

Abstract
The destruction of β cells by the islet infiltrating lymphocytes causes type 1 diabetes. Transgenic mice models expressing interferon (IFN)-β in β cells, in the non-obese diabetic (NOD) strain and in a diabetes-free, major histocompatibility complex-matched, homologous strain, the non-obese resistant (NOR) mice, developed accelerated type 1 diabetes after 3 weeks of age. Our aim was to determine if natural killer (NK) cells could affect the acceleration of the disease. We determined the amount of NK cells in the pancreas, spleen and lymph nodes from NOD rat insulin promoter (RIP)-IFN-β mice. Pancreatic cytokines were assessed by quantitative real-time polymerase chain reaction and protein arrays. To confirm the relevance of NK cells in the acceleration of autoimmune diabetes this subset was depleted with anti-asialo GM1 antibodies. An increase of intrapancreatic NK cells characterized the accelerated onset of diabetes both in NOD and NOR RIP-IFN-β transgenic models. Cytokines involved in NK function and migration were found to be hyperexpressed in the pancreas from accelerated diabetic mice. Interestingly, the depletion of NK cells in vivo abolished completely the acceleration of diabetes. NK cells connect innate to adaptive immunity and might play a role in autoimmunity. We report here that NK cells are required critically in the pancreas for accelerated diabetes. This model links inflammation to acceleration of β cell-specific autoimmunity mediated by NK cells.
Keywords: autoimmunity, diabetes, IFN-β, NK cells
Introduction
Type 1 diabetes (T1D) is an autoimmune disease caused by the destruction of the β cells [1]. The detection of type I interferon (IFN), a cytokine of natural immunity secreted typically by virally infected cells, in the pancreas of diabetic patients [2, 3] supports the importance of viruses as environmental factors in T1D. Transgenic mice expressing type I IFN in the β cells develop diabetes [4–6], suggesting mechanisms by which viral infection may trigger autoimmunity to the islets. In this context, a role for innate immunity in T1D has been proposed, and in particular for natural killer (NK) cells [7].
Natural killer cells are a form of cytotoxic lymphocyte, which constitute a major component of the innate immune system and that mediate defence against virally infected or malignant cells [8] through activating and inhibitory membrane receptors [9, 10]. Although some differences exist between human and mouse NK cell receptors [11], their main functions seem to be conserved [12]. Cytokines promote NK cell function [13], constituting a crucial step in innate and adaptive responses. In experimental T1D the lack of NK cells was associated with protective insulitis [14] and the depletion of NK cells prevented diabetes development. In humans, several alterations have been found in NK cells of diabetic patients [15].
We generated two transgenic mice models expressing IFN-β under the control of the insulin promoter, one in the non-obese diabetic (NOD) strain [NOD rat insulin promoter (RIP)-IFN-β] and the other in a diabetes-free, major histocompatibility complex (MHC)-matched, homologous strain, the non-obese resistant (NOR) mice (NOR RIP-IFN-β) [6]. Both strains develop accelerated autoimmune diabetes at 3 weeks of age. In the course of the characterization of insulitis in these models, we found a striking feature of the accelerated onset of T1D: the presence of abundant DX5/CD49b+ CD3–, basically NK cells [16]. To determine the role of these cells, anti-GM1 antibodies − a reagent used classically to deplete NK cells [17] − were administered to transgenic mice, preventing the acceleration of the disease. This finding supports the chain of events that presumably link inflammatory events to autoimmunity in the islets of predisposed mice.
Materials and methods
Mice
We used NOD and NOR transgenic mice expressing IFN-β in the β cells [6] and non-transgenic littermates as controls. Mice were kept under specific pathogen-free conditions and monitored daily for diabetes assessment, as described previously [6]. Our government's guidelines for the use and care of laboratory animals (Generalitat de Catalunya, Catalonia, Spain) were followed and the protocols were approved by our Institutional Animal Care and Use Committee.
Insulitis development
The degree of islet infiltration (insulitis) was determined as described previously [6]. Pancreases from six animals of each group were snap-frozen in an isopentane/cold acetone bath. Cryosections of 5 μm were obtained at five non-overlapping levels. The sections were stained with haematoylin and eosin (H&E) and analysed at 3, 4, 6, 9 and 12 weeks of age examining 40–100 islets per animal.
Immunohistological analysis
Consecutive pancreatic cryostat sections (5 μm) from healthy and accelerated diabetic NOD RIP-IFN-β mice were incubated sequentially with (i) goat polyclonal antibody to mouse integrin α2 (or CD49b) to NK cells (Santa Cruz Biotechnology, Santa Cruz, CA, USA); (ii) Alexa488-labelled rabbit anti-goat immunoglobulins (SBA, Birmingham, AL, USA); (iii) guinea pig anti-insulin (ICN, Eschwege, Germany); and (iv) tetramethyl rhodamine isothiocyanate-labelled goat anti-guinea pig (ICN). The preparations were analysed with a UV microscope and an image analyser (OpenLab 2·0; Improvision, Coventry, UK).
Flow cytometric analysis
Intrapancreatic lymphocytes from transgenic mice NOD RIP-IFN-β and NOR RIP-IFN-β, healthy and accelerated diabetic (3 weeks of age) and from non-transgenic littermates, healthy and late diabetic (12 weeks of age), were obtained after enzymatic digestion with collagenase, mechanical disruption and purification by discontinuous density gradient using Ficoll. Lymphocytes from spleen and pancreatic lymph nodes were isolated by mechanical disruption. Between six and 12 animals per group were analysed. Different lymphocyte subsets were detected by direct immunofluorescence staining and flow cytometry analysis with phycoerythrin (PE)-Cy5 anti-mouse CD45 (Caltag Laboratories, Burlingame, CA, USA), fluorescein isothiocyanate (FITC) anti-mouse CD4, PE anti-mouse CD8, PE anti-mouse CD19, PE anti-mouse DX5/CD49b+ and FITC anti-mouse CD3 (Pharmingen, San Diego, CA, USA). Unstained cells and cells stained with an irrelevant isotype-matched PE/FITC were used as controls. Subsets were determined by flow cytometric analysis in a fluorescence activated cell sorter (FACScan) Cell Analyser (BD, San Jose, CA, USA). Data were analysed using CellQuest software (BD).
Cytokine profile: low density protein arrays
Protein extracts were obtained from 5 μm pancreatic cryosections using the protein extraction reagent supplied in RayBioTM mouse cytokine antibody array (RayBiotech, Atlanta, GA, USA) and protease inhibitors cocktail (Calbiochem, Darmstadt, Germany). Pools of pancreatic proteins (500 μg) obtained from five mice (male), late diabetic NOD (12–14 weeks of age) and accelerated diabetic NOD RIP-IFN-β (3 weeks of age) were incubated on mouse cytokine antibody array membranes (RayBiotech). The assay was performed in triplicate using different pools of proteins. The reaction was revealed by chemiluminiscence using Hyperfilm ECL (Amersham Biosciences) in a Kodak M35X-OMAT processor (Kodak, Hemel Hewpstead, Herts, UK). The amount of cytokine was quantified by densitometry (QuantityOne 1-D analysis software; Bio-Rad, Hercules, CA, USA). The expression index was calculated normalizing the data to positive controls after background subtraction.
Laser capture microdissection and gene expression analysis
Pancreatic cryostat sections (6 μm) stained with H&E were microdissected using laser capture microdissection (LCM) (P.A.L.M® MicroBeam, P.A.L.M® Microlaser Technologies, Bernried, Germany) to obtain the endocrine tissue. The experiment was performed in triplicate in late diabetic NOD mice (12–14 weeks of age) and accelerated diabetic NOD RIP-IFN-β mice (3 weeks of age, three to five male mice per group). RNA (RNeasy Micro Kit; Qiagen, Hilden, Germany) was reverse-transcribed using random hexanucleotide primers (20 ng/μl; Promega Corp., Madrid, Spain) and SuperScript II reverse transcriptase (4 U/μl; Gibco brl, Invitrogen, Carlsbad, CA, USA). The primers used for real-time reverse transcription-polymerase chain reaction (RT-PCR) were Taqman® and probe (Assays on Demand; Applied Biosystems, Foster City, CA, USA) for chemokine (C-C motif) ligand 5 (CCL5) and CXCL10. IFN-γ primers (sense 5′-AGCGGCTGACTGAACTCAGATTGTAG-3′ and anti-sense 5′-GTCACAGTTTTCAGCTGTATAGGG-3′) were obtained from T MolBiol (Berlin, Germany). The reaction was performed using the ABI 7000 System thermocycler (Applied Biosystems). Relative values were determined by normalizing the expression for each gene of interest to the housekeeping gene 18S rRNA (sense 5′-CGGCTACCACATCCAAGGAA-3′ and anti-sense 5′-GCTGGAATTACCGCGGCT-3′) and a calibrator sample (microdissected islets from healthy NOD mice) following the 2–ΔΔCt method [18].
In vivo administration of anti-asialo GM1 polyclonal antibody
Because the NK cells from NOD mice do not express the NK1·1 epitope (CD161), the usual protocols for depletion of NK1·1+ cells could not be followed in our NOD strain. We used an anti-asialo GM1 polyclonal antibody (Cedarlane, Hornby, Ontario, Canada), a reagent used classically [17] and currently [19] to deplete NK cells. The antibody was injected intraperitoneally (300 μg in 40 μl of distilled water) in NOD RIP-IFN-β mice (n = 6; three females and three males) at 10, 14 and 18 days after birth. As control, NOD RIP-IFN-β mice (n = 7; four females and three males) were injected with 300 μg of normal rabbit serum (NRS; Calbiochem). In addition, anti-asialo GM1 anti-serum was injected in NOD wild-type mice (three females). Mice were monitored daily for glycosuria during 30 weeks.
Statistical analysis
Statistical analysis was performed to compare independent groups using the t-test when the data had a normal distribution and equal variance tests. The Mann–Whitney U-test was performed for NK/B correlation. For flow cytometry data Bonferroni's multiple comparison test was used. The log-rank test was used to compare the incidence of diabetes in treated mice and controls. Differences were considered significant when a value of P < 0·05 was reached.
Results
A transient increase of NK infiltrating the pancreas characterizes accelerated T1D in RIP-IFN-β NOD and RIP-IFN-β NOR mice
The insulitis score of the NOR RIP-IFN-β and NOD RIP-IFN-β mice at 3, 4, 6, 9 and 12 weeks of age was significantly higher (P < 0·01) than that of their non-transgenic littermates at the same age (Fig. 1a). The insulitis score of NOD RIP-IFN-β at 3 weeks of age was higher than at 4 weeks of age because this group included subjects deemed to develop T1D. Most of the islets from healthy NOD RIP-IFN-β and healthy NOR RIP-IFN-β mice were infiltrated equally, showing a similar pattern to that observed in accelerated diabetic mice. The percentage of islets with different degrees of insulitis indicated a higher insulitis in the islets from transgenic mice when compared with non-transgenic littermates of the same age (Fig. 1b). Almost half the islets from healthy transgenic animals showed moderate or severe insulitis.
Fig. 1.
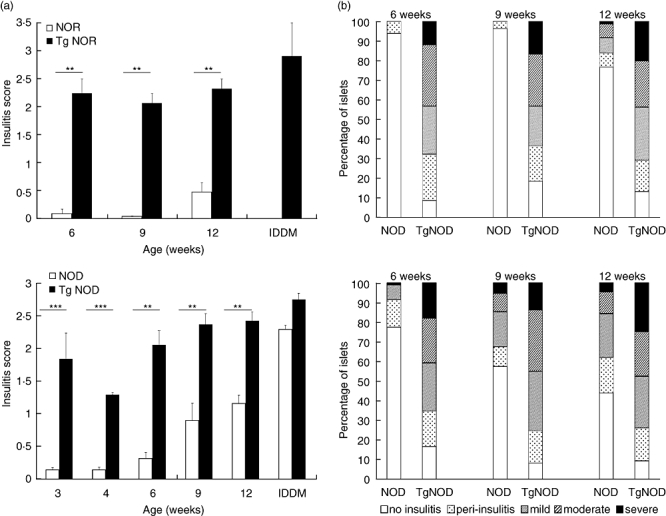
Lymphocytic infiltration in the islets from transgenic (Tg) mice expressing interferon (IFN)-β in the insulin producing cells is higher than that of their non-transgenic littermates. Pancreases from six animals of each group and age were analysed. (a) Insulitis score in non-obese resistant (NOR) (up) and non-obese diabetic (NOD) (down) strains: NOR and NOR rat insulin promoter (RIP)-IFN-β mice (Tg NOR) showed significant differences in insulitis score at 6, 9 and 12 weeks of age and at the onset of accelerated diabetes (**P < 0·01); NOD and NOD RIP-IFN-β mice (Tg NOD) showed significant differences in insulitis score at 3, 4, 6, 9 and 12 weeks of age (**P < 0·01, ***P < 0·005), but not at the onset of accelerated diabetes. Black bars correspond to transgenic mice and white bars to wild-type mice. Data are expressed as mean ± standard error. (b) Percentage of islets classified in each of the five insulitis score categories depending on severity of insulitis (white, no insulitis; dark grey, peri-insulitis; medium grey, mild; light grey, moderate; black, severe) in NOR (up) and NOD (down) strains at 6, 9 and 12 weeks of age.
A relative quantification of lymphocytes (T CD4+, T CD8+ and B) and NK (DX5/CD49b+ CD3–) cells was performed in the intrapancreatic lymphocytes, spleen and pancreatic lymph nodes of the same animal (Table 1). We found a significant increase of DX5/CD49b+ CD3– cells in the pancreas of the accelerated diabetic NOD RIP-IFN-β mice at the onset of diabetes (18 ± 1·9, percentage ± standard deviation) when compared with healthy transgenic ones (3·4 ± 0·4). The same results were found in accelerated diabetic NOR RIP-IFN-β mice when compared with healthy subjects (18 ± 2 versus 5·1 ± 1·1) (Fig. 2a). It was remarkable that this DX5/CD49b+ CD3– cell population was not increased at the late onset of diabetes in non-transgenic NOD mice (3·6 ± 0·4) when compared with healthy non-transgenic NOD mice (3·9 ± 0·9). This was a feature of the accelerated T1D and not of late diabetes at 12 weeks of age (5·4 ± 1·5). Interestingly, this fact was observed during only a very narrow time window: as soon as 24–48 h after the accelerated onset the percentage of NK cells decreased to levels similar to those of healthy mice (4·7 ± 1·3; data from six mice). This increase in NK cells was not observed in the spleen or in the pancreatic lymph nodes from mice with accelerated diabetes when compared with controls. Immunohistology of pancreases of healthy and accelerated diabetic transgenic mice showed the NK cells in the periphery of the islets (Fig. 2b): healthy mice displayed a few NK cells around the islets, whereas accelerated diabetic mice showed an accumulation of NK cells around the remaining β cells in the islets. Thus, a transient increase of NK cells in the islets is a feature of accelerated diabetes in these mice.
Table 1.
Percentage of lymphocyte subsets in the pancreas and pancreatic lymph nodes from non-obese diabetic (NOD) and non-obese resistant (NOR) mice, wild-type (Wt) and transgenic (Tg) expressing interferon-β in the β cells (mean ± standard error).
| Pancreas | Pancreatic lymph node | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Strain | Tissue | CD4 | CD8 | B | NK | CD4 | CD8 | B | NK |
| NOD mice | Wt/healthy | 20·1 ± 4·1 | 6·3 ± 1·4 | 54·6 ± 6·4 | 3·9 ± 1·0 | 54·2 ± 1·6 | 24·4 ± 2·4 | 18·4 ± 1·3 | 0·3 ± 0·1 |
| Wt/late diabetic | 21·0 ± 1·8 | 8·6 ± 0·7 | 44·7 ± 3·0 | 3·6 ± 0·4 | 51·4 ± 3·8 | 21·4 ± 2·0 | 24·4 ± 6·2 | 0·4 ± 0·1 | |
| Tg/healthy | 21·9 ± 1·0 | 6·5 ± 0·8 | 53·4 ± 1·7 | 3·4 ± 0·4 | 52·9 ± 1·6 | 23·4 ± 0·8 | 20·0 ± 1·9 | 0·7 ± 0·2 | |
| Tg/accel. diabetic | 22·7 ± 1·2 | 7·0 ± 1·0 | 17·0 ± 2·1 | 18·0 ± 1·9 | 56·8 ± 1·2 | 21·1 ± 0·6 | 18·9 ± 0·9 | 0·3 ± 0·0 | |
| Tg/late diabetic | 14·6 ± 3·7 | 6·0 ± 0·8 | 56·9 ± 9·9 | 5·4 ± 1·5 | 53·7 ± 3·1 | 23·3 ± 3·4 | 20·2 ± 5·6 | 0·2 ± 0·4 | |
| NOR mice | Wt/healthy | – | – | – | – | 52·0 ± 3·1 | 19·0 ± 1·3 | 24·3 ± 3·4 | 0·6 ± 0·1 |
| Tg/healthy | 24·3 ± 3·0 | 7·3 ± 1·1 | 39·7 ± 3·7 | 5·1 ± 1·1 | 46·2 ± 2·3 | 24·6 ± 0·9 | 26·0 ± 2·7 | 0·6 ± 0·1 | |
| Tg/accel. diabetic | 18·8 ± 1·1 | 8·7 ± 1·6 | 25·3 ± 5·2 | 18·0 ± 2·0 | 50·7 ± 2·3 | 19·9 ± 0·7 | 27·6 ± 2·5 | 0·6 ± 0·1 | |
At least six animals per group were analysed. NK, natural killer.
Fig. 2.
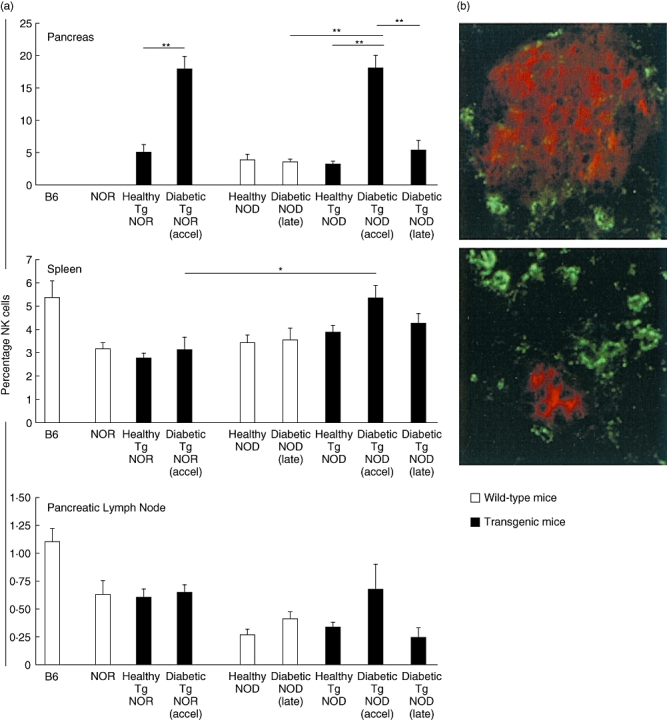
(a) Natural killer (NK) cells are increased in the pancreases from non-obese resistant rat insulin promoter interferon-β (NOR RIP-IFN-β) [transgenic (Tg) NOR] and non-obese diabetic (NOD) RIP-IFN-β (Tg NOD) mice at the onset of accelerated diabetes. Histogram of NK cells percentages in the pancreas (upper), spleen (middle) and pancreatic lymph nodes (lower) determined by fluorescence activated cell sorter (FACS) analysis in control mice B6, NOR and NOD strains, and Tg NOR and Tg NOD in different conditions of health, accelerated diabetes or late diabetes (groups of six to 12 mice per condition). Data are expressed as mean ± standard error of the percentages. *P < 0·05 and **P < 0·01. Black bars correspond to Tg mice and white bars to wild-type mice. (b) Double immunofluorescence staining of 5 μm pancreatic cryostat sections, insulin (red) and CD49b to NK cells (green) in healthy NOD RIP-IFN-β mice (upper) and in diabetic NOD RIP-IFN-β at the accelerated onset of diabetes (lower). Original magnification 400×.
The percentage of B cells in the islets correlates inversely with accelerated onset of T1D in transgenic mice
The percentage of intrapancreatic T CD4+ and T CD8+ cells did not show significant differences when comparing subjects with accelerated disease to healthy mice (Table 1). However, the proportion of B cells (CD19+) showed a marked decrease in NOD RIP-IFN-β mice at the onset of accelerated diabetes when compared with healthy mice (17 ± 2·1 versus 53·4 ± 1·7, percentage ± standard error, P < 0·01). The B cell reduction occurred within the same time window as the increase of NK cells. Moreover, 24–48 h after the onset of T1D, the percentage of B cells was up to levels of the healthy mice (41·2 ± 5·6, data from six mice), whereas the percentage of T lymphocytes CD4+ (21·1 ± 1·5) and CD8+ (7·8 ± 1·1) was not altered. This reduction of B cells was not present in NOD RIP-IFN-β mice with late onset diabetes (56·9 ± 9·9) or in healthy NOD (54·6 ± 6·4) or late diabetic NOD (44·7 ± 3·0). The index (– log %NK/%B) increases significantly in accelerated diabetes when compared with healthy transgenic mice (P < 0·01), late diabetic transgenic mice (P < 0·05) or late diabetic NOD mice (P < 0·001) (Fig. 3). Thus, accelerated diabetes correlates with the percentage of NK cells and correlates inversely with the percentage of B cells in the islets.
Fig. 3.
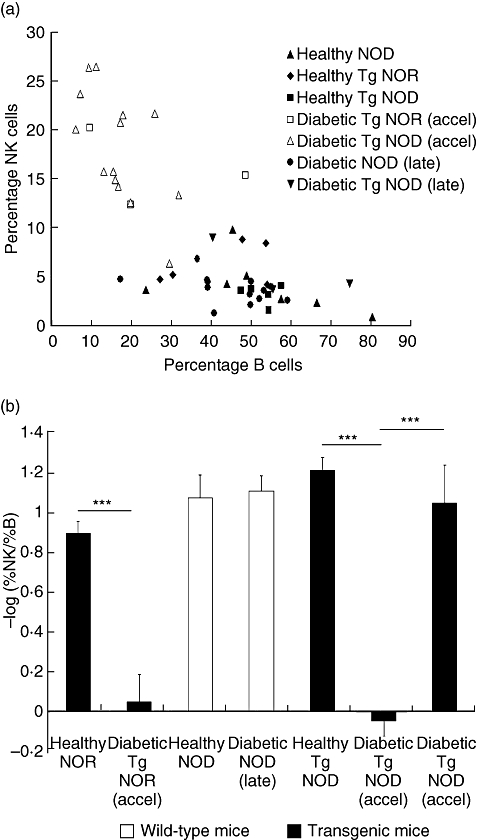
Inverse correlation between natural killer (NK) cell percentage and B cell percentage in the pancreatic infiltrates at the onset of accelerated diabetes. (a) Representation of NK cell percentage and B cell percentage in the pancreatic infiltrates: each point corresponds to an animal. White symbols correspond to subjects with accelerated diabetes; squares: non-obese resistant rat insulin promoter interferon-β (NOR RIP-IFN-β) (Tg NOR), triangle, non-obese diabetic (NOD) RIP-IFN-β (Tg NOD); black symbols: subjects with late onset of the disease [circle: NOD; inverted triangle: transgenic (Tg) NOD] and healthy mice (triangle: NOD; rhomb: Tg NOR; square: Tg NOD). (b) Histogram representing a normalized index of the quotient of the percentage of NK cells and B cells in the pancreas (–log %NK/%B) in different strains and conditions (mean ± standard error). Black bars correspond to transgenic mice and white bars to wild-type mice. Note that accelerated diabetes showed significant differences (***P < 0001) in the index (–log %NK/%B) when compared with healthy mice and diabetic mice with late onset of the disease. Determinations were performed in groups of at least three mice per condition.
Increased expression of cytokines involved in NK function and migration in the pancreas at accelerated T1D
To determine the amount of cytokines involved in NK function and migration in the islet microenvironment, the amounts of CCL5, interleukin (IL)-12, IFN-γ and CCL3 were determined using protein arrays. We found a significant increase of CCL5 (fold change 4·8) and IL-12 (fold change 2·03) in accelerated T1D transgenic mice when compared with late diabetic wild-type NOD mice (Fig. 4a). IFN-γ was 2·4-fold increased in pancreases from accelerated diabetic NOD transgenic mice when compared with late diabetic wild-type NOD mice, although the differences were not statistically significant. In addition, CCL3 also showed a tendency to increase in accelerated diabetes (not significant, data not shown). The NK attractant chemokines CXCL10, CCL5 and IFN-γ were assessed using LCM and real-time RT-PCR. Accelerated diabetic transgenic animals showed a significantly higher expression of CXCL10 (P < 0·01) (fold change 6·7) and CCL5 (P < 0·05) (fold change 3·2) in the islets when compared with late wild-type diabetic NOD mice (Fig. 4b). IFN-γ was increased moderately in accelerated diabetes when compared with late diabetes onset, although the differences were not statistically significant. Thus, IL-12, CCL5 and CXCL10 were found to be increased in the pancreas in accelerated T1D.
Fig. 4.
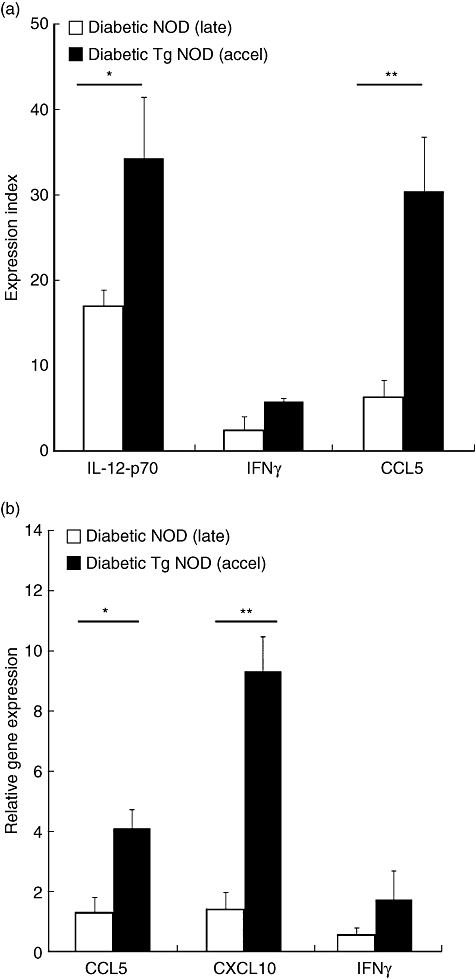
Increased expression of cytokines and chemokines in accelerated diabetes. Black bars correspond to accelerated diabetic non-obese diabetic rat insulin promoter interferon-β (NOD RIP-IFN-β) [transgenic (Tg) NOD] mice and white bars to late diabetic NOD wild-type mice. (a) Expression index, calculated normalizing the data to positive controls after background subtraction, of interleukin (IL)-12, IFN-γ and chemokine (C-C motif) ligand 5 (CCL5) protein in total pancreas from late diabetic NOD mice and accelerated diabetic NOD RIP-IFN-β (Tg NOD), determined by protein arrays. Data are expressed as mean ± standard error (s.e.) from three arrays with three different pools of proteins obtained from five animals per condition. The amounts of IL-12 (*P < 0·05) and CCL5 (**P < 0·01) were increased significantly in accelerated diabetic transgenic mice when compared with late diabetic wild-type NOD mice. IFN-γ was 2·4-fold increased in pancreases from accelerated diabetic transgenic mice when compared with late diabetic wild-type NOD mice, although differences were not statistically significant. (b) Real-time reverse transcription–polymerase chain reaction analysis for CCL5, CXCL10 and IFN-γ in microdissected islets from late diabetic NOD mice and accelerated diabetic NOD RIP-IFN-β (Tg NOD) (three mice per condition). Data are expressed as mean ± s.e. Tg NOD showed higher CCL5 (*P < 0·05) and CXCL10 (**P < 0·01) expression than NOD mice at the onset of T1D. Results were normalized to a housekeeping gene (Rn18s) and a calibrator sample (microdissected islets from healthy NOD mice) following the 2–ΔΔCt method.
In vivo administration of anti-asialo GM1 antibody inhibits accelerated diabetes
To confirm the relevance of NK cells in T1D we administered an anti-asialo GM1 antibody to NOD RIP-IFN-β mice before the onset of the disease. The depletion of NK cells abolished accelerated diabetes completely (P < 0·05) (Fig. 5). Furthermore, animals injected with NRS developed accelerated diabetes after 3 weeks of age as expected, reaching an incidence of T1D of 57% at 5 weeks of age, fitting well with the current T1D incidence in this strain. We tested this antibody in wild-type NOD mice to rule out any effect of anti-asialo GM1 in the adult onset of the disease but late diabetes onset was not altered (the three NOD mice developed diabetes), as described previously [15, 20]). Transgenic mice treated with antibodies developed late diabetes after 20 weeks of age with a similar incidence to non-NK-depleted mice: at week 30, four of six (67%) transgenic mice treated with anti-asialo GM1 were diabetic, while six of seven (86%) transgenic mice treated with NRS developed the disease. Flow cytometric analysis of intrapancreatic lymphocytes from transgenic mice treated with antibodies that developed late diabetes after 20 weeks of age showed a 4·1% of NK cells (mean of two mice), similar to the percentage of pancreatic NK cells in healthy or late diabetic mice. The pancreases of transgenic mice in which anti-asialo GM1 prevented accelerated diabetes were examined by histology at 12 weeks of age and showed no signs of destructive or severe insulitis, nor DX5/CD49b+ cells.
Fig. 5.
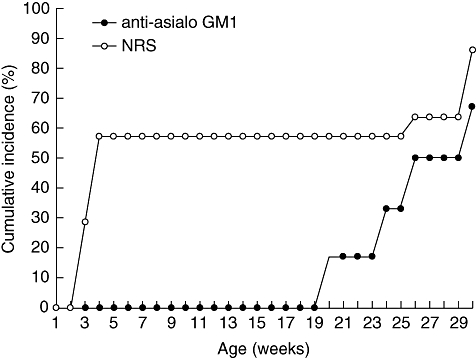
Natural killer (NK) cell depletion inhibits accelerated diabetes. Cumulative incidence of accelerated diabetes (percentage) in non-obese diabetic resistant rat insulin promoter interferon-β (NOD RIP-IFN-β) treated with anti-asialo GM1 rabbit polyclonal antibody (black circles, n = 6) and in control group treated with normal rabbit serum (white circles, n = 7).
Discussion
We report here an important increase in the population of intrapancreatic NK cells (DX5/CD49b+ CD3–) at the accelerated onset of T1D driven by IFN-β. One of the effects of IFN-β is to induce NK cell proliferation and activation, as such NK cells subsequently release inflammatory cytokines, predominantly IFN-γ[21]. The role of NK cells in accelerating T1D was demonstrated by the inactivation of these cells with anti-asialo GM1. The increased expression of NK-attractant chemokines, CCL5 and CXCL10, and cytokines involved in NK function, IL-12 and IFNγ, in the target tissue of accelerated diabetes correlates with this NK subset increase.
Natural killer cells are essential for accelerated diabetes, and their degree of involvement in insulitis composition determines the bimodal distribution of age of onset. In humans, there are also two peaks in the distribution of the age onset of T1D, the first in early childhood and the second at puberty. The clinical onset at very early age (before 5 years of age) may have different autoimmune characteristics from those diagnosed at a later age [22]. The accelerated and aggressive form of diabetes should be viewed as a different entity showing symptoms of a viral illness at diagnosis [23]. NK cells can inhibit or promote the activation of autoreactive T cells during the initiation of autoimmunity [24]. In T1D, the amounts of pancreatic NK cells distinguish innocuous and destructive forms of insulitis [14], whereas B cells seem to be a feature of benign insulitis [25]. By contrast, NK cells mediate the protective effects of complete Freund's adjuvant in the NOD mice [20]. The role of NK cells in accelerated T1D remains to be determined. One possibility is the killing of β cells by NK cells, but NOD mice have a defect in NK cell-activating receptor, NKG2D [26]. Moreover, the previously described increased expression of MHC class I molecules in the islets [6] should protect them against NK cells. The impaired NK cell response described in NOD mice might be restored by local inflammation [27], so our model could reinstate its NK-activation defect by IFN-β action. Because NOD–SCID mice − lacking T and B lymphocytes but not NK cells − expressing IFN-β in the islets did not become diabetic [6], NK cells are not enough to cause accelerated diabetes. The effect of NK cells in accelerating T1D may be due to further polarization towards a T helper type 1 (Th1) response, enhancing the cytotoxic capacity of CD8+ T cells [28], an essential subset for disease progression [29]. Another mechanism could be the activation of antigen-presenting cells by NK cells and the activation of autoreactive T cells [30]. An islet inflammation mediated mainly by NK cells has been reported recently in human T1D [31]. Type I IFN also recruits NK cells to tumours and its depletion by anti-asialo GM1 antibody abolishes the NK anti-metastatic effect [19].
Natural killer cells migrate quickly from the pancreas after the clinical onset of the disease when some β cells still remain. This dramatic change in the insulitis composition is intriguing, and it would be interesting to determine the percentage of NK cells just before the accelerated clinical onset of diabetes. This poses a difficult problem, as we cannot predict which mice will develop T1D. We suspect that in this aggressive form of autoimmune diabetes NK cell recruitment to the islets must be an acute phenomenon, in the same way that they leave the islets soon after the onset of the disease. The percentage of B cells in the islets correlates inversely with accelerated onset of T1D. This apparently contradicts the evidence that B cells are essential for the progression of diabetes in NOD mice [32]. However, B cells can perform regulatory functions by activating NK T cells, secreting anti-inflammatory cytokines and depleting cytotoxic T cells. In fact, in NOD mice, B cells down-regulate Th1 response and prevent diabetes [25] and are increased in ‘benign’ insulitis [14]. In the proinflammatory microenvironment of NOD RIP-IFN-β mice, B cells could have regulatory functions, in an attempt to protect the target tissue. However, the increase in pancreatic NK cells does not compensate fully for the percentage of decrease in B cells, and the remaining infiltrating cells could be, in part, DX5/CD49b+ CD3+ cells increased slightly at the accelerated diabetes (data not shown).
Natural killer attractant chemokines are increased in the pancreas at accelerated diabetes. CXCL10 contributes to the recruitment of NK and T cells [33, 34] and to the development of T1D [35, 36], and CCL5 is produced during viral infection in the pancreas [35]. These chemokines also recruit NK cells to the skin in psoriasis [37]. IL-12 links innate to adaptive immunity and induces IFN-γ production by T and NK cells. IFN-β shows direct Th1-inducing activity by increasing the expression of IL-12Rβ2 signalling component [38], thus enhancing the IL-12 effect in NK cells.
Anti-asialo GM1, an antibody used in many studies to deplete NK cells [17, 25], completely abrogates accelerated diabetes. This antibody could also deplete a small population of T cells and macrophages that express GM1, besides depleting the majority of NK cells. However, there is no preventive effect in NOD mice lacking NK cells in the insulitis, supporting the role of NK cells in accelerating the onset of T1D. This discrepancy reflects intrinsic differences in the immune response between accelerated and late T1D in these experimental models. The mechanism behind this effect of age at onset of diabetes is unclear and may reflect different mechanisms of autoimmunity due probably to triggering environmental factors (i.e. changes in the diet of the pups) or to the maturation of the immune system.
Our results suggest a possible mechanism of accelerated β cell destruction through IFN-β driven by NK cells, triggered by a viral infection or acute inflammation. Additional studies of the role of NK cells in accelerated T1D could help to understand its function in autoimmunity.
Acknowledgments
The authors wish to thank Dr R. Weisser, Dr A. Steinle and S. Poschel (University Clinic of Tübingen) for technical support. We are grateful to Dr M. Tarón, M. Pérez-Cano and P. Méndez (ICO) for their help in LCM and Dr J. Roca (Epidemiology Department) for statistical assistance (Hospital Germans Trias i Pujol). Special thanks go to Professor M. López-Botet (Universitat Pompeu Fabra, Barcelona) for scientific advices. Our work in this field was supported by the Fondo de Investigaciones Sanitarias (Spanish Ministry of Health, Projects 02/0107, 04/1686 and 06/0465). A. A. and R. P. were supported by the Instituto de Salud Carlos III (BEFI 01/1810 and FI05/00418). M. V. P. is supported by the Fondo de Investigaciones Sanitarias and the Health Departament, Generalitat de Catalunya.
References
- 1.Bach JF, Chatenoud L. Tolerance to islet autoantigens in type 1 diabetes. Annu Rev Immunol. 2001;19:131–61. doi: 10.1146/annurev.immunol.19.1.131. [DOI] [PubMed] [Google Scholar]
- 2.Foulis AK, Farquharson MA, Meager A. Immunoreactive alpha-interferon in insulin-secreting beta cells in type 1 diabetes mellitus. Lancet. 1987;2:1423–7. doi: 10.1016/s0140-6736(87)91128-7. [DOI] [PubMed] [Google Scholar]
- 3.Huang X, Yuang J, Goddard A, et al. Interferon expression in the pancreases of patients with type I diabetes. Diabetes. 1995;44:658–64. doi: 10.2337/diab.44.6.658. [DOI] [PubMed] [Google Scholar]
- 4.Stewart TA, Hultgren B, Huang X, Pitts-Meek S, Hully J, MacLachlan NJ. Induction of type I diabetes by interferon-alpha in transgenic mice. Science. 1993;260:1942–6. doi: 10.1126/science.8100367. [DOI] [PubMed] [Google Scholar]
- 5.Vassileva G, Chen SC, Zeng M, et al. Expression of a novel murine type I IFN in the pancreatic islets induces diabetes in mice. J Immunol. 2003;170:5748–55. doi: 10.4049/jimmunol.170.11.5748. [DOI] [PubMed] [Google Scholar]
- 6.Alba A, Puertas MC, Carrillo J, et al. IFN beta accelerates autoimmune type 1 diabetes in nonobese diabetic mice and breaks the tolerance to beta cells in nondiabetes-prone mice. J Immunol. 2004;173:6667–75. doi: 10.4049/jimmunol.173.11.6667. [DOI] [PubMed] [Google Scholar]
- 7.Bach JF, Bendelac A, Brenner MB, et al. The role of innate immunity in autoimmunity. J Exp Med. 2004;200:1527–31. doi: 10.1084/jem.20042110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moretta A, Bottino C, Mingari MC, Biassoni R, Moretta L. What is a natural killer cell? Nat Immunol. 2002;3:6–8. doi: 10.1038/ni0102-6. [DOI] [PubMed] [Google Scholar]
- 9.Kubota A, Kubota S, Lohwasser S, Mager DL, Takei F. Diversity of NK cell receptor repertoire in adult and neonatal mice. J Immunol. 1999;163:212–16. [PubMed] [Google Scholar]
- 10.Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–74. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 11.Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol. 2004;172:2731–8. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 12.Colucci F, Di Santo JP, Leibson PJ. Natural killer cell activation in mice and men: different triggers for similar weapons? Nat Immunol. 2002;3:807–13. doi: 10.1038/ni0902-807. [DOI] [PubMed] [Google Scholar]
- 13.Giancarlo B, Silvano S, Albert Z, Mantovani A, Allavena P. Migratory response of human natural killer cells to lymphotactin. Eur J Immunol. 1996;26:3238–41. doi: 10.1002/eji.1830261260. [DOI] [PubMed] [Google Scholar]
- 14.Poirot L, Benoist C, Mathis D. Natural killer cells distinguish innocuous and destructive forms of pancreatic islet autoimmunity. Proc Natl Acad Sci USA. 2004;101:8102–7. doi: 10.1073/pnas.0402065101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodacki M, Svoren B, Butty V, et al. Altered natural killer cells in type 1 diabetic patients. Diabetes. 2007;56:177–85. doi: 10.2337/db06-0493. [DOI] [PubMed] [Google Scholar]
- 16.Sawaki J, Tsutsui H, Hayashi N, et al. Type 1 cytokine/chemokine production by mouse NK cells following activation of their TLR/MyD88-mediated pathways. Int Immunol. 2007;19:311–20. doi: 10.1093/intimm/dxl148. [DOI] [PubMed] [Google Scholar]
- 17.Habu S, Fukui H, Shimamura K, et al. In vivo effects of anti-asialo GM1. I. Reduction of NK activity and enhancement of transplanted tumor growth in nude mice. J Immunol. 1981;127:34–8. [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Takehara T, Uemura A, Tatsumi T, et al. Natural killer cell-mediated ablation of metastatic liver tumors by hydrodynamic injection of IFN alpha gene to mice. Int J Cancer. 2007;120:1252–6. doi: 10.1002/ijc.22152. [DOI] [PubMed] [Google Scholar]
- 20.Lee IF, Qin H, Trudeau J, Dutz J, Tan R. Regulation of autoimmune diabetes by complete Freund's adjuvant is mediated by NK cells. J Immunol. 2004;172:937–42. doi: 10.4049/jimmunol.172.2.937. [DOI] [PubMed] [Google Scholar]
- 21.Yokoyama WM, Kim S, French AR. The dynamic life of natural killer cells. Annu Rev Immunol. 2004;22:405–29. doi: 10.1146/annurev.immunol.22.012703.104711. [DOI] [PubMed] [Google Scholar]
- 22.Komulainen J, Kulmala P, Savola K, et al. Clinical, autoimmune, and genetic characteristics of very young children with type 1 diabetes. Diabetes Care. 1999;22:1950–5. doi: 10.2337/diacare.22.12.1950. Childhood Diabetes in Finland (DiMe) Study Group. [DOI] [PubMed] [Google Scholar]
- 23.Hathout EH, Hartwick N, Fagoaga OR, et al. Clinical, autoimmune, and HLA characteristics of children diagnosed with type 1 diabetes before 5 years of age. Pediatrics. 2003;111:860–3. doi: 10.1542/peds.111.4.860. [DOI] [PubMed] [Google Scholar]
- 24.Shi FD, Van Kaer L. Reciprocal regulation between natural killer cells and autoreactive T cells. Nat Rev Immunol. 2006;6:751–60. doi: 10.1038/nri1935. [DOI] [PubMed] [Google Scholar]
- 25.Tian J, Zekzer D, Hanssen L, Lu Y, Olcott A, Kaufman DL. Lipopolysaccharide-activated B cells down-regulate Th1 immunity and prevent autoimmune diabetes in nonobese diabetic mice. J Immunol. 2001;167:1081–9. doi: 10.4049/jimmunol.167.2.1081. [DOI] [PubMed] [Google Scholar]
- 26.Ogasawara K, Hamerman JA, Hsin H, et al. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity. 2003;18:51. doi: 10.1016/s1074-7613(02)00505-8. [DOI] [PubMed] [Google Scholar]
- 27.Johansson SE, Hall H, Bjorklund J, Hoglund P. Broadly impaired NK cell function in non-obese diabetic mice is partially restored by NK cell activation in vivo and by IL-12/IL-18 in vitro. Int Immunol. 2004;16:1–11. doi: 10.1093/intimm/dxh002. [DOI] [PubMed] [Google Scholar]
- 28.Vankayalapati R, Klucar P, Wizel B, et al. NK cells regulate CD8+ T cell effector function in response to an intracellular pathogen. J Immunol. 2004;172:130–7. doi: 10.4049/jimmunol.172.1.130. [DOI] [PubMed] [Google Scholar]
- 29.Ogasawara K, Hamerman JA, Ehrlich LR, et al. NKG2D blockade prevents autoimmune diabetes in NOD mice. Immunity. 2004;20:757–67. doi: 10.1016/j.immuni.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 30.Hanna J, Gonen-Gross T, Fitchett J, et al. Novel APC-like properties of human NK cells directly regulate T cell activation. J Clin Invest. 2004;114:1612–23. doi: 10.1172/JCI22787. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Dotta F, Censini S, van Halteren AGS, et al. Coxsackie B4 virus infection of beta cells and natural killer cell Insulitis in recent-onset type 1 diabetic patients. Proc Natl Acad Sci USA. 2007;104:5115–20. doi: 10.1073/pnas.0700442104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Serreze DV, Silveira PA. The role of B lymphocytes as key antigen-presenting cells in the development of T cell-mediated autoimmune type 1 diabetes. In: Nemazee D, editor. B cell biology in autoimmunity. Basel: Karger; 2003. pp. 212–27. [DOI] [PubMed] [Google Scholar]
- 33.Krakauer M, Sorensen PS, Khademi M, Olsson T, Sellebjerg F. Dynamic T-lymphocyte chemokine receptor expression induced by interferon-beta therapy in multiple sclerosis. Scand J Immunol. 2006;64:155–63. doi: 10.1111/j.1365-3083.2006.01788.x. [DOI] [PubMed] [Google Scholar]
- 34.Lande R, Giacomini E, Grassi T, et al. IFN-alpha beta released by Micobacterium tuberculosis-infected human dendritic cells induces the expression of CXCL10: selective recruitment of NK cells and activated T cells. J Immunol. 2003;170:1174–82. doi: 10.4049/jimmunol.170.3.1174. [DOI] [PubMed] [Google Scholar]
- 35.Christen U, McGavern DB, Luster AD, von Herrath MG, Oldstone MB. Among CXCR3 chemokines, IFN-gamma-inducible protein of 10 kDa (CXC chemokine ligand (CXCL) (10) but not monokine induced by IFN-gamma (CXCL9) imprints a pattern for the subsequent development of autoimmune disease. J Immunol. 2003;171:6838–45. doi: 10.4049/jimmunol.171.12.6838. [DOI] [PubMed] [Google Scholar]
- 36.Rhode A, Pauza ME, Barral AM, et al. Islet-specific expression of CXCL10 causes spontaneous islet infiltration and accelerates diabetes development. J Immunol. 2005;175:3516–24. doi: 10.4049/jimmunol.175.6.3516. [DOI] [PubMed] [Google Scholar]
- 37.Ottaviani C, Nasorri F, Bedini C, de Pita O, Girolomoni G, Cavani A. CD56brightCD16(-) NK cells accumulate in psoriatic skin in response to CXCL10 and CCL5 and exacerbate skin inflammation. Eur J Immunol. 2006;36:118–28. doi: 10.1002/eji.200535243. [DOI] [PubMed] [Google Scholar]
- 38.Sinigaglia F, D'Ambrosio D, Panina-Bordignon P, Rogge L. Regulation of the IL-12/IL-12R axis: a critical step in T-helper cell differentiation and effector function. Immunol Rev. 1999;170:65–72. doi: 10.1111/j.1600-065x.1999.tb01329.x. [DOI] [PubMed] [Google Scholar]