet was fed which is often inherently diabetes-protective [27,28]. It is worth noting that disease incidence of control animals (less than 60% diabetic for females at 200 days of age) is also low in the more common injection studies, where D3 analogues are shown to be protective and that in such cases D3 analogue-mediated protection was never complete (7–20% diabetic at the same age) [21,37,38]. The spontaneous T1D incidence of the mice in our study was comparatively high (73% diabetic for females at 140 days of age), which may explain why 1,25D3 treatment did not reduce diabetes to previously reported levels (32% diabetic for females at 140 days of age).
The difference in hypercalcaemia and T1D incidence between mice fed the 1alphaD3 and the 1,25D3 analogues was unexpected. Our results showed that the 1alphaD3 compound was more toxic than the 1,25D3 compound, but also resulted in greater T1D protection (Table 1 and Fig. 1a). These observations are in agreement with other published data that demonstrate a greater and longer-lasting effect of 1alphaD3 over 1,25D3, in both rats and humans, for the treatment of leukaemia [39] and renal osteodystrophy [40]. 1alphaD3 was also found to be more toxic than 1,25D3 in a study where C57BL/6J mice were treated chronically with different doses of both analogues [41]. However, the majority of reports conclude that 1alphaD3 is converted immediately into the active 1,25D3 analogue by the hepatic enzyme vitamin D 25-hydroxylase and that treatment with one analogue is virtually identical to treatment with the other [24,25].
In this study we used a highly accurate competitive EIA that employs a selected monoclonal antibody that specifically recognizes plasma 1,25D3 and not 1alphaD3 (< 0·003% cross-reactivity). After a single injection of the equivalent amount of each compound, plasma 1,25D3 levels increased more gradually in the 1alphaD3 treated group (Fig. 4). Furthermore, as a percentage of controls, plasma 1,25D3 levels are higher in mice fed the 1alphaD3 compared to 1,25D3 (Fig. 1c). This suggests that the conversion of 1alphaD3 to 1,25D3 does not occur instantaneously, and the former compound is not catabolized as quickly as the latter. An increase in the half-life of the precursor compound would amplify its efficacy and could explain the greater hypercalcaemic and T1D protective effects that we observed in 1alphaD3-fed mice. It is also likely that the rate at which vitamin D 25-hydroxylase converts 1alphaD3 to 1,25D3 varies according to dietary Ca. The conversion may be less efficient after long-term treatment of animals fed diets adequate in calcium such as the one employed in our study, as opposed to the rachitic diets usually used to demonstrate the equipotency of the two analogues.
Interestingly, oral administration of 1,25D3 or 1alphaD3 to mice fed our normal-calcaemic diets containing recommended levels of vitamin D3 resulted in significant bone thickening, while for published injection studies high doses of D3 compounds are insufficient to prevent severe decalcification of bone tissue that occurs when feeding a low-calcium diet [21]. This suggests that treatment with D3 analogues without normal calcium supplementation will lead to depletion of bone calcium reserves. Future D3 compounds should be less calcaemic so that they may be added to calcium-sufficient diets.
We concede that the lower level of T1D in 1alphaD3- than 1,25D3-fed mice may simply have resulted from the fact that they were more sickly due to the toxic effects of higher plasma Ca and, hence, ate less diet, causing urine glucose levels to fall below detectable limits. Nevertheless, the cause is due more probably to differential modulation of the immune system, as when T cells from female NOD mice fed the 1alphaD3 diet for 7 weeks from weaning were transferred into normal-calcaemic NOD.scid mice, disease was also decreased compared to those receiving T cells from 1,25D3 or control-fed mice (Fig. 3). There is also a precedent that the level of plasma Ca can play an important role in modulating immunity and that the positive association between hypercalcaemia and vitamin D analogue-mediated amelioration of T1D may not be simply coincidental. An interaction between dietary Ca and 1,25D3 levels has been described for experimental autoimmune encephalitis (EAE), a mouse model for multiple sclerosis in humans [42,43]. Feeding C57BL/6 J mice low levels of 1,25D3 (6 ng/day for females) protected completely against EAE when the diet contained a high level of Ca (1%) inducing severe hypercalcaemia, while even very high levels of 1,25D3 (200 ng/day) did not elicit disease protection when dietary Ca was low (0·2%). The reason for this interaction is as yet unclear. However, as the autoimmue aetiology of T1D and EAE are similar, the possibility exists that high plasma Ca levels are also required for effective vitamin D3-analogue-mediated T1D protection and that is why the 1alphaD3 analogue was more efficient than 1,25D3 in preventing disease.
We are uncertain whether feeding the analogues affected the T cells directly and/or indirectly by targeting other cells of the immune system. Much emphasis has been placed on the effect of these compounds on APC [10,11,13]. It is unknown if D3 analogues can directly inhibit diabetogenic T cells in NOD mice. If alterations are indirect, the most probable intermediaries are DC, as there is good evidence that they present self-antigen to autoreactive T cells in a tolerogenic fashion if matured previously in the presence of 1,25D3 [10,44–46]. However, we were unable to show differences in the proliferation of a β cell-specific autoreactive CD8+ T cell clonotype (AI4) isolated from TCR transgenic mice [31] when stimulated antigenically in vitro with splenic DC purified from D3 analogue or control-fed NOD mice (data not shown). This could suggest that D3 analogue-mediated tolerance of autoreactive T-cells in vivo via DC requires additional signalling through an intermediary cell type. If so, Tregs represent a possible candidate, given that they diminish immune responses and are themselves stimulated by DC. Indeed, it was shown recently that human myeloid, but not plasmacytoid, DC purified from blood were able to induce Treg activity when treated with 1,25D3 in vitro [47]. These data are in concordance with observations that D3 analogue-induced T1D resistance in NOD mice was associated with an increased frequency of cells with a CD4+CD25+ Treg phenotype in the pancreatic lymph nodes [14]. We were unable to find a similar increase of CD4+CD25+FoxP3+ T cells in pancreatic lymph nodes from D3 analogue-fed mice, or changes in any other lymphocyte populations. However, there was an increase in T cells with a CD4+FoxP3+ Treg phenotype within the invading islet infiltrate and this was associated with a corresponding decrease in the CD4+FoxP3– T cells. It should be stressed that at this time it can only be concluded that there is a proportionate increase of T cells with a CD4+FoxP3+ Treg phenotype in the pancreatic islets of NOD mice fed either 1,25D3 or 1alphaD3. Compared to the normal chow-fed controls, cells with a Treg phenotype were increased proportionally in the islets of both 1alphaD3- and 1,25D3-fed NOD mice. Furthermore, while cells with a Treg phenotype were actually present at a higher proportional level in the islets of 1,25D3- than 1alphaD3-fed NOD mice, the latter compound provided stronger T1D resistance. Thus, it is entirely possible that cells with a Treg phenotype in the islets of 1alphaD3- and 1,25D3-fed NOD mice may differ functionally. Unfortunately, as the total number of leucocytes that could be recovered from the islets of 1alphaD3- or 1,25D3-fed NOD mice was relatively low, it was not practical to functionally compare the cells with a Treg phenotype that represented a fraction of these two populations.
In conclusion, we have demonstrated that feeding 1,25D3 and its analogue 1alphaD3, as part of a T1D supportive chow diet, is an effective means of reducing the incidence of disease in NOD mice, but the dose levels required to elicit protection induce mild to severe hypercalcaemia. When fed chronically, 1alphaD3 resulted in slightly higher plasma 1,25D3, more severe hypercalcaemia and greater T1D protection compared to the 1,25D3 compound. Although Ca toxicity may certainly have affected the development of T1D in 1alphaD3-fed mice, disease protection was clearly mediated, to some extent, through alterations in the immune system as evidenced by T lymphocyte transfer from these mice into NOD.scid recipients. Both D3 analogues appeared to increase the percentage of FoxP3+CD4+ T cells in the islet infiltrate of treated mice, indicating that these compounds may ameliorate disease by increasing the numbers of Tregs at sites of inflammation. In future, the dietary supplementation of novel low-calcaemic D3 analogues may enable their continuous delivery at levels that inhibit T1D development in susceptible humans consuming normal levels of Ca. Further work is required to determine the effect of dietary Ca level on the efficacy with which D3 analogues ameliorate disease.
Acknowledgments
This work was supported by National Institutes of Health grants DK46266 (to D. V. S.), DK51090 (to D. V. S.); Cancer Center Support Grant CA34196; as well as grants from the Juvenile Diabetes Research Foundation (to D. V. S.). J. P. Driver is the recipient of a Postdoctoral Fellowship from the Juvenile Diabetes Research Foundation.
We thank Prof. Hardy Edwards Jr, at the University of Pretoria who kindly provided the 1alphaD3 and 1,25D3 analogues. E. van Etten is a recipient of a Postdoctoral Fellowship from the Juvenile Diabetes Research Foundation. C. Mathieu is a recipient of a Clinical Research Fellowship from the Flemish Research Foundation (Fonds Voor Wetenschappelijk Onderzoek Vlaanderen, FWO).
References
- 1.Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med. 1999;5:601–4. doi: 10.1038/9442. [DOI] [PubMed] [Google Scholar]
- 2.Mathieu C, Badenhoop K. Vitamin D and type 1 diabetes mellitus: state of the art. Trends Endocrinol Metab. 2005;16:261–6. doi: 10.1016/j.tem.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 3.Brocker T, Riedinger M, Karjalainen K. Targeted expression of major histocompatibility complex (MHC) class II molecules demonstrates that dendritic cells can induce negative, but not positive selection of thymocytes in vivo. J Exp Med. 1997;185:541–50. doi: 10.1084/jem.185.3.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurts C, Kosaka H, Carbone FR, Miller JF, Heath WR. Class I-restricted cross-presentation of exogenous self-antigens leads to deletion of autoreactive CD8(+) T cells. J Exp Med. 1997;186:239–45. doi: 10.1084/jem.186.2.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kurts C, Cannarile M, Klebba I, Brocker T. Dendritic cells are sufficient to cross-present self-antigens to CD8 T cells in vivo. J Immunol. 2001;166:1439–42. doi: 10.4049/jimmunol.166.3.1439. [DOI] [PubMed] [Google Scholar]
- 6.Serreze DV. Autoimmune diabetes results from genetic defects manifest by antigen presenting cells. FASEB. 1993;7:1092–6. doi: 10.1096/fasebj.7.11.8370480. [DOI] [PubMed] [Google Scholar]
- 7.Delemarre FG, Simons PJ, de Heer HJ, Drexhage HA. Signs of immaturity of splenic dendritic cells from the autoimmune prone biobreeding rat: consequences for the in vitro expansion of regulator and effector T cells. J Immunol. 1999;162:1795–801. [PubMed] [Google Scholar]
- 8.Jansen A, van Hagen M, Drexhage HA. Defective maturation and function of antigen-presenting cells in type 1 diabetes. Lancet. 1995;345:491–2. doi: 10.1016/s0140-6736(95)90586-3. [DOI] [PubMed] [Google Scholar]
- 9.Parkkonen P, Hyoty H, Huupponen T, Leinikki P, Tuomilehto-Wolf E, Knip M. Defective HLA class II expression in monocytes of type 1 diabetic patients. APMIS. 1993;101:395–402. doi: 10.1111/j.1699-0463.1993.tb00126.x. The Childhood Diabetes Finland Study Group. [DOI] [PubMed] [Google Scholar]
- 10.Penna G, Adorini M. 1 alpha, 25-dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol. 2000;164:2405–11. doi: 10.4049/jimmunol.164.5.2405. [DOI] [PubMed] [Google Scholar]
- 11.Griffin MD, Lutz W, Phan VA, Bachman LA, McKean DJ, Kumar R. Dendritic cell modulation by 1alpha, 25 dihydroxyvitamin D3 and its analogs: a vitamin D receptor-dependent pathway that promotes a persistent state of immaturity in vitro and in vivo. Proc Natl Acad Sci USA. 2001;98:6800–5. doi: 10.1073/pnas.121172198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D'Ambosio D, Cippitelli M, Cocciolo MG, et al. Inhibition of IL-12 production by 1, 25-dihydroxyvitamin D3. Involvement of NF-kappaB downregulation in transcriptional repression of the p40 gene. J Clin Invest. 1998;101:252–62. doi: 10.1172/JCI1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Halteren AG, van Etten E, de Jong EC, Bouillon R, Roep BO, Mathieu C. Redirection of human autoreactive T cells upon interaction with dendritic cells modulated by TX527, an analog of 1, 25 dihydroxyvitamin D3. Diabetes. 2002;51:2119–25. doi: 10.2337/diabetes.51.7.2119. [DOI] [PubMed] [Google Scholar]
- 14.Gregori S, Giarratana N, Smiroldo S, Uskokovic M, Adorini L. 1a,25-dihydroxyvitamin D3 analog enhances regulatory T cells and arrests autoimmune diabetes in NOD mice. Diabetes. 2002;51:1367–74. doi: 10.2337/diabetes.51.5.1367. [DOI] [PubMed] [Google Scholar]
- 15.van Etten E, Mathieu C. Immunoregulation by 1,25-dihydroxyvitamin D3: basic concepts. J Steroid Biochem Mol Biol. 2005;97:93–101. doi: 10.1016/j.jsbmb.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 16.Lemire JM, Archer DC, Beck L, Spiegelberg HL. Immuno-suppressive actions of 1,25-dihydroxyvitamin D3; preferential inhibition of Th1 functions. J Nutr. 1995;125:1704S. doi: 10.1093/jn/125.suppl_6.1704S. [DOI] [PubMed] [Google Scholar]
- 17.Overbergh L, Decallonne B, Waer M, et al. 1a,25-dihydroxyvitamin D3 induces an autoantigen-specific T-helper 1/T-helper 2 immune shift in NOD mice immunized with GAD65 (p524–543) Diabetes. 2000;49:1301–7. doi: 10.2337/diabetes.49.8.1301. [DOI] [PubMed] [Google Scholar]
- 18.Takeuchi A, Reddy GS, Kobayashi T, Okano T, Park J, Sharma S. Nuclear factor of activated T cells (NFAT) as a molecular target for 1 alpha, 25-dihydroxyvitamin D3-mediated effects. J Immunol. 1998;160:209–18. [PubMed] [Google Scholar]
- 19.Nagpal S, Jianfen 1 Boehm MF. Vitamin D analogs; mechanism of action and therapeutic applications. Curr Med Chem. 2001;8:1661–79. doi: 10.2174/0929867013371950. [DOI] [PubMed] [Google Scholar]
- 20.Mathieu C, Laureys J, Sobis H, Vandeputte M, Waer M, Bouillon R. 1,25-dihydroxyvitamin D3 prevents insulitis in NOD mice. Diabetes. 1992;41:1491–5. doi: 10.2337/diab.41.11.1491. [DOI] [PubMed] [Google Scholar]
- 21.Mathieu C, Waer M, Casteels K, Laureys J, Bouillon R. Prevention of type I diabetes in NOD mice by nonhypercalcemic doses of a new structural analog of 1,25-dihydroxyvitamin D3, KH1060. Endocrinology. 1995;136:866–72. doi: 10.1210/endo.136.3.7867594. [DOI] [PubMed] [Google Scholar]
- 22.Ismail A, Namala R. Impaired glucose tolerance in vitamin D deficiency can be corrected by calcium. J Nutr Biochem. 2000;11:170–5. doi: 10.1016/s0955-2863(99)00090-x. [DOI] [PubMed] [Google Scholar]
- 23.Beaulieu C, Kestekian R, Havrankova J, Gascon-Barre M. Calcium is essential in normalizing intolerance to glucose that accompanies vitamin D depletion in vivo. Diabetes. 1993;42:35–43. doi: 10.2337/diab.42.1.35. [DOI] [PubMed] [Google Scholar]
- 24.Holick MF, Holick SA, Tavela T, Gallagher B, Schnoes HK, DeLuca HF. Synthesis of (6–3H)-1a-hydroxyvitamin D3 and its metabolism in vivo to (3H)-1a, 25-dihydroxyvitamin D. Science. 1975;190:576–8. doi: 10.1126/science.1188356. [DOI] [PubMed] [Google Scholar]
- 25.Biehl RR, Baker DH, DeLuca HF. Activity of various hydroxylated viatmin D3 analogs for improving phosphorus utilization in chicks receiving diets adequate in vitamin D3. Br Poult Sci. 1998;39:408–12. doi: 10.1080/00071669888971. [DOI] [PubMed] [Google Scholar]
- 26.Haussler MR, Zerwekh JE, Hesse RH, Rizzardo E, Pechet MM. Biological activity of 1a-hydroxycholecalciferol, a synthetic analog of the hormonal form of vitamin D3. Proc Natl Acad Sci USA. 1973;70:2248–52. doi: 10.1073/pnas.70.8.2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Flohe SB, Wasmuth HE, Kerad JB, et al. A wheat-based, diabetes-promoting diet induces a Th1-type cytokine bias in the gut of NOD mice. Cytokine. 2003;21:149–54. doi: 10.1016/s1043-4666(02)00486-6. [DOI] [PubMed] [Google Scholar]
- 28.Beales PE, Elliott RB, Flohe S, et al. A multi-centre, blinded international trial of the effect of A(1) and A(2) beta-casein variants on diabetes incidence in two rodent models of spontaneous type I diabetes. Diabetologia. 2002;45:1240–6. doi: 10.1007/s00125-002-0898-2. [DOI] [PubMed] [Google Scholar]
- 29.Prochazka M, Gaskins HR, Shultz LD, Leiter EH. The nonobese diabetic scid mouse: model for spontaneous thymomagenesis associated with immunodeficiency. Proc Natl Acad Sci USA. 1992;89:3290–4. doi: 10.1073/pnas.89.8.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graser RT, DiLorenzo TP, Wang F, et al. Identification of a CD8 T cell that can independently mediate autoimmune diabetes development in the complete absence of CD4 T cell helper functions. J Immunol. 2000;164:3913–18. doi: 10.4049/jimmunol.164.7.3913. [DOI] [PubMed] [Google Scholar]
- 31.DiLorenzo TP, Lieberman SM, Takaki T, et al. During the early prediabetic period in NOD mice, the pathogenic CD8+ T cell population comprises multiple antigenic specificities. Clin Immunol. 2002;105:332–41. doi: 10.1006/clim.2002.5298. [DOI] [PubMed] [Google Scholar]
- 32.Nutrition So LA. Nutrient requirements of laboratory animals. 4. Washington, DC: National Academic Press; 1995. [Google Scholar]
- 33.Serreze DV, Osborne MA, Chen YG, et al. Partial versus full allogeneic hemopoietic chimerization is a preferential means to inhibit type 1 diabetes as the latter induces generalized immunosuppression. J Immunol. 2006;177:6675–84. doi: 10.4049/jimmunol.177.10.6675. [DOI] [PubMed] [Google Scholar]
- 34.Serreze DV, Johnson EA, Chapman HD, et al. Autoreactive T cells in NOD mice can expand efficiently from a greatly reduced precursor pool. Diabetes. 2001;50:1992–2000. doi: 10.2337/diabetes.50.9.1992. [DOI] [PubMed] [Google Scholar]
- 35.Gerling IC, Serreze DV, Christianson SW, Leiter EH. Intrathymic islet cell transplantation reduces beta-cell autoimmunity and prevents diabetes in NOD/Lt mice. Diabetes. 1992;41:1672–6. doi: 10.2337/diab.41.12.1672. [DOI] [PubMed] [Google Scholar]
- 36.Pierce MA, Chapman HD, Post CM, et al. Adenovirus early region 3 antiapoptotic 10·4K, 14·5K, and 14·7K genes decrease the incidence of autoimmune diabetes in NOD mice. Diabetes. 2003;52:1119–27. doi: 10.2337/diabetes.52.5.1119. [DOI] [PubMed] [Google Scholar]
- 37.Mathieu C, Waer M, Laureys J, Rutgeerts O, Bouillon R. Prevention of autoimmune diabetes in NOD mice by 1,25 dihydroxyvitamin D3. Diabetologia. 1994;37:552–8. doi: 10.1007/BF00403372. [DOI] [PubMed] [Google Scholar]
- 38.Mathieu C, van Etten E, Decallonne B, et al. Vitamin D and 1,25-dihydroxyvitamin D3 as modulators in the immune system. J Steroid Biochem Mol Biol. 2004;89–90:449–52. doi: 10.1016/j.jsbmb.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 39.Honma Y, Hozumi M, Abe E, et al. 1 alpha,25-Dihydroxyvitamin D3 and 1 alpha-hydroxyvitamin D3 prolong survival time of mice inoculated with myeloid leukemia cells. Proc Natl Acad Sci USA. 1983;80:201–4. doi: 10.1073/pnas.80.1.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fukushima M, Niki R, Ohkawa H, et al. Comparative therapeutic effects of vitamin D3 and its derivatives on experimental renal osteodystrophy. Endocrinology. 1980;107:328–33. doi: 10.1210/endo-107-1-328. [DOI] [PubMed] [Google Scholar]
- 41.Crocker JF, Muhtadie SF, Hamilton DC, Cole DE. The comparative toxicity of vitamin D metabolites in the weanling mouse. Toxicol Appl Pharmacol. 1985;80:119–26. doi: 10.1016/0041-008x(85)90106-1. [DOI] [PubMed] [Google Scholar]
- 42.Cantorna MT, Humpal-Winter J, DeLuca HF. Dietary calcium is a major factor in 1,25-dihydroxycholecalciferol suppression of experimental autoimmune encephalomyelitis in mice. J Nutr. 1999;129:1966–71. doi: 10.1093/jn/129.11.1966. [DOI] [PubMed] [Google Scholar]
- 43.Zella JB, McCary LC, DeLuca HF. Oral administration of 1,25-dihydroxyvitamin D3 completely protects NOD mice from insulin-dependent diabetes mellitus. Arch Biochem Biophys. 2003;417:77–80. doi: 10.1016/s0003-9861(03)00338-2. [DOI] [PubMed] [Google Scholar]
- 44.Piemonti L, Monti P, Sironi M, et al. Vitamin D3 affects differentiation, maturation, and function of human monocyte-derived dendritic cells. J Immunol. 2000;164:4443–51. doi: 10.4049/jimmunol.164.9.4443. [DOI] [PubMed] [Google Scholar]
- 45.Griffin MD, Lutz WH, Phan VA, Bachman LA, McKean DJ, Kumar R. Potent inhibition of dendritic cell differentiation and maturation by vitamin D analogs. Biochem Biophys Res Commun. 2000;270:701–8. doi: 10.1006/bbrc.2000.2490. [DOI] [PubMed] [Google Scholar]
- 46.Berer A, Stockl J, Majdic O, et al. 1,25-Dihydroxyvitamin D(3) inhibits dendritic cell differentiation and maturation in vitro. Exp Hematol. 2000;28:575–83. doi: 10.1016/s0301-472x(00)00143-0. [DOI] [PubMed] [Google Scholar]
- 47.Penna G, Amuchastegui S, Giarratana N, et al. 1,25-Dihydroxyvitamin D3 selectively modulates tolerogenic properties in myeloid but not plasmacytoid dendritic cells. J Immunol. 2007;178:145–53. doi: 10.4049/jimmunol.178.1.145. [DOI] [PubMed] [Google Scholar]