

Abstract
Known genetic defects currently account for only a small proportion of patients meeting criteria for ‘probable’ or ‘possible’ common variable immunodeficiency (CVID). A 59-year-old male with a 12-year history of CVID on intravenous immunoglobulin (IVIG) is presented who developed bronchiectasis, cytopenias and malabsorption that are recognized complications of CVID. Work-up for his malabsorption suggested the possibility of Shwachman–Diamond syndrome, confirmed by mutation testing. With the identification of the molecular defect in Shwachman–Diamond syndrome (SDS), it is becoming clear that not all SDS patients have the prominent features of neutropenia or pancreatic malabsorption. A meta-analysis of published immunological defects in SDS suggests that four of 14 hypogammaglobulinaemic SDS patients meet criteria for ‘possible’ CVID. Mutations in the SBDS gene may therefore be the fifth identified molecular defect in CVID.
Keywords: common variable immunodeficiency, immunodeficiency, Shwachman–Bodian–Diamond syndrome gene, Shwachman–Diamond syndrome
Introduction
Common variable immunodeficiency (CVID) is a primary immunodeficiency disorder of unknown cause, and currently identified genetic mutations (ICOS, CD19, TACI, BAFFR) account for less than a fifth of cases [1]. The fact that some CVID patients have variable degrees of cytopenia, develop lymphoid nodular hyperplasia and subsequent lymphoma points to a failure of bone marrow B cell differentiation. Various immunological abnormalities, including low immunoglobulins and absent vaccine responses that would fit criteria for ‘probable’ or ‘possible’ CVID, have been recognized in some patients suffering from Shwachman–Diamond syndrome (SDS). This syndrome can present with a very broad array of signs, and recent advances in the genetics of SDS have demonstrated that many patients with SDS do not necessarily display the ‘classical’ features. Characteristic features include cytopenias (usually but not invariably neutropenia), skeletal defects and pancreatic insufficiency. Recent identification of the Shwachman–Bodian–Diamond syndrome (SBDS) gene at Chr7q11 has led to identification of cases without neutropenia or transient pancreatic insufficiency [2].
We report a case of CVID on intravenous immunoglobulin for 12 years with complications of lymphopenia, lymphoid nodular hyperplasia and transient pancreatic insufficiency and found to be heterozygous for a mutation in SBDS gene.
Case presentation
A 49-year-old man with no children presented initially in 1995 to the haematology department with repeated ear infections over 6 years, requiring myringotomy and grommet insertion. Over the next 3 years, he had recurrent Haemophilus influenzae pneumonia and was then found to have panhypogammaglobulinaemia (IgG 3·7 g/l which fell to 1·3 g/l, IgM < 0·2 g/l, IgA < 0·2 g/l) and lymphopenia [total lymphocyte count 0·8 × 109/l; total T cells 0·622 × 109/l (normal 0·7–2·1), CD4 T cells 0·337 × 109/l (0·3–1·4), CD8 T cells 0·233 × 109/l (0·2–0·9), CD19 B cells 0·110 × 109/l (0·1–0·5), natural killer cells 0·143 × 106/l (0·09–0·6), α/β T cell receptor (TCR) 82% and γ/δ TCR 4% of T cells]. Assessment of vaccine responses was not undertaken given the degree of his hypogammaglobulinaemia; a diagnosis of CVID was made and treatment with intravenous immunoglobulin (IVIG) was commenced. At the time of diagnosis he was noted to have short stature (150 cm), but did not have neutropenia or steatorrhoea or other features of malabsorption. Over the next 12 years, the following abnormalities developed: inflammatory nasal polyps, arthritis (knee, wrist), anaemia (haemoglobin 8·9 g/dl), eosinophilia (1·6 × 109/l) and abnormal liver function tests (alanine aminotransferase 233 μ/l and alkaline phosphatase 490 μ/l) and was shown to have bronchiectasis and fused ectopic kidneys in his right iliac fossa (Fig. 1a–d).
Fig. 1.
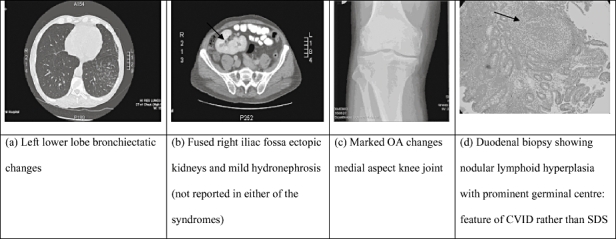
X-rays and computerized tomography/magnetic resonance imaging abnormalities and duodenal biopsy (histology) in our patient. (a) Left lower lobe bronchiectatic changes. (b) Fused right iliac fossa ectopic kidneys and mild hydronephrosis (not reported in either of the syndromes). (c) Marked osteoarthritis changes medial aspect knee joint. (d) Duodenal biopsy showing nodular lymphoid hyperplasia with prominent germinal centre − feature of common variable immunodeficiency rather than Shwachman–Diamond syndrome.
Severe malabsorption developed resulting in hypocalcaemic tetany, hypoalbuminaemia and difficulty maintaining trough IgG levels. A duodenal biopsy revealed lymphoid nodular hyperplasia (Fig. 1d) that responded poorly to steroid therapy. Faecal immunoelastase levels were abnormal (108 μg/g; normal > 200 μg/g), suggesting exocrine pancreatic insufficiency. Hydrogen breath test, sweat chloride and short synacthen tests were normal. Genetic testing for SDS were undertaken.
Materials and methods
DNA was extracted from peripheral blood [5 ml in ethylenediamine tatraacetic acid (EDTA)] using the Autopure LS™ system (Qiagen, Crawley, UK) according to the manufacturer's instructions. Analysis of the SBDS gene was then performed in two stages. The first stage of analysis involved amplification of exon 2 of the SBDS gene by polymerase chain reaction (PCR), according to the method of Boocock et al. [3]. In brief, two separate PCR reactions were set up: one using primers specific for the SBDS gene and one using ‘dual-specific’ primers that amplified exon 2 of both the SBDS gene and its pseudogene. The oligonucleotide sequences 5′−3′ of the primers for analysis of exon 2 used were as follows: forward specific, AAATGGTAAGGCAAATACGG; reverse specific, ACCAAGTTCTTTATTATTAGAAG; forward dual specificity, GGGATTTGTTGTGTCTTG; and reverse dual specificity, CTTTCCTCCAGAAAAACAGC. Each reaction contained CM129 buffer (ABgene, Epsom, UK), primers (each 10 μM) and 50–100 ng DNA. Cycling conditions were: 95°C for 15 min, followed by 30 cycles of (95°C 1 min, 55°C 1 min, 72°C 1 min), a final extension step at 72°C for 10 min, and cooled to 4°C indefinitely. Both PCR products were subjected to separate restriction endonuclease digestions with Bsu36I and Cac8I, the former detecting the mutation ca. 183_184TA > CT and the latter detecting ca. 258 + 2T > C. PCR products were subjected to electrophoresis in an agarose gel stained with ethidium bromide. The resulting products were photographed under ultraviolet light (Fig. 2).
Fig. 2.
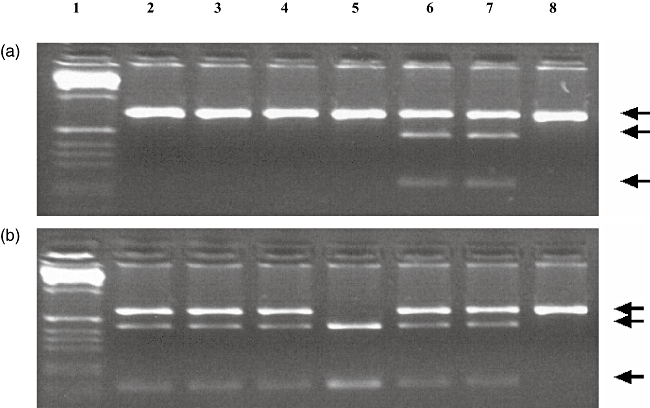
(a) Polymerase chain reaction (PCR) products generated by Shwachman–Bodian–Diamond syndrome (SBDS)-specific primers, digested with Bsu36I. The upper band represents the undigested amplimer; the lower bands are the digested products created by the presence of a Bsu36I site, arising from the ca. 183_184TA > CT mutation. Lane 1 is a 100 base pairs (bp) ladder. Lanes 2, 3, 4, 5 and 8 are negative. Lanes 6 and 7 are heterozygous for ca. 183_184TA > CT. (b) PCR products generated by SBDS-specific primers, digested with Cac8I. The upper band represents the undigested amplimer; the lower bands are the digested products created by the presence of a Cac8I site, arising from the ca. 258 + 2T > C mutation. Lane 1 is a 100 bp ladder. Lanes 2, 3, 4, 6 and 7 are heterozygous for ca. 258 + 2T > C. Lane 5 is homozygous for ca. 258 + 2T > C. Lane 8 is negative.
The second stage of analysis was to sequence the entire coding region of the gene. Exons 1–5 were amplified in 20 μl reactions using the primers outlined in Table 1. Each reaction contained CM102 buffer (ABgene), primers (10 μM) and 50 ng DNA. Cycling conditions were: 95°C for 3 min, followed by 30 cycles of (95°C for 1 min, 55°C for 1 min, 72°C for 1 min), a final extension step at 72°C for 5 min, and cooled to 4°C indefinitely. The resulting PCR product was cleaned up using AMPure™ (Agencourt, Beckman Coulter UK, High Wycombe, UK) magnetic bead technology on a Beckman NX liquid handling robot. These products were then sequenced using the Big Dye™ (Applied Biosystems, Warrington, UK) Terminator version 1·1 and N13 primers. The sequencing products were cleaned up using CleanSEQ™ magnetic bead technology (Beckman Coulter UK) on a Beckman NX liquid handling robot. All the samples were run on the ABI Prism™3730 capillary electrophoresis sequencer (Applied Biosystems).
Table 1.
Primers used in sequence analysis of exons 1–5 of the Shwachman–Bodian–Diamond syndrome (SBDS) gene. Primers are all 5′-tagged with N13 tails (small capitals).
| Primer | Oligonucleotide sequence 5′−3′ | Amplicon size |
|---|---|---|
| Exon 1 forward | GTAGCGCGACGGCCAGTTAAGCCTGCCAGACACAC | 543 |
| Exon 1 reverse | CAGGGCGCAGCGATGACCCGAACCAACCAAATAAAGA | |
| Exon 2 forward | GTAGCGCGACGGCCAGTGGGATTTGTTGTGTCTTG | 369 |
| Exon 2 reverse | CAGGGCGCAGCGATGACCTTTCCTCCAGAAAAACAGC | |
| Exon 3 forward | GTAGCGCGACGGCCAGTGCTCAAACCATTACTTACATATTGA | 462 |
| Exon 3 reverse | CAGGGCGCAGCGATGACCCAGACCCATTATTTTAATG | |
| Exon 4 forward | GTAGCGCGACGGCCAGTGCCTTCACTTTCTTCATAGT | 514 |
| Exon 4 reverse | CAGGGCGCAGCGATGACGAAAATATCTGACGTTTACAACA | |
| Exon 5 forward | GTAGCGCGACGGCCAGTGCTTGCCTCAAAGGAAGTT | 492 |
| Exon 5 reverse | CAGGGCGCAGCGATGACCACTCTGGACTTTGCATCTT |
Sequence traces were analysed with Staden sequence analysis software (http://staden.sourceforge.net).
A meta-analysis of immunological abnormalities in SDS was performed. Case series and reports of SDS were identified from medline, embase and Dialog DataStar using the following search terms: immunoglobulin, Shwachman–Diamond syndrome, Shwachman–Bodian syndrome, congenital lipomatosis of pancreas, hypogammaglobulinaemia, immunodeficiency or pancreatic insufficiency.
Results
SBDS genetic analysis
PCR products generated by SBDS-specific primers digested with Cac8I revealed the presence of ca. 258 + 2T > C mutation, while digestion of PCR products with Bsu36I did not reveal the second common ca. 183–184TA > CT mutation (Fig. 2). These gene conversion mutations on exon 2 derived from the pseudogene sequence account for 74% of SDS mutations. Owing to the autosomal recessive nature of the syndrome, entire gene sequencing of the coding region (exons 1–5) was carried out, which failed to reveal any further mutations. Our patient is thus heterozygous for the ca. 258 + 2T > C mutation of SDS.
Meta-analysis of hypogammaglobulinaemia in SDS
The results of the meta-analysis of published immunological abnormalities are tabulated on a case-by-case basis, and the resulting cohort subdivided into those meeting current European Society of Immunodeficiency (ESID) criteria for CVID, hypogammaglobulinaemia and other immunological abnormalities (Table 2). Of seven published cases and series of SDS, three patients met criteria for ‘possible’ CVID, 10 for hypogammaglobulinaemia and three had other immunological abnormalities.
Table 2.
Meta-analysis of studies in Shwachman–Diamond syndrome (SDS) patients who fulfil criteria for common variable immunodeficiency (CVID) and other immunological abnormalities.
| Disease category | Age/sex | Recurrent infections | IgG levels (g/l) | IgM levels (g/l) | IgA levels (g/l) | Vaccine responses | CD19 cells (%) | CD4/ CD8 ratio | CD16/ CD56 cells (%) | Neutrophil chemotaxis | Reference |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ‘Possible’ CVID | 7/M | Bacterial + viral | 7·0* | 0·3 | 0·8 | Absent/†† | 24 | 2·2‡ | 1·0§ | Abnormal | [4] |
| 18/M | Bacterial + viral | 4·8§ | 0·3 | 0·7 | Normal/†† | 1·2§ | 0·5§ | 1·8§ | – | [4] | |
| 41/F | Bacterial | 5·8§ (6·3–12·9 g/l) | – | – | Absent (pneumococcal) | – | Normal | – | – | [5] | |
| 59/M | Bacterial | 1·3§ | < 0·2§ | < 0·2§ | – | 1·10§ | 1·4§ | 1·43§ | – | Index case | |
| Hypogamma- globulinaemia of other types | 4 month/F 1·5/M | Bacterial Bacterial | 3·8§ (6·0–15·75 g/l) 6·8 | 0·40§ (0·7–2·3 g/l) <0·1§ | 0·07§ (0·2–1·4 g/l) 0·3 | Absent – | CD20–6 (10–30%) – | 2·0 – | 3 – | Abnormal – | [6] [7] |
| 2/M | Bacterial + viral | 6·1§,¶ | 0·3 | 0·2§ | – | – | – | – | – | [4] | |
| – | Bacterial | – | Low | – | – | Normal | Normal | – | Abnormal | [8] | |
| – | Bacterial | – | – | Low | – | Normal | Low | – | Abnormal | [8] | |
| – | Bacterial | – | – | Low | – | Normal | Normal | – | Abnormal | [8] | |
| – | Bacterial | – | Low | – | – | Normal | Normal | – | Abnormal | [8] | |
| 16/M | Bacterial | 0·36§ | 0·03§ | 0·12§ | – | – | – | – | – | [9] | |
| 3/M | Nil | 4·8§ | 0·07§ | Absent§ | – | – | – | – | – | [10] | |
| 6·5/F | Nil | 3·8§ | 0·78 | 0·18§ | – | – | – | – | – | [10] | |
| Other immunological abnormalities | 8/M | Bacterial + viral | 9·8 | 0·3 | 0·7 | –/†† | 9·3 | 2·3 | 1·5§ | Abnormal | [4] |
| 17/M | Bacterial | 8·2 | 0·4 | 11·1 | – | – | –‡ | – | Abnormal | [4] | |
| 14/F | Bacterial | 16·5 | 1·3 | 2·7 | Normal/†† | 15 | 1·0§ | 3·5§ | – | [4] |
Low IgG1 levels;
Low anti-B isohaemagglutinin levels;
low lymphocyte proliferation to concanavalin A/pokeweed mitogen;
low for age;
low IgG1 and IgG3 levels; –, not studied/available.
Discussion
This case describes a man with CVID on IVIG for 12 years who was found to have a heterozygous mutation in the SBDS gene of SDS (OMIM no. 260400). Current ESID criteria for CVID include ‘probable’ CVID in those aged > 2 years with low IgG and another low isotype level (IgA or IgM) with absent vaccine responses, and ‘possible’ CVID in those with low immunoglobulin of any isotype with absent vaccine responses [11]. The cytopenias seen in CVID patients are considered to be ‘autoimmune’, although antibodies against cellular components are not usually identified, suggesting a degree of bone-marrow suppression. SDS is a bone marrow failure disorder, and while neutropenia is the most consistent feature, additional cytopenias including aplastic anaemia can develop [12]. Bone marrow CD34+ cells in SDS are unable to form haemopoietic colonies and have high rates of apoptosis via the Fas signalling pathway [13]. Various other immunological abnormalities, such as low immunoglobulins, low T cells and natural killer (NK) cells, have been described. Features common to both CVID and SDS include the predominance of bacterial infections (Staphylococcus aureus, H. influenzae and Pseudomonas species) over fungal infections [14] and chronic diarrhoea/malabsorption, but pancreatic investigations are usually not undertaken in patients with CVID. The features of CVID and SDS are compared and contrasted in Table 3.
Table 3.
Clinical features of Shwachman–Diamond syndrome (SDS) and common variable immunodeficiency (CVID).
| SDS | CVID |
|---|---|
| Common features to both diseases | |
| Recurrent infections | Recurrent infections |
| Malabsorption due to exocrine pancreatic dysfunction (may be transient) hyperplasia, coeliac-like disease, chronic Giardia lamblia infection) | Malabsorption (inflammatory bowel disease, lymphoid nodular |
| Haematological abnormalities | Haematological abnormalities (?autoimmune) |
| •Neutropenia (intermittent/persistent) | •Cytopenias (neutropenia, lymphopenia, thrombocytopenia) |
| •Thrombocytopenia | •Anaemia (red cell aplasia) |
| •Anaemia | |
| Low immunoglobulins ± absent vaccine responses in some cases (Table 2) | Low immunoglobulins and absent vaccine responses (ESID diagnostic criteria) |
| Abnormal liver function tests (fatty liver) | Abnormal liver function tests (granulomatous CVID, rest unknown) |
| Autoimmunity (hypothyroidism) | Autoimmunity (haematological abnormalities, thyroid disease, neuropathy) |
| Malignancy | Malignancy |
| •MDS (10–44%) | •Gastric carcinoma |
| •Leukaemia (5–24%) | •Lymphoma |
| Other features | |
| Growth and skeletal abnormalities | Multi-systemic granulomatous disease (eye, lymph nodes, skin, liver, spleen, GI tract) |
| •Metaphyseal chondrodysplasia | |
| •Osteoporosis/osteomalacia | Large granular lymphocytosis |
| •Short stature | |
| •Thoracic cage defects | |
| Neurological problems | |
| •Global apraxia | |
| •Generalized weakness/hypotonia | |
| Cardiovascular problems | |
| •Myocardial fibrosis | |
| Oral/dental | |
| •Mucositis/periodontal infections | |
| •Dental dysplasia | |
| Psychological problems | |
| •Cognitive and attention deficits | |
SDS is an autosomal recessive disorder which usually presents early in life with recurrent infections, neutropenia and pancreatic insufficiency. Complications of aplastic anaemia, myelodysplastic syndromes or leukaemia occur in those who reach adult age [12]. It has an extremely heterogeneous clinical presentation, as does CVID, but 90% of patients meeting clinical criteria have mutations in the Shwachman–Bodian–Diamond syndrome gene (SBDS) (see Table 4 for summary of genetic mutations identified in CVID and SDS). The SBDS gene located on chromosome 7q11 is highly conserved in archaea, plants and eukaryotes [3, 16]. The protein is predicted to have 250 amino acids and gene conversion during meiosis with its neighbouring pseudogene, SBDSP (the duplicon of SBDS gene located 5·8Mb distally with nucleotide sequence homology of 97%), which results in the majority of mutations seen in SDS patients. Although the function of the protein remains unknown, SBDS protein shuttles in and out of the nucleolus [17] and studies in yeast homologues suggest a role in ribosomal RNA processing [18, 19]. Homozygous early truncating mutations result in complete loss in SDS function and are lethal at the embryo stage [18, 20], explaining the absence of patients with such mutations. Among the various mutations described, mutations ca. 258 + 2T > C and ca. 183_184TA > CT account for 74% of SDS mutations but the ca. 258 + 2T > C mutation alone may also result in the clinical phenotype [21]. It has been suggested that the absence of mutations in currently known genes does not necessarily preclude diagnosis if the patient fits clinical criteria [22] and equally neutropenia or pancreatic insufficiency may not be present in some patients. Phenotypic–genotypic correlations have not been found [23], and it is possible that other genes controlling RNA metabolism affect SDS function [24]. SDS shares its nucleolar and RNA involvement with other syndromes such as cartilage–hair hypoplasia (short stature and skeletal abnormalities), dyskeratosis congenita (mutations in small nucleolar and telomerase RNA) and Diamond–Blackfan anaemia (mutations in the RPS19 gene encoding ribosomal protein S19) [25].
Table 4.
Prevalence of identified genetic mutations in common variable immunodeficiency (CVID) and Shwachman–Diamond syndrome (SDS).
| Disease | Gene | Location on chromosome | Protein function | Mutations identified | Prevalence (%) | References |
|---|---|---|---|---|---|---|
| CVID | ICOS | 2q33 | T cell stimulation, isotype switching, germinal centre formation | Complete deletion of exon 2 and intron 2, or partial deletion of intron 1 and intron 3 result in partial deletion of ICOS mRNA and absent protein | 2 Deletional event during meiotic recombination | [1] |
| CVID | CD19 | 16p11.2 | Crucial role in signalling on antigen stimulation as part of B cell receptor complex | Two families with homozygous null mutations (insertion of adenine in exon 6 and deletion of guanine and adenine in exon 11) in the CD19 gene | < 1 | [1] |
| CVID | TNFRSF13C (BAFFR) | 22q13.1–31 | BAFF/BAFFR interaction: B cell survival factor, marginal zone differentiation, T cell co-stimulation | Homozygous deletion in the transmembrane region of BAFFR in one autosomal recessive CVID family | < 1 | [1] |
| CVID & IGAD | TNFRSF13B (TACI) | 17p11.2 | TACI mediates isotype switching in B cells | Missense mutation and single nucleotide insertion in one allele of TNFRSF13B | 5–10 | [1, 15] |
| SDS | SBDS | 7q11 | SBDS protein: exact function unknown; has a role in ribosome biogenesis with nucleolar and non-nucleolar functions | Two common heterozygous loss-of-function mutations apart from others [12]: ca. 258 + 2T > C: Disrupts donor splicing site and use of upstream donor site leads to frameshift mutation and premature truncation at 84th AA (84CfsX3) ca. 183_184 TA > CT: Introduction of in-frame stop codon at 62nd AA lysine (K62X); homozygous K62X null mutation is embryonically lethal | SDS: 90% CVID: unknown Recurring mutations arise from gene conversion between SBDS and SBDSP during meiotic recombination | [3, 16] |
Our patient now has normal pancreatic function, has never had neutropenia and has only the ca. 258 + 2T > C mutation. Hepatomegaly, elevated liver enzymes and malabsorption due to pancreatic insufficiency seen in SDS may improve over time in about half the patients [2, 26]. Random faecal elastase determination can provide sufficient information of pancreatic exocrine function [27] but, as adult patients with SDS may be pancreatic sufficient, the clinical criterion of exocrine pancreatic insufficiency does not appear to be essential for the diagnosis of SDS. Pancreatic insufficiency may be ameliorated by enzyme supplementation. Accordingly, genetic testing to screen for SBDS mutations could be considered in selected CVID patients with unexplained weight loss, chronic severe diarrhoea and recurrent anaemia. Abnormal neutrophil chemotaxis could also be used to support the diagnosis of SDS, as it should be normal in CVID, but this assay is not readily available in most centres and there are no external quality assurance schemes for this test. Our patient appeared to be a typical CVID patient − suggesting that other patients with these common features of CVID could also have SBDS mutations. Defects in ribosomal processing can affect cell function at various stages that explain the heterogeneity of other diseases with ribosomal defects. Given that the four established genetic defects (ICOS, CD19, TACI, BAFFR) account for < 20% of cases of CVID, screening studies of CVID patients with suggestive clinical and laboratory features for SBDS mutations may be useful in establishing whether or not SBDS is the ‘fifth’ CVID gene.
Acknowledgments
We are grateful to Dr Carol Hunt and Dr Loraine Sheehan for providing the histopathology pictures, Dr Simon Maslin for comments on the radiographic images and to Dr Georgina Hall for useful communications regarding gene sequencing in SDS.
References
- 1.Salzer U, Grimbacher B. TACItly changing tunes: farewell to a yin and yang of BAFF receptor and TACI in humoral immunity? New genetic defects in common variable immunodeficiency. Curr Opin Allergy Clin Immunol. 2005;5:496–503. doi: 10.1097/01.all.0000191887.89773.cc. [DOI] [PubMed] [Google Scholar]
- 2.Nicolis E, Bonizzato A, Assael BM, Cipolli M. Identification of novel mutations in patients with Shwachman–Diamond syndrome. Hum Mutat. 2005;25:410–18. doi: 10.1002/humu.9324. [DOI] [PubMed] [Google Scholar]
- 3.Boocock GR, Morrison JA, Popovic M, et al. Mutations in SBDS are associated with Shwachman–Diamond syndrome. Nat Genet. 2003;33:97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- 4.Dror Y, Ginzberg H, Dalal I, et al. Immune function in patients with Shwachman–Diamond syndrome. Br J Haematol. 2001;114:712–17. doi: 10.1046/j.1365-2141.2001.02996.x. [DOI] [PubMed] [Google Scholar]
- 5.Church JA. A pediatric genetic disorder diagnosed in adulthood. PLoS Med. 2006;3:e15.. doi: 10.1371/journal.pmed.0030015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kornfeld SJ, Kratz J, Diamond F, Day NK, Good RA. Shwachman–Diamond syndrome associated with hypogammaglobulinemia and growth hormone deficiency. J Allergy Clin Immunol. 1995;96:247–50. doi: 10.1016/s0091-6749(95)70014-5. [DOI] [PubMed] [Google Scholar]
- 7.Maki M, Sorto A, Hallstrom O, Visakorpi JK. Hepatic dysfunction and dysgammaglobulinaemia in Shwachman–Diamond syndrome. Arch Dis Child. 1978;53:693–4. doi: 10.1136/adc.53.8.693-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aggett PJ, Harries JT, Harvey BA, Soothill JF. An inherited defect of neutrophil mobility in Shwachman syndrome. J Pediatr. 1979;94:391–4. doi: 10.1016/s0022-3476(79)80578-8. [DOI] [PubMed] [Google Scholar]
- 9.Hudson E, Aldor T. Pancreatic insufficiency and neutropenia with associated immunoglobulin deficit. Arch Int Med. 1970;125:314–16. [PubMed] [Google Scholar]
- 10.Brueton MJ, Mavromichalis J, Goodchild MC, Anderson CM. Hepatic dysfunction in association with pancreatic insufficiency and cyclical neutropenia. Shwachman–Diamond syndrome. Arch Dis Child. 1977;52:76–8. doi: 10.1136/adc.52.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.European Society for Immunodeficiencies (ESID). CVID diagnostic criteria. [28 March 2007]. Available at: http://www.esid.org/workingparty.php?party=3&sub=2&id=73#Q2.
- 12.Dror Y, Freedman MH. Shwachman–Diamond syndrome. Br J Haematol. 2002;118:701–13. doi: 10.1046/j.1365-2141.2002.03585.x. [DOI] [PubMed] [Google Scholar]
- 13.Dror Y, Freedman MH. Shwachman–Diamond syndrome marrow cells show abnormally increased apoptosis mediated through the Fas pathway. Blood. 2001;97:3011–16. doi: 10.1182/blood.v97.10.3011. [DOI] [PubMed] [Google Scholar]
- 14.Grinspan ZM, Pikora CA. Infections in patients with Shwachman–Diamond syndrome. Pediatr Infect Dis J. 2005;24:179–81. doi: 10.1097/01.inf.0000151042.90125.f6. [DOI] [PubMed] [Google Scholar]
- 15.Castigli E, Wilson SA, Garibyan L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005;37:829–34. doi: 10.1038/ng1601. [DOI] [PubMed] [Google Scholar]
- 16.Woloszynek JR, Rothbaum RJ, Rawls AS, et al. Mutations of the SBDS gene are present in most patients with Shwachman–Diamond syndrome. Blood. 2004;104:3588–90. doi: 10.1182/blood-2004-04-1516. [DOI] [PubMed] [Google Scholar]
- 17.Austin KM, Leary RJ, Shimamura A. The Shwachman–Diamond SBDS protein localizes to the nucleolus. Blood. 2005;106:1253–8. doi: 10.1182/blood-2005-02-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shammas C, Menne TF, Hilcenko C, et al. Structural and mutational analysis of the SBDS protein family. Insight into the leukemia-associated Shwachman–Diamond syndrome. J Biol Chem. 2005;280:19221–9. doi: 10.1074/jbc.M414656200. [DOI] [PubMed] [Google Scholar]
- 19.Savchenko A, Krogan N, Cort JR, et al. The Shwachman–Bodian–Diamond syndrome protein family is involved in RNA metabolism. J Biol Chem. 2005;280:19213–20. doi: 10.1074/jbc.M414421200. [DOI] [PubMed] [Google Scholar]
- 20.Zhang S, Shi M, Hui CC, Rommens JM. Loss of the mouse ortholog of the Shwachman–Diamond syndrome gene (SBDS) results in early embryonic lethality. Mol Cell Biol. 2006;26:6656–63. doi: 10.1128/MCB.00091-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kawakami T, Mitsui T, Kanai M, et al. Genetic analysis of Shwachman–Diamond syndrome: phenotypic heterogeneity in patients carrying identical SBDS mutations. Tohoku J Exp Med. 2005;206:253–9. doi: 10.1620/tjem.206.253. [DOI] [PubMed] [Google Scholar]
- 22.Hall GW, Dale P, Dodge JA. Shwachman–Diamond syndrome: UK perspective. Arch Dis Child. 2006;91:521–4. doi: 10.1136/adc.2003.046151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuijpers TW, Alders M, Tool AT, Mellink C, Roos D, Hennekam RC. Hematologic abnormalities in Shwachman Diamond syndrome: lack of genotype–phenotype relationship. Blood. 2005;106:356–61. doi: 10.1182/blood-2004-11-4371. [DOI] [PubMed] [Google Scholar]
- 24.Menne TF, Goyenechea B, Sánchez-Puig N, et al. The Shwachman–Bodian–Diamond syndrome protein mediates translational activation of ribosomes in yeast. Nat Genet. 2007;39:486–95. doi: 10.1038/ng1994. [DOI] [PubMed] [Google Scholar]
- 25.Liu JM, Ellis SR. Ribosomes and marrow failure: coincidental association or molecular paradigm? Blood. 2006;107:4583–8. doi: 10.1182/blood-2005-12-4831. [DOI] [PubMed] [Google Scholar]
- 26.Cipolli M. Shwachman–Diamond syndrome: clinical phenotypes. Pancreatology. 2001;1:543–8. doi: 10.1159/000055858. [DOI] [PubMed] [Google Scholar]
- 27.Molinari I, Souare K, Lamireau T, et al. Fecal chymotrypsin and elastase-1 determination on one single stool collected at random: diagnostic value for exocrine pancreatic status. Clin Biochem. 2004;37:758–63. doi: 10.1016/j.clinbiochem.2004.03.010. [DOI] [PubMed] [Google Scholar]