

Abstract
The annual epidemics of respiratory syncytial virus (RSV) infection are probably explained by poor herd immunity and the existence of a dormant reservoir of virus that is activated by an unknown trigger. The virus causes particular problems in infants, the elderly and patients with chronic obstructive airways disease (COPD). During two consecutive winters, human monocyte-derived dendritic cells (DCs) were exposed on a single occasion to one of two forms of RSV labelled with a fluorescent expresser genes (rgRSV or rrRSV) during the epidemic season. The cultures were maintained for many months, with fresh DCs being added at monthly intervals. The cultures were variously exposed to 600 parts per billion (ppb) nitric oxide for 15 min, nitric oxide (NO) donors and NO inhibitors outside the RSV epidemic season. The pattern of productive infection of DCs in vitro appeared to parallel the natural epidemics, in that DCs exhibited evidence of viral replication and productive infection only as manifested by intracellular fluorescence and infection of HeLa cells during the RSV epidemic season. When the long-term cultures were exposed to the above agents outside the RSV epidemic season there was again evidence of vigorous replication and productive infection, as shown by the reappearance of fluorescence and productive infection of HeLa cells. The results indicate that RSV may remain dormant in dendritic cells for prolonged periods and that replication appears to be activated by suppression of endogenous NO production. These observations may be key to our understanding of the mechanisms contributing to the annual epidemics of RSV infection.
Keywords: dendritic cells, latency, nitric oxide, respiratory syncytial virus
Introduction
Respiratory syncytial virus (RSV) is an extraordinarily successful respiratory virus that causes annual epidemics of respiratory disease throughout the world [1–9]. Although the virus is able to reinfect individuals throughout life, its greatest impact is in the very young [1–4] and elderly [4,5] and those with chronic obstructive airways disease (COPD) [6–8]. It is the most important infectious agent affecting infants, with RSV-related infection being the most common cause for hospitalization in this age group. It is responsible for the majority of cases of acute viral bronchiolitis and pneumonia admitted to hospital. Over the past decade it has become clear that the virus also has a major impact on the elderly [9–11]. In temperate climates the annual epidemic commence in late autumn/early winter, rising rapidly to a peak and then falling away by late spring [1,2,9]. Isolation of the virus in the summer is uncommon, but has been described in patients with COPD in whom the virus was identified as commonly in summer as in winter [7]. All attempts to develop an urgently needed vaccine over the past four decades have failed, in large part because many aspects of the virus host interaction are poorly understood [10].
Epidemiological studies from North America indicate that the onset and progression of RSV epidemics is remarkably symmetrical across the continent [2]. Areas separated by thousands of miles have very similar dates for the onset, peak and disappearance of the epidemics, indicating that epidemics do not spread across the country but develop locally. The trigger for the onset of these epidemics and the factors contributing to the disappearance of RSV epidemics remain obscure. A number of proposed environmental factors such as temperature, daylight and humidity have been proposed as potential triggers, but previous studies have failed to support these suggestions [9].
An important factor in the development of the yearly epidemics appears to be the viruses ability to prevent the induction of effective long-term memory immunity leading to poor herd immunity. The mechanisms underlying this are unclear, but there are increasing data that the virus infects and influences the function of dendritic cells (DCs) [11–13], the most important antigen-presenting cells in the lung [14]. Poor herd immunity would explain why such a high percentage of the population is infected during each epidemic, but it does not explain the characteristic sudden onset and cessation of epidemics. It has been suggested that persistent infection within the airways may act as a reservoir for RSV transmission [15]. Human RSV (hRSV) has been shown to persist in the lungs of experimentally infected guinea pigs [16] and mice [17] for some weeks. However, these animals are not the natural host for the hRSV and do not develop lower airways disease when simply exposed to the virus [18]. The animal model related most closely to hRSV-induced disease in humans is natural bovine RSV (bRSV) infection in cattle, in which there is indirect evidence of persistent RSV infection [19]. Significantly, a recent study in human subjects [7] has provided evidence of persistent of RSV in the airways of COPD patients. A significant relationship was observed between persistent infection and rate of decline in lung function.
Using fluorescent labelled RSV (gift from Mark Peebles) it became apparent that the virus replicated readily within human monocyte-derived dendritic cells (MoDCs) during the RSV epidemic, as evidenced by the production of the fluorescent label within cells and transmission of labelled virus to HeLa cells. However, outside the epidemic season there was no evidence of viral replication, as no fluorescent label was evident in the DCs and there was no infection of HeLa cells exposed to the DCs. In a previous observational study we had also observed that there was an apparent correlation between ambient nitric oxide levels due to pollution, which peak in the winter, and the numbers of infants admitted to hospital with RSV bronchiolitis [9].
These observations gave rise to the hypothesis that RSV may both replicate in and persistently infect DCs and that the rate of replication may be influenced by exogenous nitric oxide. In order to test this hypothesis, a series of in vitro experiments were undertaken during two consecutive winters in which MoDCs were exposed to fluorescent-labelled RSV during the epidemic season. The appearance of fluorescence within cells was used to identify activity gene transcription of the virus; confirmation that viable virus was being produced was obtained by standard plaque assays on HeLa cells. The pattern of replication within the dendritic population during subsequent months was observed. When there was no evidence of replication the MoDC populations were exposed to exogenous nitric oxide (NO), S-nitroso-N-acetylpenicillamine (SNAP) and nitric oxide synthase inhibitors and their impact on replication of virus observed.
Materials and methods
MoDCs from a single donor were generated at 28-day intervals using a previously described method [11]. In brief, mononuclear cells were isolated by density gradient centrifugation over Histopaque 1077 and these were purified further by magnetic negative selection. To induce differentiation, the media was supplemented with 40 ng/ml granulocyte–macrophage colony-stimulating factor (GM-CSF) and 20 ng/ml interleukin (IL)-4 (Biosource Division, Invitrogen Ltd, Paisley, UK). Cells were cultured at 37°C in 7·5% CO2/92·5% air for 7 days. Because the cells were isolated from a single donor the number of cells isolated at the beginning of the two long-term cultures meant that the number of plates was limited.
Stocks of both green (rg-RSV) and red (rr-RSV) fluorescent protein-expressing laboratory strains of RSV (gifts from M. Peebles) were prepared by infection of subconfluent layers of HeLa cells (60–80%), as described previously [11]. The cells were harvested by scraping and RSV virions released by freeze/thaw and sonication. During the first winter only rg-RSV was available. When infecting the DCs during the second epidemic both forms of fluorescent virus were available.
The virus was purified using a rapid diafiltration system as described previously [20]. Briefly, a 1/20 dilution of crude RSV was added to a 1 × 106 kDa pore size Vivaspin-20TM filters prewashed with serum-free Dulbecco's modified essential medium (DMEM) and coated with 0·1% casein solution. Virus was centrifuged at 2500 g for 40 min at 4°C. Some stocks of RSV were irradiated in an ultraviolet (UV) cross-linker for 10 min and used as a negative control.
Viral titres were determined by fluorescent virus titration. Infectivity was expressed as the number of plaque-forming units formed per 1 ml of RSV (pfu/ml) [11]. MoDCs were challenged with 1 × 105 pfu/ml (equivalent to a multiplicity of infection of 0·1) RSV on day 7 of culture. Half the media was removed and replace with fresh media. The virus was then added and cells were incubated as before, with half-media changes twice weekly.
The viable status of the MoDCs was determined at 4-weekly intervals, prior to the addition of fresh MoDCs, using Hoechst 33 342 (10 μg/ml) (Sigma Chemical Company, Poole, Dorset, UK) and propidium iodide (PI) (20 μg/ml) (Molecular Probes, Cambridge Biosciences, Cambridge, UK) staining. Cells were visualized under a fluorescent microscope and the number of viable (normal nuclei, blue), apoptotic (nuclear condensation, blue or pink) and necrotic (red) cells were determined using a Whipple graticule.
In order to keep the dendritic cell cultures viable, fresh MoDCs from the same donor were then added to the remaining MoDCs at 4-week intervals. The fluorescent marker, CellTraceTM (Molecular Probes, Invitrogen Detection Technologies, Leiden, The Netherlands), was added, according to the manufacturer's instructions, to the final batch of fresh MoDCs prior to their addition to the long-term cultures in order to determine whether viral replication in response to NO was occurring in those MoDC already present in the long-term cultures or in freshly added cells.
Viral persistence in MoDCs RSV was monitored directly by washing the cells without any fixing and permeabilizing. The cells were analysed for fluorescent protein expression. For intracellular RSV antigen expression MoDCs were fixed/permeabilized with acetone : phosphate-buffered saline (PBS) (1 : 1) solution. MoDCs were washed and stained for 30 min at 4°C with 10 μg/ml monoclonal antibody (Novacastra – Antibodies, Newcastle upon Tyne, UK) specific for the phosphoprotein, fusion protein and nuclear protein of RSV. Cells were washed and labelled with a fluorescein isothiocyanate (FITC) or R-Phycoerythrin (RPE)-conjugated secondary IgG antibody (Sigma) for 30 min at 4°C. Cells were analysed immediately using a Beckman Coulter fluorescence activated cell sorter (FACSort) flow cytometer (Beckman Coulter (UK) Ltd., High Wycombe, UK) measuring fluorescence emission at 525 or 488 nm, respectively, and by fluorescent microscopy.
HeLa cell cultures were maintained in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% l-glutamine (Sigma). For plaque assays and co-cultures HeLa cells were seeded at 2·5 × 104 cells/ml in 96-well plates and incubated overnight. Serial dilutions of MoDCs ± media were added to wells in triplicate and incubated for a further 48 h. Cultures were washed gently to remove MoDCs and then analysed by fluorescent microscopy.
When there had been no evidence of viral replication for several months the long-term, RSV-infected MoDCs were exposed to one of two sources of exogenous NO. NO gas, 600 parts per billion (ppb), was pumped directly into the culture chamber for up to 2 h or the NO donor (+)-S-nitroso-N-acetylpenicillamine (SNAP) (100 μM) (Calbiochem, Merck Chemicals Ltd., Beeston, UK) added to the culture media. Because exogenous NO has been shown to inhibit intracellular NO production, an inducible nitric oxide synthase (iNOS) inhibitor NG-nitro-L-arginine methyl ester (1 mg/ml) (L-NAME) was added to the culture media of a third group of long-term cultures.
Ethical approval
Ethical approval from the South Sheffield Research Ethics Committee was obtained for obtaining blood from a single volunteer (MLE) to provide all the DCs used in this study.
Results
RSV productively infects MoDCs
RSV infection of MoDCs was established by assessing the expression of red or green fluorescent protein in MoDCs and by staining the cells for viral antigen using either fluorescent microscopy or flow cytometry analysis. In cultures of MoDCs infected with rg-RSV and rr-RSV a proportion of the cells fluoresce green and red, respectively (Figs 1a,b, 2), indicating that RSV is capable of infecting and replicating within a population of MoDCs. MoDCs that were exposed to UV-treated rg-RSV did not fluoresce and were very low positive for RSV antibody, similar to levels seen with unchallenged control cells after 8 months of culture (Fig. 2c). These data show that MoDCs are susceptible to RSV infection and that RSV is capable of replicating within them.
Fig. 1.
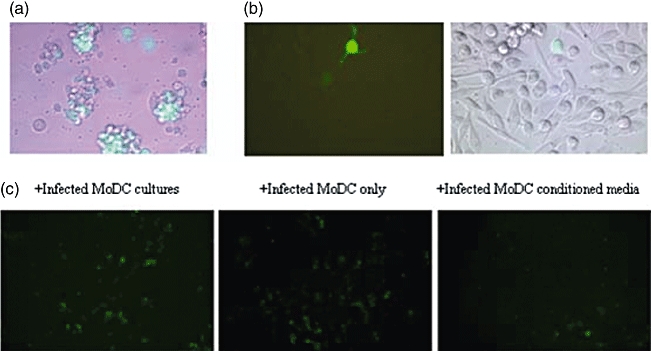
Productive infection of human monocyte-derived dendritic cells (MoDCs) by respiratory syncytial virus (RSV). MoDCs were infected with rg-RSV for 24 h and analysed using a Leica fluorescent microscope. (a) Infection of MoDC clusters with replicating rg-RSV (10× magnification). (b) Single MoDC with dendrites infected with rg-RSV (40× magnification). (c) MoDCs challenged previously with rg-RSV co-cultured with HeLa to determine productive infection. From left to right, co-cultures with unwashed rg-RSV-infected MoDCs, washed rg-RSV-infected MoDCs and rg-RSV-infected MoDC conditioned supernatant only.
Fig. 2.
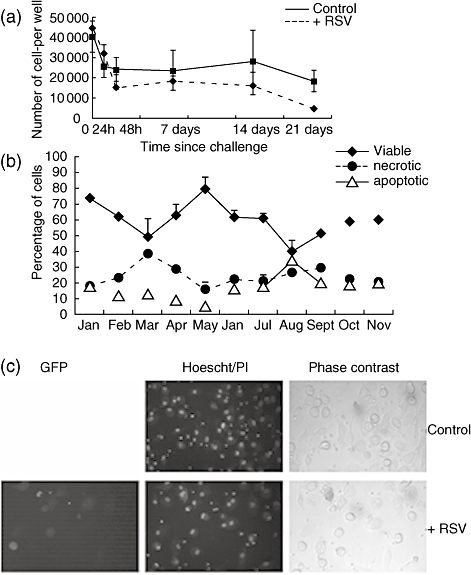
Persistent infection of monocyte-derived dendritic cells (MoDCs) by respiratory syncytial virus (RSV). MoDCs were challenged once with RSV and fresh MoDCs added at 4-weekly intervals to maintain cultures. (a) In the summer months fluorescent MoDCs could not be detected by fluorescent microscopy. However, fluorescent plaques were formed when the same MoDCs were added to HeLa cells. (b) Productive infection monitored using fluorescent plaque assays on HeLa cells. (c) The presence of RSV could also be detected using RSV antibody and flow cytometry histograms to show presence of RSV 1, 5 and 8 months after viral challenge.
To determine further if the infection of MoDCs with RSV was productive (i.e. viral particles released), washed infected MoDCs and media conditioned by infected MoDCs was added to HeLa cell cultures for 24 h. Both washed MoDCs and conditioned media were able to infect HeLa cells (Fig. 1c).
RSV persistently infects MoDCs
Long-term cultures of MoDCs infected with RSV were analysed from January 2005 to November 2005 with rg-RSV and from November 2005 to August 2006 with rr-RSV. The viable state of the long-term cultures was determined at 4-weekly intervals prior to the addition of fresh cells. The number of viable MoDCs ranged from 40% to 80% of the total population (Fig. 3a–c). It would appear that where there was a decrease in viable cell number the cells were dying through increased necrosis. The number of apoptotic cells remained below 30% for the entire culture period.
Fig. 3.
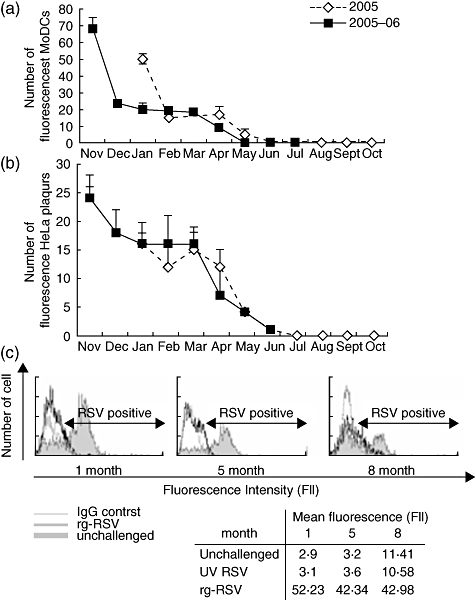
Prolonged viable state of respiratory syncytial virus (RSV)-infected monocyte-derived dendritic cells (MoDCs). The viable state of the RSV-challenged MoDCs in long-term culture was analysed by Hoechst/propidium iodide (PI) staining. (a) Graph to show percentage of viable, necrotic and apoptotis RSV-infected MoDCs over 10 months prior to the addition of fresh MoDCs at 4-weekly intervals. (b) Decreased viability of unchallenged MoDCs viability over time. (c) Hoechst/PI stain 48 h post-challenged showing increased apoptosis/necrosis (pink/red) of unchallenged cells compared to RSV-challenged cells. The error bars represent standard error of the mean.
RSV replication of infection of MoDCs was detected by fluorescent microscopy for approximately 5 months (January–May 2005 and November–April 2005–06) (Fig. 2a) Productive RSV infection of HeLa by infected MoDCs was detected for a further month in both years (Fig. 2b). Furthermore, using antibodies to RSV proteins, RSV antigens could be detected by flow cytometric analysis throughout the culture period (Fig. 2c). These data suggest that RSV infects MoDCs in vitro persistently for long periods.
Exogenous NO induces viral replication of persistently infected MoDCs
It was hypothesized that the persistent infection of DCs may allow the virus to remain dormant within the population throughout the summer and that an exogenous environmental stimulus induces viral replication during the autumn months. To test this hypothesis the long-term cultures of RSV-infected MoDCs were exposed to exogenous NO and an NO synthase inhibitor. When cells were exposed to NO or cultured in the presence of SNAP or L-NAME viral replication could be detected within 24 h (Fig. 4). Visually, there appeared to be a higher number of fluorescent MoDCs (red or green) in cultures exposed to NO compared to both SNAP and L-NAME and no fluorescence was detected in control unchallenged MoDCs. These data are consistent with the flow cytometry data, showing that RSV remains within the MoDCs throughout periods when replication cannot be detected and that exogenous NO or an iNOS inhibitor is capable of inducing the replication of this non-replicating form.
Fig. 4.
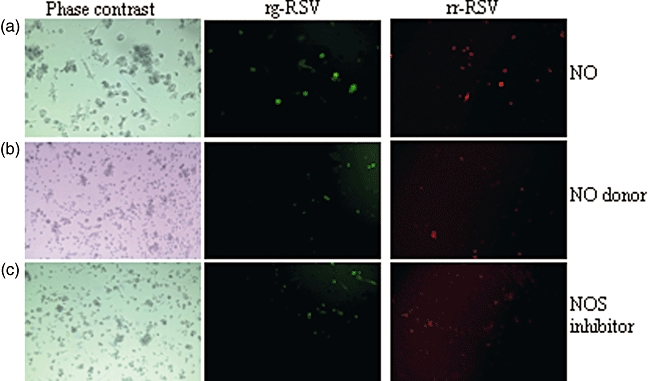
Exogenous nitric oxide induces the replication of respiratory syncytial virus (RSV) in monocyte-derived dendritic cells (MoDCs). Exogenous nitric oxide (NO) was added to long-term cultures when MoDCs containing replicating virus could no longer be detected using fluorescent microscopy. Digital images using phase contrast (left column), green (rg-RSV, middle column) and red fluorescence (rr-RSV, right column) are shown of cells exposed to (a) NO gas, (b) NO donor (S-nitroso-N-acetylpenicillamine) and (c) NG-nitro-L-arginine methyl ester (1 mg/ml) (L-NAME). Images represent typical data from the 2 years.
L-NAME induces productive replication of RSV in long-term MoDC cultures
To determine if the persistent infection of MoDCs was confined to cells isolated from ‘old’ isolation or if ‘new’ cells were infected, CellTrace was added to freshly differentiated MoDCs. The labelled MoDCs were then added to rr-RSV-infected long-term cultures in which viral replication could no longer be detected by fluorescent microscopy. The NO synthase inhibitor, L-NAME, was added 4 h later. Using fluorescent microscopy and flow cytometry viral replication was monitored at 24-h intervals after the addition of fresh MoDCs. The freshly isolated MoDCs were identified by tracking the CellTrace marker and fluoresced green; cells containing rr-RSV fluoresced red. After 24 h only a small percentage of cells in all cultures were labelled with CellTrace and fluorescing red (Table 1). These data would suggest that it is only the ‘old’ cells that are infected with virus. However, 72 h after NO exposure almost 100% of MoDCs were necrotic, as detected by Hoechst/PI staining, but interestingly the majority were fluorescing red, indicating that productive viral replication was induced or that virus was released due to cell death (data not shown).
Table 1.
Tracking of respiratory syncytial virus (RSV) infection in persistently infected monocyte-derived dendritic cells (MoDCs).
| –L-NAME | +L-NAME | |||
|---|---|---|---|---|
| Control | +RSV | Control | +RSV | |
| F12 mean fluorescence (rr-RSV) | 14·21 | 17·23 | 15·33 | 42·72 |
| F11 mean fluorescence (CellTrace) | 87 | 78 | 88 | 89 |
| Percentage of cells F11 + F12 positive (fresh cell infected with RSV) | 0·11 | 0·13 | 0·20 | 0·18 |
Freshly differentiated MoDCs were labelled with CellTrace and added to persistently infected MoDC cultures and viral replication induced by the addition of NG-nitro-L-arginine methyl ester (1 mg/ml) (L-NAME). Mean fluorescence values indicate that L-NAME induces viral replication and that after 24 h viral infection and replication in confined to the old cells.
Discussion
These experiments would indicate that human RSV is able to both infect productively and lie dormant within human DC. The time–course of alternating active replication and dormancy observed in vitro replicates the pattern of RSV infection observed in the community. The data also indicate that replication of apparently dormant virus can be triggered by exogenous sources of NO and iNOS inhibitors. In addition, it would appear that the persistence of virus in DC involves inhibition of apoptosis. In the laboratory we were able to demonstrate active replication of the virus in human DC during both the winter epidemic seasons. During each of the two summers there was no evidence of replication in the DCs, although HeLa cells exposed to the virus were able to support replication at apparently the same levels as observed during the winter. The response to exogenous NO and NOS inhibitors resulting in the rapid appearances of viral replication was also evident in both years, providing consistent results across a prolonged period of time.
The clinical significance of these results are that they imply that the virus may lie dormant between epidemics within airways DCs of previously infected individuals. The potential importance of DCs as a reservoir for the virus is highlighted by reports that indicate that DCs appear rapidly in large numbers in the airways of infected infants [21]. The increase in DC numbers does not appear to be transient, as even higher numbers of myeloid and plasmacytoid DCs were observed in the airways of these infants many weeks after the acute symptoms had resolved [21]. Similar observations have been made in an experimental rodent model [22]. Immunochemistry analysis of nasal secretions obtained from the RSV-infected infants demonstrated the presence of the RSV fusion protein in some of the DCs [21].
Our results would also suggest that it is possible that replication of the virus could be triggered by exogenous NO from environmental sources. This may occur on a population basis when ambient NO levels rise in the late autumn and winter, which may be a trigger initiating the annual epidemics. It may also occur on individual basis, as occurs in subjects who smoke, and this may explain why virus is detectable in the summer among patients with COPD.
The pattern of viral replication within DCs during the winter epidemic and dormancy in the summer was observed with two viruses over two seasons. Productive infection of DCs during the epidemic months was confirmed by plaque assay using HeLa cells which were infected readily by both exposure to cells and to the supernatant from the DC cultures. The dramatic and very rapid explosion of viral replication within MoDCs exposed to NO, an NO donor and to NOS inhibitors when the virus was apparently dormant would suggest that NO has a major role in modulation of replication. It has been shown previously that as with other respiratory viruses, RSV infection induces the production of NO by a number of cells in the airways [23–25] and that the NO produced has important anti-viral and immune modulating properties [25–29]. Several studies have indicated that endogenously generated NO has a role in limiting intracellular replication of RSV [26–28], but this was not observed with exogenous NO [26]. It has also been shown that exogenous NO can down-regulate intracellular NO production by NOS [30,31], and this is probably directly relevant to our observations. The effectiveness of the NOS inhibitor would suggest that down-regulation of intracellular NO production by exogenous sources of NO may explain how the virus is released for a period of dormancy.
It is clear from these results that viable virus remains within the MoDC population for many months, despite the absence of any discernable evidence of viral replication. This may be because of very low-grade replication leading to infection of freshly added MoDCs each month or due to prolongation of the life of infected MoDCs. While these experiments do not establish beyond doubt that the RSV infection of DCs inhibits cell death, the data suggest that this may be the case. Infected MoDCs survive for significantly longer that those not exposed to virus even in the absence of fresh DC (Fig. 3a). Much more persuasive are data presented in Table 1, which indicate that initially those cells in which replication of virus is triggered by the NO donor and NOS inhibitor are from previous cultures and not the freshly added MoDCs labelled with CellTrace.
Other viruses, such as herpes simplex (HS), are known to adopt a similar strategy with prolonged periods of dormancy [32,33]. HS inhibits apoptosis of neuronal cells, promoting its ability to lie latent within the cells for long periods. It has been shown recently that the latency associated transcript Mi RNA inhibits apoptosis by modulation of transforming growth factor (TGF)-β signalling [32]. It is also known that the measles virus, which like RSV is a paramyxovirus, can lie dormant in neurones for many years. RSV may adopt a similar approach both in DCs and possibly in other cells. The inflammatory response in the airways of infants with RSV infection is dominated by an intense neutrophil influx [34], and in vivo studies have indicated that apoptosis of neutrophils is inhibited by factors in the airways of these infants [35]. In vitro work has also indicated that granulocyte apoptosis can be inhibited by factor(s) released by infected cells [36] (Coleman, unpublished data). A recent study in a mouse model suggested that the virus may infect neurones and the authors suggested that this may contribute to persistence of the virus [37], although this study did not address the issue of cell survival. Whether the factor(s) influencing apoptosis in DCs are the same as those affecting granulocytes is unclear. An alternative explanation for RSV persistence in these experiments is that the virus replicates at extremely low levels and infects new DCs added at monthly intervals. The failure to detect any fluorescence at all and the inability to infect the HeLa cells would argue against this, but further work is required. A further potential limitation is that the work was undertaken with MoDCs from a single donor. However, it seems very unlikely that there is anything unique about this individual's cells and the findings from the first year were replicated in the experiments undertaken in the second year. Further studies replicating these results with other volunteers are under way.
As noted above, the potential implications for this work are considerable. The results could explain a number of the many unusual features that characterize the cycle of annual epidemics due to RSV. It should also lead to further insights into the impact of the virus on the immune response. Unlike other pollutants, such as PM10s and ozone, NO levels peak in the winter and fall to very low levels in the summer [38], proving a plausible link between our observations that exogenous NO can stimulate replication of dormant virus and the winter epidemics of RSV respiratory illness. While there do not appear to be major differences in the pattern of epidemics over large distances, epidemiological studies have indicated that there are important differences in the severity of disease experienced by infants from geographically distinct areas. Those from industrialized areas are more than twice as likely to be admitted to hospital as those from rural or non-industrialized areas [39,40]. Environmental monitoring indicates that NO levels in industrialized areas are considerably higher than those observed in urban areas, which in turn are higher than those in rural areas. Thus, local ambient NO levels might provide not only an explanation for the pattern and timing of the RSV season; it may also explain, in part, differences in disease severity between areas. Other factors such as overcrowding are also likely to contribute. Further evidence that environmental factors may influence disease severity comes from studies assessing the impact of indoor pollutants. Factors such as parental smoking [41,42] and the use of wood-burning stoves [43] increase both the risk and severity of RSV infections in infants. A recent study noted that in contrast to other forms of respiratory illness, the severity of RSV-related lower airways infection is associated with postnatal rather than in utero exposure [41]. In a second study there was clear evidence that hospitalization with RSV bronchiolitis was associated with acute exposure to high concentrations of cigarette smoke [42]. Evidence that pollutants may influence disease severity in RSV-related respiratory disease was also obtained in a study of older asthmatic children [44]. Increased exposure to NO2 was associated with more severe exacerbations when infected with respiratory viruses. A recent study provided evidence of persistent viral infection in patients with COPD [7]. Our results would provide a biologically plausible mechanism, with smokers exposing themselves to levels of NO in excess of those used in this experiment and for much longer periods than the 15 min of exposure used in these experiments.
In summary, we have shown that replication of RSV in human MoDCs in vitro appears to vary in parallel to the natural epidemic, with rapid productive infection during the winter and apparent dormancy during the summer. Exogenous NO can trigger replication and this appears to be due to inhibition of endogenous NO production, as suggested by the use of a NOS inhibitor. Understanding the interaction of virus both in its replicating and dormant phases on DC function and survival may lead to novel forms of treatment and, indeed, prevention.
Acknowledgments
Green fluorescent protein-expressing laboratory A2 strain of RSV (rg-RSV) (Hallak et al. 2000) and red fluorescent protein-expressing B strain (rr-RSV) were kindly donated by Dr M. Peeples. Lynsey Hobson was supported by the Sheffield Children's Hospital Research fund.
References
- 1.Everard ML. Respiratory syncytial virus bronchiolitis and pneumonia. In: Taussig L, Landau L, editors. Textbook of paediatric respiratory medicine. St Louis: Mosby; 1998. pp. 580–95. [Google Scholar]
- 2.Gilchrist S, Torok TJ, Gary HE, et al. National surveillance for respiratory syncytial virus, United States, 1985–1990. J Infect Dis. 1994;170:986–90. doi: 10.1093/infdis/170.4.986. [DOI] [PubMed] [Google Scholar]
- 3.Leader S, Kohlhase K. Recent trends in severe respiratory syncytial virus (RSV) among US infants, 1997–2000. J Pediatr. 2003;143(Suppl. 5):S127–32. doi: 10.1067/s0022-3476(03)00510-9. [DOI] [PubMed] [Google Scholar]
- 4.Fleming DM, Cross KW. Respiratory syncytial virus or influenza. Lancet. 1993;342:1507–10. doi: 10.1016/s0140-6736(05)80082-0. [DOI] [PubMed] [Google Scholar]
- 5.Falsey AR, Hennessey PA, Formica MA, et al. Respiratory syncytial virus infection in elderly and high-risk adults. N Engl J Med. 2005;352:1749–59. doi: 10.1056/NEJMoa043951. [DOI] [PubMed] [Google Scholar]
- 6.Seemungal T, Harper-Owen R, Bhowmik A, et al. Respiratory viruses, symptoms, and inflammatory markers in acute exacerbations and stable chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2001;164:1618–23. doi: 10.1164/ajrccm.164.9.2105011. [DOI] [PubMed] [Google Scholar]
- 7.Wilkinson TM, Donaldson GC, Johnston SL, et al. Respiratory syncytial virus, airway inflammation, and FEV1 decline in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173:871–6. doi: 10.1164/rccm.200509-1489OC. [DOI] [PubMed] [Google Scholar]
- 8.Falsey AR, Formica MA, Hennessey PA, et al. Detection of respiratory syncytial virus in adults with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173:639–43. doi: 10.1164/rccm.200510-1681OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhatt JM, Everard ML. Do environmental pollutants influence the onset of respiratory syncytial virus epidemics or disease severity? Paediatr Respir Rev. 2004;5:333–8. doi: 10.1016/j.prrv.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Moore ML, Peebles RS., Jr. Respiratory syncytial virus disease mechanisms implicated by human, animal model, and in vitro data facilitate vaccine strategies and new therapeutics. Pharmacol Ther. 2006;112:405–24. doi: 10.1016/j.pharmthera.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 11.Jones A, Morton I, Hobson L, et al. Differentiation and immune function of human dendritic cells following infection by respiratory syncytial virus. Clin Exp Immunol. 2006;143:513–22. doi: 10.1111/j.1365-2249.2005.03004.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bartz H, Turkel O, Hoffjan S, et al. Respiratory syncytial virus decreases the capacity of myeloid dendritic cells to induce interferon-gamma in naive T cells. Immunology. 2003;109:49–57. doi: 10.1046/j.1365-2567.2003.01629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guerrero-Plata A, Casola A, Suarez G, et al. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am J Respir Cell Mol Biol. 2006;34:320–9. doi: 10.1165/rcmb.2005-0287OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Heer HJ, Hammad H, Kool M, Lambrecht BN. Dendritic cell subsets and immune regulation in the lung. Semin Immunol. 2005;17:295–303. doi: 10.1016/j.smim.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Tripp RA. The brume surrounding respiratory syncytial virus persistence. Am J Respir Crit Care Med. 2004;169:778–9. doi: 10.1164/rccm.2401013. [DOI] [PubMed] [Google Scholar]
- 16.Hegele RG, Hayashi S, Bramley AM, Hogg JC. Persistence of respiratory syncytial virus genome and protein after acute bronchiolitis in guinea pigs. Chest. 1994;105:1848–54. doi: 10.1378/chest.105.6.1848. [DOI] [PubMed] [Google Scholar]
- 17.Schwarze J, O'Donnell DR, Rohwedder A, Openshaw PJ. Latency and persistence of respiratory syncytial virus despite T cell immunity. Am J Respir Crit Care Med. 2004;169:801–5. doi: 10.1164/rccm.200308-1203OC. [DOI] [PubMed] [Google Scholar]
- 18.Everard ML. What link between early respiratory viral infections and atopic asthma. Lancet. 1999;354:527–8. doi: 10.1016/S0140-6736(99)00159-2. [DOI] [PubMed] [Google Scholar]
- 19.Van der Poel WH, Langedijk JP, Kramps JA, et al. Serological indication for persistence of bovine respiratory syncytial virus in cattle and attempts to detect the virus. Arch Virol. 1997;142:1681–96. doi: 10.1007/s007050050189. [DOI] [PubMed] [Google Scholar]
- 20.Bataki EL, Evans GS, Everard ML. Respiratory syncytial virus neutrophil activation. Clin Exp Immunol. 2005;140:470–7. doi: 10.1111/j.1365-2249.2005.02780.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gill MA, Palucka AK, Barton T, et al. Mobilization of plasmacytoid and myeloid dendritic cells to mucosal sites in children with respiratory syncytial virus and other viral respiratory infections. J Infect Dis. 2005;191:1105–15. doi: 10.1086/428589. [DOI] [PubMed] [Google Scholar]
- 22.Beyer M, Bartz H, Horner K, et al. Sustained increases in numbers of pulmonary dendritic cells after respiratory syncytial virus infection. J Allergy Clin Immunol. 2004;113:127–33. doi: 10.1016/j.jaci.2003.10.057. [DOI] [PubMed] [Google Scholar]
- 23.Kao YJ, Piedra PA, Larsen GL, Colasurdo GN. Induction and regulation of nitric oxide synthase in airway epithelial cells by respiratory syncytial virus. Am J Respir Crit Care Med. 2001;163:532–9. doi: 10.1164/ajrccm.163.2.9912068. [DOI] [PubMed] [Google Scholar]
- 24.Tsutsumi H, Takeuchi R, Ohsaki M, et al. Respiratory syncytial virus infection of human respiratory epithelial cells enhances inducible nitric oxide synthase gene expression. J Leukoc Biol. 1999;66:99–104. [PubMed] [Google Scholar]
- 25.Xu W, Zheng S, Dweik RA, Erzurum SC. Role of epithelial nitric oxide in airway viral infection. Free Radic Biol Med. 2006;41:19–28. doi: 10.1016/j.freeradbiomed.2006.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ali-Ahmad D, Bonville CA, Rosenberg HF, Domachowske JB. Replication of respiratory syncytial virus is inhibited in target cells generating nitric oxide in situ. Front Biosci. 2003;8:a48–53. doi: 10.2741/986. [DOI] [PubMed] [Google Scholar]
- 27.Stark JM, Khan AM, Chiappetta CL, et al. Immune and functional role of nitric oxide in a mouse model of respiratory syncytial virus infection. J Infect Dis. 2005;191:387–95. doi: 10.1086/427241. [DOI] [PubMed] [Google Scholar]
- 28.Colasurdo GN, Fullmer JJ, Elidemir O, et al. Respiratory syncytial virus infection in a murine model of cystic fibrosis. J Med Virol. 2006;78:651–8. doi: 10.1002/jmv.20589. [DOI] [PubMed] [Google Scholar]
- 29.Bove PF, van der Vliet A. Nitric oxide and reactive nitrogen species in airway epithelial signaling and inflammation. Free Radic Biol Med. 2006;41:515–27. doi: 10.1016/j.freeradbiomed.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 30.Morin C, Fessi H, Devissaguet JP, et al. Factors influencing macrophage activation by muramyl peptides: inhibition of NO synthase activity by high levels of NO. Biochim Biophys Acta. 1994;1224:427–32. doi: 10.1016/0167-4889(94)90278-x. [DOI] [PubMed] [Google Scholar]
- 31.Lu MP, Du LZ, Gu WZ, Chen XX. Nitric oxide inhalation inhibits inducible nitric oxide synthase but not nitrotyrosine formation and cell apoptosis in rat lungs with meconium-induced injury. Acta Pharmacol Sin. 2005;26:1123–9. doi: 10.1111/j.1745-7254.2005.00153.x. [DOI] [PubMed] [Google Scholar]
- 32.Gupta A, Gartner JJ, Sethupathy P, et al. Anti-apoptotic function of a microRNA encoded by the HSV-1 latency-associated transcript. Nature. 2006;442:82–5. doi: 10.1038/nature04836. [DOI] [PubMed] [Google Scholar]
- 33.Sullivan CS, Ganem D. MicroRNAs and viral infection. Mol Cell. 2005;20:3–7. doi: 10.1016/j.molcel.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 34.Everard ML, Swarbrick A, Wrightham M, et al. Analysis of cells obtained by bronchial lavage of infants with respiratory syncytial virus infection. Arch Dis Child. 1994;71:428–32. doi: 10.1136/adc.71.5.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans GS, Jones A, Qui JM, et al. Neutrophil survival is prolonged in the airways of healthy infants and infants with RSV bronchiolitis. Eur Respir J. 2002;20:651–7. doi: 10.1183/09031936.02.00278902. [DOI] [PubMed] [Google Scholar]
- 36.Lindemans CA, Coffer PJ, Schellens IM, et al. Respiratory syncytial virus inhibits granulocyte apoptosis through a phosphatidylinositol 3-kinase and NF-kappaB-dependent mechanism. J Immunol. 2006;176:5529–37. doi: 10.4049/jimmunol.176.9.5529. [DOI] [PubMed] [Google Scholar]
- 37.Li XQ, Fu ZF, Alvarez R, et al. Respiratory syncytial virus (RSV) infects neuronal cells and processes that innervate the lung by a process involving RSV G protein. J Virol. 2006;80:537–40. doi: 10.1128/JVI.80.1.537-540.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams ML. Patterns of air pollution in developed countries. In: Holgate ST, Samet JM, Koren HS, Maynard RL, editors. Air pollution and health. San Diego, CA: Academic Press; 1999. pp. 83–104. [Google Scholar]
- 39.Report to the Medical Research Council Subcommittee on Respiratory Syncytial Virus Vaccines. Respiratory syncytial virus infection: admissions to hospital in industrial, urban, and rural areas. BMJ. 1978;2:796–8. doi: 10.1136/bmj.2.6140.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brandenburg AH, Jeannet P-Y, Steensel-Moll HA, et al. Local variability in respiratory syncytial virus disease severity. Arch Dis Child. 1997;77:410–14. doi: 10.1136/adc.77.5.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bradley JP, Bacharier LB, Bonfiglio J, et al. Severity of respiratory syncytial virus bronchiolitis is affected by cigarette smoke exposure and atopy. Pediatrics. 2005;115:e7–14. doi: 10.1542/peds.2004-0059. [DOI] [PubMed] [Google Scholar]
- 42.Gurkan F, Kiral A, Dagli E, Karakoc F. The effect of passive smoking on the development of respiratory syncytial virus bronchiolitis. Eur J Epidemiol. 2000;16:465–8. doi: 10.1023/a:1007658411953. [DOI] [PubMed] [Google Scholar]
- 43.Jeena PM, Ayannusi OE, Annamalai K, et al. Risk factors for admission and the role of respiratory syncytial virus-specific cytotoxic T-lymphocyte responses in children with acute bronchiolitis. S Afr Med J. 2003;93:291–4. [PubMed] [Google Scholar]
- 44.Chauhan AJ, Inskip HM, Linaker CH, et al. Personal exposure to nitrogen dioxide (NO2) and the severity of virus-induced asthma in children. Lancet. 2003;361:1939–44. doi: 10.1016/S0140-6736(03)13582-9. [DOI] [PMC free article] [PubMed] [Google Scholar]