

Abstract
Factor H is an abundant plasma glycoprotein that plays a critical role in the regulation of the complement system in plasma and in the protection of host cells and tissues from damage by complement activation. Several recent studies have described the association of genetic variations of the complement factor H gene (CFH) with atypical haemolytic uraemic syndrome (aHUS), age-related macular degeneration (AMD) and membranoproliferative glomerulonephritis (MPGN). This review summarizes our current knowledge of CFH genetics and examines the CFH genotype–phenotype correlations that are helping to understand the molecular basis underlying these renal and ocular pathologies.
Keywords: age-related macular degeneration (AMD), factor H, haemolytic–uraemic syndrome (HUS), membranoproliferative glomerulonephritis type II (MPGN2), RCA
Complement activation and regulation
Complement is a major component of innate immunity, with crucial roles in microbial killing, apoptotic cell clearance and immune complex handling. Activation of complement by foreign surfaces (alternative pathway; AP), antibody (classical pathway; CP) or mannan (lectin pathway; LP) causes target opsonization, leucocyte recruitment and cell lysis. The critical steps in complement activation are the formation of unstable protease complexes, named C3-convertases (AP, C3bBb; CP/LP, C2aC4b) and the cleavage of C3 to generate C3b. Convertase-generated C3b can form more AP C3-convertase, providing exponential amplification to the initial activation. Binding of C3b to the C3-convertases generates the C5-convertases with the capacity to bind and cleave C5, initiating formation of the lytic membrane attack complex (MAC).
Nascent C3b binds indiscriminately to pathogens and adjacent host cells. To prevent damage to self and to avoid wasteful consumption of components, complement is under the control of multiple regulatory proteins that limit complement activation, either by inactivating C3b or C4b, by dissociating the C3/C5 convertases or by inhibiting the MAC formation. In health, activation of C3 in the blood is kept at a low level and deposition of C3b and further activation of complement is limited to the surface of pathogens [1].
Complement regulation by factor H
Factor H is a relatively abundant plasma protein that is essential to maintain complement homeostasis and to restrict the action of complement to activating surfaces. Factor H binds to C3b, accelerates the decay of the alternative pathway C3-convertase (C3bBb) and acts as a co-factor for the factor I-mediated proteolytic inactivation of C3b [2–4]. Factor H regulates complement both in fluid phase and on cellular surfaces. However, while factor H binds and inactivates C3b promptly in fluid phase, the inactivation of surface-bound C3b by factor H is dependent on the chemical composition of the surface to which C3b is bound. In the presence of polyanions such as sialic acids, glycosaminoglycans or sulphated polysaccharides (heparins), the affinity of factor H for surface-bound C3b increases as a consequence of the simultaneous recognition of both polyanionic molecules and bound C3b by the same factor H molecule [5–7].
Factor H and factor H-related proteins
Factor H is a single polypeptide chain glycoprotein (155 kDa; ε = 1·95 M−1 cm−1) composed of 20 repetitive units of 60 amino acids [8], named short consensus repeats (SCR) or complement control protein modules (CCP), arranged in a continuous fashion. The factor H molecule includes different interaction sites for C3b and polyanions which delineate distinct functional domains at the N- and C-termini (Fig. 1). The C3b binding site in SCR1-4 is the only site essential for the factor I co-factor activity of factor H. Similarly, the C3b/polyanions-binding site located within SCR19–20 is the most important site for preventing alternative pathway activation on host cells (reviewed in [21]). Factor H is produced constitutively by the liver [22,23] and is found in human plasma at concentrations of 116–562 μg/ml [24]. Extrahepatic synthesis of factor H also occurs in a wide variety of cell types, such as retinal pigment epithelial cells, peripheral blood lymphocytes, myoblasts, rhabdomyosarcoma cells, fibroblasts, umbilical vein endothelial cells, glomerular mesangial cells, neurones, glia cells, etc. [25–27]. The extrahepatic production of factor H is interpreted as a mechanism to increase the local concentration, which could be advantageous for the protection of host cells from complement activation in sites of infection or inflammation.
Fig. 1.
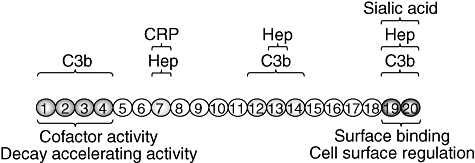
Functional domains in complement factor H. Factor H has three C3b-binding sites, short consensus repeats (SCR)1–4, SCR12–14 and SCR19–20, respectively [9–13]. Similarly, a total of three separate binding sites for heparin and sialic acid have been identified in SCR7, SCR13 and SCR19–20, respectively [14–17]. The critical sites for co-factor activity/decay accelerating activity and cell surface regulation at the N- and C-termini, respectively, are indicated. In addition to C3b and polyanion binding sites, there are other domains in factor H that have been shown to interact with plasma proteins or with micoorganisms and that are interesting because of their potential relevance in pathology. In this regard, it has been shown that factor H binds to C-reactive protein (CRP) which may help to counteract and inhibit the CRP-dependent alternative pathway activation induced by damaged tissue [18,19]. The heparin- and CRP-binding sites in SCR7 are overlapping sites in which one substrate inhibits the binding of the others [20].
In human plasma there are six proteins that are structurally related and cross-react immunochemically with factor H. FHL-1 is the product of the alternative splicing of the gene encoding factor H (CFH) [9,23,28–30]. In addition, five proteins related to factor H are encoded by the five genes CFHR1, CFHR2, CFHR3, CFHR4 and CFHR5 linked closely to CFH [31–34]. These proteins are also probably synthesized in the liver, but their concentrations are much lower than that of factor H. The functional properties of CFHR1, -2, -3, -4 and -5 are not defined fully. They are all composed of SCRs with different degrees of identity with SCRs in factor H [33–37].
The CFH gene
CFH is a member of the regulator of complement activation (RCA) gene cluster on chromosome 1q32 (Fig. 2) [21,39]. CFH comprises 23 exons and spans over 94 kb of genomic DNA [37,40]. The first exon encodes the 5′ untranslated region of the mRNA and the N-terminal 18 amino acids that organize the signal peptide. Each SCR in factor H is encoded by a single exon except for SCR2, that is encoded by exons 3 and 4. Exon 10 does not contribute to the factor H transcript. It is used exclusively in the alternative transcript that codes for the FHL-1 molecule. Exon 10 encodes the last four amino acids (Ser-Phe-Leu-Thr) and the 3′ untranslated region of FHL-1 [28].
Fig. 2.
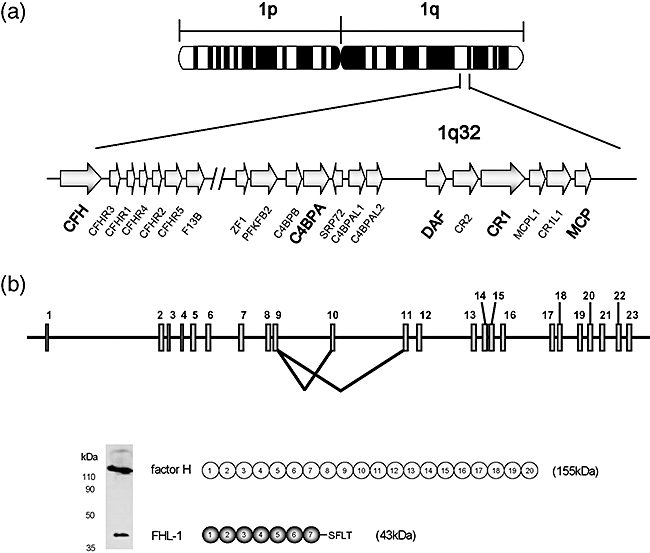
Chromosomal location and structure of the factor H gene. (a) The human regulator of complement activation (RCA) gene cluster in 1q32. The human RCA gene cluster spans a total of 21·45 cM and includes more than 60 genes of which 15 are complement-related genes. All of the complement-related genes are arranged in tandem within two groups. The two groupings are a telomeric 900 kb-long DNA segment which contains the C4BPB, C4BPA, C4BPAL1, C4BPAL2, DAF, CR2, CR1, MCPL1, CR1L1 and MCP genes and a centromeric 650 kb-long DNA segment that contains CFH, CFHR3, CFHR1, CFHR4, CFHR2 and CFHR5, as well as the gene coding for the B subunit of the coagulation factor XIII, F13B. These two gene groups are separated by 14·59 cM, a large amount of DNA-containing genes that are unrelated to complement and that have very diverse functions [38]. It is generally accepted that these complement regulatory genes share a common ancestor from which they originated by multiple events of gene duplication. (b) Structure of CFH, showing a diagram of the 23 exons and the two alternative splicing products of the CFH gene. Exon 10 does not contribute to the factor H transcript but it is utilized for the FHL-1 molecule. The figure also shows a Western blot, using a monoclonal antibody (35H9) that recognizes both factor H and FHL-1, to illustrate the relative amounts of these proteins in normal human plasma.
The single nucleotide polymorphism (SNP) database at the National Center for Biotechnology Information (NCBI) lists a total of 569 SNPs in the human CFH gene region (locus ID: 3075). Of these, roughly a dozen are located in the CFH proximal promoter region or result in an amino acid substitution in the CFH coding sequence. The potential functional implications of some of these CFH polymorphisms will be discussed later in the context of the CFH–disease associations. Of interest, however, is the observation that there is very strong linkage disequilibrium (LD) in the CFH genomic region, which reduces the genetic variability within this region to the combination of four SNP–haplotype blocks spanning the CFH and CFHR1–5 genes [41].
Levels of factor H in human plasma vary widely (116–562 μg/ml) in the population. This variation is not a consequence of CFH null alleles, which are extremely rare, but the result of the combined effect of genetic and environmental factors. Using variance-component methods [42] it was determined that factor H plasma levels show an age-dependent increase and are decreased in smokers [24]. Most important, these studies showed that 63% of the variation in plasma levels of factor H is determined genetically [heritability (h2) = 0·63 ± 0·07; P < 0·0001]. A genome-wide screen in order to identify genes regulating the factor H trait provided suggestive evidence of linkage to three genomic regions (1q32, 2p21–24 and 15q22–24) [24] and more recently we have obtained evidence that demonstrates the existence of low expression CFH alleles [43]. It is therefore likely that genetic variations in both cis- and trans-regulatory elements contribute to the variation in the levels of expression of factor H.
Rearrangements in the CFH genomic region
In close proximity to the CFH gene there are the genes CFHR3, CFHR1, CFHR4, CFHR2 and CFHR5 encoding the five factor H-related proteins (Fig. 2). Sequence analyses of the CFH–CFHR1–5 gene region demonstrated the existence of a number of large genomic duplications including different exons of the CFH and CFHR1–5 genes (Fig. 3a). These duplications range in size from 1·2 to 38 kb and present a pairwise nucleotide identity from 85% to 97% [37]. Low-copy repeats, or segmental duplications, such as these in the CFH–CFHR1–5 gene region, are highly dynamic regions in the genome and a potential source of additional genetic variation in the CFH and CFHR1–5 genes through mechanisms of gene conversion and non-homologous recombination.
Fig. 3.
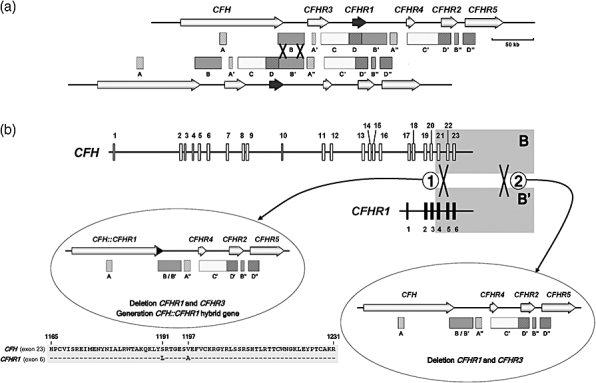
The complement factor H gene (CFH)–CFHR1–5 gene subregion of the regulator of complement activation (RCA) gene cluster. (a) Genomic organization of the CFH and CFHR1–5 genes and location of low copy number repeats. Arrows represent the genes with their names above. The boxes underneath indicate the sequence repeats. Low copy repeats are named with the same letter (i.e. A, A′, A′′). A grey colour-code is used to identify the different repeats. The figure shows two chromosomes aligned by the B and B′ repeats to illustrate the CFH–CFHR1 genomic that occurred through non-homologous recombination between a 23 kb-long repeat region in the 3′ end of the CFH and CFHR1 genes, labelled B and B′, respectively. (b) Deletion of the CFHR1 and CFHR3 genes and generation of a CFH::CFHR1 hybrid gene. The rearrangement marked 2 is relatively common in multiple African and European populations. This rearrangement involves non-homologous recombination between the B and B′ homologous regions downstream of the CFH and CFHR1 genes and results in no sequence-modification of the CFH gene. A second rearrangement (labelled 1) is much less frequent and found associated exclusively with atypical haemolytic uraemic syndrome (aHUS). Non-homologous cross-over in this case occurred between the B and B′ homologous regions in intron 21 of CFH and intron 4 of CFHR1 and results in generation of a hybrid CFH::CFHR1 gene. The hybrid gene consists of the first 21 exons of CFH [encoding SCRs 1–18 of CFH] and the last two exons of CFHR1 (encoding SCR4 and 5 of CFHR1) [44]. Amino acid sequences of CFH exon 23 and CFHR1 exon 6 are aligned to illustrate two amino acid differences between them (S1191L/V1197A) that are present in the protein product of the hybrid gene. These amino acid changes in SCR20 are associated with aHUS and are identical to those present in the CFH mutant protein, also associated with aHUS, that generates by gene conversion [45].
Several examples of gene conversion events between exon 23 of CFH and the homologous exon 6 of CFHR1 have been documented recently [37,45]. Similarly, there is also robust evidence of major rearrangements in the CFH–CFHR1–5 gene region that result in the deletion of the CFHR1 and CFHR3 genes [41,46] and, occasionally, also in the generation of CFH::CFHR1 hybrid genes [44] (see Fig. 3 for details). These CFH–CFHR1–5 rearrangements can be identified easily by MLPA (multiplex ligation-dependent probe amplification) technologies [44] or by Western blot in the case of homozygote carriers [46]. Chromosomes carrying both the deletion of the CFHR1 and CFHR3 genes and a CFH::CFHR1 hybrid gene are rare and associated specifically with aHUS [44]. In contrast, the deletion of the CFHR1 and CFHR3 genes (without rearrangements in CFH) is a common genetic polymorphism included in a single extended CFH haplotype that associates with both lower risk to age-related macular degeneration (AMD) [41,47] and increased risk to aHUS [46]. Deletion of the CFHR1 and CFHR3 genes is not a recurrent phenomenon, but the result of a single rearrangement event that became fixed in the human population a long time ago [47]. Additional rearrangements are likely to occur in the CFH–CFHR1–5 region. For instance, there is one involving an unequal cross-over between homologous regions in the 3′ end of the CFHR3 and CFHR4 that specifically removes the CFHR1 and CFHR4 genes [48].
Factor H disease associations
Several reports in recent years have stablished that membranoproliferative glomerulonephritis type II (MPGN2) [49–52], atypical haemolytic uraemic syndrome (aHUS) [37,53–55] and AMD [27,56–58] are associated with mutations or polymorphisms in the CFH gene. The data available support the hypothesis that AP dysregulation is a unifying pathogenetic feature of these diverse conditions. They also illustrate a remarkable genotype–phenotype correlation in which distinct genetic variations at CFH predispose specifically to aHUS, AMD or MPGN2. As described below, functional characterization of these CFH genetic variations is helping to understand the molecular basis underlying these pathologies.
Membranoproliferative glomerulonephritis
Membranoproliferative glomerulonephritis (MPGN) is an uncommon cause of chronic nephritis characterized by proliferation of mesangial and endothelial cells and by thickening of the peripheral capillary walls (due to subendothelial immune and/or intramembranous dense deposits) that on light microscopy present a double-contour appearance. MPGN may be secondary to autoimmune diseases, chronic infections and malignancies or idiopathic, which accounts for approximately 5% of primary renal causes of nephrotic syndrome and affects predominantly children and young adults (6–30 years). Three distinct types of primary (idiopathic) MPGN have been described based on immunofluorescence staining, ultrastructural appearance and complement profiles. The light microscopy features and clinical presentation are similar among the three types. Hypocomplementaemia is a characteristic finding with all types of MPGN, although the three types present different mechanisms of complement activation. Types I (MPGN1) and III (MPGN3) are variants of immune-complex-mediated disease, whereas type II (MPGN2) has no known association with immune complexes [59].
MPGN2 is very rare. Its morphological hallmark is the presence of dense deposits within the glomerular basement membrane (GBM), as resolved by electron microscopy. The chemical composition of these dense deposits is unknown. Notably, immunoglobulin (IgG) is absent from them and other regions of the glomerulus, which excludes a role for immune complexes in dense deposits formation. MPGN2 is associated with complement abnormalities that lead to intense deposition of C3c in GBM deposits and persistent reduction of C3 serum levels. Among the different factors associated with these complement abnormalities are factor H deficiencies due to mutations in the CFH gene. Approximately half a dozen MPGN patients are described in the literature in which the deficiency of factor H, both heterozygous and homozygous, has been associated with the development of the disease. In all but one of these MPGN cases the factor H deficiency is caused by mutations in CFH that result in truncations or amino acid substitutions that impair secretion of factor H into the circulation [49,60,61] (Fig. 4). The exception is ΔK224, a CFH mutation that results in the deletion of a lysine residue at position 224 [51]. K244 is located in SCR4 within the complement regulatory region of factor H (Fig. 1). Consistent with the location of the mutation, functional studies of factor H ΔK224 have shown that binding to C3b is weak and that both factor I-mediated C3b co-factor activity and AP C3-convertase decay-accelerating activity of factor H ΔK224 are reduced markedly. In contrast, and as expected from an intact C-terminal domain, the mutant factor H ΔK224 protein shows normal binding to heparin, C3d and human umbilical vein endothelial cells [51]. The co-existence of a functionally inactive factor H (homozygous ΔK224 mutation), or a factor H deficiency, with a complement activator such as C3NeF, probably exacerbates a situation of chronic complement activation and results in the complete consumption of C3 in plasma that characterizes MPGN patients [51].
Fig. 4.
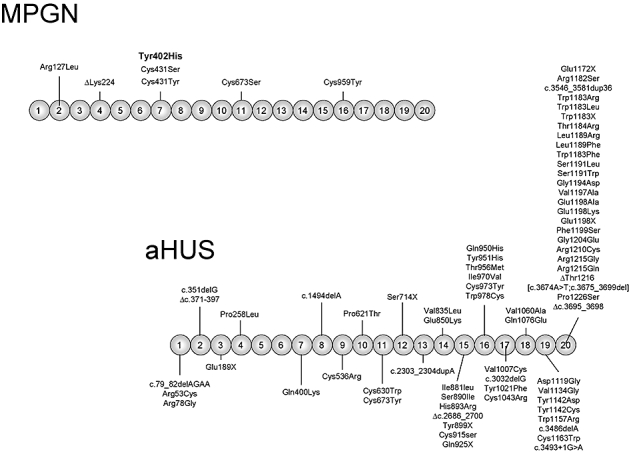
Complement factor H gene (CFH) mutations in MPGN and atypical haemolytic uraemic syndrome (aHUS) patients. The figure shows a diagram of the structure of human factor H with the 20 SCRs. The location of the missense mutations characterized thus far in membranoproliferative glomerulonephritis (MPGN) and aHUS patients is indicated. The position of the Tyr402His polymorphisms associated strongly with predisposition to AMD is highlighted in bold. Note that mutations associated with aHUS are clustered in the C-terminus, the region of factor H that is critical for the control of C3b deposited on cell surfaces.
In addition to mutations in CFH, deficiencies of factor H due to inhibitory autoantibodies have also been reported in some MPGN patients, which also lead to accumulation of the AP C3-convertase, chronic C3 consumption and hypocomplementaemia [62].
Altogether, these genetic and functional data illustrate that those alterations that decrease factor H in plasma, or eliminate its complement regulatory activity, lead to unrestricted activation of the AP of complement, causing damage to glomerular cells and deposition of complement product in the GBM. The severe dysregulation of the AP of complement activation observed in MPGN2 is consistent with animal data that present this renal phenotype. In the pig, factor H deficiency results in a progressive glomerulonephritis, similar to human MPGN2, which leads to renal failure [63]. Similarly, the factor H knock-out mice develop spontaneously a glomerulonephritis that also resembles human MPGN2 [64]. These factor H-deficient animals have been very useful to demonstrate that the uncontrolled activation of C3 in plasma that results from the lack of factor H is essential for the development of MPGN2 [64]. Further studies are, however, necessary to unravel the precise molecular events that end up in MPGN2.
Although the majority of patients with MPGN2 do not have disease-causing mutations in CFH, some common alleles of CFH (and also of CFHR5) have been found to be increased significantly among these patients, supporting further the critical role of genetic variations in the CFH genomic region in the pathogenesis of MPGN2. One of these polymorphisms conferring predisposition to MPGN2 is the His402 allele of CFH [65,66], a major predisposing factor to AMD (see below).
Haemolytic uraemic syndrome (HUS)
HUS is characterized by thrombocytopenia, Coomb's test negative microangiopathic haemolytic anaemia and acute renal failure. The typical form of HUS follows a diarrhoeal prodrome and is associated with 0157:H7 Esherichia coli infections. However, 5–10% of HUS patients lack an association with infection. This atypical form of HUS (aHUS) occurs in both adults and in very young children and has the poorest long-term prognosis. Recurrences in aHUS are common with a mortality rate that approaches 30%. aHUS has an incidence of about 2/106 per year and a prevalence of 1/105 children in the whole of the European Union.
Endothelial cell injury appears to be the primary event in the pathogenesis of HUS. The microvascular lesion of HUS consists of vessel wall thickening with endothelial swelling and detachment from the basement membrane. The endothelial damage triggers a cascade of events that result in the formation of platelet-fibrin hyaline microthrombi that occlude arterioles and capillaries. A hallmark of HUS is the presence of schistocytes (fragmented cells) that generate as the red blood cells traverse these partially occluded microvessels [67].
aHUS is associated with mutations or polymorphisms in the genes encoding the complement regulatory proteins factor H (CFH) [37,44,53–55,68,69], membrane co-factor protein (MCP) [70–73] and factor I (CFI) [74,75] and with mutations in the complement activating components factor B (CFB) [76] and C3 genes [77]. Importantly, mutations in the complement regulators factor H, MCP and factor I are loss-of-function mutations [73,74,78] while mutations in the complement activator factor B are gain-of-function mutations [76]. These data establish unequivocally the critical role of complement AP dysregulation in the pathogenesis of aHUS and illustrate that complement dysregulation may result from either a defect in the regulatory proteins or an abnormally increased activity of the alternative complement pathway activators.
Missense mutations in the C-terminal region of factor H are the most prevalent genetic alterations among aHUS patients (Fig. 4). In a significant number of patients mutations in the C-terminal region of factor H are the result of gene conversion events between exon 23 of CFH and the homologous exon 6 of CFHR1 [45] or of genomic rearrangements creating CFH::CFHR1 hybrid genes [44]. An updated record of all mutations associated with aHUS can be found at the FH–HUS database (http://www.fh-hus.org/).
In contrast with CFH mutations associated with MPGN2, CFH mutations associated with aHUS rarely result in hypocomplementaemia or decreased factor H plasma levels. Most aHUS-associated factor H mutant proteins express normally and present normal co-factor activity for the factor I-mediated proteolytic inactivation of C3b in plasma [37,78]. As indicated, aHUS-associated CFH mutations cluster in the C-terminus of the protein, a region that is critical to control activation of complement on cell surfaces. Consistent with this location, carriers of these CFH mutations express factor H molecules that present normal regulatory activity in plasma but a limited capacity to protect cells from complement lysis [69,78,79]. These findings fit well with the identification of aHUS-associated loss-of-function mutations in MCP and CFI for the reason that the MCP and factor I mutations also lead to decreased protection of host cells from complement lysis without affecting significantly complement homeostasis in plasma [80]. The combination of both an active complement system in plasma and a defective protection of cellular surfaces portrays aHUS as a situation of ‘autolesion’ caused by the uncontrolled activation of complement on cell surfaces. By decreasing concentrations of factor H or factor I in plasma, or MCP on cell surfaces, aHUS-associated mutations predispose to disease. In a situation that triggers complement activation, deposition and amplification of C3b on the microvasculature cellular surfaces cannot be controlled and results in tissue damage and destruction. This is clearly distinct from the lack of complement regulation in plasma, leading to complete C3 consumption and severe hypocomplementaemia, that characterizes MPGN2 patients with factor H deficiency due to mutations in CFH.
While mutations at the C-terminus of factor H are distinctive of aHUS, there are some aHUS patients with partial factor H deficiency due to mutations in the CFH gene [81–84]. These individuals develop aHUS and not MPGN2 mainly because partial factor H deficiencies, like mutations in SCR19-20, affect primarily the control of complement activation on cellular surfaces [85]. In addition, genetic and environmental factors may provide a ‘context’ that influences the pathological outcome for some of these factor H deficiencies. Thus, concurrence of factor H deficiencies with other mutations that decrease protection to host cells have been described in aHUS [37,78], whereas the coincidence of factor H deficiencies with strong complement activators such as C3NeF may be critical in MPGN2 [51].
Mutations in CFH, MCP, CFI, CFB and C3 reveal the molecular defect in approximately 50% of the aHUS patients. To identify additional aHUS susceptibility factors, the complement regulator genes have been analysed further in genetic association studies [53,70]. These and subsequent replication studies [66,71,86] unravelled two relatively frequent CFH and MCP alleles (CFH–H3 and MCPggaac haplotypes) that were significantly more frequent in aHUS patients (either with or without CFH, MCP, CFI or CFB mutations) than in controls. Moreover, in a significant number of aHUS families where CFH, MCP, CFI or CFB mutations segregated with the phenotype aHUS, it could be shown that the proband had inherited the allele carrying the mutation from one parent and an allele carrying the disease-associated CFH and/or MCP haplotype from the other parent. Most interestingly, the healthy CFH, MCP, CFI or CFB mutation carriers in these families did not inherit the aHUS-associated CFH and MCP polymorphisms [53,76,87].
Both CFH–H3 and MCPggaac haplotypes include SNPs located in the promoter region of CFH and MCP that have potential functional implications in the expression of factor H and MCP [53,70]. Although additional studies are needed to fully characterize functionally these CFH and MCP haplotypes, the association of CFH–H3 and MCPggaac with aHUS is extremely important because it indicates that common variations at the CFH and MCP genes predispose to aHUS in the absence of mutations in CFH, MCP, CFI or CFB and that even in carriers of mutations in these genes, these CFH and MCP variations may be needed for full manifestation of the disease. In fact, it is now well established that concurrence of different susceptibility alleles greatly influences predisposition to aHUS and provides an explanation for the incomplete penetrance of aHUS (close to 50%) in carriers of mutations in CFH, MCP, CFI and CFB [70,76,87]. In the Spanish aHUS cohort (n = 98 unrelated patients), seven patients (7%) carry more than one mutation in the complement genes (CFH, MCP, CFI and CFB). In addition, in the 28 patients with mutations in CFH, MCP, CFI and CFB, the allele frequencies of CFH–H3 and MCPggaac haplotypes is increased from 0·19 to 0·34 [controls versus aHUS; P = 0·012; odds ratio (OR), 95% confidence interval (CI) = 2·27 (1·21–4·27)] and from 0·29 to 0·45 [controls versus aHUS; P = 0·027; OR, 95% CI) = 2·77 (1·54–4·95)], respectively (Goicoechea de Jorge et al., unpublished).
Recently, two additional polymorphisms in the CFH genomic region have been reported to influence predisposition to aHUS. The CFH–H1 haplotype was found to be associated with lower risk of aHUS [66]. Similarly, homozygosity for the deletion of the CFHR1 and CFHR3 genes associated strongly with increased risk of aHUS [46]. Moreover, it has been shown that approximately 10% of aHUS patients, mostly children, present auto-antibodies to factor H and that these antibodies have functional consequences similar to those caused by the mutations in the C-terminal region of factor H [88,89]. Interestingly, individuals presenting anti-factor H antibodies are, with very few exceptions, homozygous for the deletion of the CFHR1 and CFHR3 genes, which make unclear whether the deletion of these genes and the presence of auto-antibodies are independent risk factors for aHUS [48,90,91].
In conclusion, genetic and functional analyses have established that aHUS involves alternative complement dysregulation and probably develops as a consequence of defective protection of cellular surfaces from complement activation due to an improper function of complement regulatory proteins. Multiple hits, involving plasma and membrane-associated complement regulatory proteins, as well as complement activators, are probably required to cause dysregulation and impair protection to host tissues significantly. Environmental factors that activate complement probably modulate genetic predisposition and are also very important in aHUS. Infection, immunosuppressive drugs, cancer therapies, oral contraceptives, pregnancy or childbirth are important factors that trigger aHUS in a significant number of patients. In carriers of multiple strong aHUS risk factors the contribution of the environment is probably minor. On the other hand, strong environmental factors may compensate for low genetic predisposition, which perhaps helps to explain the severe or fatal outcome of a small percentage of individuals with the more common diarrhoea-associated typical HUS.
AMD
AMD is one of the most common causes of visual disability in the elderly in developed countries. The hallmark of early-stage disease is the development of drusen, lipoproteinaceous deposits localized between the retinal pigment epithelium (RPE) and Bruch's membrane. Later, an extensive atrophy of the RPE and overlaying photoreceptor cells (geographical atrophy; GA) or aberrant choroidal angiogenesis is observed. This choroidal neovascularization (CNV) under the macular area is the leading cause for blindness. Although the pathogenesis of AMD is still unclear, it has been proposed that inflammatory response play an important role in the development of AMD [92].
AMD is a multi-factorial disease, influenced by age, ethnicity and a combination of environmental and genetic risk factors. Genetic predisposition in AMD has been suggested based on familial segregation and twin studies, involving several candidate genes such as ABCA4, APOE, FBLN5, ELOVL4 and TLR4. However, the individual contribution of these genes to overall AMD prevalence appears relatively minor [93]. Two major AMD susceptibility loci (1q31, CFH, and 10q26, LOC387715/HTRA1) that contribute independently to AMD disease risk have been identified recently by candidate region linkage studies and whole genome association analyses [27,56–58,94].
The most studied SNP at the CFH locus is rs1061170, which causes a Tyr402His amino acid substitution in complement factor H. Several independent studies have shown that the allele 402His confers a significantly increased risk to AMD in many different populations with an OR between 2·1 and 7·4 [27,56–58]. Interestingly, the frequency of the 402His allele varies greatly between populations, which may contribute to the observed differences in the incidence of AMD among different ethnic groups [47].
The Tyr402His polymorphism lies in the SCR7 of factor H, within the cluster of positively charged amino acids implicated in the binding of heparin, C-reactive protein (CRP) and M protein [20] (Fig. 1). Structural studies have shown that the substitution occurs towards the centre of SCR7 and that the 3D structures of both allotypes are otherwise identical [95]. In vitro functional studies with factor H recombinant fragments indicate that the substitution of Tyr for His at position 402 alters the binding specificity of SCR7 for different glycosaminoglycans [96,97] and decreases its binding to retinal pigment epithelial cells [98], although the physiological relevance of these observations is still unclear. It has also been reported that the Tyr402His polymorphism influences the binding of factor H to C-reactive protein [98–100], but the difficulties in replicating these data question the implication of the Tyr402His polymorphism in C-reactive protein binding [101,102].
Within the CFH gene, downstream of the SNP-haplotype block including the Tyr402His polymorphism, there is a second SNP LD group containing three polymorphisms, a synonymous SNP in exon 11 (rs2274700) and two intronic SNPs (rs1410996 and rs7535263), showing a stronger association with disease susceptibility than the Tyr402His variant [103,104]. Although these polymorphisms and the His402 variant form part of an extended CFH haplotype, they were described as independent predisposition variants that may be important in regulating the expression of CFH, or other nearby complement genes or both [103].
In addition to these CFH variants conferring increased risk to AMD, two common extended haplotypes in CFH gene [27,41,104] and two common SNPs in complement factor B [105] have been described associated with lower risk to AMD.
CFH haplotype H2, decreased markedly in AMD [27], is also decreased in MPGN2 and aHUS [66] (Fig. 5). CFH haplotype H2 carries the Ile62 factor H variant within the C3b binding site in the N-terminal region (SCR1–4) that is essential for the factor I-mediated co-factor and decay-accelerating activities of factor H (Fig. 1). Substitution of Val for Ile at position 62 may increase the factor H regulatory activity [106] and thus confer lower risk to AMD, MPGN2 and aHUS by reducing AP activation.
Fig. 5.
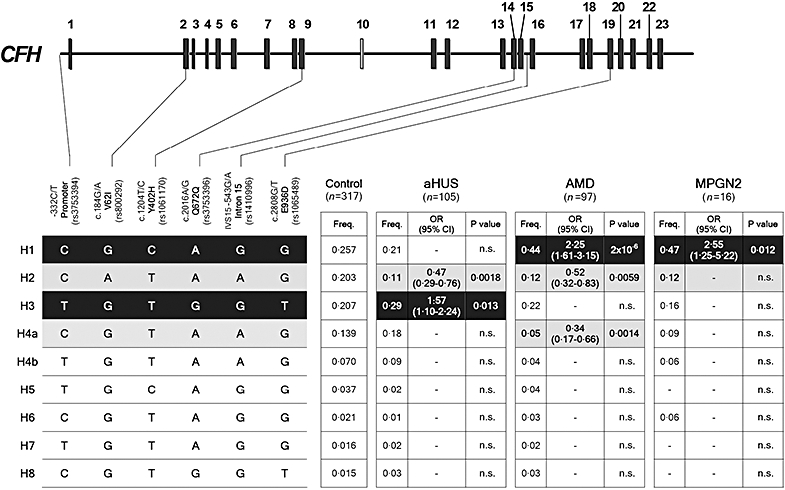
Complement factor H gene (CFH) haplotypes and their association with disease. Schematic illustration of the CFH exon structure showing the location of the six single nucleotide polymorphisms (SNPs) included in these studies. These SNPs represent a minimal informative set for genetic variation within the CFH gene. Haplotype frequencies in the control and patient cohorts were estimated using the expectation maximization (EM) algorithm implemented by the SNPStats software (available on-line at: http://bioinfo.iconcologia.net/SNPstats). CFH haplotypes with a frequency > 1% are shown. The frequency of each CFH haplotype was compared between the controls and the atypical haemolytic uraemic syndrome, age-related macular degeneration and membranoproliferative glomerulonephritis type II cohorts and the P-values and the odds ratios (OR) were calculated. Risk haplotypes are shaded black, while protective haplotypes are shaded in grey. P-values were derived using the two-sided Fisher's exact test. OR and 95% confidence intervals are shown. The nucleotide and amino acid numbering are referred to the translation start site (A in ATG is +1; Met is +1) as recommended by the Human Genome Variation Society. This figure is an updated version of that published in Pickering et al. [66].
CFH haplotype H4 is also associated with decreased risk of AMD [41,66], but not to MPGN2 or aHUS [66] (Fig. 5). Interestingly, this CFH haplotype is also unique because it carries a deletion of the CFHR1 and CFHR3 genes [41]. Although it has been indicated that CFHR1 and CFHR3 proteins have the potential to compete with factor H for C3b binding [41], the potential benefit of the absence of CFHR1 and CFHR3 proteins is puzzling, in particular because it has been reported recently that the deletion of CFHR1 and CFHR3 genes is associated with increased risk to aHUS [46].
Two factor B polymorphisms were identified that were protective for AMD [105], one of which (32Q) had been reported previously to reduce the haemolytic activity of factor B [107] and the other, in the signal peptide, was suggested to modulate secretion of factor B. The observed protection in each case was ascribed to a reduced activity of the complement alternative pathway.
The identification of CFH as a major susceptibility locus for AMD and the characterization of multiple genetic variants in the CFH–CFHR1–5 and CFB genomic regions conferring risk or protection to AMD indicate that the complement system plays a significant role in AMD pathogenesis. Further studies are, however, needed to determine the functional consequences of the CFH variants associated with AMD and to identify the molecular events influenced by the complement system in the pathogenesis of AMD. In the meantime, definition of the AMD at-risk or protective factors associated with the CFH, LOC387715/HTRA1 and CFB genes has allowed the development of risk models for AMD [87,94,104] that should be very helpful to delineate the individual risk to develop AMD, facilitating the implementation of preventive therapeutics.
CFH genotype–phenotype correlations
AMD [27,56–58], aHUS [37,53–55] and MPGN2 [49,50,52,103] are distinct pathological entities that are all associated with mutations and polymorphisms in the gene (CFH), which support the hypothesis that AP dysregulation is a unifying pathogenic feature of these diverse conditions. However, there are differences in the CFH genetic variants conferring risk or protection to one or other disease, indicating the existence of a peculiar genotype–phenotype relationship. AMD and MPGN2 share pathological similarities with accumulation of complement-containing debris within the eye and kidney, respectively. Indeed, AMD-like pathology is well-recognized in patients with MPGN2 [108]. The hallmark of AMD is drusen, complement-containing material that accumulates beneath the retinal pigmented epithelium [92], while in MPGN2 accumulation of C3 and electron-dense material is seen along the GBM [59]. In contrast to these ‘debris-associated’ conditions, aHUS is characterized by renal endothelial injury and thrombosis (thrombotic microangiopathy) resulting in haemolytic anaemia, thrombocytopenia and renal failure.
Consistent with these pathological differences, we have discussed earlier in this review that the complete factor H deficiency in humans, pigs and mice is associated with MPGN2, while aHUS-associated CFH mutations cluster within the carboxy-terminal SCR of the protein. In addition, CFH association data derived from a comparative genetic analysis, using a minimal set of informative CFH SNPs, in subjects with aHUS, AMD and MPGN2 from a single population showed no overlapping between CFH at-risk polymorphisms for aHUS and AMD (or MPGN2) [66] (Fig. 5). Recently, this CFH genotype–phenotype correlation has been established formally in a murine model. Factor H-deficient mice (Cfh–/–) develops MPGN2 as a consequence of the massive activation of C3 [64]. These mice present very low levels of C3 and complement activities in plasma. Introduction into Cfh–/– mice of a transgenic factor H molecule (FHΔ16−20) that mimics the human aHUS-associated mutations restored the C3 levels and the complement activity in the plasma of these factor H-deficient animals. As a result, Cfh–/–FHΔ16−20 animals switch their disease phenotype from MPGN2 to aHUS [66]. These data validate the previous hypothesis [37,78], establishing definitively that the combination of active complement in plasma with a decreased protection of cell surfaces leads to aHUS.
The challenge now is to understand the functional consequences of all the genetic variations in the complement genes associated with high and low risk to aHUS, MPGN2 and AMD and to determine how they influence the complex interplay of regulators and activators in the homeostasis of the complement system, in the elimination of cellular debris and in the protection of the host cells.
Concluding remarks
We have reviewed recent advances in the genetics of factor H and summarized overwhelming evidence that associates different genetic variants of factor H with ocular and renal disease. The data available support a strong genotype–phenotype correlation between CFH and these conditions suggesting that, despite a common link involving complement dysregulation, there are distinct functional alterations in factor H that are essential in the pathogenesis of these disorders. It is now well established that mutations or polymorphisms altering the C3b/polyanions binding site located at the C-terminal region of factor H are associated strongly with aHUS. Specifically, these mutations impair the capacity of factor H to protect host cells. Accordingly, aHUS is emerging as a paradigm of disease resulting from the inefficient protection of the host cellular surfaces from complement activation. On the other hand, mutations that disrupt the plasma activities of factor H so that it fails to control complement activation result in a massive activation of C3 that causes MPGN2. AMD associates strongly and specifically with a common extended CFH haplotype carrying the 402His polymorphism, but the molecular bases of this association are controversial and still unclear. There are also common CFH haplotypes that associate specifically with lower risk to AMD. Understanding the functional consequences of the different CFH genetic variants should help to determine the molecular events that are critical in the pathogenesis of AMD. These studies, and the generation of animal models for the different disease-associated CFH genetic variants, should guide the future development of effective aHUS, MPGN and AMD therapeutics.
Acknowledgments
S. R. de C. is supported by funds provided by the Spanish Ministerio de Educación y Cultura (SAF2005-00913).
References
- 1.Law SKA, Reid KBM. Complement. 2. Oxford: IRL Press; 1995. [Google Scholar]
- 2.Pangburn MK, Schreiber RD, Müller-Eberhard HJ. Human complement C3b inactivator: isolation, characterization, and demonstration of an absolute requirement for the serum protein beta1H for cleavage of C3b and C4b in solution. J Exp Med. 1977;146:257–70. doi: 10.1084/jem.146.1.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiler JM, Daha MR, Austen KF, Fearon DT. Control of the amplification convertase of complement by the plasma protein beta1H. Proc Natl Acad Sci USA. 1976;73:3268–72. doi: 10.1073/pnas.73.9.3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whaley K, Ruddy S. Modulation of the alternative complement pathways by beta 1 H globulin. J Exp Med. 1976;144:1147–63. doi: 10.1084/jem.144.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fearon DT. Regulation by membrane sialic acid of beta1H-dependent decay-dissociation of amplification C3 convertase of the alternative complement pathway. Proc Natl Acad Sci USA. 1978;75:1971–5. doi: 10.1073/pnas.75.4.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kazatchkine MD, Fearon DT, Austen KF. Human alternative complement pathway: membrane-associated sialic acid regulates the competition between B and beta1 H for cell-bound C3b. J Immunol. 1979;122:75–81. [PubMed] [Google Scholar]
- 7.Pangburn MK, Schreiber RD, Muller-Eberhard HJ. C3b deposition during activation of the alternative complement pathway and the effect of deposition on the activating surface. J Immunol. 1983;131:1930–5. [PubMed] [Google Scholar]
- 8.Ripoche J, Day AJ, Harris TJ, Sim RB. The complete amino acid sequence of human complement factor H. Biochem J. 1988;249:593–602. doi: 10.1042/bj2490593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuhn S, Skerka C, Zipfel PF. Mapping of the complement regulatory domains in the human factor H-like protein 1 and in factor H1. J Immunol. 1995;155:5663–70. [PubMed] [Google Scholar]
- 10.Alsenz J, Schulz TF, Lambris JD, Sim RB, Dierich MP. Structural and functional analysis of the complement component factor H with the use of different enzymes and monoclonal antibodies to factor H. Biochem J. 1985;232:841–50. doi: 10.1042/bj2320841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon DL, Kaufman RM, Blackmore TK, Kwong J, Lublin DM. Identification of complement regulatory domains in human factor H. J Immunol. 1995;155:348–56. [PubMed] [Google Scholar]
- 12.Jokiranta TS, Zipfel PF, Hakulinen J, et al. Analysis of the recognition mechanism of the alternative pathway of complement by monoclonal anti-factor H antibodies: evidence for multiple interactions between H and surface bound C3b. FEBS Lett. 1996;393:297–302. doi: 10.1016/0014-5793(96)00905-2. [DOI] [PubMed] [Google Scholar]
- 13.Prodinger WM, Hellwage J, Spruth M, Dierich MP, Zipfel PF. The C-terminus of factor H: monoclonal antibodies inhibit heparin binding and identify epitopes common to factor H and factor H-related proteins. Biochem J. 1998;331:41–7. doi: 10.1042/bj3310041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blackmore TK, Hellwage J, Sadlon TA, et al. Identification of the second heparin-binding domain in human complement factor H. J Immunol. 1998;160:3342–8. [PubMed] [Google Scholar]
- 15.Blackmore TK, Sadlon TA, Ward HM, Lublin DM, Gordon DL. Identification of a heparin binding domain in the seventh short consensus repeat of complement factor H. J Immunol. 1996;157:5422–7. [PubMed] [Google Scholar]
- 16.Pangburn MK, Atkinson MA, Meri S. Localization of the heparin-binding site on complement factor H. J Biol Chem. 1991;266:16847–53. [PubMed] [Google Scholar]
- 17.Ram S, Sharma AK, Simpson SD, et al. A novel sialic acid binding site on factor H mediates serum resistance of sialylated neisseria gonorrhoeae. J Exp Med. 1998;187:743–52. doi: 10.1084/jem.187.5.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaplan MH, Volanakis JE. Interaction of C-reactive protein complexes with the complement system. I. Consumption of human complement associated with the reaction of C-reactive protein with pneumococcal C-polysaccharide and with the choline phosphatides, lecithin and sphingomyelin. J Immunol. 1974;112:2135–47. [PubMed] [Google Scholar]
- 19.Mold C, Kingzette M, Gewurz H. C-reactive protein inhibits pneumococcal activation of the alternative pathway by increasing the interaction between factor H and C3b. J Immunol. 1984;133:882–5. [PubMed] [Google Scholar]
- 20.Giannakis E, Jokiranta TS, Male DA, et al. A common site within factor H SCR 7 responsible for binding heparin, C-reactive protein and streptococcal M protein. Eur J Immunol. 2003;33:962–9. doi: 10.1002/eji.200323541. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez de Cordoba S, Esparza-Gordillo J, Goicoechea de Jorge E, Lopez-Trascasa M, Sanchez-Corral P. The human complement factor H: functional roles, genetic variations and disease associations. Mol Immunol. 2004;41:355–67. doi: 10.1016/j.molimm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 22.Morris KM, Aden DP, Knowles BB, Colten HR. Complement biosynthesis by the human hepatoma-derived cell line HepG2. J Clin Invest. 1982;70:906–13. doi: 10.1172/JCI110687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwaeble W, Zwirner J, Schulz TF, Linke RP, Dierich MP, Weiss EH. Human complement factor H: expression of an additional truncated gene product of 43 kDa in human liver. Eur J Immunol. 1987;17:1485–9. doi: 10.1002/eji.1830171015. [DOI] [PubMed] [Google Scholar]
- 24.Esparza-Gordillo J, Soria JM, Buil A, et al. Genetic and environmental factors influencing the human factor H plasma levels. Immunogenetics. 2004;56:77–82. doi: 10.1007/s00251-004-0660-7. [DOI] [PubMed] [Google Scholar]
- 25.Chen M, Forrester JV, Xu H. Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp Eye Res. 2007;84:635–45. doi: 10.1016/j.exer.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 26.Friese MA, Hellwage J, Jokiranta TS, et al. FHL-1/reconectin and factor H: two human complement regulators which are encoded by the same gene are differently expressed and regulated. Mol Immunol. 1999;36:809–18. doi: 10.1016/s0161-5890(99)00101-7. [DOI] [PubMed] [Google Scholar]
- 27.Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227–32. doi: 10.1073/pnas.0501536102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Estaller C, Schwaeble W, Dierich M, Weiss EH. Human complement factor H: two factor H proteins are derived from alternatively spliced transcripts. Eur J Immunol. 1991;21:799–802. doi: 10.1002/eji.1830210337. [DOI] [PubMed] [Google Scholar]
- 29.Misasi R, Huemer HP, Schwaeble W, Sölder E, Larcher C, Dierich MP. Human complement factor H: an additional gene product of 43 kDa isolated from human plasma shows cofactor activity for the cleavage of the third component of complement. Eur J Immunol. 1989;19:1765–8. doi: 10.1002/eji.1830190936. [DOI] [PubMed] [Google Scholar]
- 30.Zipfel PF, Skerka C. FHL-1/reconectin: a human complement and immune regulator with cell-adhesive function. Immunol Today. 1999;20:135–40. doi: 10.1016/s0167-5699(98)01432-7. [DOI] [PubMed] [Google Scholar]
- 31.McRae JL, Cowan PJ, Power DA, et al. Human factor H-related protein 5 (FHR-5). A new complement-associated protein. J Biol Chem. 2001;276:6747–54. doi: 10.1074/jbc.M007495200. [DOI] [PubMed] [Google Scholar]
- 32.Skerka C, Hellwage J, Weber W, et al. The human factor H-related protein 4 (FHR-4). A novel short consensus repeat-containing protein is associated with human triglyceride-rich lipoproteins. J Biol Chem. 1997;272:5627–34. doi: 10.1074/jbc.272.9.5627. [DOI] [PubMed] [Google Scholar]
- 33.Zipfel PF, Jokiranta TS, Hellwage J, Koistinen V, Meri S. The factor H protein family. Immunopharmacology. 1999;42:53–60. doi: 10.1016/s0162-3109(99)00015-6. [DOI] [PubMed] [Google Scholar]
- 34.Zipfel PF, Skerka C. Complement factor H and related proteins: an expanding family of complement-regulatory proteins? Immunol Today. 1994;15:121–6. doi: 10.1016/0167-5699(94)90155-4. [DOI] [PubMed] [Google Scholar]
- 35.Díaz-Guillén MA, Rodríguez de Córdoba S, Heine-Suñer D. A radiation hybrid map of complement factor H and factor H-related genes. Immunogenetics. 1999;49:549–52. doi: 10.1007/s002510050534. [DOI] [PubMed] [Google Scholar]
- 36.McRae JL, Murphy BE, Eyre HJ, Sutherland GR, Crawford J, Cowan PJ. Location and structure of the human FHR-5 gene. Genetica. 2002;114:157–61. doi: 10.1023/a:1015114512924. [DOI] [PubMed] [Google Scholar]
- 37.Perez-Caballero D, Gonzalez-Rubio C, Gallardo ME, et al. Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome. Am J Hum Genet. 2001;68:478–84. doi: 10.1086/318201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodriguez de Cordoba S, Diaz-Guillen MA, Heine-Suñer D. An integrated map of the human regulator of complement activation (RCA) gene cluster on 1q32. Mol Immunol. 1999;36:803–8. doi: 10.1016/s0161-5890(99)00100-5. [DOI] [PubMed] [Google Scholar]
- 39.Rodriguez de Cordoba S, Lublin DM, Rubinstein P, Atkinson JP. Human genes for three complement components that regulate the activation of C3 are tightly linked. J Exp Med. 1985;161:1189–95. doi: 10.1084/jem.161.5.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Male DA, Ormsby RJ, Ranganathan S, Giannakis E, Gordon DL. Complement factor H: sequence analysis of 221 kb of human genomic DNA containing the entire fH, fHR-1 and fHR-3 genes. Mol Immunol. 2000;37:41–52. doi: 10.1016/s0161-5890(00)00024-9. [DOI] [PubMed] [Google Scholar]
- 41.Hughes AE, Orr N, Esfandiary H, Diaz-Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age-related macular degeneration. Nat Genet. 2006;38:1173–7. doi: 10.1038/ng1890. [DOI] [PubMed] [Google Scholar]
- 42.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tortajada A, Hakobyan S, Goicoechea de Jorge E, et al. Factor H allele-specific quantification in Tyr402His heterozygotes reveals the existence of low-expression alleles associated with atypical haemolytic uraemic syndrome. Mol Immunol. 2007;44:3925. [Google Scholar]
- 44.Venables JP, Strain L, Routledge D, et al. Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PLoS Med. 2006;3:e431. doi: 10.1371/journal.pmed.0030431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heinen S, Sanchez-Corral P, Jackson MS, et al. De novo gene conversion in the RCA gene cluster (1q32) causes mutations in complement factor H associated with atypical hemolytic uremic syndrome. Hum Mutat. 2006;27:292–3. doi: 10.1002/humu.9408. [DOI] [PubMed] [Google Scholar]
- 46.Zipfel PF, Edey M, Heinen S, et al. Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet. 2007;3:e41. doi: 10.1371/journal.pgen.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hageman GS, Hancox LS, Taiber AJ, et al. Extended haplotypes in the complement factor H (CFH) and CFH-related (CFHR) family of genes protect against age-related macular degeneration: characterization, ethnic distribution and evolutionary implications. Ann Med. 2006;38:592–604. [PMC free article] [PubMed] [Google Scholar]
- 48.Martínez-Barricarte B, Goicoechea de Jorge E, Recalde S, et al. Complement factor H haplotypes and copy number variations of the factor H-related genes in renal and ocular disorders. Mol Immunol. 2007;44:3919–20. [Google Scholar]
- 49.Dragon-Durey M-A, Fremeaux-Bacchi V, Loirat C, et al. Heterozygous and homozygous factor H deficiencies associated with hemolytic uremic syndrome or membranoproliferative glomerulonephritis: report and genetic analysis of 16 cases. J Am Soc Nephrol. 2004;15:787–95. doi: 10.1097/01.asn.0000115702.28859.a7. [DOI] [PubMed] [Google Scholar]
- 50.Levy M, Halbwachs-Mecarelli MC, Guber G, et al. H deficiency in two brothers with atypical dense intramembraneous deposit disease. Kidney Int. 1986;30:949–56. doi: 10.1038/ki.1986.278. [DOI] [PubMed] [Google Scholar]
- 51.Licht C, Heinen S, Jozsi M, et al. Deletion of Lys224 in regulatory domain 4 of factor H reveals a novel pathomechanism for dense deposit disease (MPGN II) Kidney Int. 2006;70:42–50. doi: 10.1038/sj.ki.5000269. [DOI] [PubMed] [Google Scholar]
- 52.López-Larrea C, Dieguez MA, Enguix A, Dominguez O, Marin B, Gomez E. A familial deficiency of complement factor H. Biochem Soc Trans. 1987;15:648–9. [Google Scholar]
- 53.Caprioli J, Castelletti F, Bucchioni S, et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C-257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet. 2003;12:3385–95. doi: 10.1093/hmg/ddg363. [DOI] [PubMed] [Google Scholar]
- 54.Richards A, Buddles MR, Donne RL, et al. Factor H mutations in hemolytic uremic syndrome cluster in exons 18–20, a domain important for host cell recognition. Am J Hum Genet. 2001;68:485–90. doi: 10.1086/318203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Warwicker P, Goodship THJ, Donne RL, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998;53:836–44. doi: 10.1111/j.1523-1755.1998.00824.x. [DOI] [PubMed] [Google Scholar]
- 56.Edwards AO, Ritter R, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005;308:421–4. doi: 10.1126/science.1110189. [DOI] [PubMed] [Google Scholar]
- 57.Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005;308:419–21. doi: 10.1126/science.1110359. [DOI] [PubMed] [Google Scholar]
- 58.Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005;308:385–9. doi: 10.1126/science.1109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Appel GB, Terence CH, Hageman G, et al. Membranoproliferative glomerulonephritis type II (dense deposit disease): an update. J Am Soc Nephrol. 2005;16:1392–403. doi: 10.1681/ASN.2005010078. [DOI] [PubMed] [Google Scholar]
- 60.Ault BH, Schmidt BZ, Fowler NL, et al. Human factor H deficiency. Mutations in framework cysteine residues and block in H protein secretion and intracellular catabolism. J Biol Chem. 1997;272:25168–75. doi: 10.1074/jbc.272.40.25168. [DOI] [PubMed] [Google Scholar]
- 61.Zipfel PF, Heinen S, Jozsi M, Skerka C. Complement and diseases: defective alternative pathway control results in kidney and eye diseases. Mol Immunol. 2006;43:97–106. doi: 10.1016/j.molimm.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 62.Jokiranta TS, Solomon A, Pangburn MK, Zipfel PF, Meri S. Nephritogenic {lambda} light chain dimer: a unique human miniautoantibody against complement factor H. J Immunol. 1999;163:4590–6. [PubMed] [Google Scholar]
- 63.Høgåsen K, Jansen JH, Mollnes TE, Hovdenes J, Harboe M. Hereditary porcine membranoproliferative glomerulonephritis type II is caused by factor H deficiency. J Clin Invest. 1995;95:1054–61. doi: 10.1172/JCI117751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pickering MC, Cook HT, Warren J, et al. Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet. 2002;31:424–8. doi: 10.1038/ng912. [DOI] [PubMed] [Google Scholar]
- 65.Abrera-Abeleda MA, Nishimura C, Smith JLH, et al. Variations in the complement regulatory genes factor H (CFH) and factor H related 5 (CFHR5) are associated with membranoproliferative glomerulonephritis type II (dense deposit disease) J Med Genet. 2006;43:582–9. doi: 10.1136/jmg.2005.038315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pickering MC, Goicoechea de Jorge E, Martinez-Barricarte R, et al. Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med. 2007;204:1249–56. doi: 10.1084/jem.20070301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:1035–50. doi: 10.1681/ASN.2004100861. [DOI] [PubMed] [Google Scholar]
- 68.Caprioli J, Bettinaglio P, Zipfel PF, et al. The molecular basis of familial hemolytic uremic syndrome: mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J Am Soc Nephrol. 2001;12:297–307. doi: 10.1681/ASN.V122297. [DOI] [PubMed] [Google Scholar]
- 69.Manuelian T, Hellwage J, Meri S, et al. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J Clin Invest. 2003;111:1181–90. doi: 10.1172/JCI16651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Esparza-Gordillo J, Goicoechea de Jorge E, Buil A, et al. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet. 2005;14:703–12. doi: 10.1093/hmg/ddi066. [DOI] [PubMed] [Google Scholar]
- 71.Fremeaux-Bacchi V, Kemp EJ, Goodship JA, et al. The development of atypical haemolytic–uraemic syndrome is influenced by susceptibility factors in factor H and membrane cofactor protein: evidence from two independent cohorts. J Med Genet. 2005;42:852–6. doi: 10.1136/jmg.2005.030783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Noris M, Brioschi S, Caprioli J, et al. Familial haemolytic uraemic syndrome and an MCP mutation. Lancet. 2003;362:1542–7. doi: 10.1016/S0140-6736(03)14742-3. [DOI] [PubMed] [Google Scholar]
- 73.Richards A, Kemp EJ, Liszewski MK, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2003;100:12966–71. doi: 10.1073/pnas.2135497100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fremeaux-Bacchi V, Dragon-Durey MA, Blouin J, et al. Complement factor I: a susceptibility gene for atypical haemolytic uraemic syndrome. J Med Genet. 2004;41:e84. doi: 10.1136/jmg.2004.019083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kavanagh D, Kemp EJ, Mayland E, et al. Mutations in complement factor I predispose to development of atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:2150–5. doi: 10.1681/ASN.2005010103. [DOI] [PubMed] [Google Scholar]
- 76.Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, et al. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2007;104:240–5. doi: 10.1073/pnas.0603420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fremeaux-Bacchi V, Regnier C, Blouin J, et al. Protective or aggressive: paradoxical role of C3 in atypical hemolytic uremic syndrome. Mol Immunol. 2007;44:172. [Google Scholar]
- 78.Sánchez-Corral P, Pérez-Caballero D, Huarte O, et al. Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am J Hum Genet. 2002;71:1285–95. doi: 10.1086/344515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sanchez-Corral P, Gonzalez-Rubio C, Rodriguez de Cordoba S, Lopez-Trascasa M. Functional analysis in serum from atypical hemolytic uremic syndrome patients reveals impaired protection of host cells associated with mutations in factor H. Mol Immunol. 2004;41:81–4. doi: 10.1016/j.molimm.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 80.Atkinson JP, Liszewski MK, Richards A, Kavanagh D, Moulton EA. Hemolytic uremic syndrome. An example of insufficient complement regulation on self-tissue. Ann NY Acad Sci. 2005;1056:144–52. doi: 10.1196/annals.1352.032. [DOI] [PubMed] [Google Scholar]
- 81.Ohali M, Shalev H, Schlesinger M, et al. Hypocomplementemic autosomal recessive hemolytic uremic syndrome with decreased factor H. Pediatr Nephrol. 1998;12:619–24. doi: 10.1007/s004670050515. [DOI] [PubMed] [Google Scholar]
- 82.Pichette V, Querin S, Schurch W, Brun G, Lehner-Netsch G, Delage JM. Familial hemolytic–uremic syndrome and homozygous factor H deficiency. Am J Kidney Dis. 1994;24:936–41. doi: 10.1016/s0272-6386(12)81065-1. [DOI] [PubMed] [Google Scholar]
- 83.Rougier N, Kazatchkine MD, Rougier JP, et al. Human complement factor H deficiency associated with hemolytic uremic syndrome. J Am Soc Nephrol. 1998;9:2318–26. doi: 10.1681/ASN.V9122318. [DOI] [PubMed] [Google Scholar]
- 84.Thompson RA, Winterborn MH. Hypocomplementaemia due to a genetic deficiency of beta 1H globulin. Clin Exp Immunol. 1981;46:110–19. [PMC free article] [PubMed] [Google Scholar]
- 85.Schreiber RD, Pangburn MK, Lesavre PH, Müller-Eberhard HJ. Initiation of the alternative pathway of complement: recognition of activators by bound C3b and assembly of the entire pathway from six isolated proteins. Proc Natl Acad Sci USA. 1978;75:3948–52. doi: 10.1073/pnas.75.8.3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Neumann HPH, Salzmann M, Bohnert-Iwan B, et al. Haemolytic uraemic syndrome and mutations of the factor H gene: a registry-based study of German speaking countries. J Med Genet. 2003;40:676–81. doi: 10.1136/jmg.40.9.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Esparza-Gordillo J, Goicoechea de Jorge E, Abarrategui Garrido C, et al. Insights into hemolytic uremic syndrome: segregation of three independent predisposition factors in a large, multiple affected pedigree. Mol Immunol. 2006;43:1769–75. doi: 10.1016/j.molimm.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 88.Dragon-Durey M-A, Loirat C, Cloarec S, et al. Anti-factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:555–63. doi: 10.1681/ASN.2004050380. [DOI] [PubMed] [Google Scholar]
- 89.Jozsi M, Strobel S, Dahse HM, et al. Anti factor H autoantibodies block C-terminal recognition function of factor H in hemolytic uremic syndrome. Blood. 2007;110:1516–18. doi: 10.1182/blood-2007-02-071472. [DOI] [PubMed] [Google Scholar]
- 90.Dragon-Durey M-A, Loirat C, Ranchin B, et al. Development of anti-factor H auto-antibodies genetically predisposed in atypical HUS. Mol Immunol. 2007;44:3922. [Google Scholar]
- 91.Józsi M, Strobel S, Zipfel SLH, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency and affect recognition functions. Mol Immunol. 2007;44:3024. [Google Scholar]
- 92.Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–31. doi: 10.1016/s0002-9394(02)01624-0. [DOI] [PubMed] [Google Scholar]
- 93.Scholl HP, Fleckenstein M, Charbel Issa P, Keilhauer C, Holz FG, Weber BH. An update on the genetics of age-related macular degeneration. Mol Vis. 2007;13:196–205. [PMC free article] [PubMed] [Google Scholar]
- 94.Rivera A, Fisher SA, Fritsche LG, et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet. 2005;14:3227–36. doi: 10.1093/hmg/ddi353. [DOI] [PubMed] [Google Scholar]
- 95.Herbert AP, Deakin JA, Schmidt CQ, et al. Structure shows glycosaminoglycan- and protein-recognition site in factor H is perturbed by age-related macular degeneration-linked SNP. J Biol Chem. 2007;282:18960–8. doi: 10.1074/jbc.M609636200. [DOI] [PubMed] [Google Scholar]
- 96.Clark SJ, Higman VA, Mulloy B, et al. His-384 allotypic variant of factor H associated with age-related macular degeneration has different heparin binding properties from the non-disease-associated form. J Biol Chem. 2006;281:24713–20. doi: 10.1074/jbc.M605083200. [DOI] [PubMed] [Google Scholar]
- 97.Prosser BE, Johnson S, Roversi P, et al. Structural basis for complement factor H linked age-related macular degeneration. J Exp Med. 2007;204:2277–83. doi: 10.1084/jem.20071069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Skerka C, Lauer N, Weinberger AAWA, et al. Defective complement control of factor H (Y402H) and FHL-1 in age-related macular degeneration. Mol Immunol. 2007;44:3398–406. doi: 10.1016/j.molimm.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 99.Laine M, Jarva H, Seitsonen S, et al. Y402H polymorphism of complement factor H affects binding affinity to C-reactive protein. J Immunol. 2007;178:3831–6. doi: 10.4049/jimmunol.178.6.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sjoberg AP, Trouw LA, Clark SJ, et al. The factor H variant associated with age-related macular degeneration (His-384) and the non-disease-associated form bind differentially to C-reactive protein, fibromodulin, DNA, and necrotic cells. J Biol Chem. 2007;282:10894–900. doi: 10.1074/jbc.M610256200. [DOI] [PubMed] [Google Scholar]
- 101.Fernández-Alonso C, Goicoechea de Jorge E, Jiménez M, Rodriguez de Cordoba S, Germán R. Analytical ultracentrifugation analysis of the human complement factor H variants 402His and 402Tyr. Mol Immunol. 2007;44:3982. [Google Scholar]
- 102.Hakobyan S, Harris CL, van den Berg C, Pepys MB, Morgan BP. Binding of factor H to C-reactive protein occurs only when the latter has undergone non-physiologic denaturation. Mol Immunol. 2007;44:3983–4. [Google Scholar]
- 103.Li M, Atmaca-Sonmez P, Othman M, et al. CFH haplotypes without the Y402H coding variant show strong association with susceptibility to age-related macular degeneration. Nat Genet. 2006;38:1049–54. doi: 10.1038/ng1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Maller J, George S, Purcell S, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006;38:1055–9. doi: 10.1038/ng1873. [DOI] [PubMed] [Google Scholar]
- 105.Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006;38:458–62. doi: 10.1038/ng1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kuttner-Kondo L, Muqim N, Crabb JW, Hollyfield JG, Medof ME. Effects on factor H function of polymorphisms linked to age-related macular degeneration. Mol Immunol. 2007;44:201. [Google Scholar]
- 107.Lokki ML, Koskimies SA. Allelic differences in hemolytic activity and protein concentration of BF molecules are found in association with particular HLA haplotypes. Immunogenetics. 1991;34:242–6. doi: 10.1007/BF00215259. [DOI] [PubMed] [Google Scholar]
- 108.Mullins RF, Aptsiauri N, Hageman GS. Structure and composition of drusen associated with glomerulonephritis: implications for the role of complement activation in drusen biogenesis. Eye. 2001;15:390–5. doi: 10.1038/eye.2001.142. [DOI] [PubMed] [Google Scholar]