

Abstract
The time course of inhibitory postsynaptic currents (IPSCs) reflects GABAA receptor deactivation, the process of current relaxation following transient activation. Fast desensitization has been demonstrated to prolong deactivation, and these processes have been described as being ‘coupled’. However, the relationship between desensitization and deactivation remains poorly understood. We investigated the ‘uncoupling’ of GABAA receptor macroscopic desensitization and deactivation using experimental conditions that affected these two processes differently. Changing agonist affinity preferentially altered deactivation, changing agonist concentration preferentially altered macroscopic desensitization, and a pore domain mutation prolonged deactivation despite blocking fast desensitization. To gain insight into the mechanistic basis for coupling and uncoupling, simulations were used to systematically evaluate the interplay between agonist affinity, gating efficacy, and desensitized state stability in shaping macroscopic desensitization and deactivation. We found that the influence of individual kinetic transitions on macroscopic currents depended not only on model connectivity, but also on the relationship among transitions within a given model. In addition, changing single rate constants differentially affected macroscopic desensitization and deactivation, thus providing parsimonious kinetic explanations for experimentally observed uncoupling. Finally, these findings permitted development of an algorithmic framework for kinetic interpretation of experimental manipulations that alter macroscopic current properties.
Transient synaptic release of GABA onto clusters of postsynaptic αβγ GABAA receptors represents a major mechanism of inhibition in the brain. The time course of GABAA receptor synaptic currents influences the complex behaviour of inhibitory circuits, and many clinically useful drugs that potentiate GABAA receptor function act by prolonging IPSCs. Because IPSC duration depends primarily on postsynaptic GABAA receptor properties, the kinetic principles governing channel behaviour have been the focus of active experimental investigation (Twyman et al. 1990; Jones & Westbrook, 1995; Galarreta & Hestrin, 1997; Haas & Macdonald, 1999; Bai et al. 1999; Mozrzymas et al. 2003). In particular, GABAA receptor deactivation, the process by which activated receptors relax toward the resting state, shapes the decay rate (and thus charge transfer) of IPSCs. This process can be studied experimentally by activating GABAA receptors with brief pulses of high concentration GABA and taking the ensuing deactivation time course as a model of synaptic currents.
Early work by Jones & Westbrook (1995, 1996) demonstrated that the fast phase of macroscopic desensitization played an important physiological role in augmenting IPSC duration, as the underlying desensitized state provided a surrogate high affinity conformation that prolonged the time during which an activated receptor could re-open. The importance of fast desensitization in shaping IPSCs has been validated by subsequent studies (Galarreta & Hestrin, 1997; Mozrzymas et al. 2003), and extended to include possible roles for slower phases of desensitization (Overstreet et al. 2000; Bianchi & Macdonald, 2002). This phenomenon of desensitization–deactivation ‘coupling’, defined as increased macroscopic desensitization in the context of prolonged deactivation (or decreased macroscopic desensitization in the context of accelerated deactivation), has been observed with changes in subunit composition (Haas & Macdonald, 1999; Bianchi et al., 2001; Lagrange et al. 2007), allosteric modulators (Bianchi et al. 2002; Feng & Macdonald, 2004), post-translational modifications (Hinkle & Macdonald, 2003), and disease-related mutations (Buhr et al. 2002).
While it is clear that desensitized states prolong the time course of deactivation, other receptor conformations (both conducting and non-conducting) are predicted to serve similar roles. Indeed, for Markovian models of ligand-gated ion channels, every agonist bound state delays unbinding in non-cyclic kinetic schemes (for example, Twyman et al. 1990; Celentano & Wong, 1994; Jones & Westbrook, 1995; Haas & Macdonald, 1999). This delay is unrelated to microscopic affinity (which also affects unbinding); occupancy of any kinetic state not associated with a binding/unbinding step is said to ‘trap’ agonist (Bianchi & Macdonald, 2001a). The functional consequence of trapping is that additional transitions among open and closed states occur prior to agonist unbinding, the macroscopic correlate of which is a prolonged time course of deactivation. Although cyclic models have been proposed for cys-loop receptors (for example, Grosman & Auerbach, 2001; Scheller & Forman, 2002; Chang et al. 2002) and are not predicted to exhibit trapping per se, open and desensitized states will nonetheless prolong deactivation if they are high affinity states (low unbinding rate constants).
How other receptor conformations influence macroscopic desensitization, however, has received less attention (Mozrzymas et al. 2003). In many experimental circumstances, desensitization and deactivation appeared to be ‘uncoupled’ (Bianchi & Macdonald, 2001b; Scheller & Forman, 2002; Fisher, 2004; Mercik et al. 2006; Barberis et al. 2007), such that decreased extents of macroscopic desensitization were associated with prolonged time courses of deactivation (or increased extents of macroscopic desensitization with accelerated deactivation time courses). This suggested that the kinetic determinants of macroscopic desensitization were distinct from those of deactivation, despite the known sensitivity of both processes to all rate constants in the gating scheme (Mozrzymas et al. 2003). To explore the kinetic basis for coupling and uncoupling (purely phenomenological descriptions of macroscopic currents), we focused on experimental manipulations that affected desensitization and deactivation differently: GABAA receptor agonists of different affinity, a range of concentrations of GABA application, and a pore domain mutation that blocked fast desensitization. In addition, using kinetic simulations, we investigated the relative roles of agonist affinity, efficacy, desensitized state stability, and model connectivity in shaping macroscopic current properties. Analysis of simulated currents generated from a spectrum of rate constants and gating schemes provided mechanistic insight into experimental observations of coupling and uncoupling, and established a preliminary framework for interpretation of changes in macroscopic current properties due to the effects of mutations or allosteric modulators.
Methods
Cell culture and expression of recombinant GABAA receptors
The cDNAs encoding rat α1, α6, β1, β3, γ2L, δ and α1(L245S) GABAA receptor subunits were subcloned into the pCMVNeo vector. Human embryonic kidney cells (HEK293T; a gift from P. Connely, COR Therapeutics, San Francisco, CA, USA) were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum and penicillin–streptomycin at 37°C in 5% CO2–95% air. Experiments shown in Fig. 2 were conducted using mouse L929 fibroblasts. Cells were transfected with 4 μg of each subunit plasmid and 1–2 μg of pHOOK (Invitrogen, Carlsbad, CA) for immunomagnetic bead selection of transfected cells using a modified calcium phosphate technique (Fisher et al. 1997). The next day, cells were replated and recordings were made at room temperature 18–30 h later.
Figure 2.

Macroscopic desensitization and deactivation of α1β3γ2L receptor currents evoked by different GABA concentrations A, representative traces from a single cell exposed to increasing concentrations of GABA are labelled with concentration in μm. The 30 μm trace is staggered horizontally for clarity. B, the extent of macroscopic desensitization, measured as percentage current loss during the GABA pulse, is plotted for each concentration. C, deactivation currents expanded from the traces shown in panel A. The traces are staggered horizontally to illustrate their similar time courses. D, the weighted deactivation time constant is shown for 8 concentrations of GABA (4–6 s applications each). Only the deactivation following the 300 nm concentration was significantly faster than the others.
Electrophysiology
On recording days, transfected fibroblasts were bathed in an external solution consisting of (mm): NaCl 142; KCl 8; MgCl2 6; CaCl2 1; Hepes 10; glucose 10 (pH adjusted to 7.4 with NaOH, ∼330 mosmol l−1). For whole cell studies, thin-walled borosilicate glass electrodes (World Precision Instruments, Pittsburgh, PA, USA) were pulled with a P-2000 laser puller (Sutter Instrument Co., San Rafael, CA, USA) and heat-polished to resistances of 0.5–1.5 MΩ. The internal solution consisted of (mm): KCl 153; MgCl2 1; MgATP 2; Hepes 10; EGTA 5 (pH adjusted to 7.3 with KOH prior to ATP addition, ∼300 mosmol l−1). ATP was added on the day of recording. This combination of internal and external solutions produced a chloride equilibrium potential near 0 mV. For excised patch experiments, thick-walled borosilicate glass was used with resistances of 5–15 MΩ after heat-polishing. Electrodes used for single channel recordings were coated with Q-Dope. Cells and patches were voltage-clamped using an Axopatch 200A or 200B amplifier (Axon Instruments, Union City, CA, USA). GABA was applied (via gravity) with a rapid perfusion system consisting of pulled multibarrel square glass connected to a Warner Perfusion Fast-Step (Warner Instrument Corp., Hamden, CT, USA), or ‘theta’ glass mounted on a piezo-electric translator (Burleigh Instruments, Victor, NY, USA). The solution exchange time for excised patch experiments was 400 μs or less, as determined after each recording by blowing off the patch and then stepping a dilute external solution across the open electrode tip. For whole cell experiments, solution flow rates were decreased by lowering the height of the source solutions so that open tip exchange times were ∼1 ms, although slower exchange probably occurred around intact cells. All chemicals were from Sigma-Aldrich.
Analysis of currents
Currents were low-pass filtered at 2 kHz by the internal 4-pole Bessel filter of the amplifier, digitized at 10–20 kHz, and analysed using the pCLAMP8 or 9 software suite (Axon Instruments). For experiments using outside out patches, multiple applications were made at 30–90 s intervals and averaged prior to kinetic analysis. The desensitization and deactivation time courses were fitted using the Levenberg-Marquardt least squares method with multiple component exponential functions of the form  , where n is the number of exponential components, a is the relative (fractional) amplitude of the component at time = 0, t is time, and τ is the time constant. The number of components was incremented until additional components did not significantly improve the fit as determined by an F test performed on residuals. Fitting the decay during 400 ms and 6 s applications sometimes revealed three or four exponential components, respectively. To simplify comparisons, no more than two or three components, respectively, were considered for analysis. A weighted summation of biphasic deactivation time courses (afτf+asτs) was used. Three component fits were not considered. Single channel recordings were obtained after 3–4 min of incubation in 1 mm GABA. Currents were idealized using a 50% threshold method with the Fetchan program of the pCLAMP suite, and binned histograms were fitted with multiple exponential functions using Interval5 software (Dr B. Pallotta, University of North Carolina, Chapel Hill). Only main conductance state openings (approximately 2 pA at −75 mV) were included in the analysis. However, brief openings that reached threshold were accepted in the idealization. All patches contained more than one channel, as indicated by overlapped openings. Although these openings were ignored in the idealization, the presence of multiple openings complicates the analysis of closed times (artificially shortened), opening frequency (artificially increased), and open probability (artificially increased). Numerical data were expressed as means ±s.e.m. Statistical significance, using Student's t test (paired or unpaired as appropriate) was taken as P < 0.05.
, where n is the number of exponential components, a is the relative (fractional) amplitude of the component at time = 0, t is time, and τ is the time constant. The number of components was incremented until additional components did not significantly improve the fit as determined by an F test performed on residuals. Fitting the decay during 400 ms and 6 s applications sometimes revealed three or four exponential components, respectively. To simplify comparisons, no more than two or three components, respectively, were considered for analysis. A weighted summation of biphasic deactivation time courses (afτf+asτs) was used. Three component fits were not considered. Single channel recordings were obtained after 3–4 min of incubation in 1 mm GABA. Currents were idealized using a 50% threshold method with the Fetchan program of the pCLAMP suite, and binned histograms were fitted with multiple exponential functions using Interval5 software (Dr B. Pallotta, University of North Carolina, Chapel Hill). Only main conductance state openings (approximately 2 pA at −75 mV) were included in the analysis. However, brief openings that reached threshold were accepted in the idealization. All patches contained more than one channel, as indicated by overlapped openings. Although these openings were ignored in the idealization, the presence of multiple openings complicates the analysis of closed times (artificially shortened), opening frequency (artificially increased), and open probability (artificially increased). Numerical data were expressed as means ±s.e.m. Statistical significance, using Student's t test (paired or unpaired as appropriate) was taken as P < 0.05.
Simulations
Kinetic modelling was carried out with Berkeley Madonna 3.1 (http://www.berkeleymadonna.com), a differential equation solver, using the fourth order Runge–Kutta method with a time interval of 10–100 μs.
Results
Agonists with different affinities for GABAA receptors altered deactivation without altering macroscopic desensitization
We tested the hypothesis that deactivation could be altered independently of macroscopic desensitization using ligands with higher (muscimol) or lower (THIP) affinity than GABA for GABAA receptors. Since muscimol (Twyman & Macdonald, unpublished observation) and THIP (Mortensen et al. 2004) evoked single channel currents similar to those evoked by GABA, we predicted that macroscopic desensitization would be similar for all three agonists when applied at nearly saturating concentrations, as activated receptors would transition within the same di-liganded portion of the receptor gating scheme. In contrast, we expected that deactivation would be markedly different for each of the three agonists, as the unbinding rate plays an important role in determining how quickly activated receptors return to the resting state (Jones et al. 1998; Li & Pearce, 2000).
Macroscopic desensitization of α1β1γ2L receptor currents was investigated in response to rapid application of EC-equivalent concentrations of each agonist, approximated from whole cell concentration–response curves generated using a Y-tube perfusion system (not shown). Outside-out patches were obtained from transfected L929 fibroblasts, and agonists were applied rapidly with the concentration-jump technique (see Methods). GABA- and THIP-evoked currents desensitized to similar extents during 400 ms applications (66.1 ± 6.1%; n = 6, for 100 μm GABA; 59.6 ± 5.0%; n = 11, for 10 mm THIP) (Fig. 1Aa and Ba). Also, 8 of 11 patches exposed to THIP exhibited a biphasic desensitization time course that included a fast (∼20 ms or less) phase. The rate and extent of the fast phase of desensitization was similar between the biphasic currents evoked by GABA (12.1 ± 3.1 ms; 51.1 ± 6.7%; n = 6) and THIP (11.2 ± 2.2 ms; 49.7 ± 6.0%; n = 11). Currents evoked by muscimol (30 μm) activated more slowly (5.7 ± 0.9 ms; n = 4) than those evoked by THIP (2.2 ± 0.4 ms; n = 11) or GABA (1.8 ± 0.2 ms; n = 6) (not shown). However, the extent of desensitization (53.2 ± 4.9%; n = 4) and the rate and relative proportion of fast desensitization (17.4 ± 5.6 ms; 44.5 ± 4.5%; n = 4) of biphasic currents activated by muscimol were not different from those observed for GABA or THIP (Fig. 1Ca).
Figure 1.

Macroscopic desensitization and deactivation of α1β1γ2L GABAA receptor currents evoked by different agonists Currents were obtained from outside-out membrane patches using concentration jumps of 400 or 5 ms duration into equivalent concentrations of GABA (Aa and b), THIP (Ba and b), or Muscimol (Ca and b). The currents displayed were obtained from different patches. The horizontal scale bar in panel Aa applies to panels A–C. The agonist pulse duration is shown by a filled bar above each trace. Quantification of macroscopic desensitization is given in the text. D, weighted time constants of deactivation following brief pulses of 1 mm THIP (T; filled bar), 100 μm GABA (G; grey bar), and 30 μm Muscimol (M; open bar). E, deactivation currents from patches containing α1β3δ (top trace) and α6β3δ receptors (bottom trace) following a 5 ms pulse of 1 mm GABA (filled bar). The horizontal scale bar applies to both traces.
In contrast, the deactivation time course depended strongly on agonist affinity (Fig. 1Aa, 1Ba, Ca). To further explore this difference, brief (5 ms) applications of each agonist were applied (Fig. 1Ab, 1Bb and 1Cb). For muscimol, some of these applications were made using a longer duration (30 ms) to ensure that peak current was obtained. Since the resulting deactivation time courses were similar between these two conditions, the results were pooled. The weighted time constants of current deactivation were 10.5 ± 0.9 ms (n = 5) for THIP, 61.3 ± 6.8 ms (n = 5) for GABA, and 198.1 ± 48.0 ms (n = 6) for muscimol (Fig. 1D). Whereas brief GABA- and muscimol-evoked currents decayed biphasically, in 4 of 5 THIP-evoked currents, the decay was dominated by a fast phase (> 95%). These results were similar to those of Jones et al. (1998), who reported that currents evoked by muscimol and the low affinity agonist β-alanine had longer and shorter deactivation time courses, respectively, than those evoked by GABA, despite identical desensitization kinetics.
This apparent uncoupling of desensitization and deactivation demonstrated that the unbinding rate constant could dominate the deactivation time course, even in the presence of fast desensitization. To explore this observation, deactivation time courses were measured following activation of different receptor isoforms by the same agonist (note that changes in agonist affinity can also be related to subunit composition). While kinetic models of α6 subunit-containing αβδ receptors are unavailable, their low GABA EC50 compared to α1 subunit-containing receptors has been attributed to increased affinity (Saxena & Macdonald, 1996; Fisher et al. 1997; Fisher, 2004). When excised patches containing α1β3δ or α6β3δ receptors were exposed to brief (5 ms) pulses of GABA (1 mm), those containing the α6 subunit had markedly slower deactivation (Fig. 1E). Neither receptor isoform, however, exhibited fast desensitization (Bianchi et al. 2002). Taken together, these data suggest that fast desensitization was neither necessary nor sufficient for prolonged deactivation. In addition, the data also raised the more general questions: under what conditions could other rate constants in the gating scheme dominate the deactivation time course, and in what context would this also be manifested as desensitization-deactivation uncoupling?
Different agonist concentrations altered macroscopic desensitization without affecting deactivation
One interpretation of the apparent increase in macroscopic desensitization observed with increasing agonist concentrations (Fig. 2A) is that desensitized states are preferentially accessed during activation with high agonist concentrations. However, the slow activation observed with low agonist concentrations may mask the manifestation of an inherently concentration-independent desensitization process (Celentano & Wong, 1994), similar to slow agonist perfusion (Bianchi & Macdonald, 2002). Indeed, ‘predesensitization’ has been shown using preincubations of low GABA concentrations prior to test jumps into a saturating GABA concentration (Overstreet et al. 2000; Lagrange et al. 2007). If desensitized states were accessible at low GABA concentrations, similar deactivation time courses would be predicted over a broad range of GABA concentrations despite variable macroscopic desensitization (i.e. uncoupling). In contrast, if desensitized state accessibility decreased with lower GABA concentrations, the observed loss of macroscopic desensitization would be associated with an accelerated deactivation time course (i.e. coupling).
Macroscopic desensitization was measured during 4–6 s applications of different GABA concentrations to cells expressing α1β3γ2L receptors (Fig. 2A). Although the extent of desensitization varied from 0% to 80% over this concentration range (Fig. 2B), deactivation time courses were indistinguishable at concentrations from 1 mm (near-saturating) to 1 μm (approximately EC10) (Fig. 2C and D), providing an additional experimental example of desensitization-deactivation uncoupling. The deactivation time course was significantly faster only following the 300 nm GABA applications (Fig. 2D), possibly caused by a significant mono-liganded component at this low concentration. Similar results were obtained in excised patches, eliminating the possibility that our findings were an artifact of the slower solution exchange characteristic of whole-cell recording (online supplemental material, Supplemental Fig. 1). This suggested that the underlying kinetic processes shaping deactivation were unchanged over a wide range of GABA concentrations, and thus, that accessibility of desensitized states was not compromised. As in the previous section, this demonstrated that prolonged deactivation was possible even in the absence of fast macroscopic desensitization.
A channel mutation blocked macroscopic desensitization without accelerating deactivation
Mutations of the conserved pore-lining TM2 9′leucine in the cys-loop family of receptors to the polar residues serine or threonine have been shown to alter macroscopic current properties via an increase in single channel mean open time (Revah et al. 1991; Yakel et al. 1993; Filatov & White, 1995; Thompson et al. 1999). Specifically, during rapid kinetic studies of the α1(L9′T)β2γ2L receptor mutation, the extent of macroscopic desensitization was found to decrease despite a prolonged deactivation time course (Scheller & Forman, 2002). To better understand the mechanistic basis for this desensitization–deactivation uncoupling, we analysed the kinetic properties of a similar mutation in the GABAA receptor α1 subunit.
α1(L9′S)β3γ2L receptors exhibited a left-shifted whole cell GABA EC50 of ∼300 nm (Fig. 3A), as well as bicuculline-sensitive spontaneous activity (not shown), consistent with previous studies (Labarca et al. 1995; Chang & Weiss, 1999; Thompson et al. 1999; Bianchi & Macdonald, 2001b; Scheller & Forman, 2002). During a 6 second application of GABA to excised outside-out patches, macroscopic desensitization of α1(L9′S)β3γ2L currents was slower and less extensive than that of α1β3γ2L currents (Fig. 3B). In contrast to α1β3γ2L currents, which desensitized with three exponential components (Fig. 3C, filled symbols), α1(L9′S)β3γ2L currents desensitized with a single exponential component (Fig. 3C, grey squares), similar to the results of Scheller & Forman (2002). The single α1(L9′S)β3γ2L desensitization time constant (2098 ± 435 ms; n = 4) was not different from the longest wild-type desensitization time constant (τ3; 2395 ± 184 ms; n = 9), but its relative contribution was increased from 37.0 ± 3.0% to 57.5 ± 6.3%.
Figure 3.

Characterization of α1(L9′S)β3γ2L GABAA receptor currents A, concentration–response curves obtained for α1β3γ2L (filled circles) and α1(L9′S)β3γ2L (grey triangles) receptors. Representative current traces from individual cells are shown at concentrations of 3, 10, and 100 μm for α1β3γ2L receptors, and 0.03, 0.1, and 10 μm for α1(L9′S)β3γ2L receptors. Vertical scale bars are 1.0 nA and 50 pA for α1β3γ2L and α1(L9′S)β3γ2L receptors, respectively. Horizontal scale bars are 3 s for both isoforms. B, currents evoked by 6 s applications of 1 mm GABA to outside out patches containing α1β3γ2L (black trace) and α1(L9′S)β3γ2L (grey trace) receptors. Traces were normalized to peak and slightly staggered horizontally for comparison. C, scatter plot of the time constants obtained from exponential fitting of the desensitization time course. Three time constants were required to fit the time course of α1β3γ2L receptor desensitization (filled symbols), while α1(L9′S)β3γ2L receptor desensitization was mono-exponential (grey symbols). Note the logarithmic axis. D and E, α1β3γ2L (black traces) and α1(L9′S)β3γ2L (grey traces) receptor currents were evoked by 400 ms (D) and 5 ms (E) concentration jumps into 1 mm GABA. F, activation rates (left), as indicated by the 10–90% rise time observed for 400 ms concentration jumps, are shown for both isoforms. The extent of macroscopic desensitization is plotted as the loss of current during the 400 ms application (right). G, the weighted deactivation time constant was shown for α1β3γ2L (black bars) and α1(L9′S)β3γ2L (grey bars) receptor currents following 5 ms (left) and 400 ms (right) application durations. Filled squares in D indicate isoform colour for bar graphs in F and G.
Shorter duration pulses of GABA (1 mm) were used to investigate the effects of the α1(L9′S) subunit mutation on deactivation (Fig. 3D, 400 ms; Fig. 4E, 5 ms). The 10–90% rise times were significantly increased by the α1(L9′S) subunit mutation from 0.71 ± 0.07 ms to 1.51 ± 0.19 ms (Fig. 3F, left). For 400 ms pulses, the extent of desensitization was minimal for α1(L9′S)β3γ2L compared to wild-type receptors (Fig. 3F, right), while the deactivation time courses of α1β3γ2L (238.7 ± 14.7 ms, n = 6) and α1(L9′S)β3γ2L 181.8 ± 21.9 ms, n = 9 currents were similar. Consistent with prior reports, this uncoupling manifested as the loss of fast desensitization without accelerated deactivation time course. After 5 ms pulses, deactivation was faster than after 400 ms pulses for both mutant and wild-type receptors, although the time course was significantly longer for the mutant (114.0 ± 9.4 ms) in comparison to wild-type (68.4 ± 10.9 ms) receptors (Fig. 3G). Thus, with longer GABA exposure, prolongation of deactivation for the wild-type current (∼300% increase) was greater than for the mutant current (∼40% increase) (Fig. 3G).
Figure 4.

Single channel analysis of α1(L9′S)β3γ2L receptors A, representative α1β3γ2L receptor single channel events recorded from an outside-out patch voltage clamped at −75 mV are shown. A portion of the continuous trace (open bar) is expanded below. Channel openings are visible as downward deflections from the baseline current. B, continuous trace showing α1(L9′S)β3γ2L receptor single channel events for comparison. C and D, open time histograms for WT (C) and L9′S mutant (D) single channel recordings. Open durations were fitted best with three exponential functions in each case (overlapping curves in each plot). The sum of the three exponentials is shown as the top curve in each plot.
The dependence of the deactivation time course on the duration of GABA exposure has been suggested to reflect increasing occupancy of desensitized states (Jones & Westbrook, 1995). Thus, the failure to observe such an increase with the α1(L9′S) mutation, particularly given the absence of fast macroscopic desensitization, could indicate failure to access desensitized state(s). However, this would be expected to accelerate deactivation, while in this case, deactivation was prolonged following synaptic pulse durations. We hypothesized that increased efficacy could account for the constellation of findings, as this would be consistent with both prolonged deactivation after brief pulses and decreased macroscopic desensitization. Moreover, since deactivation following prolonged agonist exposure is dominated by the ability of long-lived desensitized states to delay agonist unbinding, this would also be consistent with a similar deactivation time course following long applications.
Changes in gating efficacy accounted for slow deactivation despite reduced desensitization of α1(L9′S)β3γ2L receptor currents
Although it has been proposed that increased spontaneous openings necessarily reflects increased efficacy of liganded gating (Grosman & Auerbach, 2000; Scheller & Forman, 2002), spontaneous openings may involve a different reaction pathway from the gating of liganded receptors, thus necessitating single channel analysis. To test the hypothesis that prolonged deactivation after brief pulses (despite absent fast desensitization) in α1(L9′S) mutants was due to increased gating efficacy, we obtained single channel recordings from α1β3γ2L and α1(L9′S)β3γ2L receptor channels (Fig. 4A and B). Both mean open time (2.5 versus 1.8 ms) and burst duration (18.6 versus 3.7 ms) were significantly increased by the mutation. Open duration histograms for α1β3γ2L and α1(L9′S)β3γ2L receptor single channel currents were both fitted best by three exponential functions (Fig. 4C and D). The increases in mean open and burst durations were accounted for in part by longer time constants associated with the two longest open components (O2 and O3). Opening frequency, open probability and closed time distributions were not investigated, since the patches used for analysis contained multiple channels (overlapped openings). While development of an exhaustive kinetic scheme of the mutated receptors was not possible with this limited data set, the results suggested that increased efficacy could explain prolonged deactivation despite reduced macroscopic desensitization, and supported the idea that multiple rate constants in the gating scheme (not just the agonist binding and unbinding rates) could mediate desensitization–deactivation uncoupling.
Kinetic modelling was used to explore the role of microscopic kinetic parameters on macroscopic desensitization and deactivation
Although comprehensive kinetic models have been developed that account for both the microscopic and macroscopic properties of GABAA receptors (Haas & Macdonald, 1999; Fig. 5A), simple models are often sufficient to illustrate the salient features of GABA-evoked currents such as rapid activation, extensive macroscopic desensitization, and prolonged deactivation. In addition, with fewer free parameters, they facilitate systematic exploration of relationships between individual rate constants and macroscopic current properties. Historically, two simple models have been commonly used to describe the behaviour of ligand-gated ion channels. Katz and colleagues described cholinergic responses with a linear scheme of binding, isomerization to the open state, and subsequent entry into the desensitized state (Katz & Thesleff, 1957; Fig. 5B). Jones & Westbrook (1995) explained GABAA receptor desensitization using a branched arrangement of states, which at high agonist concentration, reduces to the scheme shown in Fig. 5C. In both cases, the single non-conducting state not associated with a binding step was designated the desensitized (D) state. This was supported by the fact that no combination of rate constants supported macroscopic desensitization in the absence of this state (macroscopic desensitization is impossible for C–C–O arrangements; not shown).
Figure 5.

Kinetic models of ligand-gated ion channel function A, comprehensive kinetic scheme for the α1β3γ2L receptor isoform taken from Haas & Macdonald (1999). For simplicity, the two distal ‘intraburst’ closed states connected to each open state were omitted from the display. Closed (C), open (O), and desensitized (Df, ‘fast’; Di, ‘intermediate’; Ds, ‘slow’) states were reversibly interconnected. The microscopic transitions associated with agonist binding were labelled kon and koff for association and dissociation rate constants, respectively. Each agonist binding step was taken to be equivalent and independent in this scheme (the first binding and unbinding rates were therefore multiplied by 2). [G], concentration of GABA. B, 4-state kinetic model, in linear arrangement. A single binding step connects C1 and C2. The O state is accessed from C2, and is arranged in series with the D state. C, 4-state kinetic model, in branched arrangement. D and O states are arranged in parallel, each being directly accessible following sojourns in the ligand-bound closed state. All rate constants referred to in the text and in subsequent figures have units of s−1, except for the binding rate, kon, which is multiplied by the concentration of ligand, and thus has units of s−1m−1.
To gain insight into the mechanistic basis for coupling and uncoupling, simulations were conducted to explore the effects of agonist affinity, agonist concentration, open state efficacy, and desensitized state stability (defined as the ratio of the entry rate, δ, to the exit rate, ρ) on macroscopic desensitization and deactivation. While the comprehensive α1β3γ2L receptor model was used to recapitulate our experimental findings, the simple four-state models were used to systematically evaluate the impact of each kinetic parameter on macroscopic currents. By covarying rate constants, the kinetic conditions for which deactivation was either coupled to or uncoupled from macroscopic desensitization were determined. In this manner, we elucidated patterns relevant to the experimental setting, where neither the kinetic model nor the relevant underlying transitions are typically known.
The relationships among agonist unbinding, macroscopic desensitization, and deactivation
Our experimental observations using agonists of different affinity suggested that unbinding, the terminating step in the relaxation of activated receptors to the resting state, could have a dominant influence on deactivation independent of the rate or extent of macroscopic desensitization. This form of desensitization-deactivation uncoupling was further examined using the comprehensive α1β3γ2L receptor model (Fig. 5A) (Haas & Macdonald, 1999). For a 30-fold increase (Fig. 6Aa) or decrease (Fig. 6Ab) in koff, the peak current amplitude and shape of macroscopic desensitization were nearly overlapping during activation by a near-saturating GABA concentration, while deactivation differed markedly (current with the ‘wild-type’koff is marked with an asterisk). Additional increases in koff resulted in decreased peak amplitude, attributable to substantial shifts in GABA EC50 that rendered 1 mm GABA subsaturating and thus slowed macroscopic activation (which also resulted in decreased apparent desensitization; not shown). The same phenomena were also observed for alterations in koff using the 4-state branched and linear models (not shown). These results confirmed that deactivation could be markedly altered by changes in unbinding that produced little effect on macroscopic desensitization, similar to our experimental observations with currents evoked by agonists of different affinity (Fig. 1; Jones et al. 1998).
Figure 6.

Predicted effects of altering microscopic unbinding on macroscopic desensitization and deactivation A, simulated responses to 400 ms applications of near-saturating GABA (1 mm) were overlapped for various values of koff using the comprehensive α1β3γ2L receptor model (Fig. 5A). In the left panel, koff= 5100, 1700, 510, and 170 s−1. In the right panel, koff= 170, 51, 17, and 5.1 s−1. Currents were not normalized. Insets show the first 10 ms of the overlapped traces to illustrate the effect of altering koff on activation rate and peak current amplitude. *Indicates ‘wild-type’, where koff= 170 s−1. B, simulated current responses to 5 ms applications of GABA (1 mm) for wild-type (*) and either increasing (Ba; koff varied from 170 to 170 000 s−1) or decreasing (Bb; koff varied from 170 to 0 s−1) unbinding rates. For the two fastest koff traces, the GABA concentration was raised to 20 mm to overcome the extremely low affinity. C, in the left panel, simulated responses of the α1β3γ2L model to 5 or 1000 ms pulses (black lines, open circles) are overlaid with a simulated response to a 5 ms pulse when koff was set to zero (grey line, filled circle). In the right panel, the response of the same model, but without D states, to a 1 s application of 1 mm GABA (black line, open circle) is overlaid with a 5 ms application for which koff was set to zero (grey line, filled circle). The small amount of macroscopic desensitization that persisted even in the absence of desensitized states was due to C4 serving as a ‘branched’ D state relative to O2.
Macroscopic desensitization sets a boundary condition for deactivation time course
To investigate the relationship between koff and deactivation under synaptic conditions, currents evoked by brief GABA pulses were simulated using the α1β3γ2L model in the context of increasing (Fig. 6Ba) or decreasing (Fig. 6Bb) koff. The resulting deactivation currents were multiphasic, with the fast phase demonstrating less sensitivity to changes in koff than the slower phases (Fig. 6Ba and Bb). Overall, both slow and fast phases of deactivation became faster with increasing koff until approaching a limit at higher koff values (Fig. 6Ba). Similarly, with decreasing koff, both slow and fast phases of deactivation became slower until approaching a limit at lower koff values (Fig. 6Bb). The observation of a fast limit under conditions of extremely rapid unbinding was not surprising, since open receptors must close before unbinding, in which case, open durations (or in the case of more complex models, burst/cluster durations) become rate limiting. The basis for the slow limit, however, was less clear. Given that the slowly desensitizing α6β3δ isoform also deactivated slowly (Fig. 1E), we hypothesized that macroscopic desensitization served to limit the deactivation time course. Although this initially seemed counterintuitive, additional simulations revealed a simple kinetic basis for this phenomenon.
The simulated response to a 5 ms pulse of 1 mm GABA using the kinetic scheme of Fig. 5A and the default value for koff (Fig. 6Ca, left open circle) was compared to the response of the same brief pulse in the extreme case when koff was set to zero (Fig. 6Ca, filled circle). As expected, when unbinding could not occur, the deactivation time course was markedly prolonged. Interestingly, when the response to a 1 s application of 1 mm GABA was overlaid on these responses (Fig. 6Ca, right open circle), it was found to follow a time course identical to that of deactivation following the 5 ms pulse when koff was set to zero (Fig. 6Ca, filled circle). The overlap between deactivation following the 5 ms pulse (where unbinding was impossible) and macroscopic desensitization during the 1 s pulse (where unbinding was possible, but functionally irrelevant given the near-saturating agonist concentration) occurred simply because receptors were fully liganded in both conditions, and as a result, behaved in the same manner. Similar constraints on the deactivation time course by macroscopic desensitization were also evident with the four-state models (not shown).
The idea that the time course of deactivation could not be slower than macroscopic desensitization was illustrated further by repeating the simulations of Fig. 6Ca in the absence of all three D states. We compared the currents generated by a 1 s pulse of 1 mm GABA using the default value for koff (Fig. 6Cb, open circle) with a 5 ms pulse where koff was set to zero (Fig. 6Cb, filled circle). Again, deactivation following the brief pulse followed the same time course as that of macroscopic desensitization during the 1 s pulse. However, compared to deactivation in the context of all three D states, the overall time course was markedly prolonged (compare Fig. 6Ca to Fig. 6Cb; filled circles). Thus, when unbinding was impossible, desensitization-deactivation uncoupling always occurred, as decreased macroscopic desensitization allowed for prolonged deactivation, while increased macroscopic desensitization forced deactivation to accelerate.
It should be noted that the constraint imposed by macroscopic desensitization on deactivation actually depended on the duration of agonist application. For example, the constraint imposed on the fast phase of deactivation was limited to brief applications, because macroscopic desensitization manifested a fast phase selectively in this time domain. In other words, only if an agonist pulse terminated prior to the onset of a given phase of macroscopic desensitization could that phase constrain deactivation. Thus, while deactivation following brief pulses was constrained by all phases of macroscopic desensitization, deactivation following prolonged pulses, where macroscopic fast desensitization had already occurred, was only constrained by slower phases of desensitization.
Although the extreme case of irreversible binding was used to illustrate the complex interplay between macroscopic desensitization and deactivation, the interpretations are applicable to the continuum of rate constants likely to occur biologically. The implication of fast macroscopic desensitization in shaping the fast phase of deactivation is of particular physiological relevance since rapid desensitization has been widely observed in native and recombinant GABAA receptor currents (Celentano & Wong, 1994; Jones & Westbrook, 1995; Tia et al. 1997; Haas & Macdonald, 1999; Li & Pearce, 2000; Burkat et al. 2001; Scheller & Forman, 2002; Yang et al. 2002; Mozrzymas et al. 2003). In summary, the simulations suggested that the effect of agonist affinity on deactivation was not actually independent of macroscopic desensitization. Instead, agonist affinity determined the ‘position’ of deactivation between two limits: a fast limit dictated by channel gating, and a slow one imparted by the shape of macroscopic desensitization.
The relationships among agonist concentration, macroscopic desensitization, and deactivation
Higher concentrations of GABA evoke faster and more extensively desensitizing currents (Fig. 2A), but it is important to distinguish between two possible explanations for this phenomenon: increased D state occupancy, or improved resolution of the concentration-independent desensitization process. While the experimental observation of similar deactivation time courses for currents evoked by concentrations above ∼EC20 (3 μm) suggested that relative occupancy of microscopic states was similar over this range of concentrations, we further explored this hypothesis using kinetic simulations. Using the α1β3γ2L model (Fig. 5A), currents evoked by four different agonist concentrations were simulated (Fig. 7A). Total D state occupancy (Df+ Di+ Ds) is represented in the upward traces, while total O state occupancy (O1+ O2+ O3) is represented in the downward traces (i.e. the currents). When the current evoked by 10 μm GABA (∼EC30) was compared with the current evoked by 1 mm GABA (∼EC100), the extent of macroscopic desensitization was markedly different, though the fractional occupancy of D states was similar (Fig. 7A). This clearly demonstrated that increasing concentrations of GABA did not produce increasing extents of macroscopic desensitization due to increased accessibility of D states. Equilibrium occupancy of O states was also similar in this concentration range, indicating that increasing the GABA concentration did not substantially affect the relative distribution of receptors among fully-liganded states. Consistent with this result, simulated deactivation was faster only following applications of very low GABA concentrations, (Fig. 7B). Interestingly, the simulated current generated by 0.3 μm GABA (Fig. 7A, smallest current) was associated with several-fold higher probability of D state occupancy (Fig. 7A, smallest of the upward traces) than of O state occupancy, despite the extent of macroscopic desensitization being < 1%. Taken together, these findings demonstrate that the extent of macroscopic desensitization is a poor predictor of desensitized state occupancy, as has previously been suggested (Mozrzymas et al. 2003).
Figure 7.

Predicted concentration dependence of macroscopic desensitization and deactivation A, the sum of desensitized state (upward traces) and open state (downward traces) occupancies are shown for 4 agonist concentrations (1, 3, 10, and 1000 μm) using the comprehensive α1β3γ2L receptor model from Fig. 5A. The top of the upward traces and the bottom of the downward traces corresponds to 1000 μm. The calibration bars apply to both sets of traces. The vertical calibration represents a probability of receptor occupancy of 0.2. Note that the currents (downward traces) and the D state probability curves (upward traces) represent the combined occupancy of all 3 O and D states, respectively. B, weighted deactivation time constant of simulated currents generated with the α1β3γ2L model over a range of GABA concentrations applied for 400 ms. For each concentration, simulations were generated using the ‘wild-type’ rate constants (open circles), setting the entry rate into Df to zero (filled circles), setting the entry rates into both Di and Ds to zero (grey circles), and setting entry rates into all 3 desensitized states to zero (x). The labels to the right of the circles indicated the desensitized states that remained accessible for a set of simulations. The longer deactivation predicted by the model relative to the experimental data may be attributed to a long time constant that is unlikely to be resolved experimentally because its small amplitude would be difficult to distinguish from baseline noise.
To investigate the kinetic basis for this concentration relationship, we measured deactivation over this concentration range for simulations in which one or more D states were eliminated. Eliminating Df accelerated deactivation similarly at each concentration (Fig. 7B, filled circles) relative to the intact scheme (Fig. 7B, open circles), suggesting that this state contributed to deactivation similarly at all tested concentrations. Eliminating the two slower D states, however, accelerated deactivation and blunted the concentration dependence (Fig. 7B, grey circles), suggesting that receptors activated by very low agonist concentrations were unable to achieve sufficient occupancy of these slowly equilibrating states. Because low concentrations forced receptors to spend more time in mono- and unliganded states, receptors had fewer opportunities to enter slowly equilibrating D states (which were only accessible to di-liganded receptors). In support of this slow equilibration idea, increasing the application duration (> 25 s) for low concentrations (300 nm and 1 μm GABA) allowed receptors time to equilibrate among slower D states, significantly prolonging the time course of deactivation (not shown). In other words, increasing the fractional occupancy of slowly equilibrating D states (Di and Ds) could be accomplished either by increasing the concentration of agonist for a given duration application, or by increasing the application duration for low concentrations.
The relationship between macroscopic desensitization and the stability of open and desensitized states
Although macroscopic desensitization is not possible without D states, extracting microscopic information about D states from current traces remains a significant challenge, as other states are known to contribute to macroscopic current shape (Mozrzymas et al. 2003; Celentano & Hawkes, 2004). We therefore examined the extent of macroscopic desensitization (percent of current lost) for branched and linear four-state models (Fig. 5), over a range of entry and exit rate constants from O and D states spanning two orders of magnitude (60 s−1 to 6000 s−1). Plotting the extent of macroscopic desensitization (on the ordinate) against each pair of rate constants yielded a ‘landscape’ illustrating the dependence of macroscopic desensitization on the relative stability (defined as the ratio of the entry rate to the exit rate) of O (β/α) and D (δ/ρ) states (Fig. 8). For example, in Fig. 8A and B, the extent of macroscopic desensitization of the branched model was evaluated over a range of β and α values (right and left corners represented the highest and lowest O state stabilities, respectively), in the context of either more (Fig. 8A) or less (Fig. 8B) D state stability. Note that for these simplified schemes, the percentage of current lost was considered a suitable index of macroscopic desensitization, as faster time constants correlated in most cases with more extensive desensitization (not shown).
Figure 8.
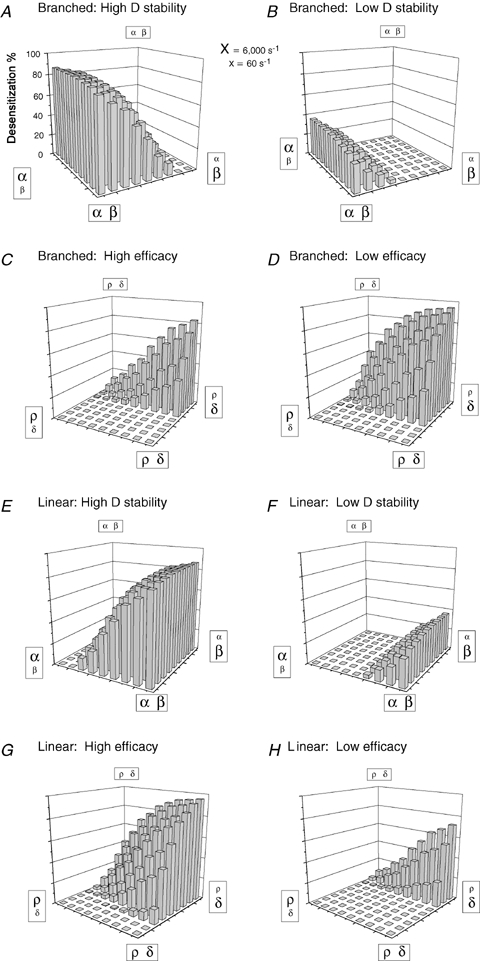
Dependence of the extent of macroscopic desensitization on the stability of open and desensitized states for branched and linear kinetic schemes Landscape plots illustrate the extent of macroscopic desensitization (Z axis) for branched (A–D) and linear (E–H) kinetic schemes over a range of β/α and δ/ρ combinations. The Z axis labels of A apply to all 8 plots. Each grid shows the 81 combinations of rate constants used to generate the simulated currents (not shown), the values of which are indicated in the 4 corners of each plot by variable letter sizes. Large letters correspond to 6000 s−1, small letters correspond to 60 s−1, and the rate constant values in between were 3000, 2000, 1000, 600, 300, 200 and 100 s−1 (not labelled). The top 4 landscapes were generated using the branched 4-state model (Figure 5C), and the bottom 4 were generated using the linear 4-state model (Fig. 5B). For all panels, kon= 106 s−1m−1, koff= 300 s−1, and the GABA concentration was 1 mm. For panels in which the O state transitions were varied (A, B, E and F), the D state rate constants were δ= 1.0 and ρ= 0.1 for ‘high D stability’ panels, and δ= 0.6 and ρ= 0.6 for ‘low D stability’ panels. For panels in which the D state transitions were varied, the O state rate constants were β= 3.0 and α= 0.6 for ‘high efficacy’ panels and β= 0.3 and α= 2.0 for ‘low efficacy’ panels. Note that in simulations of the linear kinetic scheme, efficacy was actually changing in the δ×ρ grids because open duration was determined by the reciprocal sum of the two ‘closing’ rate constants, α and δ.
When efficacy (O state stability; β/α) was varied in the branched model, the extent of macroscopic desensitization was greatest for high values of α (brief openings), and least when α was low (long openings) (Fig. 8A and B). Increasing β (higher opening frequency) reduced the extent of macroscopic desensitization, while decreasing β (lower opening frequency) increased the extent. Although varying β had much less of an effect than varying α, these landscape patterns were consistent with the idea that increasing efficacy caused the branched model to shift towards a C–C–O arrangement, which cannot macroscopically desensitize (not shown). When D state stability was varied in the branched model, the extent of macroscopic desensitization was greatest for high values of δ and low values of ρ (Fig. 8C and D). Overall, the high efficacy condition (Fig. 8C) displayed less macroscopic desensitization than the low efficacy condition (Fig. 8D), again reflecting the inability of C–C–O arrangements to macroscopically desensitize. Note, however, that high values of δ were not always associated with macroscopic desensitization, even for low efficacy receptors (Fig. 8D). Indeed, macroscopic desensitization was only possible when α was greater than ρ. When this condition was violated, neither increasing δ nor decreasing β restored macroscopic desensitization, even when this drove D state fractional occupancy above 90% (not shown).
Varying efficacy in the linear model produced patterns of macroscopic desensitization distinct from those of the branched model (Fig. 8E and F). The extent of macroscopic desensitization was greatest when efficacy was highest and was more sensitive to changing β than α. In addition, while both models predicted extensive macroscopic desensitization for high values of δ and low values of ρ (Fig. 8C, D, G and H), high efficacy receptors in the linear model were more susceptible to changes in D state stability than low efficacy receptors (Fig. 8G and H). Interestingly, the rate constant relationships that determined whether macroscopic desensitization occurred were also different. Unlike the branched model, macroscopic desensitization was possible in the linear model only when β was greater than ρ. When this condition was violated, altering other rate constants could not restore macroscopic desensitization, even when this yielded D state fractional occupancies above 90% (not shown).
These results demonstrated that tremendous variation in macroscopic desensitization could arise despite constant D state stability (note the constant δ/ρ ratio along the diagonal between the nearest and furthest corners of Fig. 8D). Conversely, the landscapes demonstrated regions of negligible macroscopic desensitization despite marked changes of D state stability. Thus, current shape was a poor predictor of D state fractional occupancy, suggesting that failure to observe macroscopic desensitization should not, by itself, be considered evidence against the presence of D states. Although macroscopic currents are shaped by all rate constants in the gating scheme (Mozrzymas et al. 2003), these results also suggest that only certain rate constants determine whether macroscopic desensitization can occur.
The relationship between deactivation and the stability of O and D states
Similar to the approach taken in the previous section to determine the relationship between individual rate constants and the extent of macroscopic desensitization, landscape plots were generated from weighted deactivation time courses following simulated 100 ms agonist pulses for branched (Fig. 9A–D) and linear (Fig. 9E–H) schemes. For the branched model, increasing efficacy prolonged deactivation while decreasing efficacy accelerated deactivation, in the context of both high (Fig. 9A) and low (Fig. 9B) D state stabilities. These deactivation landscapes, however, trended in the opposite direction compared to their corresponding desensitization landscapes (compare Fig. 9A and B to Fig. 8A and B), thus revealing a pattern of uncoupling: regions with the slowest deactivation showed the least macroscopic desensitization, while those with the fastest deactivation showed the most macroscopic desensitization. Figure 9C and D illustrate branched model deactivation when D state stability was varied in the context of high and low efficacy. Deactivation was slowest for the most stable D state conditions and fastest for the least stable D state conditions for both high and low efficacy conditions, matching the relationship between deactivation and efficacy (Fig. 9A and B). These deactivation landscapes trended in the same direction as their corresponding desensitization landscapes (compare Fig. 9C and D to Fig. 8C and D), thus illustrating desensitization–deactivation coupling. Note, however, that deactivation varied substantially even for regions that lacked macroscopic desensitization.
Figure 9.

Dependence of deactivation on the stability of the open and desensitized states for branched and linear schemes Landscape plots illustrate the weighted deactivation time constant (Z axis) for branched (A–D) and linear (E–H) kinetic schemes over a range of β/α and δ/ρ combinations. The Z axis labels in A apply to all 8 panels. See the legend of Fig. 8 for plot descriptions and rate constants.
Similar to the branched model, increasing and decreasing O or D state stabilities in the linear model corresponded to longer and shorter deactivation time courses, respectively (Fig. 9E–H). This suggested that unlike macroscopic desensitization, the effect of stabilizing O and D states on deactivation was independent of model connectivity, consistent with the idea that weighted deactivation time courses simply reflect the mean time receptors are agonist bound, which increases when O and/or D states are stabilized. Exceptions to this relationship between time bound and deactivation include irreversible microscopic desensitization, and small amplitude components not resolvable through baseline noise (neither case is considered here).
There were, however, notable quantitative differences between branched and linear deactivation landscapes. Compared to the branched model (Fig. 9A–D to Fig. 9E–H), stabilizing O and/or D states in the linear model resulted in slower deactivation (Fig. 9E–H, right corners). This was related to O and D states being directly interconnected, which allowed transitions without any chance of unbinding. It should also be noted that while deactivation was typically biphasic for both models, the magnitude and relative contribution of each phase depended greatly on state connectivity. This was particularly evident for deactivation following brief pulses (Supplemental Fig. 2), which are constrained by macroscopic desensitization (Fig. 6). Thus, while deactivation in the linear model had the most prominent fast phase when efficacy was high (which corresponded to extensive macroscopic desensitization; Fig. 8), the opposite was true for the branched model.
Interestingly, all deactivation landscapes obtained with the linear model trended in the same direction as their corresponding desensitization landscapes, suggesting that linear arrangements primarily supported desensitization–deactivation coupling (compare Fig. 9E–H with Fig. 8E–H). As with the branched model, however, changes in deactivation were observed even when corresponding grid positions lacked macroscopic desensitization, providing additional landscape regions of apparent uncoupling. These results suggest that neither coupling nor uncoupling was the rule per se for GABAA receptors. Instead, it appears that both phenomena are theoretically possible for any given kinetic scheme, the result of desensitization and deactivation having markedly different kinetic determinants.
Discussion
Coupling and uncoupling of macroscopic desensitization and deactivation
It is common practice to use the term ‘coupled’ to describe correlated observations, such as that of fast desensitization with slow deactivation. We described several experimental conditions that differed from the typical correlation of fast desensitization with slow deactivation, or of slow desensitization with fast deactivation. Such ‘uncoupling’ resulted from macroscopic desensitization and deactivation being differentially sensitive to changes in certain rate constants, and consequently, able to vary independently. We found the kinetic determinants of macroscopic desensitization to be complex, depending not only on the relationship between subsets of rate constants, but also on the specific connectivity of states.
While we limited our simulations to non-cyclic gating schemes, coupling and uncoupling are also theoretically possible for schemes containing cyclic features (Scheller & Forman, 2002), as macroscopic desensitization and deactivation have different kinetic determinants under these circumstances as well. We did not, however, systematically evaluate the behaviour of cyclic schemes, as they introduced a higher level of kinetic complexity to the simulations (for example, maintaining microscopic reversibility precluded alterations of individual rate constants). In addition, our prior data suggested that unbinding does not occur from fully liganded open or pre-open states (Bianchi & Macdonald, 2001a), which argues against the presence of cyclic features.
The relationship between macroscopic current shape and microscopic rate constants
Our simulations emphasized that macroscopic phenomena could not be attributed to individual states or rate constants; rather, they were shaped by all transitions within a given scheme, as well as the connectivity of states (Mozrzymas et al. 2003). As a result, the terminology used to describe an experimental observation may inappropriately suggest relevance to a microscopic process. For this reason, Colquhoun (1998) has argued against using ‘affinity’ or even ‘apparent affinity’ interchangeably with EC50. Similarly, the phenomenological description of fading current during continued agonist application as ‘desensitization’ implies relevance to microscopic D states; however, our simulations indicated that macroscopic desensitization provides little or no information about the fractional occupancy or stability of D states. Even ‘flat’ currents do not preclude existence of D states; indeed, the fractional occupancy of D states can actually exceed that of O states without causing macroscopic densitization (Fig. 7). Moreover, describing macroscopic desensitization requires observation of an initial peak amplitude, which may imply that microscopic desensitization is a kinetic process mechanistically preceded by channel opening (Colquhoun & Hawkes, 1995). Our simulations, however, demonstrated that D states may achieve substantial occupancy before currents reach peak, and therefore, before any loss of macroscopic current can be measured.
The influence of agonist affinity on macroscopic desensitization and deactivation
Both the extent and time course of macroscopic desensitization were relatively insensitive to changes in agonist affinity in the setting of a near-saturating GABA concentration. This occurred because the effective binding rate (kon×[GABA]) was orders of magnitude higher than the unbinding rate (koff), and as a result, changes in affinity could not affect the relative distribution of receptors in the gating scheme (which were essentially fully liganded). In contrast, deactivation was highly sensitive to agonist affinity, as the unbinding rate was an important determinant of the rate at which receptors returned to the resting state. While this represented one example of apparent uncoupling between desensitization and deactivation, it should be noted that altering affinity in the context of subsaturating agonist concentrations causes desensitization–deactivation coupling. Under these conditions, increasing agonist affinity not only prolongs deactivation, but also increases the extent of macroscopic desensitization (as if a higher concentration of agonist was applied; see Fig. 7).
Although the unbinding rate played a dominant role in shaping deactivation under certain circumstances, it was clear that multiple microscopic parameters contributed to the deactivation time course, including the unbinding rate constant, the entry and exit rates from O and D states, and the gating scheme connectivity. Interestingly, macroscopic desensitization constrained the effect of agonist affinity on deactivation by providing a slow limit to its time course. This constraint had particular relevance to brief (such as synaptic) pulses, where the fast phase of deactivation could not be slower than the fast phase of macroscopic desensitization. An unexpected consequence of this finding was that deactivation of currents lacking fast macroscopic desensitization should theoretically be unconstrained (Figs 2E and 6D), a concept that may be important for understanding the kinetic basis for prolonged deactivation in the slowly desensitizing but high affinity α4βδ and α6βδ isoforms. Thus, not only can prolonged deactivation occur without macroscopic desensitization, but the presence of fast macroscopic desensitization may actually accelerate early phases of deactivation.
The impact of altered gating efficacy on macroscopic desensitization and deactivation
Whether unbinding from the O state is assumed to be impossible (non-cyclic schemes) or less probable than unbinding from other states (cyclic schemes), increasing efficacy in branched arrangements is predicted to prolong deactivation (Fig. 9A and B). Since increased O state occupancy shifts the gating scheme towards the C–C–O arrangement (which cannot macroscopically desensitize), this reduces the extent of macroscopic desensitization (Fig. 8A and B), thereby providing an example of uncoupling. In the linear model, however, the concept of efficacy is more complex. Increasing efficacy can technically occur in two ways (since there are two routes for channel closure): increasing β/α or decreasing δ/ρ. Increasing β/α prolongs deactivation because of increased time spent in both O and D states. Increasing efficacy via decreasing δ/ρ, however, increases O state occupancy while decreasing D state occupancy. In our simulations, the changes in D state occupancy dominated, probably due to the more distal positioning of the D state relative to the unbinding step. Thus, unlike in the branched model, increasing efficacy in the linear scheme can either prolong (via β/α) or accelerate (via δ/ρ) deactivation. Either way, altered efficacy in the linear scheme is always associated with coupling. If efficacy is increased via increasing β/α, deactivation is prolonged and the extent of macroscopic desensitization is increased (Figs 8E and F and 9E and F). If efficacy is increased via decreasing δ/ρ, deactivation is accelerated and the extent of macroscopic desensitization is decreased (Figs 8G and H and 9G and H). Therefore, uncoupling in the context of altered efficacy necessitates a branched arrangement.
Several experimental observations argue against a purely linear kinetic arrangement for GABAA receptors. Barbiturates prolong IPSC duration via increased gating efficacy (Twyman et al. 1989) while decreasing the extent of macroscopic desensitization (Feng et al. 2004), consistent with a branched arrangement. Increased efficacy in the context of uncoupling due to mutations in the pore domain supports this idea (Figs 3 and 4). Although altered efficacy may be only one of several kinetic alterations caused by the α1(L9′S) mutation, preliminary simulations using the α1β3γ2L model (Fig. 5), whose O and D states are in branched arrangement, confirm that increasing efficacy can recapitulate the decreased macroscopic desensitization, prolonged deactivation following brief but not long applications, and slower rise time (Supplemental Fig. 3).
Predictive kinetic value of observed changes in macroscopic desensitization and deactivation
Although the complex kinetic basis underlying current shape essentially precludes direct extraction of microscopic parameters, the differential sensitivities of macroscopic desensitization and deactivation to various kinetic parameters can help distinguish between model arrangements and guide mechanistic interpretations (Fig. 10). For example, given an experimental observation of decreased macroscopic desensitization (from a mutation or allosteric modulator), changes in deactivation could be used to constrain the number of potentially responsible microscopic transitions. If deactivation was prolonged (uncoupling), this would be consistent with increased efficacy in a branched model, a prediction testable using single channel recording. However, if deactivation was accelerated (coupling), this could result from destabilization of the D state in either kinetic scheme or from decreased efficacy in the linear scheme. Comparing peak current amplitudes would then be useful, since destabilization of the D state and decreased efficacy should have opposite effects (increased and decreased peak current amplitudes, respectively). If peak current amplitude was increased, single channel studies could then be employed to distinguish between D state destabilization in branched and linear schemes. If the D state was destabilized in the branched model, then efficacy should be unchanged. If, however, the D state was destabilized in the linear model, then efficacy would be expected to increase.
Figure 10.

Flowchart for interpreting changes in GABAA receptor current kinetics The flowchart depicts an algorithmic approach for interpreting the effect of a kinetic perturbation (due to a mutation or allosteric modulator) on GABAA receptor macroscopic currents. By analysing macroscopic current properties (‘Des’, extent of desensitization; ‘Deact τ’, weighted deactivation time constant) and single channel kinetic properties, both the relevant microscopic transition (β/α, δ/ρ, kon/koff) and the arrangement of O and D states (branched or linear) can be determined assuming a simple 4-state kinetic scheme. The abbreviations ‘↑ Des’, ‘↓ Des’, and ‘= Des’ refer to increased, decreased, and unchanged extents of macroscopic desensitization, respectively, when receptors are activated by a nearly saturating GABA concentration. ‘↑ Deact τ’ and ‘↓ Deact τ’ refer to prolonged and shortened time courses of deactivation, respectively. *Indicates that for linear models, changes in either β/α or δ/ρ will change efficacy, since receptors can open and close via two independent pathways.
Single channel studies might also be helpful for distinguishing branched and linear arrangements in the special case of unchanged desensitization but altered deactivation, an example of uncoupling presumably reflecting altered microscopic affinity. In this case, evaluation of the concentration sensitivity of the closed time distribution can expose different arrangements of D states. Only linear arrangements can yield a fixed (concentration-independent) time constant, since only then do a subset of all closures correspond with certainty to sojourns in a single closed state (Colquhoun & Hawkes, 1982). In contrast, branched D states are directly connected with the pre-open state, which is directly connected with the unbound state. Thus, individual closures may include transitions among all three closed states, causing the time constants to depend on a complex relationship between all rate constants connecting these states. Since one of these rates (the effective binding rate) depends on the agonist concentration, all time constants will be concentration sensitive, even though entry and exit rates from the D state are not themselves concentration dependent. It should be noted that while this approach is valid in theory, in practice, even branched arrangements can appear to have fixed time constants when D states are very long-lived. Nonetheless, if all time constants in the closed time distribution vary with concentration, then it can be concluded that all non-conducting states in the gating scheme are connected.
Despite the mentioned limitations, the goal of this algorithmic approach was to collapse the spectrum of potential explanations for a given macroscopic observation into a smaller and potentially testable set of hypotheses. Although the observations outlined here represent characterization of simplified models that are likely to underestimate the complexity of GABAA receptor kinetics, it is clear that even in these simplified cases, macroscopic current interpretation can be complex or even counterintuitive. The likelihood that GABAA receptors obey even more complex kinetic rules emphasizes the necessity for establishing more quantitative frameworks to transition from macroscopic observations to microscopic models.
Acknowledgments
This work was supported by NIH R01-NS 33300 to R.L.M., NIH-NINDS NS 045950 to J.L.F., and by the PHS Award T32-GM07347 from the National Institute of General Medical Studies to the Vanderbilt Medical Scientist Training Program.
Supplementary material
Online supplemental material for this paper can be accessed at:
http://jp.physoc.org/cgi/content/full/jphysiol.2007.142364/DC1
and
http://www.blackwell-synergy.com/doi/suppl/10.1113/jphysiol.2007.142364
References
- Bai D, Pennefather PS, MacDonald JF, Orser BA. The general anesthetic propofol slows deactivation and desensitization of GABAA receptors. J Neurosci. 1999;19:10635–10646. doi: 10.1523/JNEUROSCI.19-24-10635.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barberis A, Mozrzymas JW, Ortinski PI, Vicini S. Desensitization and binding properties determine distinct α1β2γ2 and α3β2γ2 GABAA receptor-channel kinetic behavior. Eur J Neurosci. 2007;25:2726–2740. doi: 10.1111/j.1460-9568.2007.05530.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi MT, Haas KF, Macdonald RL. Structural determinants of fast desensitization and desensitization-deactivation coupling in GABAA receptors. J Neurosci. 2001;21:1127–1136. doi: 10.1523/JNEUROSCI.21-04-01127.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi MT, Haas KF, Macdonald RL. α1 and α6 subunits specify distinct desensitization, deactivation and neurosteroid modulation of GABAA receptors containing the δ subunit. Neuropharmacol. 2002;43:492–502. doi: 10.1016/s0028-3908(02)00163-6. [DOI] [PubMed] [Google Scholar]
- Bianchi MT, Macdonald RL. Agonist trapping by GABAA receptor channels. J Neurosci. 2001a;21:9083–9091. doi: 10.1523/JNEUROSCI.21-23-09083.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi MT, Macdonald RL. Mutation of the 9′ leucine in the GABAA receptor γ2L subunit produces an apparent decrease in desensitization by stabilizing open states without altering desensitized states. Neuropharmacology. 2001b;41:737–744. doi: 10.1016/s0028-3908(01)00132-0. [DOI] [PubMed] [Google Scholar]
- Bianchi MT, Macdonald RL. Slow phases of GABAA receptor desensitization: structural determinants and possible relevance for synaptic function. J Physiol. 2002;544:3–18. doi: 10.1113/jphysiol.2002.020255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhr A, Bianchi MT, Baur R, Courtet P, Pignay V, Boulenger JP, Gallati S, Hinkle DJ, Macdonald RL, Sigel E. Functional characterization of the new human GABAA receptor mutation β3 (R192H) Hum Genet. 2002;111:154–160. doi: 10.1007/s00439-002-0766-7. [DOI] [PubMed] [Google Scholar]
- Burkat PM, Yang J, Gingrich KJ. Dominant gating governing transient GABAA receptor activity: a first latency and P (o/o) analysis. J Neurosci. 2001;21:7026–7036. doi: 10.1523/JNEUROSCI.21-18-07026.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celentano JJ, Hawkes AG. Use of the covariance matrix in directly fitting kinetic parameters: application to GABAA receptors. Biophys J. 2004;87:276–294. doi: 10.1529/biophysj.103.036632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celentano JJ, Wong RKS. Multiphasic desensitization of the GABAA receptor in outside-out patches. Biophys J. 1994;66:1039–1050. doi: 10.1016/S0006-3495(94)80885-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Ghansah E, Chen Y, Ye J, Weiss DS. Desensitization mechanism of GABA receptors revealed by single oocyte binding and receptor function. J Neurosci. 2002;22:7982–7990. doi: 10.1523/JNEUROSCI.22-18-07982.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Weiss DS. Allosteric activation mechanism of the α1β2γ2 γ-aminobutyric acid type A receptor revealed by mutation of the conserved M2 leucine. Biophys J. 1999;77:2542–2551. doi: 10.1016/s0006-3495(99)77089-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D. Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol. 1998;125:924–947. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Hawkes AG. On the stochastic properties of bursts of single ion channel openings and of clusters of bursts. Philos Trans R Soc Lond B Biol Sci. 1982;300:1–59. doi: 10.1098/rstb.1982.0156. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Hawkes AG. Desensitization of N-methyl-D-aspartate receptors: a problem of interpretation. Proc Natl Acad Sci U S A. 1995;92:10327–10329. doi: 10.1073/pnas.92.22.10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng HJ, Bianchi MT, Macdonald RL. Pentobarbital differentially modulates α1β3δ and α1β3γ2L GABA-A receptor currents. Mol Pharmacol. 2004;66:988–1003. doi: 10.1124/mol.104.002543. [DOI] [PubMed] [Google Scholar]
- Feng HJ, Macdonald RL. Multiple actions of propofol on αβγ and αβδ GABAA receptors. Mol Pharmacol. 2004;66:1517–1524. doi: 10.1124/mol.104.003426. [DOI] [PubMed] [Google Scholar]
- Filatov GN, White MW. The role of conserved leucines in the M2 domain of the acetylcholine receptor in channel gating. Mol Pharmacol. 1995;48:379–384. [PubMed] [Google Scholar]
- Fisher JL. The α1 and α6 subunit subtypes of the mammalian GABAA receptor confer distinct channel gating kinetics. J Physiol. 2004;561:433–448. doi: 10.1113/jphysiol.2003.051839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher JL, Zhang J, Macdonald RL. The role of α1 and α6 subtype amino-terminal domains in allosteric regulation of γ-aminobutyric acid A receptors. Mol Pharmacol. 1997;52:714–724. doi: 10.1124/mol.52.4.714. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Hestrin S. Properties of GABAA receptors underlying inhibitory synaptic currents in neocortical pyramidal neurons. J Neurosci. 1997;17:7220–7227. doi: 10.1523/JNEUROSCI.17-19-07220.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosman C, Auerbach A. Kinetic, mechanistic, and structural aspects of unliganded gating of acetylcholine receptor channels. J Gen Physiol. 2000;115:621–635. doi: 10.1085/jgp.115.5.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosman C, Auerbach A. The dissociation of acetylcholine from open nicotinic receptor channels. Proc Natl Acad Sci U S A. 2001;98:14102–14107. doi: 10.1073/pnas.251402498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas KF, Macdonald RL. GABAA receptor subunit γ2 and δ subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol. 1999;514:27–45. doi: 10.1111/j.1469-7793.1999.027af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle DJ, Macdonald RL. B subunit phoshorylation selectively increases fast desensitization and prolongs deactivation of α1β1γ2L and α1β3γ2L GABAA receptor currents. J Neurosci. 2003;23:11698–11710. doi: 10.1523/JNEUROSCI.23-37-11698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Sahara Y, Dzubay JA, Westbrook GL. Defining affinity with the GABAA receptor. J Neurosci. 1998;18:8590–8604. doi: 10.1523/JNEUROSCI.18-21-08590.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL. The impact of receptor desensitization on fast synaptic transmission. Trends Neurosci. 1996;19:96–101. doi: 10.1016/s0166-2236(96)80037-3. [DOI] [PubMed] [Google Scholar]
- Katz B, Thesleff S. A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol. 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labarca C, Nowak MW, Zhang H, Tang L, Deshpande P, Lester HA. Channel gating governed symmetrically by conserved leucine residues in the M2 domain of nicotinic receptors. Nature. 1995;376:514–516. doi: 10.1038/376514a0. [DOI] [PubMed] [Google Scholar]
- Lagrange AH, Botzolakis EJ, Macdonald RL. Enhanced macroscopic desensitization shapes the response of α4 subtype-containing GABAA receptors to synaptic and extrasynaptic GABA. J Physiol. 2007;578:655–676. doi: 10.1113/jphysiol.2006.122135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Pearce R. Effects of halothane on GABAA receptor kinetics: evidence for slowed agonist unbinding. J Neurosci. 2000;20:899–907. doi: 10.1523/JNEUROSCI.20-03-00899.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercik K, Pytel M, Cherubini E, Mozrzymas JW. Effect of extracellular pH on recombinant α1β2γ2 and α1β2 GABAA receptors. Neuropharmacology. 2006;51:305–314. doi: 10.1016/j.neuropharm.2006.03.023. [DOI] [PubMed] [Google Scholar]
- Mortensen M, Kristiansen U, Ebert B, Frolund B, Krogsgaard-Larsen P, Smart TG. Activation of single heteromeric GABAA receptor ion channels by full and partial agonists. J Physiol. 2004;557:389–413. doi: 10.1113/jphysiol.2003.054734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozrzymas JW, Barberis A, Mercik K, Zarnowska ED. Binding sites, singly bound states, and conformational coupling shape GABA-evoked currents. J Neurophysiol. 2003;89:871–883. doi: 10.1152/jn.00951.2002. [DOI] [PubMed] [Google Scholar]
- Overstreet LS, Jones MV, Westbrook GL. Slow desensitization regulates the availability of synaptic GABAA receptors. J Neurosci. 2000;20:7914–7921. doi: 10.1523/JNEUROSCI.20-21-07914.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revah F, Bertrand D, Galzi JL, Devillers-Thiery A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux JP. Mutations in the channel domain alter desensitization of a neuronal nicotinic receptor. Nature. 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- Saxena NC, Macdonald RL. Properties of putative cerebellar gamma-aminobutyric acid A receptor isoforms. Mol Pharmacol. 1996;49:567–579. [PubMed] [Google Scholar]
- Scheller M, Forman SA. Coupled and uncoupled gating and desensitization effects by pore domain mutations in GABAA receptors. J Neurosci. 2002;22:8411–8421. doi: 10.1523/JNEUROSCI.22-19-08411.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson S-A, Smith MZ, Wingrove PB, Whiting PJ, Wafford KA. Mutation at the putative GABAA ion channel gate reveals changes in allosteric modulation. Br J Pharmacol. 1999;127:1349–1358. doi: 10.1038/sj.bjp.0702687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tia S, Wang JF, Kotchabhakdi N, Vicini S. Distinct deactivation and desensitization kinetics of recombinant GABAA receptors. Neuropharmacology. 1997;35:1375–1382. doi: 10.1016/s0028-3908(96)00018-4. [DOI] [PubMed] [Google Scholar]
- Twyman RE, Rogers CJ, Macdonald RL. Differential regulation of γ-aminobutyric acid receptor channels by diazepam and phenobarbital. Ann Neurol. 1989;25:213–220. doi: 10.1002/ana.410250302. [DOI] [PubMed] [Google Scholar]
- Twyman RE, Rogers CJ, Macdonald RL. Intraburst kinetic properties of the GABAA receptor main conductance state of mouse spinal cord neurones in culture. J Physiol. 1990;423:193–219. doi: 10.1113/jphysiol.1990.sp018018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakel JL, Lagrutta A, Adelman JP, North RA. Single amino acid substitution affects desensitization kinetics of the 5-hydroxytryptamine type 3 receptor expressed in Xenopus oocytes. Proc Natl Acad Sci U S A. 1993;90:5030–5033. doi: 10.1073/pnas.90.11.5030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang P, Jones BL, Henderson LP. Mechanisms of anabolic androgenic steroid modulation of α1β3γ2L GABAA receptors. Neuropharmacology. 2002;43:619–633. doi: 10.1016/s0028-3908(02)00155-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.