

Abstract
Decoy receptor 3 (DcR3/TR6) is a decoy receptor for the Fas ligand (FasL) and can inhibit FasL-induced apoptosis. It has been reported recently that DcR3 can induce T cell activation via co-stimulation of T cells, suggesting that DcR3 may be involved in the pathophysiology of autoimmune diseases. This study aims to analyse the serum DcR3 in patients with systemic lupus erythematosus (SLE) and to investigate the role of DcR3 in the pathogenesis of SLE. Significantly elevated serum DcR3 was observed in SLE patients, and the mean serum DcR3 level was significantly higher for those with active disease [SLE disease activity index (SLEDAI) ≥ 10] compared with that in patients with inactive disease (SLEDAI < 10). In addition to reducing activation-induced cell death in activated T cells via neutralization of the FasL, soluble DcR3–Fc enhanced T cell proliferation and increased interleukin-2 and interferon-γ production via co-stimulation of T cells. Moreover, enhanced T cell reactivity to DcR3-induced co-stimulation was demonstrated in lymphocytes from patients with SLE, suggesting the elevated serum DcR3 may associate with enhanced T cell activation in vivo. These findings are the first to demonstrate that serum DcR3 concentrations are increased in SLE patients, and this may imply a possible role of DcR3 in the pathogenesis of SLE via enhanced T cell hyperreactivity and reduced apoptosis in activated T cells.
Keywords: apoptosis, DcR3, SLE, T cell activation
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disease with a broad spectrum of clinical and immunological abnormalities. Numerous aberrations of the immune system have been reported in SLE and have been implicated in the pathogenesis of this autoimmune disease. Included in these abnormalities are defects in apoptosis leading to the accumulation of autoreactive T and B cells [1] and changes in the maturation and function of antigen-presenting cells such as dendritic cells, leading to an enhanced presentation of autoantigens to CD4 T cells and to expansion of autoreactive T cells [2]. In addition, enhanced reactivity of T cells to autoantigens and aberrant signal transduction have been implicated in the pathogenesis of SLE [3–5].
Decoy receptor 3 (DcR3/TR6) is a decoy receptor for the Fas ligand (FasL) and can inhibit FasL-induced apoptosis. DcR3 lacks an apparent transmembrane domain in its sequence, and is a secreted protein belonging to the tumour necrosis factor receptor family [6, 7]. DcR3 has three known ligands: FasL, LIGHT and TL1A [6–9]. DcR3 can bind to the FasL and inhibit the interaction between Fas and FasL. Consequently, FasL-induced apoptosis of lymphocytes and of several tumour cell lines can be repressed by DcR3 [7]. DcR3 can also bind to LIGHT to protect against LIGHT-mediated apoptosis [6, 8, 10]. The third known ligand of DcR3 is TL1A, which is a new member of the tumour necrosis factor family, and is expressed predominantly on endothelial cells [9]. DcR3 can repress TL1A-augmented lymphokine secretion and the graft-versus-host response [9]. In addition to these ligands, it has been reported recently that DcR3 may bind to and cross-link proteoglycans to induce monocyte adhesion [11].
The DcR3 gene has been reported to be amplified and overexpressed in primary lung and colon tumours and other cancers [7, 12–15]. Because DcR3 seems to function in a manner similar to soluble Fas (sFas) in terms of binding FasL and competing with membrane Fas, overproduction of DcR3 may be involved in the acquisition of autoimmunity. There are several indications that DcR3 may be involved in the pathophysiology of autoimmune diseases. It has been reported that sFas levels are elevated in SLE [16, 17], rheumatoid arthritis (RA) and other rheumatic diseases [18]. It has been demonstrated that the DcR3 gene is overexpressed significantly in peripheral blood mononuclear cells (PBMCs) of SLE patients [19]. These findings may indicate the biological role of DcR3 molecules in autoimmunity. Overexpression and overproduction of DcR3 may overcome Fas/FasL binding; therefore, the self-recognizing clones may be protected from activation-induced cell death (AICD) mediated by the Fas/FasL interaction.
It has been reported recently that DcR3 can induce T cell activation via co-stimulation of mouse and human T cells [20, 21]. These findings suggest the possibility that DcR3-induced co-stimulation may trigger activation of human T cells in vivo. Enhanced reactivity of T cells to autoantigens via co-stimulation might be implicated in the pathogenesis of SLE [4, 5]. In the present study, we analysed serum DcR3 levels in patients with SLE and compared them with levels in healthy controls. The correlation between the serum levels of the DcR3 and disease activity was analysed.
Materials and methods
Subjects
One hundred and fifty-two patients with SLE were studied (15 men and 137 women, with a mean age of 39·2 ± 11·4 years and a mean SLE disease activity index (SLEDAI) [22] of 8·2 ± 5·98. All met the criteria of the American College of Rheumatologists for a diagnosis of SLE. Forty healthy age- and gender-matched volunteers and 122 RA patients were used as controls. All studies which included samples involving humans were carried out in accordance with institutional guidelines.
Enzyme-linked immunosorbent assay for DcR3 protein
Serum levels of DcR3 were determined by a validated sandwich solid-phase enzyme linked immunosorbent assay (ELISA) (Anawrahta Biotech, Taipei, Taiwan). A monoclonal antibody (mAb) specific for DcR3 was coated onto the wells of microtitre strips. An aliquot of 100 μl of undiluted serum was pipetted in duplicate into the wells. During the first incubation, the DcR3 antigen and a biotinylated mAb specific for DcR3 were incubated simultaneously for 3 h at room temperature. After washing, the enzyme streptavidin–polyhorseradish peroxidase was added. After incubation for 30 min and washing to remove any unbound enzyme, a substrate solution was added to the bound enzyme to induce a coloured reaction product. The optical density (OD), reflecting the intensity of the product, was measured in a spectrophotometer with 450 nm as the primary wavelength and 650 nm as the reference wavelength. Linear calibration curves were made using standard DcR3 provided with the kit. The amount of DcR3 in the serum samples was determined by extrapolating OD values to DcR3 concentrations using the calibration curves.
Lymphocyte isolation
Heparinized peripheral venous blood was obtained from study subjects. T cells were separated by Rosette separation (Stem Cell Technologies, Vancouver, Canada). Briefly, non-T cells are selected by a tetrameric antibody mixture against CD14, CD16, CD19, CD56 and glyA and bound to erythrocytes. These complexes are separated from the T cells by a Ficoll-Paque gradient. The purity of human T cells isolated was >97% as tested with an anti-CD3 mAb by flow cytometry.
Expression and purification of the recombinant DcR3 fusion protein, DcR3–Fc and Fas–Fc
The recombinant sFas–Fc fusion protein was prepared as described previously [23]. To generate soluble recombinant DcR3–Fc fusion molecules, the coding sequence for the extracellular domain of human DcR3 was isolated by reverse-transcription-polymerase chain reaction, and the amplified product was ligated in-frame into the BamHI-cut pUC19–immunoglobulin G1 (IgG1)–Fc vector containing the human IgG1 Fc coding sequence. The fusion gene was then subcloned into the pBacPAK9 vector (Clontech, Palo Alto, CA, USA). The DcR3–Fc fusion protein was recovered from the filtered supernatants of the recombinant virus-infected Sf21 cells using protein G-Sepharose beads (Pharmacia, Piscataway, NJ, USA). The bound DR4–Fc protein was eluted with glycine buffer (pH 3) and dialysed into phosphate-buffered saline (PBS).
Apoptosis assay for activation-induced T cell death
Human T cells were isolated from peripheral blood using Rosette separation (Stem Cell Technologies), stimulated with phytohaemagglutinin (PHA; 1 μg/ml) for 24 h and cultures in the presence of interleukin (IL)-2 (1 ng/ml) for 5 days at 37°C. The activated T cells were then restimulated with plate-bound anti-CD3 in the absence or presence of soluble DcR3–Fc (0–40 μg/ml) or control human IgG1 (20 μg/ml) on day 7. Apoptosis was determined by annexin V–fluorescein isothiocyanate/phycoerythrin (FITC/PI) double staining 16 h later. For assaying T cell apoptosis, cells were washed once with cold PBS and then resuspended in 1× binding buffer (140 mM NaCl and 2·5 mM CaCl2 in 10 mM HEPES/NaOH; pH 7·4) at a concentration of 106 cells/ml. For each sample, 3 × 105 cells/100 μl was transferred to a 1·5-ml tube containing 5 μl annexin V–FITC and 10 μl PI and incubated at room temperature for 10 min in the dark, followed by the addition of another 200 μl of 1× binding buffer. Samples were analysed by flow cytometry.
T cell proliferation assay
For assaying T cell proliferation with DcR3-induced co-stimulation, isolated human T cells (2 × 105 cells/well) were cultured for 72 h in 96-well flat-bottomed microtitre plates precoated with anti-human CD3 (500 ng/ml) and soluble DcR3–Fc recombinant protein (10 μg/ml) or human IgG1 (10 μg/ml; Sigma, St Louis, MO, USA). The cultures were pulsed with [3H]-thymidine (1·0 μCi/well) 18 h before harvesting the cells, and [3H]-thymidine incorporation was measured in a Microbeta Plus liquid scintillation counter (Wallac, Gaithersburg, MD, USA). Cultures were run in triplicate, and each experiment was repeated at least three times.
Cytokine assays
To trigger the activation of T cells via DcR3, purified human T cells (2 × 105 cells/well) were stimulated with a suboptimal concentration of plate-bound anti-CD3 mAb (500 ng/ml) and soluble DcR3–Fc fusion protein (10 μg/ml) or human IgG1 (10 μg/ml; Sigma) in 96-well flat-bottomed microtitre plates. Cell culture supernatants were collected and levels of IL-2, interferon (IFN)-γ and IL-4 were quantified using commercial ELISA kits (Endogen, Woburn, MA, USA), according to the vendor's instructions.
Statistical analysis
Data are presented as the mean ± standard error of the mean. Differences in mean DcR3 levels between the patient groups and normal controls were determined by analysis of variance and by χ2 test. The results of T cell proliferation and cytokine production were evaluated by Student's t-test. A P-value of <0·05 was regarded as statistically significant. Experiments were performed more than three times with T cells derived from a single donor, and a representative data set is presented. A similar result in each figure was obtained with T cells purified from a second independent donor. The results of the T cell proliferation response to DcR3 in T cells from SLE patients and normal healthy subjects were evaluated using the non-parametric Mann–Whitney rank sum test. A P-value of <0·05 was regarded as statistically significant.
Results
Elevated serum DcR3 levels in SLE patients
Samples from 152 SLE patients, 40 normal health controls and 122 RA patients were collected, and the serum DcR3 levels were detected using a DcR3 ELISA kit. Characteristics of the SLE patients are given in Table 1. As shown in Fig. 1, significantly elevated serum DcR3 was observed in SLE patients when compared with normal healthy controls (P < 0·001) and RA patients (P < 0·005). The average DcR3 concentration in all SLE patients tested was 20·81 ng/ml, in normal donors tested was 5·66 ng/ml and in RA patients tested was 7·92 ng/ml. Serum DcR3 concentrations in patients with RA did not differ from those in healthy volunteers (P > 0·05).
Table 1.
Characteristics of patients with systemic lupus erythematosus (SLE).
| Variable | Active (n = 77) (SLEDAI ≥ 10) | Inactive (n = 75) (SLEDAI < 10) | P-value |
|---|---|---|---|
| Age (years) | 38·6 ± 12·8 | 40·2 ± 11·6 | 0·06 |
| Male/female | 8/69 | 7/68 | 0·36 |
| Duration of lupus (months) | 48 ± 16 | 42 ± 18 | 0·12 |
| Anti-dsDNA (IU/ml) | 162·6 ± 57·6 | 45·35 ± 4·6 | 0·015* |
| SLEDAI | 13·42 ± 1·65 | 4·86 ± 0·87 | 0·0001* |
| C3 (mg/dl) | 56·4 ± 12·2 | 67·6 ± 10·6 | 0·032* |
| C4 (mgd/l) | 9·2 ± 4·6 | 12·4 ± 5·8 | 0·075 |
| Creatinine (mg/dl) | 1·38 ± 0·58 | 1·24 ± 0·46 | 0·24 |
| Leucocytes (×109/l) | 3·82 ± 0·50 | 4·76 ± 0·50 | 0·15 |
| DcR3 (ng/ml) | 27·8 ± 6·2 | 13·1 ± 4·7 | 0·0035* |
| Renal disease | 36 | 8 | |
| Central nervous system disease | 8 | – | |
| Serositis | 14 | 3 | |
| Haematological disease | 27 | 6 | |
| Polyarthritis | 46 | 18 | |
| Skin involvement | 24 | 12 | |
| Fever | 25 | 2 |
Significant P-values. Values are mean ± standard error of the mean or n, unless stated. SLEDAI: SLE disease activity index.
Fig. 1.
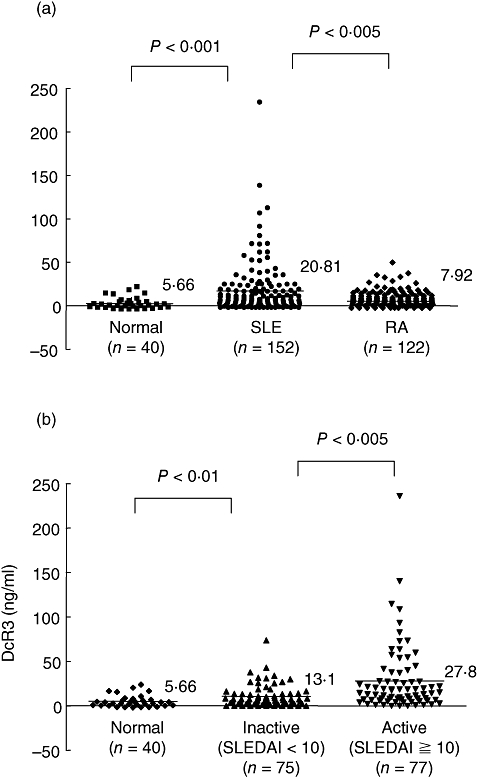
Elevated serum decoy receptor 3 (DcR3) concentrations (ng/ml) in systemic lupus erythematosus (SLE) patients. (a) Samples from 152 SLE patients, 40 health controls and 122 rheumatoid arthritis (RA) patients were collected, and the serum DcR3 levels were detected with a DcR3 enzyme-linked immunosorbent assay kit. The horizontal lines give the mean DcR3 value. Statistical analysis by the Mann–Whitney rank sum test revealed significantly elevated serum DcR3 in SLE patients when compared with healthy controls (P < 0·001) and RA patients (P < 0·005). (b) Serum DcR3 concentrations in SLE patients with active [SLE disease activity index (SLEDAI) ≥ 10; n = 77] and inactive disease (SLEDAI < 10; n = 75). Statistical analysis with the Mann–Whitney rank sum test revealed a significant difference between the two groups (P < 0·005).
To evaluate whether DcR3 levels are influenced by gender, age or both, we determined the correlation between the DcR3 level and age, and we analysed the difference in DcR3 values between men and women in the control group. There was no difference in mean DcR3 concentrations between men (n = 15; 21·8 ± 5·1 ng/ml) and women (n = 137; 20·2 ± 4·7 ng/ml) (P = 0·24). There was also no correlation between age and DcR3 concentration. To evaluate further whether serum DcR3 levels correlated with SLE disease activity, we analysed the serum DcR3 levels of patients with active (SLEDAI ≥ 10) and inactive diseases (SLEDAI < 10). The results are shown in Fig. 1; the mean serum DcR3 concentration was 13·1 ± 4·7 ng/ml for patients with inactive disease and 27·8 ± 6·2 ng/ml for those with active disease. The difference between the two groups was significant (P < 0·005). We examined the association between the level of serum DcR3 and specific clinical features. No clear relationship was found between serum DcR3 concentrations and renal, cerebral, cutaneous, serosal, haematological or musculoskeletal disease manifestations. Furthermore, there was no significant correlation between the DcR3 concentration and serological indices of disease activity such as anti-dsDNA and complement levels and no correlation with C reactive protein levels.
Inhibition of AICD by soluble DcR3 in human T cells
Because DcR3 is the decoy receptor for FasL and is able to inhibit FasL-mediated cell death, this raises the possibility that high levels of serum DcR3 in SLE patients may attenuate the AICD in activated T cells. Therefore, we investigated the role of DcR3 in inhibition of AICD in human T cells. We set up an in vitro system to assay AICD in primary human T cells [24]. We purified human T cells from PBMCs, followed by stimulation with PHA for 24 h and culturing in the presence of IL-2 for 5 days (long-term activated T cells). The long-term activated T cells were then restimulated with plate-bound anti-CD3 mAb on day 7 to induce AICD in the presence or absence of soluble DcR3–Fc or Fas–Fc. The results in Fig. 2 demonstrate that soluble DcR3–Fc protein reduced apoptosis significantly in activated T cells induced by restimulation with the anti-CD3 mAb when compared with the control human IgG1. These results indicate that DcR3 can inhibit AICD in activated T cells. This effect is similar to that of sFas–Fc, suggesting the DcR3-induced inhibition of AICD is via binding to FasL to inhibit Fas-mediated apoptosis in activated T cells (Fig. 2).
Fig. 2.
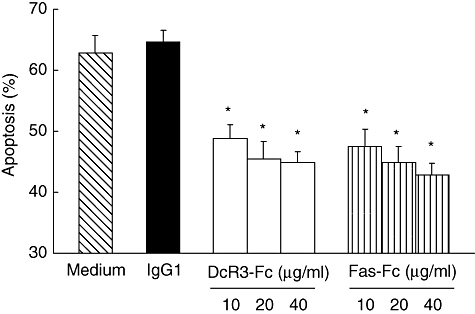
Soluble decoy receptor 3 (DcR3)–Fc inhibits activation-induced cell death (AICD) in human T cells. Human T cells were isolated from peripheral blood followed by stimulation with phytohaemagglutinin for 24 h, and after being cultured in the presence of interleukin-2 for 5 days the long-term activated T cells were restimulated with plate-bound anti-CD3 monoclonal antibody on day 7 to induce AICD in the presence or absence of soluble DcR3–Fc proteins (0–40 μg/ml), Fas–Fc proteins (0–40 μg/ml) or soluble immunoglobin G1 (IgG1) (10 μg/ml) as the control (*P < 0·01 by Student's t-test when compared with medium alone samples). The experiments were performed more than three times, and a representative data set is presented. A similar result was obtained with T cells purified from two other independent donors.
Soluble DcR3 protein induces co-stimulation of human T cells
In addition to inhibition of apoptosis via binding to FasL, the elevated serum DcR3 proteins may have been associated with T cell hyperreactivity in SLE. In order to demonstrate whether soluble DcR3 can induce T cell activation, we purified T cells from human PBMCs and stimulated them with recombinant DcR3–Fc proteins in vitro. The results in Fig. 3 demonstrate that soluble DcR3–Fc enhanced human T cell proliferation in conjunction with a suboptimal concentration of anti-CD3 in a dose-dependent manner. Human IgG1 was used as the control. In contrast to DcR3–Fc, the sFas–Fc was not able to induce T cell proliferation, indicating that this effect is not due to binding to FasL on T cell surface. To confirm this observation further, we evaluated the T cell proliferation response to DcR3–Fc and anti-CD3 in 20 healthy subjects. Similar results were obtained for each group. These results indicate that soluble DcR3–Fc induces co-stimulation of human T cells.
Fig. 3.
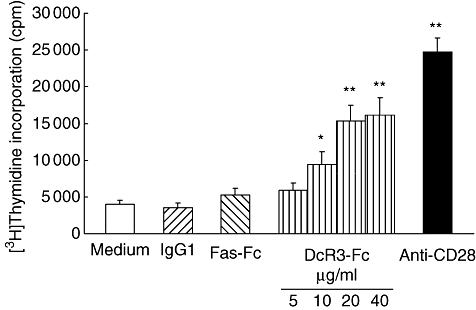
Soluble decoy receptor 3 (DcR3)–Fc enhances human T cell proliferation in conjunction with suboptimal concentration of immobilized anti-CD3. Purified human T cells from peripheral blood mononuclear cells were cultured for 72 h in 96-well flat-bottomed microtitre plates precoated with an anti-human CD3 monoclonal antibody (mAb) (500 ng/ml) and soluble DcR3–Fc recombinant protein (5–40 μg/ml), soluble Fas–Fc recombinant protein (10 μg/ml) anti-CD28 mAb (1 μg/ml) or human immunoglobulin G1 (IgG1) (10 μg/ml) as controls. The cultures were pulsed with [3H]-thymidine (1·0 μCi/well) 18 h before harvesting the cells, and [3H]-thymidine incorporation was measured. The mean ± standard deviation of the counts per minute value from triplicate samples is shown. The experiments were performed more than three times, and a representative data set is presented. Statistical analysis by Student's t-test revealed significant differences between human immunoglobulin 1- and soluble DcR3–Fc-treated samples (*P < 0·05; **P < 0·01). A similar result was obtained with T cells purified from 20 other independent healthy donors.
We then investigated cytokine production when T cells are activated by DcR3. To investigate the cytokine production profiles of T cells activated by DcR3, we measured IL-2 as well as IFN-γ and IL-4 in supernatants released from cultures co-stimulated by anti-CD3 and DcR3–Fc. The results in Fig. 4 demonstrate that the production of IL-2 by T cells was enhanced significantly in the presence of soluble DcR3 compared with human IgG1. Similarly, the secretion of IFN-γ by T cells was also enhanced significantly when triggered by soluble DcR3 (Fig. 4). In contrast to the Th2 cytokine, IL-4, secretion was not elevated significantly compared with IFN-γ when stimulated by soluble DcR3.
Fig. 4.
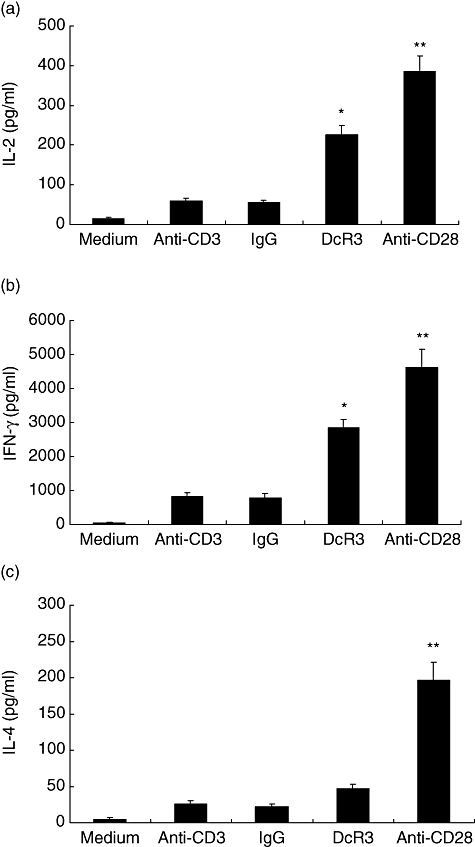
Soluble decoy receptor 3 (DcR3)–Fc induces cytokine production in human T cells. (a) Soluble DcR3–Fc enhances interleukin (IL)-2 production in human T cells. Human T cells purified from peripheral blood mononuclear cells (PBMCs) were cultured with a plate-bound anti-CD3 monoclonal antibody (mAb) (500 ng/ml) plus soluble human immunoglobulin G1 (IgG1) (10 μg/ml), anti-CD28 mAb (1 μg/ml) or soluble DcR3–Fc (10 μg/ml) for 24 h. Supernatants were harvested and assayed for their cytokine production in IL-2 by enzyme-linked immunosorbent assay (ELISA). Statistical analysis by Student's t-test revealed significant differences between the soluble human IgG1- and soluble DcR3–Fc-treated samples (*P < 0·05). (b) and (c) Soluble DcR3–Fc enhances interferon (IFN)-γ production in human T cells. Human T cells purified from PBMCs were cultured with a plate-bound anti-CD3 mAb (500 ng/ml) plus human IgG1 (10 μg/ml) or soluble DcR3–Fc (10 μg/ml) for 3 days. Supernatants were harvested and assayed for their IFN-γ (b) and IL-4 (c) production by ELISA (*P < 0·05, **P < 0·01 when compared with immobilized human IgG1-treated samples).
Enhanced T cell reactivity to DcR3-induced co-stimulation in lymphocytes from patients with SLE
Our results demonstrate that DcR3 induces activation of human T cells in conjunction with signals from the T cell receptor (TCR). This raises the possibility that DcR3-induced co-stimulation of T cells may occur in human T cell activation, leading to enhanced reactivity to low-affinity self-antigens in autoreactive T cells. Therefore, we investigated DcR3-induced co-stimulation of T cells in patients with SLE. The results in Fig. 5 demonstrate that a significantly higher proliferation response to DcR3-induced co-stimulation was observed in T cells isolated from patients with SLE compared with normal healthy subjects control (P < 0·05). We examined further the correlation between the DcR3-induced co-stimulation of T cells and the SLEDAI score to clarify whether the DcR3-induced co-stimulation of T cells reflects disease activity in SLE. There was no strong correlation between these values (data not shown). Taken together, our results indicate that T cells from SLE patients showed enhanced reactivity to DcR3-induced co-stimulation compared with normal controls.
Fig. 5.
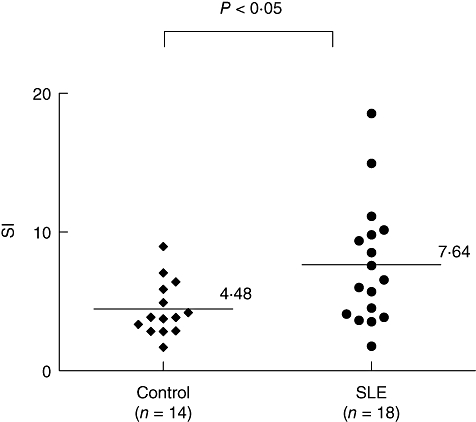
Enhanced T cell reactivity to decoy receptor 3 (DcR3)-induced co-stimulation in T cells from patients with systemic lupus erythematosus (SLE). Purified T cells from peripheral blood mononuclear cells of SLE patients were cultured for 72 h in 96-well flat-bottomed microtitre plates precoated with an anti-human CD3 monoclonal antibody (500 ng/ml) and soluble DcR3–Fc recombinant protein (10 μg/ml) or human immunoglobulin G1 (IgG1) (10 μg/ml) as controls. The cultures were pulsed with [3H]-thymidine (1·0 μCi/well) 18 h before harvesting the cells, and [3H]-thymidine incorporation was measured. The stimulation index (SI) was calculated as the ratio of counts per minute (cpm) of DcR3–Fc to that of IgG1. The SI was calculated according to the formula: cpm of T cells stimulated with anti-CD3 and DcR3–Fc/cpm of T cells stimulated with anti-CD3 and IgG1. Background cpm obtained with T cells in medium alone was negligible. Each symbol represents one donor (n = 18 for SLE and n = 14 for normal healthy subjects). The mean SI obtained with these donors is indicated (P < 0·05 by the Mann–Whitney rank sum test).
Discussion
Our study demonstrates significantly elevated serum DcR3 levels in patients with SLE. The mean serum DcR3 level was significantly higher for those with active disease compared with that in patients with inactive disease. Overexpression of the DcR3 gene has been shown previously in PBMCs derived from silicosis and SLE patients [19], but this is the first report to demonstrate that serum DcR3 protein is elevated in SLE patients, and is associated with disease activity. It is unclear, however, whether overproduction of the DcR3 protein in SLE merely accompanies the alteration in Fas-related molecules or precedes the clinical onset of autoimmune abnormalities. Overproduction of DcR3 has been reported in ulcerative colitis lamina propria T cells [25] and acutely inflamed intestinal epithelia [26]. These findings may indicate the biological role of DcR3 molecules in autoimmunity. However, as the increase in DcR3 only reflects T cell activation so far, there is no evidence to suggest that this phenomenon precedes the onset of autoimmune disease. Our study demonstrates that serum DcR3 concentrations are increased in SLE patients. This seems to be disease-specific and could indicate a role of DcR3 in SLE pathophysiology.
In this study, we have also shown that triggering of T cells by soluble DcR3–Fc, in conjunction with a suboptimal level of the anti-CD3 mAb, induced a maximal proliferation response and enhanced IL-2 as well as IFN-γ secretion in activated human T cells. Moreover, T cells from SLE patients demonstrated a significantly enhanced proliferation response to DcR3 compared with controls. The results indicated that in addition to its role in inhibiting apoptosis by binding to FasL and LIGHT, DcR3 itself can enhance T cell proliferation after TCR engagement. The phenomenon of DcR3 co-stimluation via engagement with LIGHT has been reported in murine and human T cells [20, 21]; however, other mechanisms for potential co-stimulation have been shown recently: DcR3 can bind to and cross-link proteoglycans on the cell surface [11]. Moreover, it was shown that Fas can bind to FasL to induce co-stimulation of T cells [27, 28]. Our results demonstrate that sFas–Fc was not able to induce T cell proliferation, indicating that this effect is not due to binding to FasL on the T cell surface (Fig. 3). It is likely that the DcR3–Fc-induced T cell proliferation is via interaction between DcR3 and LIGHT. Previous studies have demonstrated that DcR3 could bind to LIGHT and to transduce co-stimulatory signals into both human and murine T cells [20, 21]. It is still not clear whether there is any particular T cell subset responding to DcR3-induced co-stimulation, and what effector function will develop after triggering by DcR3.
Our results also demonstrate that DcR3–Fc can reduce AICD in activated human T cells and this effect is similar to that of sFas–Fc, indicating that the DcR3-induced suppression of AICD in T cells is due probably to binding to FasL to inhibit FasL-mediated apoptosis in activated T cells (Fig. 2). All these observations suggest that DcR3 may promote survival of activated T cells in SLE. Recent studies have demonstrated that co-stimulatory signals might promote survival of T cells after activation by antagonizing Fas-mediated apoptosis, leading to expansion of antigen-specific T cell clones. Our results demonstrate that in addition to reduced apoptosis in activated T cells, soluble DcR3–Fc could also induce T cell activation via enhanced reactivity to antigens. This raises the possibility that enhanced T cell reactivity to autoantigens via DcR3 might be implicated in the pathogenesis of SLE. Patients with SLE display major alterations in T cell reactivity to autoantigens, and the systemic autoimmune response that characterizes SLE might be explained in part by enhanced T cell activation. The enhanced activation of T cells to low-affinity autoantigens via DcR3 co-stimulation could thus initiate the expansion of autoreactive T cells, followed by differentiation of autoantibody-producing B cells. Therefore, DcR3-induced T cell co-stimulation might activate human T cells in vivo and enhance reactivity to autoantigens in SLE. The results of increased T cell proliferation after DcR3 engagement may also reflect the hyperreactivity of SLE T cells rather than a specific role for DcR3. These results indicate that there is increased activation of SLE T cells compared with that in normal controls after TCR stimulation.
In conclusion, in this study we have shown elevated serum DcR3 levels in patients with SLE. Levels in patients with active disease were higher than those in patients with inactive disease. Elevated serum DcR3 levels in SLE patients imply a possible role of DcR3 in the pathogenesis of SLE via enhanced hyperreactivity of T cells; meanwhile, it could promote the survival of activated T cells via inhibition of apoptosis.
Acknowledgments
We thank Dr Men-Luh Yen for the critical review of the manuscript and Ms S.-Y. Chou for assistance. This project was supported by grants from the National Health Research Institute, Taiwan (NHRI-EX95–9532S1), the National Science Council, Taiwan (NSC95-2320-B-002–026; NSC95-2321-B-002–025) and McKay Memorial Hospital (MMH-9415).
References
- 1.Nataga S, Suda T. Fas and Fas ligand: lpr and gld mutations. Immunol Today. 1995;6:39–43. doi: 10.1016/0167-5699(95)80069-7. [DOI] [PubMed] [Google Scholar]
- 2.Blanco P, Palucka AK, Gill M, Pascual V, Banchereau J. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science. 2001;294:1540–3. doi: 10.1126/science.1064890. [DOI] [PubMed] [Google Scholar]
- 3.Tsokos GC, St Liossis C. Immune cell signaling defects in lupus: activation, anergy and death. Immunol Today. 1999;20:119–24. doi: 10.1016/s0167-5699(98)01395-4. [DOI] [PubMed] [Google Scholar]
- 4.Kammer GM, Perl A, Richardson BC, Tsokos GC. Abnormal T cell signal transduction in systemic lupus erythematosus. Arthritis Rheum. 2002;46:1139–54. doi: 10.1002/art.10192. [DOI] [PubMed] [Google Scholar]
- 5.Tsai HF, Lai JJ, Chou AH, Wang TF, Wu CS, Hsu PN. Induction of costimulation of human CD4 T cells by tumor necrosis factor-related apoptosis-inducing ligand: possible role in T cell activation in systemic lupus erythematosus. Arthritis Rheum. 2004;50:629–39. doi: 10.1002/art.20038. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J, Salcedo TW, Wan X, et al. Modulation of T-cell responses to alloantigens by TR6/DcR3. J Clin Invest. 2001;107:1459–68. doi: 10.1172/JCI12159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pitti RM, Marsters SA, Lawrence DA, et al. Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature. 1998;396:699–703. doi: 10.1038/25387. [DOI] [PubMed] [Google Scholar]
- 8.Yu KY, Kwon B, Ni J, Zhai Y, Ebner R, Kwon BS. A newly identified member of tumor necrosis factor receptor superfamily (TR6) suppresses LIGHT-mediated apoptosis. J Biol Chem. 1999;274:13733–6. doi: 10.1074/jbc.274.20.13733. [DOI] [PubMed] [Google Scholar]
- 9.Migone TS, Zhang J, Luo X, et al. TL1A is a TNF-like ligand for DR3 and TR6/DcR3 and functions as a T cell costimulator. Immunity. 2002;16:479–92. doi: 10.1016/s1074-7613(02)00283-2. [DOI] [PubMed] [Google Scholar]
- 10.Gill RM, Hunt JS. Soluble receptor (DcR3) and cellular inhibitor of apoptosis-2 (cIAP-2) protect human cytotrophoblast cells against LIGHT-mediated apoptosis. Am J Pathol. 2004;165:309–17. doi: 10.1016/S0002-9440(10)63298-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang YC, Chan YH, Jackson DG, Hsieh SL. The glycosaminoglycan-binding domain of decoy receptor 3 is essential for induction of monocyte adhesion. J Immunol. 2006;176:173–80. doi: 10.4049/jimmunol.176.1.173. [DOI] [PubMed] [Google Scholar]
- 12.Yamana K, Bilim V, Hara N, et al. Prognostic impact of FAS/CD95/APO-1 in urothelial cancers: decreased expression of Fas is associated with disease progression. Br J Cancer. 2005;93:544–51. doi: 10.1038/sj.bjc.6602732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li H, Zhang L, Lou H, et al. Overexpression of decoy receptor 3 in precancerous lesions and adenocarcinoma of the esophagus. Am J Clin Pathol. 2005;124:282–7. doi: 10.1309/XK59-4E4B-5WU8-2QR6. [DOI] [PubMed] [Google Scholar]
- 14.Bai C, Connolly B, Metzker ML, et al. Overexpression of M68/DcR3 in human gastrointestinal tract tumors independent of gene amplification and its location in a four-gene cluster. Proc Natl Acad Sci USA. 2000;97:1230–5. doi: 10.1073/pnas.97.3.1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu Y, Han B, Sheng H, et al. Clinical significance of detecting elevated serum DcR3/TR6/M68 in malignant tumor patients. Int J Cancer. 2003;105:724–32. doi: 10.1002/ijc.11138. [DOI] [PubMed] [Google Scholar]
- 16.Jodo S, Kobayashi S, Kayagaki N, et al. Serum levels of soluble Fas/APO-1 (CD95) and its molecular structure in patients with systemic lupus erythematosus (SLE) and other autoimmune diseases. Clin Exp Immunol. 1997;107:89–95. doi: 10.1046/j.1365-2249.1997.d01-901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tokano Y, Miyake S, Kayagaki N, et al. Soluble Fas molecule in the serum of patients with systemic lupus erythematosus. J Clin Immunol. 1996;16:261–5. doi: 10.1007/BF01541390. [DOI] [PubMed] [Google Scholar]
- 18.Nozawa K, Kayagaki N, Tokano Y, Yagita H, Okumura K, Hasimoto H. Soluble Fas (APO-1, CD95) and soluble Fas ligand in rheumatic diseases. Arthritis Rheum. 1997;40:1126–9. doi: 10.1002/art.1780400617. [DOI] [PubMed] [Google Scholar]
- 19.Otsuki T, Tomokuni A, Sakaguchi H, et al. Over-expression of the decoy receptor 3 (DcR3) gene in peripheral blood mononuclear cells (PBMC) derived from silicosis patients. Clin Exp Immunol. 2000;119:323–7. doi: 10.1046/j.1365-2249.2000.01132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wan X, Zhang J, Luo H, et al. A TNF family member LIGHT transduces costimulatory signals into human T cells. J Immunol. 2002;169:6813–21. doi: 10.4049/jimmunol.169.12.6813. [DOI] [PubMed] [Google Scholar]
- 21.Shi G, Luo H, Wan X, Salcedo TW, Zhang J, Wu J. Mouse T cells receive costimulatory signals from LIGHT, a TNF family member. Blood. 2002;100:3279–86. doi: 10.1182/blood-2002-05-1404. [DOI] [PubMed] [Google Scholar]
- 22.Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. SLE. tCoPSi. Derivation of the SLEDAI: a disease activity index for lupus patients. Arthritis Rheum. 1992;35:630–40. doi: 10.1002/art.1780350606. [DOI] [PubMed] [Google Scholar]
- 23.Hsu PN, Lin HH, Tu CF, et al. Expression of human Fas ligand on mouse beta islet cells does not induce insulitis but is insufficient to confer immune privilege for islet grafts. J Biomed Sci. 2001;8:262–9. doi: 10.1007/BF02256600. [DOI] [PubMed] [Google Scholar]
- 24.Schmitz I, Krueger A, Baumann S, Schulze-Bergkamen H, Krammer PH, Kirchhoff S. An IL-2-dependent switch between CD95 signaling pathways sensitizes primary human T cells toward CD95-mediated activation-induced cell death. J Immunol. 2003;171:2930–6. doi: 10.4049/jimmunol.171.6.2930. [DOI] [PubMed] [Google Scholar]
- 25.Fayad R, Brand MI, Stone D, Keshavarzian A, Qiao L. Apoptosis resistance in ulcerative colitis: high expression of decoy receptors by lamina propria T cells. Eur J Immunol. 2006;36:2215–22. doi: 10.1002/eji.200535477. [DOI] [PubMed] [Google Scholar]
- 26.Kim S, Fotiadu A, Kotoula V. Increased expression of soluble decoy receptor 3 in acutely inflamed intestinal epithelia. Clin Immunol. 2005;115:286–94. doi: 10.1016/j.clim.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 27.Suzuki I, Fink PJ. Maximal proliferation of cytotoxic T lymphocytes requires reverse signaling through Fas ligand. J Exp Med. 1998;187:123–8. doi: 10.1084/jem.187.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun M, Ames KT, Suzuki I, Fink PJ. The cytoplasmic domain of Fas ligand costimulates TCR signals. J Immunol. 2006;177:1481–91. doi: 10.4049/jimmunol.177.3.1481. [DOI] [PubMed] [Google Scholar]