

Abstract
Superantigens (SAgs) are potent stimulators of T cells bearing specific Vβ T cell receptors (TCR) and may play a role in the pathogenesis of Kawasaki syndrome (KS), although despite 15 years of intense study this area remains controversial. Because SAgs can cause Vβ restricted T cell activation in the absence of Vβ skewing the aims of this study were to describe a flow cytometric protocol to study both CD4 and CD8 Vβ repertoires, and CD69 expression across the CD4 and CD8 Vβ repertoire in children with KS. Sixteen children with KS were studied. There was no significant increase in overall peripheral blood CD4 or CD8 T cell activation as determined by CD69 expression. However, Vβ restricted CD4 and/or CD8 activation was observed in eight of 11 (72%) of the KS patients, a finding not observed in healthy controls. Thirteen of 16 (81%) of the KS patients had evidence of either Vβ skewing (particularly CD4 Vβ2 and Vβ5·1) and/or Vβ restricted activation. Three patients had Vβ restricted activation in the absence of skewing. We suggest that these preliminary observations highlight the many layers of complexity when considering T cell activation in KS, which could explain some of the conflicting studies regarding peripheral blood T cell activation and Vβ skewing. It is likely that in order to move forward with this debate a combination of detailed microbiological, immunological and molecular techniques applied to individual patients will be required ultimately to prove or refute the SAg hypothesis of KS.
Keywords: child, Kawasaki syndrome, superantigen, T cells, vasculitis
Introduction
Kawasaki syndrome (KS) is associated with the development of systemic vasculitis complicated by coronary and peripheral arterial aneurysms, and myocardial infarction in some patients [1]. It has superseded rheumatic fever, in that KS is now the most common cause of acquired heart disease in children in the United Kingdom and the United States [1,2]. It is generally accepted that an as yet undefined infectious trigger in a genetically predisposed individual results in the disease. Despite intensive research into the illness the cause remains unknown, and although there have been significant improvements in diagnosis and treatment of children with the disease there are still a number of important unanswered questions regarding aetiopathogenesis.
Superantigens (SAgs) are one of the environmental factors that have been proposed to modulate a number of autoimmune diseases, including vasculitis [3,4]. The most compelling evidence for involvement of SAg in the pathogenesis of vasculitis relates to KS, but at the same time this hypothesis has provided the most controversy [5,6]. SAgs are a class of immunostimulatory proteins of bacterial or viral origin with the ability to activate large fractions (5–30%) of the T cell population, and are responsible for human toxic shock syndrome and some forms of gastroenteritis [7,8].
There are similarities between the clinical and immunological features of KS and the superantigen toxin-mediated staphylococcal and streptococcal toxic shock syndromes, and scarlet fever [9–13]. Although there is evidence supporting a role for superantigens (SAgs) in KS, so far this has been conflicting. Several key observations in patients with KS would be consistent with the SAg hypothesis and include: percentage skewing of the peripheral blood T cell Vβ repertoire during the acute and subacute phases of the disease, particularly of T cells expressing Vβ2 [3,9,10,13–16]; increased carriage of toxic shock syndrome toxin 1 (TSST-1) producing Staphylococcus aureus, and Streptococcal pyrogenic exotoxin types B and C (SPEB/ SPEC) producing group A Streptococci in patients with KS [17]; and most recently seroconversion of immunoglobulin M (IgM) antibodies against SAgs of Staph. aureus and S. pyogenes during the course of KS [18].
Another important observation is that T cells infiltrating the walls of coronary arterial aneurysms and the intestinal mucosa of patients with KS show a skewed T cell Vβ repertoire, with increased numbers of cells expressing Vβ2 [13,19], again compatible with a SAg-driven process resulting in sequestration of activated T cells into lesional vascular tissue.
SAgs are capable of inducing endothelial activation and injury. We have shown previously that Class II major histocompatibilty complex (MHC)-positive endothelial cells operate as competent superantigen-presenting cells for CD4 and CD8 lymphocytes in vitro[7]. Dual signalling between endothelial cells and T cells results in Vβ-restricted activation and adherence to endothelium, resulting in endothelial cell activation and injury. Thus SAgs could result mechanistically in severe endothelial and vascular injury, and could account in part for the vascular injury associated with superantigen-mediated diseases such as toxic shock syndrome. Vascular sequestration of Vβ-restricted activated T cells by this mechanism could explain why T cell activation in the peripheral blood of children with KS during the acute phase is relatively low, whereas lesional KS tissue has high numbers of CD3 positive T cells expressing a restricted Vβ T cell receptor pattern, particularly high numbers of CD3Vβ2 cells [13,19]. Moreover, the finding of high surface expression of MHC class II on endothelial cells from a fatal case of KS could be compatible with this proposed model of SAg-induced vascular injury [20].
However, there are a number of studies that contest the SAg hypothesis in KS, including the observation that peripheral blood Vβ T cell receptor skewing has been an inconsistent observation [21–24]. Furthermore, Rowley et al. reported three fatal cases of KD and observed IgA plasma cell infiltration into the vascular wall during the acute phase of the illness. By examining the clonality of this IgA response using reverse trranscription–polymerase chain reaction (RT–PCR) in lesional vascular tissue they observed that the IgA response was oligoclonal, suggesting a conventional Ag, possibly intracellular, rather than a SAg-driven process [25]. These observations led the authors of these studies to suggest that there is a common infectious aetiology for KS, perhaps viral, and they suggest that this may cast doubt on the SAg hypothesis in KS.
If SAgs are involved in the pathogenesis of KS, why is the classical immunological footprint of Vβ T cell receptor skewing in peripheral blood not seen in all cases? Before the SAg hypothesis is discounted on this basis it is important to consider that SAgs cause sequential activation, then expansion and subsequent deletion of T cells expressing specific Vβ subunits of the T cell receptor [26]. Thus, depending on the timing of blood sampling in relation to disease onset, and factors including the persistence of concomitant infection at the time of KS, it could be possible to have SAg-induced Vβ restricted T cell activation in the absence of percentage skewing of the Vβ repertoire. Indeed, no study to date has examined peripheral blood for specific Vβ T cell activation as opposed to percentage skewing in patients with KS, an approach that has been applied in the past to other infectious diseases [27,28].
We hypothesized that if SAgs are involved in KS then we may be able to detect Vβ restricted T cell activation with or without Vβ skewing. The aims of this study were therefore to assess T cell activation profiles in more detail in patients with KS using a flow cytometric protocol which examined both CD4 and CD8 Vβ restricted activation in addition to Vβ skewing.
Materials and methods
KS patients
KS patients were enrolled prospectively from multiple district general hospitals in the Greater London area from September 2001–August 2003, co-ordinated by the London Kawasaki disease research group and with multi-regional clinical ethics approval. Informed consent was obtained from the parents of all children studied. Sixteen patients with complete KS were studied. There were nine males and seven females, mean age 2·7 years (range 0·25–6·9 years). All patients had complete KS as defined by presence of fever for 5 or more days plus four of five of the following criteria: bilateral non-purulent injection; oral mucosal changes (strawberry tongue and/or red cracked lips); cervical lymphadenopathy (greater than or equal to 1·5 cm), polymorphous rash; and typical changes of the hands or feet (indurative oedema, palmar/plantar erythema or peeling).
Echocardiography was undertaken in all cases by a paediatric cardiologist as part of routine clinical care. Coronary artery lesions (CALs) were defined as coronary artery internal diameter > 2 standard deviations (s.d.) above the mean for age adjusted body surface area (BSA) [2]. Coronary artery ectasia was defined as internal vessel diameter > 2 s.d. but < 4 mm absolute diameter [2]. Coronary aneurysms were defined as internal diameters > 4 mm with giant aneurysms defined as internal diameter > 8 mm.
All patients had blood sampled prior to treatment with intravenous immunoglobulin (IVIG 2 g/kg over 12 h) and aspirin. Patients were sampled on median day 10 of illness (range 6–22 days), with the first day of fever defining day 1 of illness. Five of 16 had coronary artery aneurysms (31%); one had coronary and axillary artery aneurysms. Two of 16 were IVIG resistant and required a second dose of IVIG.
Healthy controls and other childhood controls
A total of 31 healthy control children were included in the study. Twenty of these controls were described previously in our paper examining Vβ repertoires in children with vasculitis [3]. Of the 11 additional controls included in this study, there was insufficient blood available to undertake all aspects of T cell analysis described: hence 11 of the healthy control children were used to compare overall CD4 or CD8 CD69 expression. Ten of these 11 controls had Vβ repertoire studies undertaken and were added to the previous 20 children we had described. Six of these 11 controls had both Vβ repertoire and Vβ-restricted activation assessed. The demographics of the overall control group and subgroups are summarized in Table 1.
Table 1.
Demographics of the control children.
| Control group used for study | Number of controls studied | Mean age (years) (range) | Sex (M/F) |
|---|---|---|---|
| Total controls | 31 | 8·58 (0·5–19) | 17M/14F |
| CD4 and CD8 Vβ repertoire | 30 | 8·8 (0·5–19) | 16M/14F |
| CD4 and CD8 CD69 expression | 11 | 4·3 (1–9) | 6M/5F |
| CD4 and CD8 differential Vβ CD69 expression | 6 | 3·2 (1–5) | 4M/2F |
F, female; M, male.
In addition to the six healthy control children, four disease controls of median age 11 years (range 1–12 years) with sepsis (n = 2); enteropathy (n = 1), and eczema (n = 1) were also assessed for Vβ restricted activation and Vβ skewing.
Preparation of peripheral blood mononuclear cells (PBMC) for flow cytometry
Blood (5–10 ml) was collected into sterile universal bottles containing 40 μl of preservative-free heparin (1000 U/ml). PBMCs were separated from whole blood by LymphoprepTM (Nycomed, Roskilde, Denmark) centrifugation, and either prepared directly for flow cytometry or frozen suspended in fetal calf serum with 10% dimethyl sulphoxide (Sigma, St Louis, MO, USA). PBMCs were plated onto U-bottomed 96-well plates at a concentration of 1–2 × 106 cells per ml, and incubated for 30 min at 4°C using fluorochrome-conjugated monoclonal antibodies to CD3 (phycoerythrin, mouse IgG1, 1 : 20 dilution; Becton Dickinson, San Jose, CA, USA), CD4 (Quantum RedTM, mouse IgG1, 1 : 20 dilution; Sigma) or CD8 (Quantum RedTM, mouse IgG2a, 1 : 20 dilution; Sigma), and the following different T cell Vβ families [all fluorescein isothiocyanate (FITC) conjugated, 1 : 10 dilution; Immunotech Marseille, France]: Vβ1 (rat IgG1; clone BL37·2), Vβ2 (mouse IgG1; clone MPB2D5), Vβ3 (mouse IgM; clone CH92), Vβ5·1 (mouse IgG2a; clone IMMU157), Vβ8·1 and 8·2 (mouse IgG2a; clone 56C5) and Vβ12 (mouse IgG2a; clone VER2·32·1. All antibody dilutions were made with 0·01 M phosphate-buffered saline (PBS) with 0·1% sodium azide and checked by plotting a dilution curve, the relevant dilution for each antibody corresponding to the shoulder of the curve. Each antibody was checked against an appropriate isotype-control antibody, as per the manufacturer's recommendation. Following incubation for 30 min, PBMCs were washed three times with 0·01 M PBS with 0·1% sodium azide.
Flow cytometric protocol for analysis of Vβ repertoires Vβ restricted activation
Three-colour flow cytometry was performed on a fluorescence activated cel sorter (FACScalibur) flow cytometer (Becton Dickinson) with optimal compensation set for green, orange and far-red fluorescence. T cells were identified by double-gating for forward- and light-scatter characteristics of live cells in a lymphocyte gate checked with CD3 staining. The CD4+ or CD8+ populations and the percentage of cells bearing different Vβ gene products were calculated subsequently using quadrants set on dot-plots, with markers for positivity defined using isotype-control antibodies. Cells double-positive for CD4 or CD8 and the corresponding Vβ family were then gated and a histogram of CD69 surface staining expressed both as median fluorescence index (MFI) and percentage positivity based on a marker constructed using an isotype control antibody, as per the manufacturer's recommendations. In this way it was possible to derive both the percentage of each Vβ family studied within the CD4 or CD8 T cells, as well as the differential CD69 expression across the Vβ families studied. A total of 20 000–40 000 gated lymphocyte events were stored for each Vβ family. We validated this flow cytometry protocol by incubating PBMCs in vitro for 4 h with either TSST-1 10 ng/ml or SEB 100 ng/ml and confirming the expected pattern of Vβ restricted CD69 expression (Vβ2 for TSST-1; Vβ3 and Vβ12 for SEB) in the absence of Vβ skewing, as described previously by our group [7]. Figure 1 illustrates this gating protocol using PBMCs from a healthy adult control patient with or without incubation with SEB 100 ng/ml for 4 h in vitro (data for CD4 Vβ3 only shown), and Fig. 2 demonstrates representative results from two of the KS patients using this protocol. The response of healthy adult PBMCs (n = 3) to 4-h in vitro stimulation with conventional antigen (tetanus toxoid) did not cause detectable Vβ-restricted activation using this flow cytometric protocol (data not shown).
Fig. 1.
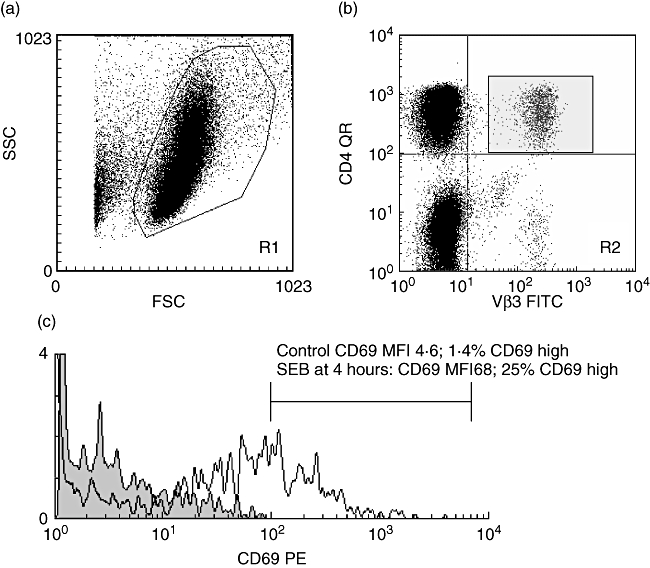
Flow cytometric protocol for the assessment of Vβ restricted CD 69 expression. (a) A forward- (FSC) and side-scatter (SSC) plot was used to define the lymphocyte gate (checked by back-gating on CD3; data not shown) region 1 (R1). (b) A dot-plot gated on R1 was then used to plot CD4 or CD8 against Vβ 1, 2, 3, 5·1, 8 or 12 (data for CD4 Vβ3 only shown). Region 2 (R2, in this example CD4 Vβ3) was then gated and a histogram plotted (c) gated on R1 and R2 of CD69 expression. In this experiment, the CD4Vβ3 response to 4 h incubation in vitro with the superantigen Staphylococcal enterotoxin B (100 ng/ml) resulted in an increase in CD69 expression from a median fluorescence index (MFI) of 4·6 (grey fill) to an MFI of 68 (or a percentage increase in CD69 high cells of 1·4% to 25%). Using this flow cytometric approach it was thus possible to derive both the percentage of each Vβ family within the CD4 or CD8 T cell subpopulation, and the CD69 expression across the CD4 and Cd8 Vβ repertoire. Abbreviations: QR, quantum red; PE, phycoerythrin; FITC, fluorescein isothiocyanate.
Fig. 2.
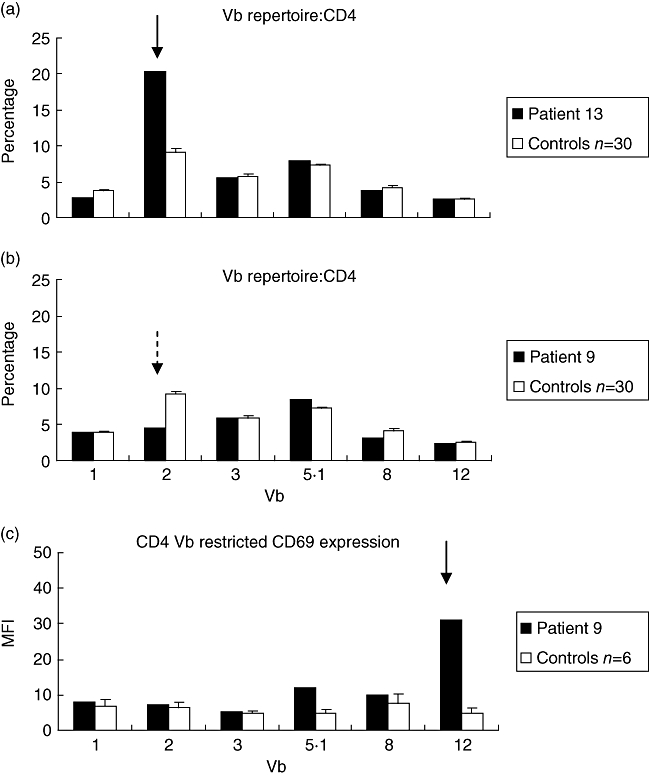
Vβ repertoire skewing and Vβ restricted CD69 expression in KS patients. (a) shows the CD4 Vβ repertoire from patient 13 sampled on day 21 of the illness with expansion of CD4 Vβ2 (solid arrow). (b) shows the CD4 Vβ repertoire from patient 9 sampled on day 11 of the illness. While there was a decrease in CD4 Vβ2 (dotted arrow) in this patient this did not fall below 2 standard deviations of the control mean and hence was not classified as a major deletion. Further examination of the pattern of CD4 Vβ restricted CD69 expression, (c), revealed up-regulated CD69 CD4 Vβ12 (solid arrow) with a greater than fivefold shift in median fluorescence index defining Vβ restricted CD69 expression.
Statistics
Assessment of skewing of the T cell Vβ repertoire in the KS patients was performed by comparison of mean percentages for each Vβ family with 30 healthy control children (including our previously published normative data in 20 healthy control children [3]) using non-paired t-tests having confirmed normality of the data. For comparison, T cell Vβ skewing was also assessed using previously published arbitrarily defined definitions of T cell Vβ expansions and deletions, with the proportion of children in each group having an expansion or deletion compared using the two-sample test of proportion. Thus, a Vβ expansion was defined as a value more than the control group mean plus 2 s.d for an individual Vβ family, and a deletion was a value less than the control group mean minus 2 s.d. [3]. Vβ-restricted activation of a particular Vβ-family was defined when the CD69 MFI was greater than five times the median of healthy control values.
Statistical significance was defined at P < 0·05, and is implied in the results wherever differences are highlighted. Error bars in figures are standard error of the mean (s.e.m.). All statistics were performed using Microsoft Excel 2003 and spss version 14·0 for Windows.
Results
CD4 and CD8 CD69 expression
Peripheral blood T cell (CD4 or CD8) CD69 expression was not increased significantly in the KS patients (n = 16) compared with healthy control children (n = 11) in agreement with previous reports [29–32] (Fig. 3).
Fig. 3.
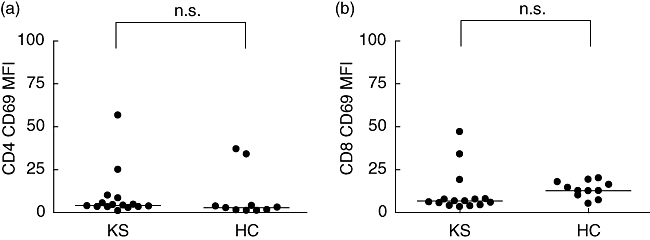
CD4 (a) and CD8 (b) CD69 expression in peripheral blood lymphocytes in Kawasaki syndrome (KS) patients (n = 16) and healthy control (HC) children (n = 11); n.s.: non-significant.
Skewing of the CD4 and CD8 T cell Vβ repertoire
There was significantly increased mean CD4 Vβ2 (P = 0·027) and CD4 Vβ5·1 (P = 0·05) in the 16 KS patients compared to 30 healthy paediatric controls (20 of whom we have described previously [3]) (Fig. 4). Figure 2a demonstrates the skewed Vβ repertoire in patient 13. CD4Vβ3 was lower in the KS patients, although this did not reach statistical significance (P = 0·055). There were no significant differences in the CD8 Vβ repertoires between the KS patients and controls. Vβ skewing affected predominantly the CD4 population of lymphocytes. Ten of 16 KS patients had CD4Vβ expansions or deletions compared with six of 30 healthy controls (P < 0·01). Two of 16 of the KS patients had a CD8 Vβ expansion or deletion compared with five of 30 of the controls [P = not significant (n.s.)]. Table 2 summarizes the expansions and deletions of the Vβ family in individual patients.
Fig. 4.
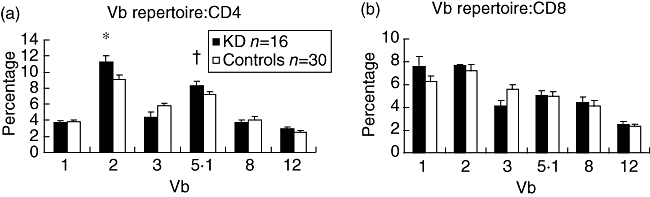
CD4 and CD8 Vβ repertoire in Kawasaki syndrome patients (n = 16) and healthy control children (n = 30). *P = 0·027; +P = 0·05 (non-paired t-tests). Error bars represent standard error of the mean (s.e.m.).
Table 2.
Vβ skewing and Vβ-restricted CD69 expression in Kawasaki syndrome (KS) patients. Individual KS patient T cell phenotype summarizing the presence (Vβ family given) or absence (−) of expansions or deletions (skewing), and also Vβ restricted CD4 and CD8 activation defined as greater than five times increase in CD69 median fluorescence.
| Vβ expansion | Vβ deletion | Vβ-specific activation | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Patient number | Age (year) | Day of illness | CD4 | CD8 | CD4 | CD8 | CD4 | CD8 | CAL |
| 1 | 0·6 | 6 | 5·1, 12 | − | − | − | − | − | − |
| 2 | 2·5 | 14 | − | 1 | − | − | − | 8 | − |
| 3 | 0·8 | 22 | 2 | − | 3 | − | 2 | − | − |
| 4 | 6·3 | ? | − | − | 3 | − | 8 | 5·1, 12 | − |
| 5 | 6·3 | 22 | − | − | − | − | − | − | − |
| 6 | 6·9 | 19 | − | − | − | − | − | − | − |
| 7 | 0·8 | 6 | − | − | − | − | 3, 8, 12 | 12 | − |
| 8 | 1·1 | 10 | − | − | 3 | − | − | 8 | − |
| 9 | 6 | 11 | − | − | − | − | 12 | − | + |
| 10 | 2·0 | 7 | 5·1 | − | − | − | 12 | 12 | − |
| 11 | 1·8 | 21 | − | − | 3 | − | n.d. | n.d. | − |
| 12 | 5·5 | ? | 12 | − | − | − | n.d. | n.d. | − |
| 13 | 2·5 | 21 | 2 | 8 | − | − | n.d. | n.d. | + |
| 14 | 0·25 | 6 | − | − | − | − | 2 | 2 | + |
| 15 | 0·25 | 10 | 5·1 | − | − | − | n.d. | n.d. | + |
| 16 | 0·25 | 10 | 2, 5·1 | − | − | − | n.d. | n.d. | + |
Eight of 11 of the KS patients had Vβ restricted activation versus none of six childhood controls (P < 0·01). This was not linked obviously to age, day of illness or to the presence or absence of coronary artery lesions (CAL). CAL, coronary artery lesion; n.d., not done; present (+); absent (−).
CD4 and CD8 Vβ-restricted CD69 expression
Eight of 11 (72%) of the KS patients had Vβ restricted activation (defined as greater than a fivefold shift in CD69 median fluorescence) versus none of six controls (P < 0·01). Vβ-specific activation affected both the CD4 and CD8 lymphocyte populations. This was not linked obviously to age, day of illness or to the presence or absence of CALs. Table 2 details the CD4 and CD8 Vβ families affected in individual patients. CD69 expression was increased on CD4 lymphocytes in the following Vβ families: Vβ12 (three of 11 patients), Vβ2 (two of 11 patients) and Vβ8 (two of 11 patients) and Vβ3 (one of 11 patients). CD69 expression was increased on CD8 lymphocytes in Vβ12 (three of 11 patients), Vβ8 (two of 11 patients), Vβ2 (one of 11 patients) and Vβ5·1 (one of 11 patients). Three of 11 patients demonstrated Vβ specific activation in the absence of skewing (patients 7, 9 and 14; Table 2), affecting CD4 Vβ3, 8 and 12 and CD8 Vβ12 (patient 7); CD4 Vβ12 (patient 9, Fig. 2b and c); and CD4 and CD8 Vβ2 (patient 14).
Vβ restricted activation was not observed in any of the four disease control children studied (data not shown).
Discussion
Since the early 1990s there have been conflicting results from several studies describing T cell Vβ skewing in patients with KS, other studies not confirming this observation. Thus the SAg debate remains unresolved. The data presented in our study suggest that the peripheral T cell response to SAgs is more complex than merely the presence or absence of Vβ skewing, and could begin to provide insight into this contentious area.
In agreement with previous reports, we demonstrated overall increased percentage CD4 Vβ2 in KS patients. We also documented Vβ5·1 skewing (P = 0·05) in the KS patients (albeit to a lesser degree than Vβ2), to the best of our knowledge a novel observation that will require further validation in a larger group of patients. Both these Vβ families are known to be responsive to common staphylococcal and streptococcal SAgs [17,33–35]. In a recent study Benseler et al. demonstrated that, in contrast to the historical notion that KS was excluded if infection was identified, approximately 33% of children with typical KS had an identifiable infection at the time of diagnosis [36]. Moreover, the most common infection in that study was group A Streptococcus, which could be consistent with our observation of Vβ2 and Vβ5·1 skewing in a proportion of our patients (Table 2).
Also shown in Table 2, however, we observed other CD4 and CD8 expansions or deletions within individual patients. Our study, and previous studies, are unlikely to have been powered to detect differences relating to multiple Vβ families and this could account partly for some of the conflicting observations relating to previous reports.
In a limited number of patients (n = 11), we attempted to define further the T cell activation profile in KS. We (and others) have demonstrated that SAgs could produce Vβ restricted activation of T cells in the absence of skewing, suggesting that the absence of peripheral T cell Vβ skewing may not preclude SAg involvement. To test this hypothesis in KS we designed a flow cytometric protocol that would enable analysis of Vβ skewing and Vβ restricted activation in individual KS patients (Fig. 1). This approach has not been studied in KS, although preliminary work has been reported relating to T cell responses to Shigella antigen [27], and to the possible link between coxsackievirus B4 and type 1 diabetes mellitus [28].
In keeping with previous reports we did not observe a significant increase in overall CD4 or CD8 T cell activation based on CD69 expression in the KS patients. However, a more detailed analysis of the pattern of CD69 expression across the CD4 and CD8 Vβ repertoire revealed that in eight of 11 patients there was evidence of Vβ restricted activation that was not apparent when considering only overall CD4 or CD8 CD69 expression. Overall, there was inconsistent overlap between the specific Vβ family skewed and activated, and in three patients (patients 7, 9 and 14) Vβ restricted activation occurred in the absence of skewing. This finding was not observed in healthy controls, or in our small number of disease control children.
Varela-Calvino et al. have suggested that such Vβ-specific C69 expression is also observed in a limited number of patients with new-onset type 1 diabetes mellitus, and suggest that this may not be due necessarily to a superantigenic effect [28]. This preliminary finding, and the implication that conventional antigens may cause Vβ restricted activation and/or skewing, if confirmed could have implications for our understanding of the findings presented in our study, and also could reconcile a number of seemingly conflicting observations in KS patients: for example, the observation of T cell Vβ skewing observed in many patients and the oligoclonal IgA responses in lesional KS tissue observed by Rowley et al. [25].
In summary, the present study, based on a limited number of KS patients, showed no significant increase in overall peripheral blood CD4 or CD8 T cell activation. Vβ skewing was observed and affected predominantly CD4 lymphocytes. Vβ restricted CD4 and/or CD8 activation was observed in eight of 11 (72%) of the KS patients, a finding not observed in any of the healthy controls. Overall, 13 of 16 (81%) of the KS patients had evidence of either Vβ skewing and/or Vβ restricted activation (Table 2). We suggest that while these preliminary observations will require validation in a bigger cohort of patients, these data do, however, highlight the many layers of complexity when considering T cell activation in KS which could explain some of the conflicting studies regarding peripheral blood T cell activation and Vβ skewing. An important limitation of the present study was that, unfortunately, we were unable (for logistical reasons) to study the KS patients longitudinally from early disease onset (late presentation was relatively common), an approach we feel would be important, as temporal changes of Vβ T cell distributions in KS have been documented by others [10].
Thus, in order to prove or refute the SAg hypothesis we suggest that detailed studies of individual patients may be ultimately more informative than summated data from cohorts of KS patients attempting to implicate any single SAg or individual Vβ-responding family. For example, the combination of commensal culture including PCR to define SAg production from any cultured organisms; IgM seroconversion to SAgs; peripheral T cell Vβ skewing and/or activation; and (where possible) targeted tissue biopsy to look for lesional Vβ infiltration in individual patients could provide a more robust approach to solve the SAg debate once and for all.
Acknowledgments
We would like to acknowledge the support of the London Kawasaki disease Research Group in helping with patient recruitment. This study was funded in part by the Charlotte Parkinson Research Fund; and the John Herring and friends Trust Fund.
References
- 1.Brogan PA, Bose A, Burgner D, et al. Kawasaki disease: an evidence based approach to diagnosis, treatment, and proposals for future research. Arch Dis Child. 2002;86:286–90. doi: 10.1136/adc.86.4.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newburger JW, Takahashi M, Gerber MA, et al. Diagnosis, treatment, and long-term management of Kawasaki disease: a statement for health professionals from the Committee on Rheumatic Fever, Endocarditis and Kawasaki Disease, Council on Cardiovascular Disease in the Young, American Heart Association. Circulation. 2004;110:2747–71. doi: 10.1161/01.CIR.0000145143.19711.78. [DOI] [PubMed] [Google Scholar]
- 3.Brogan PA, Shah V, Bagga A, Klein N, Dillon MJ. T cell Vbeta repertoires in childhood vasculitides. Clin Exp Immunol. 2003;131:517–27. doi: 10.1046/j.1365-2249.2003.02081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen Tervaert JW, Popa ER, Bos NA. The role of superantigens in vasculitis. Curr Opin Rheumatol. 1999;11:24–33. doi: 10.1097/00002281-199901000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Barron KS. Kawasaki disease: etiology, pathogenesis, and treatment. Cleve Clin J Med. 2002;69(Suppl. 2):SII69–78. doi: 10.3949/ccjm.69.suppl_2.sii69. [DOI] [PubMed] [Google Scholar]
- 6.Rowley AH, Shulman ST. New developments in the search for the etiologic agent of Kawasaki disease. Curr Opin Pediatr. 2007;19:71–4. doi: 10.1097/MOP.0b013e328012720f. [DOI] [PubMed] [Google Scholar]
- 7.Brogan PA, Shah V, Klein N, Dillon MJ. Vbeta-restricted T cell adherence to endothelial cells: a mechanism for superantigen-dependent vascular injury. Arthritis Rheum. 2004;50:589–97. doi: 10.1002/art.20021. [DOI] [PubMed] [Google Scholar]
- 8.Li H, Llera A, Malchiodi EL, Mariuzza RA. The structural basis of T cell activation by superantigens. Annu Rev Immunol. 1999;17:435–66. doi: 10.1146/annurev.immunol.17.1.435. [DOI] [PubMed] [Google Scholar]
- 9.Abe J, Kotzin BL, Jujo K, et al. Selective expansion of T cells expressing T-cell receptor variable regions V beta 2 and V beta 8 in Kawasaki disease. Proc Natl Acad Sci USA. 1992;89:4066–70. doi: 10.1073/pnas.89.9.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curtis N, Zheng R, Lamb JR, Levin M. Evidence for a superantigen mediated process in Kawasaki disease. Arch Dis Child. 1995;72:308–11. doi: 10.1136/adc.72.4.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Curtis N. Kawasaki disease and toxic shock syndrome − at last the etiology is clear? Adv Exp Med. 2004;549:191–200. doi: 10.1007/978-1-4419-8993-2_26. [DOI] [PubMed] [Google Scholar]
- 12.Leung DYM, Meissner HC, Fulton DR, Abe J, Quimby F, Schlievert PM. Superantigen hypothesis of Kawasaki syndrome. In: Kato H, editor. Kawasaki Disease. Amsterdam: Elsevier; 1995. pp. 120–126. [Google Scholar]
- 13.Leung DYM, Giorno RC, Kazemi LV, Flynn PA, Busse JB. Evidence for superantigen involvement in cardiovascular injury due to Kawasaki syndrome. J Immunol. 1995;155:5018–21. [PubMed] [Google Scholar]
- 14.Abe J, Kotzin BL, Meissner C, et al. Characterization of T cell repertoire changes in acute Kawasaki disease. J Exp Med. 1993;177:791–6. doi: 10.1084/jem.177.3.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reichardt P, Lehmann I, Sierig G, Borte M. Analysis of T-cell receptor V-beta 2 in peripheral blood lymphocytes as a diagnostic marker for Kawasaki disease. Infection. 2002;30:360–4. doi: 10.1007/s15010-002-3063-4. [DOI] [PubMed] [Google Scholar]
- 16.Nomura Y, Masuda K, Shinkoda Y, et al. Twenty-five types of T-cell receptor Vbeta family repertoire in patients with Kawasaki syndrome. Eur J Pediatr. 1998;157:981–6. doi: 10.1007/s004310050982. [DOI] [PubMed] [Google Scholar]
- 17.Leung DY, Meissner HC, Shulman ST, et al. Prevalence of superantigen-secreting bacteria in patients with Kawasaki disease. J Pediatr. 2002;140:742–6. doi: 10.1067/mpd.2002.123664. [DOI] [PubMed] [Google Scholar]
- 18.Matsubara K, Fukaya T, Miwa K, et al. Development of serum IgM antibodies against superantigens of Staphylococcus aureus and Streptococcus pyogenes in Kawasaki disease. Clin Exp Immunol. 2006;143:427–34. doi: 10.1111/j.1365-2249.2006.03015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yamashiro Y, Nagata S, Oguchi S, Shimizu T. Selective increase of V beta 2+ T cells in the small intestinal mucosa in Kawasaki disease. Pediatr Res. 1996;39:264–6. doi: 10.1203/00006450-199602000-00013. [DOI] [PubMed] [Google Scholar]
- 20.Terai M, Kohno Y, Namba M, et al. Class II major histocompatibility antigen expression on coronary arterial endothelium in a patient with Kawasaki disease. Hum Pathol. 1990;21:231–4. doi: 10.1016/0046-8177(90)90135-r. [DOI] [PubMed] [Google Scholar]
- 21.Mancia L, Wahlstrom J, Schiller B, et al. Characterization of the T-cell receptor V-beta repertoire in Kawasaki disease. Scand J Immunol. 1998;48:443–9. doi: 10.1046/j.1365-3083.1998.00415.x. [DOI] [PubMed] [Google Scholar]
- 22.Nishiyori A, Sakaguchi M, Kato H, et al. Toxic shock syndrome toxin-1 and Vb 2 expression on T cells in Kawasaki disease. In: Kato H, editor. Kawasaki Disease. Amsterdam: Elsevier; 1995. pp. 139–43. [Google Scholar]
- 23.Pietra BA, De Inocencio J, Giannini EH, Hirsch R. TCR V beta family repertoire and T cell activation markers in Kawasaki disease. J Immunol. 1994;153:1881–8. [PubMed] [Google Scholar]
- 24.Sakaguchi M, Kato H, Nishiyori A, Sagawa K, Itoh K. Characterization of CD4+ T helper cells in patients with Kawasaki disease (KD): preferential production of tumour necrosis factor-alpha (TNF-alpha) by V beta 2– or V beta 8– CD4+ T helper cells. Clin Exp Immunol. 1995;99:276–82. doi: 10.1111/j.1365-2249.1995.tb05545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rowley AH, Shulman ST, Spike BT, Mask CA, Baker SC. Oligoclonal IgA response in the vascular wall in acute Kawasaki disease. J Immunol. 2001;166:1334–43. doi: 10.4049/jimmunol.166.2.1334. [DOI] [PubMed] [Google Scholar]
- 26.Shoukry NH, Lavoie PM, Thibodeau J, D'Souza S, Sekaly RP. MHC class II-dependent peptide antigen versus superantigen presentation to T cells. Hum Immunol. 1997;54:194–201. doi: 10.1016/s0198-8859(97)00074-8. [DOI] [PubMed] [Google Scholar]
- 27.Islam D, Wretlind B, Lindberg AA, Christensson B. Changes in the peripheral blood T-Cell receptor V beta repertoire in vivo and in vitro during shigellosis. Infect Immun. 1996;64:1391–9. doi: 10.1128/iai.64.4.1391-1399.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varela-Calvino R, Sgarbi G, Wedderburn LR, Dayan CM, Tremble J, Peakman M. T cell activation by coxsackievirus B4 antigens in type 1 diabetes mellitus: evidence for selective TCR Vbeta usage without superantigenic activity. J Immunol. 2001;167:3513–20. doi: 10.4049/jimmunol.167.6.3513. [DOI] [PubMed] [Google Scholar]
- 29.Furukawa S, Matsubara T, Tsuji K, Motohashi T, Okumura K, Yabuta K. Serum soluble CD4 and CD8 levels in Kawasaki disease. Clin Exp Immunol. 1991;86:134–9. doi: 10.1111/j.1365-2249.1991.tb05785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furukawa S, Matsubara T, Tsuji K, Okumura K, Yabuta K. Transient depletion of T cells with bright CD11a/CD18 expression from peripheral circulation during acute Kawasaki disease. Scand J Immunol. 1993;37:377–80. doi: 10.1111/j.1365-3083.1993.tb02567.x. [DOI] [PubMed] [Google Scholar]
- 31.Matsubara T, Ichiyama T, Furukawa S. Immunological profile of peripheral blood lymphocytes and monocytes/macrophages in Kawasaki disease. Clin Exp Immunol. 2005;141:381–7. doi: 10.1111/j.1365-2249.2005.02821.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Terai M, Kohno Y, Niwa K, Toba T, Sakurai N, Nakajima H. Imbalance among T-cell subsets in patients with coronary arterial aneurysms in Kawasaki disease. Am J Cardiol. 1987;60:555–9. doi: 10.1016/0002-9149(87)90304-3. [DOI] [PubMed] [Google Scholar]
- 33.Leung DY, Meissner HC, Fulton DR, Murray DL, Kotzin BL, Schlievert PM. Toxic shock syndrome toxin-secreting Staphylococcus aureus in Kawasaki syndrome. Lancet. 1993;342:1385–8. doi: 10.1016/0140-6736(93)92752-f. [DOI] [PubMed] [Google Scholar]
- 34.Orwin PM, Leung DY, Tripp TJ, et al. Characterization of a novel staphylococcal enterotoxin-like superantigen, a member of the group V subfamily of pyrogenic toxins. Biochemistry. 2002;41:14033–40. doi: 10.1021/bi025977q. [DOI] [PubMed] [Google Scholar]
- 35.Watanabe-Ohnishi R, Low DE, McGeer A, et al. Selective depletion of V beta-bearing T cells in patients with severe invasive group A streptococcal infections and streptococcal toxic shock syndrome. J Infect Dis. 1995;171:74–84. doi: 10.1093/infdis/171.1.74. Ontario Streptococcal Study Project. [DOI] [PubMed] [Google Scholar]
- 36.Benseler SM, McCrindle BW, Silverman ED, Tyrrell PN, Wong J, Yeung RS. Infections and Kawasaki disease: implications for coronary artery outcome. Pediatrics. 2005;116:e760–6. doi: 10.1542/peds.2005-0559. [DOI] [PubMed] [Google Scholar]