

Abstract
When the length of the myocardium is increased, a biphasic response to stretch occurs involving an initial rapid increase in force followed by a delayed slow increase called the slow force response (SFR). Confirming previous findings involving angiotensin II in the SFR, it was blunted by AT1 receptor blockade (losartan). The SFR was accompanied by an increase in reactive oxygen species (ROS) of ∼30% and in intracellular Na+ concentration ([Na+]i) of ∼2.5 mmol l−1 over basal detected by H2DCFDA and SBFI fluorescence, respectively. Abolition of ROS by 2-mercapto-propionyl-glycine (MPG) and EUK8 suppressed the increase in [Na+]i and the SFR, which were also blunted by Na+/H+ exchanger (NHE-1) inhibition (HOE642). NADPH oxidase inhibition (apocynin or DPI) or blockade of the ATP-sensitive mitochondrial potassium channels (5HD or glybenclamide) suppressed both the SFR and the increase in [Na+]i after stretch, suggesting that endogenous angiotensin II activated NADPH oxidase leading to ROS release by the ATP-sensitive mitochondrial potassium channels, which promoted NHE-1 activation. Supporting the notion of ROS-mediated NHE-1 activation, stretch increased the ERK1/2 and p90rsk kinases phosphorylation, effect that was cancelled by losartan. In agreement, the SFR was cancelled by inhibiting the ERK1/2 signalling pathway with PD98059. Angiotensin II at a dose that mimics the SFR (1 nmol l−1) induced an increase in ·O2− production of ∼30–40% detected by lucigenin in cardiac slices, an effect that was blunted by losartan, MPG, apocynin, 5HD and glybenclamide. Taken together the data suggest a pivotal role of mitochondrial ROS in the genesis of the SFR to stretch.
During myocardial stretch, as the length of the cardiac muscle increases, a corresponding rapid then slow increase in twitch force occurs (Parmley & Chuck, 1973; Allen & Kentish, 1985). The rapid change in force that forms the basis of the Frank–Starling mechanism is believed to be due to a sudden increase in myofilament Ca2+ responsiveness. The slow force response (SFR) was first described by Parmley & Chuck (1973) as an increase in contractility that developed over approximately 10 min after the initial rapid change. The SFR may possibly explain the Anrep effect which was first described by von Anrep (1912). The Anrep effect is the response to an elevation in aortic pressure, which involves an initial increase in ventricular volume followed by a subsequent decrease towards the original volume. Sarnoff et al. (1960) called this phenomenon ‘pressure-induced homeometric autoregulation’ and showed that catecholamines did not have a significant role in this mechanism. Interestingly, these authors defined ‘homeometric autoregulation’ as a phenomenon that occurred in an organ that was not attributable to nerves or chemical influences originating in the vicinity of that organ. Based on these observations, it was hypothesized that an autocrine/paracrine mechanism may be activated after stretching of the cardiac muscle.
The SFR has been detected in isolated myocytes (White et al. 1995), in strips of cardiac muscle (Parmley & Chuck, 1973; Kentish & Wrzosek, 1998; Alvarez et al. 1999; Pérez et al. 2001) as well as in isolated blood perfused hearts (Tucci et al. 1984; Burkhoff et al. 1991) and anaesthetized dogs (Lew, 1993). However, the mechanism by which the SFR develops is controversial. Based on pharmacological interventions, we have previously proposed that the myocardial stretch releases preformed angiotensin II (Ang II), which induces the release/formation of endothelin (ET) through AT1 receptors (Alvarez et al. 1999; Pérez et al. 2001). Ang II/ET activates intracellular signalling pathways that lead to activation of the Na+/H+ exchanger (NHE-1), and an increase in intracellular Na+ concentration ([Na+]i) followed by an increase in the Ca2+ transient through the Na+/Ca2+ exchanger (NCX). Our hypothesis concerning the role of the increase in [Na+]i in driving the reverse NCX has been confirmed by several authors (von Lewinski et al. 2004; Calaghan & White, 2004; Luers et al. 2005), but the mechanism that leads to this increase in [Na+]i has been challenged (Isenberg et al. 2005; Calaghan & White, 2004). If our hypothesis is correct, then the SFR is the result of a signalling cascade triggered by AT1 receptor activation.
Recent studies have suggested that reactive oxygen species (ROS) may act as intracellular signalling markers of Ang II/ET-1 in the myocardium (Sugden & Clerk, 2006) as well as after myocardial stretch (Pimentel et al. 2006). In addition, we have recently reported that in isolated cat myocytes, 1 nmol l−1 Ang II increased sarcomere shortening by approximately 30% and exerted their action entirely through an autocrine crosstalk with endogenous ET-1 (Cingolani et al. 2006). Furthermore, this effect was accompanied by an increase in ROS production and inhibited by the prevention of ROS formation. Here, we explore the possible participation of ROS in the generation of the SFR during myocardial stretch.
Methods
All procedures followed during this investigation conform to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication no. 85–23, revised 1996) and to the guidelines laid down by the Animal Welfare Committee of the La Plata School of Medicine. Cats (body weight 3–4 kg) were anaesthetized by intraperitoneal injection of sodium pentobarbitone (35 mg (kg body weight)−1). The chests were opened to excise the heart when deep anaesthesia was reached, verified by the loss of the corneal reflex and appearance of slow deep diaphragmatic breathing. Animals were provided by a local supplier (San Cayetano, Monte Grande, Argentina). Forty-six cats were used in the study. Each heart was used to provide papillary muscles as well as cardiac tissue slices.
Isolated papillary muscles
Isolation of cat papillary muscles
Papillary muscles from the right ventricle were used to assess the SFR to stretch as previously described (Pérez et al. 2001). Briefly, the muscles were mounted in a perfusion chamber placed on the stage of an inverted microscope (Olympus) for epifluorescence measurements and superfused at a constant rate (5 ml min−1) with a CO2/HCO3− -buffered solution containing (mmol l−1): NaCl 128.3, KCl 4.5, CaCl2 1.35, NaHCO3 20.23, MgSO4 1.05, glucose 11.0 and equilibrated with 5% CO2–95% O2 (pH ∼7.40). The possible participation of cathecholamines released by the nerve endings was prevented by adrenergic receptor blockade with 1.0 μmol l−1 prazosin plus 1.0 μmol l−1 atenolol. The muscles were paced at 0.2 Hz at a voltage 10% over threshold and maintained at 30°C, and isometric contractions were recorded. Cross-sectional area (calculated as 0.75 of the product of thickness by width) was used to normalize force records obtained with a silicon strain gauge (model AEM 801, SensoNor). The slack length of each muscle was determined after mounting, and then the muscles were progressively stretched to the length at which they developed maximal twitch force (Lmax). After a few minutes at Lmax, they were shortened to obtain the 95% of the maximal twitch force (length that approximated 98% of Lmax and referred to as L98). Then, the muscles were shortened to 92% of Lmax (L92) and maintained at this length until the beginning of the experimental protocol, during which the muscles were abruptly stretched from L92 to L98. In those experiments with pharmacological interventions, the drugs were added ∼20 min before the stretch. None of the drugs changed basal contractility significantly.
Determination of [Na+]i by epifluorescence
The papillary muscles were loaded with sodium-binding benzofuran isophthalate (SBFI, AM form). The esters were daily prepared as 1 mmol l−1 solution in dimethylsulfoxide (DMSO) plus Pluronic F-127 (20% w/w in DMSO) at a ratio of 4 : 1 to help disperse the indicators in the loading medium. Loading time lasted 2 h (SBFI final concentration 10 μmol l−1) at room temperature (22–24°C). The excitation wavelengths were 340 and 380 nm for SBFI and the emitted fluorescence was monitored after passage through a 535 ± 5 nm filter. The ratio 340/380 (SBFI) of emitted fluorescence was performed off-line after subtraction of the corresponding autofluorescence value at each wavelength. The values of [Na+]i were estimated from ‘in vivo’ calibrations of SBFI fluorescence performed at the end of each experiment according to Harootunian et al. (1989) with 2.0 μmol l−1 gramicidin, 5.0 μmol l−1 monensin and 0.05 mmol l−1 ouabaine.
Reactive oxygen species measurements
Intracellular ROS production was measured in isolated papillary muscles by epifluorescence of dichloride-hydrofluorescein diacetate (H2DCFDA) (Keller et al. 2004). The muscles were incubated with 20 μmol l−1 H2DCFDA at room temperature for 1 h. The muscles were excited at 495 nm and the emitted light collected after passage through a 535 ± 5 nm filter. Before obtaining the signal, background fluorescence was subtracted.
Cardiac tissue slices
Preparation of cardiac slices
Hearts were quickly removed from previously anaesthetized animals. The hearts were perfused through the aorta with Krebs–Ringer buffer pH 7.4 to eliminate the blood, and tissue slices from the left ventricle (1 × 5 mm) were cut and kept at 4°C until assayed. Assay buffer consisted of Krebs-Hepes buffer of the following composition (mmol l−1): 118.3 NaCl; 4.7 KCl; 1.8 CaCl2; 1.2 MgSO4; 1.0 K2HPO4; 25 NaHCO3; 11 glucose; 20 Hepes (pH 7.4 after 1.5 h aeration with 95% O2–5% CO2 at 37°C). Cardiac tissue were incubated in the assay buffer during 30 min in the presence of different drugs in a metabolic incubator under 95% O2–5% CO2 at 37°C before measurements of ·O2− production.
Measurements of superoxide anion (·O2−) production
We used lucigenin-enhanced chemiluminescence to measure ·O2− production by cat cardiac tissue in Krebs-Hepes buffer with 5 μmol l−1 lucigenin. The chemiluminescence in arbitrary units (AU) was recorded with a luminometer (Chameleon, Hidex) during 30 s each with 4.5 min interval during 30 min. The lucigenin-containing assay buffer with tissue slices minus background as well as responses to the different drugs assayed were reported. ·O2− production was normalized to milligrams dry weight tissue per minute. Control tissue slices without any pharmacological intervention produced low levels of ·O2− which was only slightly above background. Although the lucigenin enhanced chemiluminescence method does not allow accurate detection of basal ·O2− production (Dikalov et al. 2007), its sensitivity was enough to detect a dose-dependent increase in ·O2− production above the background after 1 and 100 nmol l−1 Ang II (see Results). When necessary the increase in ·O2− production was expressed as a percentage of basal value after 15 min of each intervention.
Determination of extracellular signal-regulated protein kinases (ERK1/2) and p90 ribosomal S6 kinase (p90rsk) phosphorylation after stretch
At the end of the protocols, the superfusion solution was quickly removed and papillary muscles were homogenized in lysis buffer: 300 mmol l−1 sacarose; 1 mmol l−1 DTT; 4 mmol l−1 EGTA, protease inhibitors cocktail (Complete Mini Roche); 20 mmol l−1 Tris-HCl, pH 7.4. After a brief centrifugation the supernatant was kept and protein concentration determined by the Bradford method. Samples were denatured and equal amounts of protein subjected to PAGE and electrotransferred to PVDF membranes. After blocking with non-fat dry milk membranes were incubated overnight either with anti-P-p90rsk or anti P-ERK1/2 polyclonal antibodies (Santa Cruz Biotechnology). A peroxidase-conjugated anti-rabbit IgG (Santa Cruz Biotechnology) was used as secondary antibody and finally bands were visualized using the ECL-Plus chemiluminescence detection system (Amersham). Autoradiograms were analysed by densitometric analysis (Scion Image).
Chemicals
All drugs used in the present study were of analytical grade. Ang II, lucigenin, 5-hydroxydecanoate (5HD), diphenyleneiodonium chloride (DPI), N-2-mercap-topropionyl-glycine (MPG) and polyethylene glycol-superoxide dismutase (PEG-SOD) were purchased from Sigma; glybenclamide was from RBI; apocynin was from Fluka; losartan was from Merck; HOE642 was from Aventis; EUK8 was from Calbiochem; PD98059 was from Biosource. Either assay buffer or dimethyl sulfoxide (DMSO) was used to prepare the drugs. For the superoxide assay the final DMSO concentration was kept < 0.5%, which did not interfere with the lucigenin signal.
Statistics
Data are expressed as means ±s.e.m. Differences between initial phase and poststretch records, comparison of ·O2− production after 15 min of incubation with the different drugs and ERK1/2 and p90rsk phosphorylation under the different conditions were assessed by one-way ANOVA followed by Student–Newman–Keuls test. Two-way ANOVA was used to compare post-stretch data in the absence and presence of drugs and the time course of different curves in the lucigenin experiments. P < 0.05 was considered significant.
Results
Figure 1A shows an original record of twitch-developed force from a cat papillary muscle that was subjected to an increase in length from L92 to L98. It can be seen that the SFR developed during the 10–15 min following the initial rapid increase in force. The SFR, depicted as a percentage of the initial rapid phase, was suppressed by inhibition of AT1 but not AT2 receptors (Fig. 1B). Previous studies from our laboratory have suggested that the SFR is the result of an autocrine/paracrine loop beginning with the release of Ang II and ending with an increase in the Ca2+ transient through the NCX, without changes in the myofilament Ca2+ responsiveness during its development (for review see Cingolani et al. 2005). The activation of reverse NCX that follows an increase in [Na+]i and is responsible for an increase in the Ca2+ transient has been described by several authors including ourselves for myocardial stretch (Alvarez et al. 1999; Pérez et al. 2001; von Lewinski et al. 2004; Calaghan & White, 2004; Luers et al. 2005) and for other experimental conditions (Weisser-Thomas et al. 2003; Pérez et al. 2003; Aiello et al. 2005; Bers et al. 2006). Interestingly, a mathematical model that analysed the potential contribution of sarcolemmal ion fluxes to the development of the SFR predicted that this route was a suitable mechanism for regulating the increase in Ca2+ entry into the cells after stretch, without involvement of the sarcoplasmic reticulum (Bluhm et al. 1998).
Figure 1.
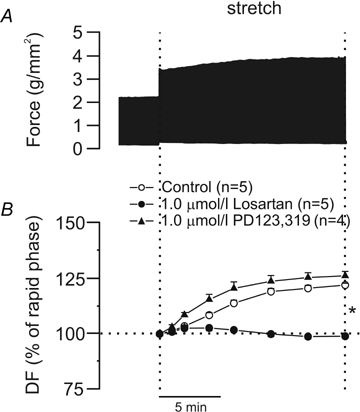
A, original force record from a papillary muscle subjected to an increase in length from L92 to L98; the biphasic response to stretch can be seen. B, suppression of the SFR (expressed as a percentage of the initial rapid phase) after AT1 but not AT2 receptor blockade (losartan and PD123,319, respectively). DF, developed force. *P < 0.05 control and PD123,319 versus Losartan.
The magnitude of the SFR can be mimicked by exogenous addition of 1.0 nmol l−1 Ang II and inhibition of the ET receptors abolishes this inotropic effect (Pérez et al. 2003). However, AT1 receptor blockade failed to abolish the inotropic response to an equipotent dose of exogenous ET-1 (Perez et al. 2003). Thus, the crosstalk between both peptides appears to be Ang II to ET and not the other way around. In addition, we have reported an increase in ET-1 mRNA after treatment with 1 nmol l−1 Ang II as well as the presence of preproET, bigET and ET in adult cat isolated myocytes (Cingolani et al. 2006).
Recently, we emphasized the role of ROS formation in activation of the intracellular signalling pathway triggered by physiological doses of Ang II/ET in the myocardium (Cingolani et al. 2006; Villa-Abrille et al. 2006). Since the SFR appears to be due to these peptides, we examined the effects of stretch upon ROS formation on cat papillary muscles by H2DCFDA fluorescence. Figure 2A shows that stretch, in addition to its mechanical effect, induced an increase in intracellular ROS formation of approximately 30% above baseline levels. Furthermore, scavenging of ROS by MPG (2.0 mmol l−1) or EUK8 (50 μmol l−1) inhibited both the stretch-induced increase in ROS (Fig. 2A) and the SFR (Fig. 2B). We also found that the scavenging of ROS inhibited the increase in [Na+]i that occurs in response to the stretch (Fig. 2C). Figure 3 shows inhibition of both the increase in [Na+]i and the SFR following NHE-1 blockade with HOE642 (1 μmol l−1).
Figure 2.
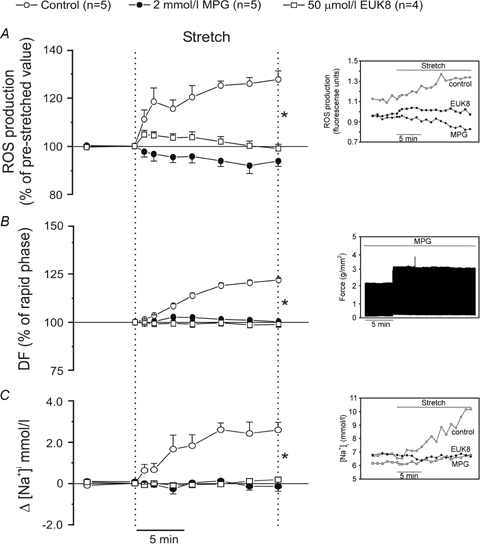
A, myocardial stretch induced an intracellular ROS increase of ∼30% above the baseline levels that was cancelled by the ROS scavengers MPG and EUK8. B, MPG and EUK8 also cancelled the SFR (expressed as percent of the initial rapid phase). C, furthermore, ROS scavenging also blunted the stretch-induced increase in [Na+]i. Insets show original raw data for ROS production (A), force (B) and intracellular [Na+] (C). DF, developed force. *P < 0.05 control versus MPG and EUK8.
Figure 3.
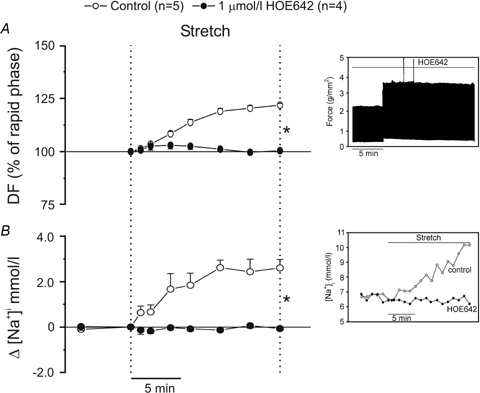
NHE-1 blockade with HOE642 cancelled both the SFR (expressed as a percentage of the initial rapid phase) (A) and the increase in [Na+]i (B) Insets show original raw data for force (A) and intracellular [Na+] (B). DF, developed force. *P < 0.05 control versus HOE642.
Previous studies have described a phenomenon called ‘ROS-induced ROS-release’, by which a small amount of ROS (not detected by the usual techniques) activates a larger ROS production from the mitochondria (Zorov et al. 2000; Kimura et al. 2005a). Based on the assumptions that Ang II is involved in the SFR to stretch and activates NADPH oxidase (NOX) (Lavigne et al. 2001; Kimura et al. 2005b), we hypothesized that activation of NOX after stretch would produce a small amount of ·O2−, which may open the ATP-sensitive mitochondrial potassium (mKATP) channels and produce a larger amount of ·O2− responsible for generating the SFR. Although there is no direct evidence to demonstrate that activation of NOX is required to open mKATP channels, previous studies showing that reconstituted mKATP channels are opened by ·O2− give support to this proposed mechanism (Zhang et al. 2001). Furthermore, other reports have proposed that an interaction between mKATP channels and NHE-1 inhibition may be involved in protection against myocardial ischaemia (Miura et al. 2001; Fantinelli et al. 2006). Therefore, if our assumptions are correct, the SFR should be abolished by either NOX inactivation or blockade of mKATP channels. As shown in Fig. 4A, the SFR was abolished after inhibition of NOX with apocynin or DPI or after blockade of mKATP channels with 5HD or glybenclamide. The NHE-1-induced increase in [Na+]i that accompanies the SFR was also abolished by these interventions (Fig. 4B).
Figure 4.
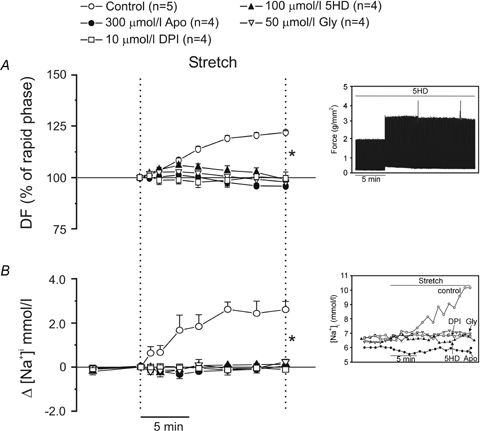
A, NOX inhibition by apocynin (Apo) or DPI as well as mKATP channel blockade with 5HD or glybenclamide (Gly) abolished the SFR (expressed as a percentage of the initial rapid phase). B, these interventions also cancelled the NHE-1-induced increase in [Na+]i that accompanied the SFR. Insets show original raw data for force (A) and intracellular [Na+] (B). DF, developed force. *P < 0.05 control versus all other groups.
Previous studies have shown that the sarcoplasmic reticulum is not involved in the generation of the SFR (Bluhm & Lew, 1995; Hongo et al. 1995; Kentish & Wrzosek, 1998). Based on these studies and on the findings reported here, it would be reasonable to conclude that mitochondrial ·O2− or H2O2 production after stretch may lead to phosphorylation and activation of the NHE-1. It has previously been shown that activation of the NHE-1 after stretch is followed by an increase in [Na+]i without significant changes in intracellular pH when bicarbonate is present in the medium, due to simultaneous activation of the Na+-independent Cl−/HCO3− exchanger (Alvarez et al. 1999; Pérez et al. 2001). ROS-mediated activation of the NHE-1 is reportedly due to kinase-mediated phosphorylation of the cytosolic tail of the exchanger, with MEK, ERK1/2 and p90rsk kinases being the most likely candidates that mediate this effect (Rothstein et al. 2002). With respect to these findings, RAS-dependent activation of these kinases has been recently reported after stretch in neonatal cardiomyocytes (Pimentel et al. 2006). Here, we detect a significant increase in ERK1/2 and p90rsk phosphorylation after 15 min of stretch (Fig. 5A). This effect was abolished by treatment with 1 μmol l−1 losartan (Fig. 5A). In addition, we show that inhibition of MEK (a kinase that is upstream of ERK1/2 and downstream of RAS) by PD98059 (50 μmol l−1) also abolished the SFR (Fig. 5B).
Figure 5.
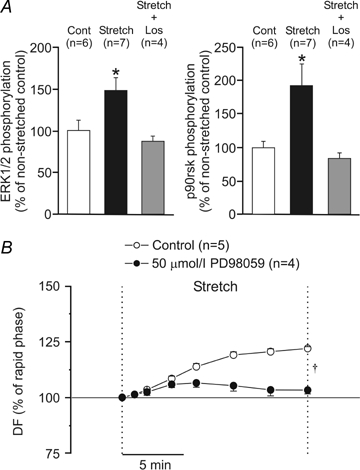
A, myocardial stretch significantly increased ERK1/2 and p90rsk phosphorylation, an effect that was cancelled by losartan (Los). B, inhibition of MEK (a kinase upstream ERK1/2 and downstream RAS) by PD98059 cancelled the SFR (expressed as a percentage of the initial rapid phase). DF, developed force. *P < 0.05 versus non-stretched control (cont); †P < 0.05 control versus PD98059.
Taken together, these data appear to support the hypothesis that the stretch triggers the release/formation of Ang II/ET, which in an autocrine/paracrine manner leads to NOX activation, ·O2− production, opening of mKATP channels and increased mitochondrial ·O2− production. The mitochondrial ·O2− or H2O2 (after dismutation) may activate the ERK1/2–p90rsk pathway leading to phosphorylation and activation of the NHE-1, thereby increasing [Na+]i and generating the SFR through the NCX. It is known that the increase in [Na+]i can induce an increase in intracellular Ca2+ levels through the NCX as a result of a decrease in Ca2+ efflux (decreased forward mode) and/or an increase in Ca2+ entry (increased reverse mode). If our hypothesis is correct, then small doses of exogenous Ang II should increase ·O2− production in a NOX and mKATP channel-dependent manner. In addition, this response should be inhibited by losartan, apocynin, 5HD and glybenclamide. To test this hypothesis, we examined the effect of exogenous Ang II on ·O2− production (determined by the lucigenin chemiluminescence method) in cat myocardial slices (Fig. 6). As shown in Fig. 6A, Ang II (1 and 100 nmol l−1) induced a dose-dependent increase in ·O2− production which was blocked by losartan (1 μmol l−1) and MPG (2 mmol l−1). These compounds did not affect basal background levels (Fig. 6B). Figure 7 shows that at this dose the Ang II-induced increase in the rate of ·O2− production was NOX dependent since it was suppressed by apocynin (300 μmol l−1). The NOX-dependent increase in ·O2− production was also abolished by mKATP channel blockade with 5HD (100 μmol l−1) or glybenclamide (50 μmol l−1).
Figure 6.
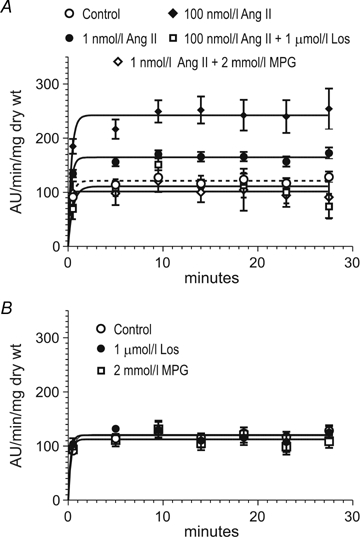
A, dose-dependent effect of 1 nmol l−1 (n = 34) and 100 nmol l−1 Ang II (n = 14) on superoxide generation by myocardial slices. Ang II (1 and 100 nmol l−1) significantly increased superoxide production above the control lucigenin level (tissue minus background) (P < 0.05). The effect of 100 nmol l−1 Ang II was abolished in the presence of losartan (Los, n = 5) and MPG (n = 3) blunted the superoxide production induced by 1 nmol l−1 Ang II. B, luminescence values of the tissue in the absence of drugs (Control, n = 32), and in the presence of Los (n = 8) and MPG (n = 7). Note that the basal value was unaffected by the scavenger MPG, demonstrating that it was not due to a basal ·O2− production by the tissue.
Figure 7.
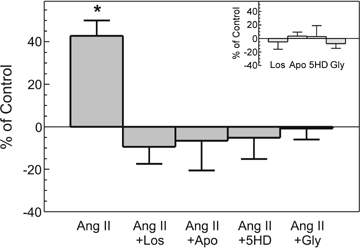
Superoxide production induced by 1 nmol l−1 Ang II (n = 34) in the absence and presence of 1 μmol l−1 losartan (Los, n = 8), 300 μmol l−1 apocynin (Apo, n = 7), 100 μmol l−1 5HD (n = 10) and 50 μmol l−1 glybenclamide (Gly, n = 6). Since superoxide production stabilized after 15 min (see Fig. 6), the data obtained at this time are expressed as a percentage of control values without additions. Inset shows the lack of effect of all drugs used upon the basal measurement. *P < 0.05 versus control.
Since changes in ROS levels were measured by H2DCFDA fluorescence, we were unable to determine whether the intracellular mediator that led to the activation of kinases responsible for the NHE activation was ·O2− or H2O2. NOX-generated ·O2− is very unstable and rapidly converted into H2O2 by the superoxide dismutase (SOD), a more stable membrane permeant ROS, widely recognized as a signalling molecule. In the myocardium, dismutation is performed by a membrane bound extracellular SOD as well as by cytoplasmic CuZn and mitochondrial Mn isoforms of SOD. Therefore, the ·O2− detected in cardiac slices may be easily converted into H2O2 at different cellular locations upstream or downstream of the mitochondria.
If the intracellular signal activating the SFR is H2O2 and not ·O2−, then dismutation of ·O2− to H2O2 would presumably enhance this effect. In contrast, the SFR would decrease if the intracellular signalling pathway involved ·O2−. Figure 8 shows that when the papillary muscles were stretched in the presence of PEG-SOD (100 units ml−1), the SFR almost doubled compared with controls. Although addition of PEG-SOD produced a negative inotropic effect in three out of six muscles, the SFR was always of the same order of magnitude, i.e. approximately 45% higher than the initial rapid phase. The larger SFR under these conditions suggests that dismutation of intracellular ·O2− to H2O2 induced the positive inotropic effect. Experiments by Sabri et al. (1998) and Rothstein et al. (2002) indicated that H2O2 is the intracellular signal leading to the activation of kinases that phosphorylate the NHE.
Figure 8.
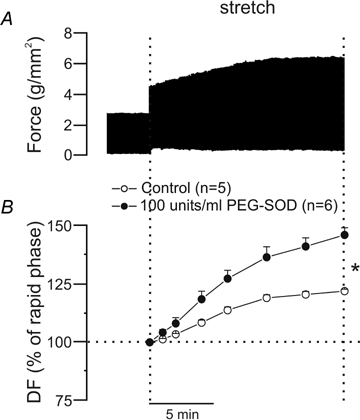
A, original force record from a papillary muscle subjected to an increase in length from L92 to L98 in the presence of 100 units ml−1 of PEG-SOD; there is a greater SFR than that shown under control conditions (see Fig. 1). As stated in the main text, although addition of PEG-SOD produced a negative inotropic effect in three out of six muscles, the SFR was always of the same order of magnitude. The original record shown here belongs to a muscle in which no decrease in force after PEG-SOD addition was observed. B, averaged results for the SFR (expressed as a percentage of the initial rapid phase) under control conditions and after preincubation with PEG-SOD. DF, developed force. *P < 0.05 control versus PEG-SOD.
Discussion
During the last few years, the view about the deleterious effect of ROS has changed with the recognition that both ·O2− and H2O2 can play important roles in signal transduction through specific modification of signalling proteins. Here, we present evidence that a known physiological phenomenon, the Anrep effect, which can be reproduced in myocardial strips and is represented by the SFR, is the result of mitochondrial-derived ROS targeting the NHE-1. Previous reports by our group (Alvarez et al. 1999; Pérez et al. 2001) and others (Yamazaki et al. 1998; Calaghan & White, 2004; von Lewinski et al. 2004) have proposed that the NHE-1 has a role in the development of the SFR after myocardial stretch.
In this study, we show that the SFR and the increase in [Na+]i that follows myocardial stretch can be abolished by ROS scavenging. In addition, we report that myocardial stretch induced an AT1-dependent increase in ERK1/2 and p90rsk phosphorylation. These findings indirectly support the theory that the myocardial stretch activates the NHE-1. We also show that doses of Ang II that mimic the SFR increase ·O2− production in a NOX-dependent manner. Our data suggest that small and/or localized amounts of ·O2− produced after NOX activation by Ang II induce a greater increase in ·O2− production as a consequence of mKATP channels opening, since this effect was not observed in the presence of the mKATP channel blockers, 5HD and glybenclamide. Furthermore, 5HD and glybenclamide also abolished the SFR and NHE-1 mediated increase in [Na+]i. Our findings suggest that activation of the NHE-1 probably occurred after ERK1/2 and p90rsk phosphorylation, since the increased phosphorylation of both kinases after stretch was abolished by inhibition of the AT1 receptors.
The activation of NHE-1 by H2O2 is reportedly the result of kinases that lead to phosphorylation of the cytosolic tail of the exchanger (Sabri et al. 1998; Rothstein et al. 2002). MEK, ERK1/2 and p90rsk kinases are all suitable candidates that could potentially mediate this effect. Interestingly, a recent study in neonates reported RAS-dependent activation of these kinases after stretch (Pimentel et al. 2006). Sabri et al. (1998) showed that NHE activity was stimulated after neonatal rat ventricular myocytes were briefly exposed to H2O2 and that this effect was completely abolished by pretreatment with the MEK inhibitor, PD98059. Furthermore, it has been reported that H2O2-induced Ca2+ overload was partially mediated by NHE-1 activation after phosphorylation of the exchanger by the ERK1/2 MAP kinase pathway in neonatal rat ventricular myocytes (Rothstein et al. 2002). Taken together, our data and these previously reported findings support the chain of events schematized in Fig. 9.
Figure 9.
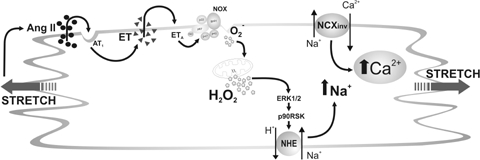
Schematic proposed mechanism leading to the SFR Myocardial stretch induces release of preformed Ang II, which through AT1 receptors promotes release/formation of ET. ET, by activation of NOX generates ·O2− which by a ‘ROS-induced ROS-release’ mechanism produces a greater amount of ·O2− from the mitochondria, which activates the ERK1/2–p90rsk cascade leading to NHE-1 phosphorylation, increasing [Na+]i through the NHE-1 and Ca2+ through the NCX. The fact that a low dose of Ang II induces its effects entirely by endogenous ET encourages us to propose ET and not Ang II as the activator of NOX. However, we cannot rule out the possibility of some additional direct action of Ang II on NOX at higher Ang II doses.
Many studies have described the release of Ang II after stretch. Sadoshima et al. (1993) were the first to demonstrate that Ang II was the autocrine/paracrine mediator of stretch-induced hypertrophy in neonatal cardiomyocytes. At the same time, Ito et al. (1993) found that Ang II promoted the release/formation of ET-1 in neonatal cardiomyocytes. These observations suggested that ET-1 was an autocrine/paracrine factor in the mechanism of Ang II-induced cardiac hypertrophy. In addition, it was also reported that stretch induced a rise in the ET-1 concentration in the culture medium of neonates (Yamazaki et al. 1996). Interestingly, Leri et al. (1998) showed that stretch induced release of Ang II in adult cardiomyocytes, whereas Leskinen et al. (1997) demonstrated pharmacologically that Ang II and ET were autocrine/paracrine mediators released after volume expansion of the adult rat heart. Here, we show that in the adult multicellular cat papillary muscle, increased phosphorylation of kinases known to be activated by Ang II/ET that target the NHE-1 occurred after myocardial stretch in an AT1 receptor-dependent manner and through a mechanism that appears to involve mitochondrial ROS. Taken together our data suggest a pivotal role of mitochondrial ROS in the genesis of the SFR to stretch.
Acknowledgments
This work was supported in part by grants PIP 5141 from Consejo Nacional de Investigaciones Científicas y Técnicas and PICT 25475 from Agencia Nacional de Promoción Científica of Argentina to Dr Horacio E. Cingolani. C. D. Garciarena, R. A. Dulce and A. M. Yeves, are Fellows of Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina. I. L. Ennis, H. E. Cingolani, G. Chiappe de Cingolani and N. G. Pérez are Established Investigators of CONICET, Argentina.
References
- Aiello EA, Villa-Abrille MC, Dulce RA, Cingolani HE, Pérez NG. Endothelin-1 stimulates the Na+/Ca2+ exchanger reverse mode through intracellular Na+ (Na+i)-dependent and Na+i-independent pathways. Hypertension. 2005;45:288–293. doi: 10.1161/01.HYP.0000152700.58940.b2. [DOI] [PubMed] [Google Scholar]
- Allen DG, Kentish JC. The cellular basis of the length-tension relationship in cardiac muscle. J Mol Cell Cardiol. 1985;17:821–840. doi: 10.1016/s0022-2828(85)80097-3. [DOI] [PubMed] [Google Scholar]
- Alvarez BV, Pérez NG, Ennis IL, Camilión de Hurtado MC, Cingolani HE. Mechanisms underlying the increase in force and calcium transient that follows stretch of cardiac muscle: a possible explanation of the Anrep effect. Circ Res. 1999;85:716–722. doi: 10.1161/01.res.85.8.716. [DOI] [PubMed] [Google Scholar]
- Bers DM, Despa S, Bossuyt J. Regulation of Ca2+ and Na+ in normal and failing cardiac myocytes. Ann N Y Acad Sci. 2006;1080:165–177. doi: 10.1196/annals.1380.015. [DOI] [PubMed] [Google Scholar]
- Bluhm WF, Lew WY. Sarcoplasmic reticulum in cardiac length-dependent activation in rabbits. Am J Physiol Heart Circ Physiol. 1995;269:H965–H972. doi: 10.1152/ajpheart.1995.269.3.H965. [DOI] [PubMed] [Google Scholar]
- Bluhm WF, Lew WY, Grafinkel A, McCulloch AD. Mechanism of length history-dependent tension in an ionic model of the cardiac myocyte. Am J Physiol Heart Circ Physiol. 1998;274:H1032–H1040. doi: 10.1152/ajpheart.1998.274.3.H1032. [DOI] [PubMed] [Google Scholar]
- Burkhoff D, de Tombe PP, Hunter WC, Kass DA. Contractile strength and mechanical efficiency of left ventricle are enhanced by physiological afterload. Am J Physiol Heart Circ Physiol. 1991;260:H569–H578. doi: 10.1152/ajpheart.1991.260.2.H569. [DOI] [PubMed] [Google Scholar]
- Calaghan SC, White E. Activation of Na+-H+ exchange and stretch-activated channels underlies the slow inotropic response to stretch in myocytes and muscle from the rat heart. J Physiol. 2004;559:205–214. doi: 10.1113/jphysiol.2004.069021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani HE, Pérez NG, Aiello EA, Camilión de Hurtado MC. Intracellular signalling following myocardial stretch: An autocrine/paracrine loop. Regulatory Peptides. 2005;128:211–220. doi: 10.1016/j.regpep.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Cingolani HE, Villa-Abrille MC, Cornelli M, Nolly A, Ennis IL, Garciarena C, Suburo AM, Torbidoni V, Correa MV, Camilion de Hurtado MC, Aiello EA. The positive inotropic effect of angiotensin II. Role of endothelin-1 and reactive oxygen species. Hypertension. 2006;47:727–734. doi: 10.1161/01.HYP.0000208302.62399.68. [DOI] [PubMed] [Google Scholar]
- Dikalov S, Griendling KK, Harrison DG. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49:717–727. doi: 10.1161/01.HYP.0000258594.87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantinelli JC, Cingolani HE, Mosca SM. Na+/H+ exchanger inhibition at the onset of reperfusion decreases myocardial infarct size: role of reactive oxygen species. Cardiovasc Pathol. 2006;15:179–184. doi: 10.1016/j.carpath.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Harootunian AT, Kao JPY, Eckert BK, Tsien RY. Fluorescence ratio imaging of cytosolic free Na+ in individual fibroblasts and lymphocytes. J Biol Chem. 1989;264:19449–19457. [PubMed] [Google Scholar]
- Hongo K, White E, Orchard CH. Effect of stretch on contraction and the Ca2+ transient in ferret ventricular muscles during hypoxia and acidosis. Am J Physiol Cell Physiol. 1995;269:C690–C697. doi: 10.1152/ajpcell.1995.269.3.C690. [DOI] [PubMed] [Google Scholar]
- Isenberg G, Kondratev D, Dyachenko V, Kazanski V, Gallitelli MF. Isolated cardiomyocytes: Mechanosesitivity of action potential, membrane current and ion concentration. In: Kamkin A, Kisileva I, editors. Mechanosensitivity in cells and tissues. Moscow: Academia; 2005. pp. 126–164. [PubMed] [Google Scholar]
- Ito H, Hirata Y, Adachi S, Tanaka M, Tsujino M, Koike A, Nogami A, Murumo F, Hiroe M. Endothelin-1 is an autocrine/paracrine factor in the mechanism of angiotensin II-induced hypertrophy in cultured rat cardiomyocytes. J Clin Invest. 1993;92:398–403. doi: 10.1172/JCI116579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Mohamed A, Drose S, Brandt U, Fleming I, Brandes RP. Analysis of dichlorodihydrofluorescein and dihydrocalcein as probes for the detection of intracellular reactive oxygen species. Free Radic Res. 2004;38:1257–1267. doi: 10.1080/10715760400022145. [DOI] [PubMed] [Google Scholar]
- Kentish JC, Wrzosek A. Changes in force and cytosolic Ca2+ concentration after length changes in isolated rat ventricular trabeculae. J Physiol. 1998;506:431–444. doi: 10.1111/j.1469-7793.1998.431bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Abe Y. Mitochondrial-derived reactive oxygen species and vascular MAP kinases. Hypertension. 2005a;45:438–444. doi: 10.1161/01.HYP.0000157169.27818.ae. [DOI] [PubMed] [Google Scholar]
- Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Suzuki T, Maeta H, Abe Y. Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension. 2005b;45:860–866. doi: 10.1161/01.HYP.0000163462.98381.7f. [DOI] [PubMed] [Google Scholar]
- Lavigne MC, Malech HL, Holland SM, Leto TL. Genetic demonstration of p47phox-dependent superoxide anion production in murine vascular smooth muscle cells. Circulation. 2001;104:79–84. doi: 10.1161/01.cir.104.1.79. [DOI] [PubMed] [Google Scholar]
- Leri A, Claudio PP, Li Q, Wang X, Reiss K, Wang S, Malhotra A, Kajstura J, Anversa P. Stretch-mediated release of angiotensin II induces myocyte apoptosis by activating p53 that enhances the local renin-angiotensin system and decreases the Bcl-2-to-Bax protein ratio in the cell. J Clin Invest. 1998;101:1326–1342. doi: 10.1172/JCI316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leskinen H, Vuolteenaho O, Ruskoaho H. Combined inhibition of endothelin and angiotensin II receptors blocks volume load-induced cardiac hormone release. Circ Res. 1997;80:114–123. doi: 10.1161/01.res.80.1.114. [DOI] [PubMed] [Google Scholar]
- Lew WYW. Mechanisms of volume-induced increase in left ventricular contractility. Am J Physiol Heart Circ Physiol. 1993;265:H1778–H1786. doi: 10.1152/ajpheart.1993.265.5.H1778. [DOI] [PubMed] [Google Scholar]
- Luers C, Fialka F, Elgner A, Zhu D, Kockskamper J, von Lewinski D, Pieske B. Stretch-dependent modulation of [Na+]i, [Ca2+]i, and pHi in rabbit myocardium – a mechanism for the slow force response. Cardiovasc Res. 2005;68:454–463. doi: 10.1016/j.cardiores.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Miura T, Liu Y, Goto M, Tsuchida A, Miki T, Nakano A, Nishino Y, Ohnuma Y, Shimamoto K. Mitochondrial ATP-sensitive K+ channels play a role in cardioprotection by Na+-H+ exchange inhibition against ischemia/reperfusion injury. J Am Coll Cardiol. 2001;37:957–963. doi: 10.1016/s0735-1097(00)01183-9. [DOI] [PubMed] [Google Scholar]
- Parmley WW, Chuck L. Length-dependent changes in myocardial contractile state. Am J Physiol. 1973;224:1195–1199. doi: 10.1152/ajplegacy.1973.224.5.1195. [DOI] [PubMed] [Google Scholar]
- Pérez NG, de Hurtado MC, Cingolani HE. Reverse mode of the Na+-Ca2+ exchange after myocardial stretch: underlying mechanism of the slow force response. Circ Res. 2001;88:376–382. doi: 10.1161/01.res.88.4.376. [DOI] [PubMed] [Google Scholar]
- Pérez NG, Villa-Abrille MC, Aiello EA, Dulce RA, Cingolani HE, Camilión de Hurtado MC. A low dose of angiotensin II increases inotropism through activation of reverse Na+/Ca2+ exchange by endothelin release. Cardiovasc Res. 2003;60:589–597. doi: 10.1016/j.cardiores.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Pimentel DR, Adachi T, Ido Y, Heibeck T, Jiang B, Lee Y, Melendez JA, Cohen RA, Colucci WS. Strain-stimulated hypertrophy in cardiac myocytes is mediated by reactive oxygen species-dependent Ras S-glutathiolation. J Mol Cell Cardiol. 2006;41:613–622. doi: 10.1016/j.yjmcc.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Rothstein EC, Byron KL, Reed RE, Fliegel L, Lucchesi PA. H2O2-induced Ca2+ overload in NRVM involves ERK1/2 MAP kinases: role for an NHE-1-dependent pathway. Am J Physiol Heart Circ Physiol. 2002;283:H598–H605. doi: 10.1152/ajpheart.00198.2002. [DOI] [PubMed] [Google Scholar]
- Sabri A, Byron KL, Samarel AM, Bell J, Lucchesi PA. Hydrogen peroxide activates mitogen-activated protein kinases and Na+/H+ exchange in neonatal rat cardiac myocytes. Circ Res. 1998;82:1053–1062. doi: 10.1161/01.res.82.10.1053. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Xu Y, Slayter HS, Izumo S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75:977–984. doi: 10.1016/0092-8674(93)90541-w. [DOI] [PubMed] [Google Scholar]
- Sarnoff SJ, Mitchell JH, Gilmore JP, Remensnyder JP. Homeometric autoregulation in the heart. Circ Res. 1960;8:1077–1091. doi: 10.1161/01.res.8.5.1077. [DOI] [PubMed] [Google Scholar]
- Sugden PH, Clerk A. Oxidative stress and growth-regulating intracellular signalling pathways in cardiac myocytes. Antioxid Redox Signal. 2006;8:2111–2124. doi: 10.1089/ars.2006.8.2111. [DOI] [PubMed] [Google Scholar]
- Tucci PJ, Bregagnollo EA, Spadaro J, Cicogna AC, Ribeiro MC. Length dependence of activation studied in the isovolumic blood-perfused dog heart. Circ Res. 1984;55:59–66. doi: 10.1161/01.res.55.1.59. [DOI] [PubMed] [Google Scholar]
- Villa-Abrille MC, Cingolani HE, Garciarena CD, Ennis IL, Aiello EA. Angiotensin II-induced endothelin-1 release in cardiac myocytes. Medicina (B Aires) 2006;66:229–236. [PubMed] [Google Scholar]
- von Anrep G. On the part played by the suprarenals in the normal vascular reactions on the body. J Physiol. 1912;45:307–317. doi: 10.1113/jphysiol.1912.sp001553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lewinski D, Stumme B, Fialka F, Luers C, Pieske B. Functional relevance of the stretch-dependent slow force response in failing human myocardium. Circ Res. 2004;94:1392–1398. doi: 10.1161/01.RES.0000129181.48395.ff. [DOI] [PubMed] [Google Scholar]
- Weisser-Thomas J, Piacentino V, Gaughan JP, Margulies K, Houser SR. Calcium entry via Na/Ca exchange during the action potential directly contributes to contraction of failing human ventricular myocytes. Cardiovasc Res. 2003;57:974–985. doi: 10.1016/s0008-6363(02)00732-0. [DOI] [PubMed] [Google Scholar]
- White E, Boyett MR, Orchard CH. The effects of mechanical loading and changes of length on single guinea-pig ventricular myocytes. J Physiol. 1995;482:93–107. doi: 10.1113/jphysiol.1995.sp020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Komuro I, Kudoh S, Zou Y, Nagai R, Aikawa R, Uozumi H, Yazaki Y. Role of ion channels and exchangers in mechanical stretch-induced cardiomyocyte hypertrophy. Circ Res. 1998;82:430–437. doi: 10.1161/01.res.82.4.430. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Komuro I, Kudoh S, Zou Y, Shiojima I, Hiroi Y, Mizuno T, Maemura K, Kurihara H, Aikawa R, Takano H, Yazaki Y. Endothelin-1 is involved in mechanical stress-induced cardiomyocyte hypertrophy. J Biol Chem. 1996;271:3221–3228. doi: 10.1074/jbc.271.6.3221. [DOI] [PubMed] [Google Scholar]
- Zhang DX, Chen YF, Campbell WB, Zou AP, Gross GJ, Li PL. Characteristics and superoxide-induced activation of reconstituted myocardial mitochondrial ATP-sensitive potassium channels. Circ Res. 2001;89:1177–1183. doi: 10.1161/hh2401.101752. [DOI] [PubMed] [Google Scholar]
- Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]